
Abstract
Aims:
Accumulation of reactive oxygen species (ROS) in hepatocytes in response to excessive nutrients and the ensuing liver damages caused by ROS constitute a key pathophysiological event in nonalcoholic steatohepatitis (NASH). In the present study, we investigated the epigenetic mechanism underlying ROS production in NASH pathogenesis.
Results:
NASH was induced by feeding the mice with a methionine-and-choline-deficient (MCD) diet for 4 weeks. Compared with the control mice (wild type [WT]), mice with hepatocyte-specific deletion of Brg1 (HepcKO), a core component of the mammalian chromatin remodeling complex, developed a less severe form of NASH when fed on the MCD diet. Importantly, ROS levels were attenuated in HepcKO mice as opposed to WT mice. Brahma-related gene 1 (Brg1) deficiency downregulated the transcription of NADPH oxidases (NOX1, NOX2, and NOX4) both in vivo and in vitro. Mechanistically, Brg1 deletion rendered a more repressive chromatin structure surrounding the NOX promoters as characterized by reduced levels of acetylated histones. In addition, Brg1 interacted with the histone H4K16 acetyltransferase males absent on the first (MOF) to activate NOX transcription. MOF knockdown by small interfering RNA or pharmaceutical inhibition by MG149 suppressed NOX transcription and ameliorated ROS levels.
Innovation:
Our data highlight a novel epigenetic mechanism through which Brg1 and MOF cooperate to regulate ROS production in hepatocytes in response to pro-NASH stimuli.
Conclusion:
A cross talk between Brg1 and MOF epigenetically activates NOX transcription and elevates ROS synthesis contributing to NASH pathogenesis.
Introduction
Nonalcoholic fatty liver disease (NAFLD) represents a prototypical human pathology acutely impacted by the external environment: poor dietary choices combined with a lack of physical activity as a result of fast-pacing lifestyle inevitably lead to a disruption of the internal metabolism in the liver (15, 45). By definition, NAFLD encompasses a series of interconnected yet distinctive pathological stages that can, short of effective intervention, progress from simple steatosis to nonalcoholic steatohepatitis (NASH) to cirrhosis and hepatocellular carcinoma (38). A host of risk factors have been identified for NAFLD, including obesity, hyperglycemia, type 2 diabetes, and hypertriglyceridemia, either independently or collaboratively promoting its development (18). Decades of rigorous efforts aiming at unveiling the mechanisms underlying NAFLD pathogenesis have engendered multiple groundbreaking findings but at the same time left many critical questions unanswered (29).
NADPH oxidase-mediated synthesis of reactive oxygen species (ROS) in the liver contributes to nonalcoholic steatohepatitis (NASH) pathogenesis. The present investigation has unveiled a previously unrecognized epigenetic mechanism through which Brahma-related gene 1 (Brg1) and males absent on the first (MOF) cooperate to regulate ROS production in hepatocytes in response to pro-NASH stimuli. Screening for small molecules that inhibit Brg1/MOF activity or disrupt Brg1–MOF interaction might yield novel therapeutic strategies against NASH, and, more broadly, ROS-related diseases.
A burst in the levels of intracellular reactive oxygen species (ROS) is considered a host defense mechanism to combat invading pathogens (33). When produced in excess, however, ROS accumulation poses significant threats to liver homeostasis. Several lines of evidence support a cause/effect relationship between ROS overproduction and NAFLD deterioration. Seki et al. have shown that compared with healthy individuals, patients with NASH exhibit significant lipid peroxidation, a proxy for ROS levels, in the liver (37). In addition, an independent study has found that there is a correlation between NAFLD severity and mitochondrial dysfunction, a major culprit for excessive ROS production (34). On the contrary, antioxidant regimens seem to have some protective effects in NASH patients although the outcomes are far from convincingly conclusive (13, 24).
In mammals, intracellular ROS synthesis is dictated by a diverse range of enzymes of which the NADPH oxidase (NOX) family of proteins plays a key role (6). Emerging evidence has implicated NOX proteins in the pathogenesis of liver diseases. For instance, mice with deficiency in either NOX1 or NOX2 or NOX4 are protected from CCl4-induced liver injury and fibrosis (27, 32). The specific NOX1/NOX4 inhibitor GKT137831 is able to attenuate liver fibrosis in mouse models (2). Importantly, a recent investigation by Torok and colleagues has shown that GKT137831 could effectively block the progression of NASH in mice (8).
Despite the critical roles NOX proteins play in liver pathophysiology, little is known regarding their epigenetic regulation in the context of NASH pathogenesis. In mammals, the epigenetic machinery is intimately involved in the regulation of human diseases. Brahma-related gene 1 (Brg1) is the core component of the chromatin remodeling complex (47). Brg1 regulates transcription by communicating with other epigenetic factors, including histone modifying enzymes (1, 28, 44, 46). Males absent on the first (MOF), also called MYST1, is a histone H4K16 acetyltransferase that has been previously shown to play a key role in carcinogenesis (4). Here we present evidence that Brg1 is essential for ROS generation in hepatocytes in vivo and in vitro by recruiting MOF to program the epigenetic activation of NOX gene transcription. Our data thus unveil a previously unknown role for Brg1 in NASH pathogenesis.
Results
Hepatocyte-specific Brg1 deletion in mice attenuates methionine-and-choline-deficient diet-induced NASH
Previously we have demonstrated, using a lentivirus-delivered, short hairpin RNA (shRNA)-mediated knockdown system, that systemic Brg1 depletion attenuates NASH development in mice (42). We sought to determine whether Brg1 deficiency in hepatocyte would be sufficient to impede NASH progression. To this end, Smarc4 f/f mice were crossed with Alb-Cre mice to obtain hepatocyte conditional Brg1 knockout (HepcKO) mice. When fed on a methionine-and-choline-deficient (MCD) diet for 4 weeks, HepcKO mice exhibited diminished steatotic injury compared with wild-type (WT) littermates as evidenced by plasma ALT levels (Fig. 1A) and liver weight versus body weight ratio (Supplementary Fig. S1). Hematoxylin and eosin (H&E) staining showed that hepatocyte ballooning was decreased in HepcKO mice (Fig. 1B). Immunohistochemical staining suggested that there were fewer F4/80+ infiltrates in the livers of HepcKO mice (Fig. 1C). Quantitative polymerase chain reaction (qPCR) analyses confirmed that proinflammatory mediators were suppressed in HepcKO mice (Supplementary Fig. S2). In addition, steatosis was less prominent in HepcKO as demonstrated by Oil Red O staining (Fig. 1D). Chemiluminescence measurements provided corroborating evidence that both total cholesterol and triglyceride levels were downregulated as a result of Brg1 deletion (Supplementary Fig. S3). Cumulatively, HepcKO mice fed with the MCD diet earned a significantly smaller NAFLD activity score (NAS) as opposed to WT mice (Fig. 1E). Together, these data illustrate that hepatocyte-Brg1 may provide a driving force for NASH pathogenesis in mice.

Brg1 regulates ROS levels in hepatocytes
Since ROS accumulation plays a critical role in NASH pathogenesis, we evaluated the effects of Brg1 deficiency on ROS levels in the liver. As shown in Figure 2A, there was augmented dihydroethidium (DHE) staining in the livers of WT mice fed on the MCD diet compared with the normal diet; HepcKO mice displayed diminished ROS levels compared with the WT mice. Furthermore, treatment with palmitate (PA) stimulated ROS synthesis in primary hepatocytes isolated from WT mice but not as much in hepatocytes from HepcKO mice (Fig. 2B and Supplementary Fig. S4).

We then asked whether Brg1 deficiency could be equally effective in suppressing ROS production in another mouse model of NASH. To this end, WT and HepcKO mice were fed on a high-fat diet (HFD) for 16 weeks. Brg1 deletion significantly attenuated HFD-induced liver injury as indicated by ALT measurements (Supplementary Fig. S5A). Importantly, Brg1 deficiency dampened hepatic ROS levels in HFD-fed mice as evidenced by diminished DHE staining (Supplementary Fig. S5B). Therefore, Brg1 could indeed influence ROS production both in vivo and in vitro.
Next, we assessed the possibility that Brg1 modulates ROS production via transcriptional regulation of NOX genes. Expression of NOX1, NOX2, and NOX4, but not NOX3, was upregulated in the livers of MCD-fed WT mice at both mRNA (Fig. 3A) and protein (Fig. 3B) levels; the induction of NOX proteins was mostly abrogated in HepcKO mice. Similarly, we observed that there was attenuated induction of NOX expression in the livers of HFD-fed HepcKO mice compared with HFD-fed WT mice (Supplementary Fig. S6). In addition, PA failed to stimulate the expression of NOX1, NOX2, and NOX4 in primary hepatocytes isolated from HepcKO mice (Fig. 3C, D). Reporter assay indicated that Brg1 enhanced the activation of NOX promoters by PA in hepatocytes (Supplementary Fig. S7). In contrast, knockdown of Brg1 by shRNA dampened PA-induced transactivation of NOX promoters (Supplementary Fig. S8). Of key importance, PA promoted the recruitment of Brg1 to the NOX promoter regions in hepatocytes (Fig. 3E). We also found enhanced Brg1 occupancy on the NOX promoter regions in the livers of MCD-fed mice (Supplementary Fig. S9) and in the livers of HFD-fed mice (Supplementary Fig. S10). Together, we conclude that Brg1 may be essential for NOX transcription in the context of NASH pathogenesis.
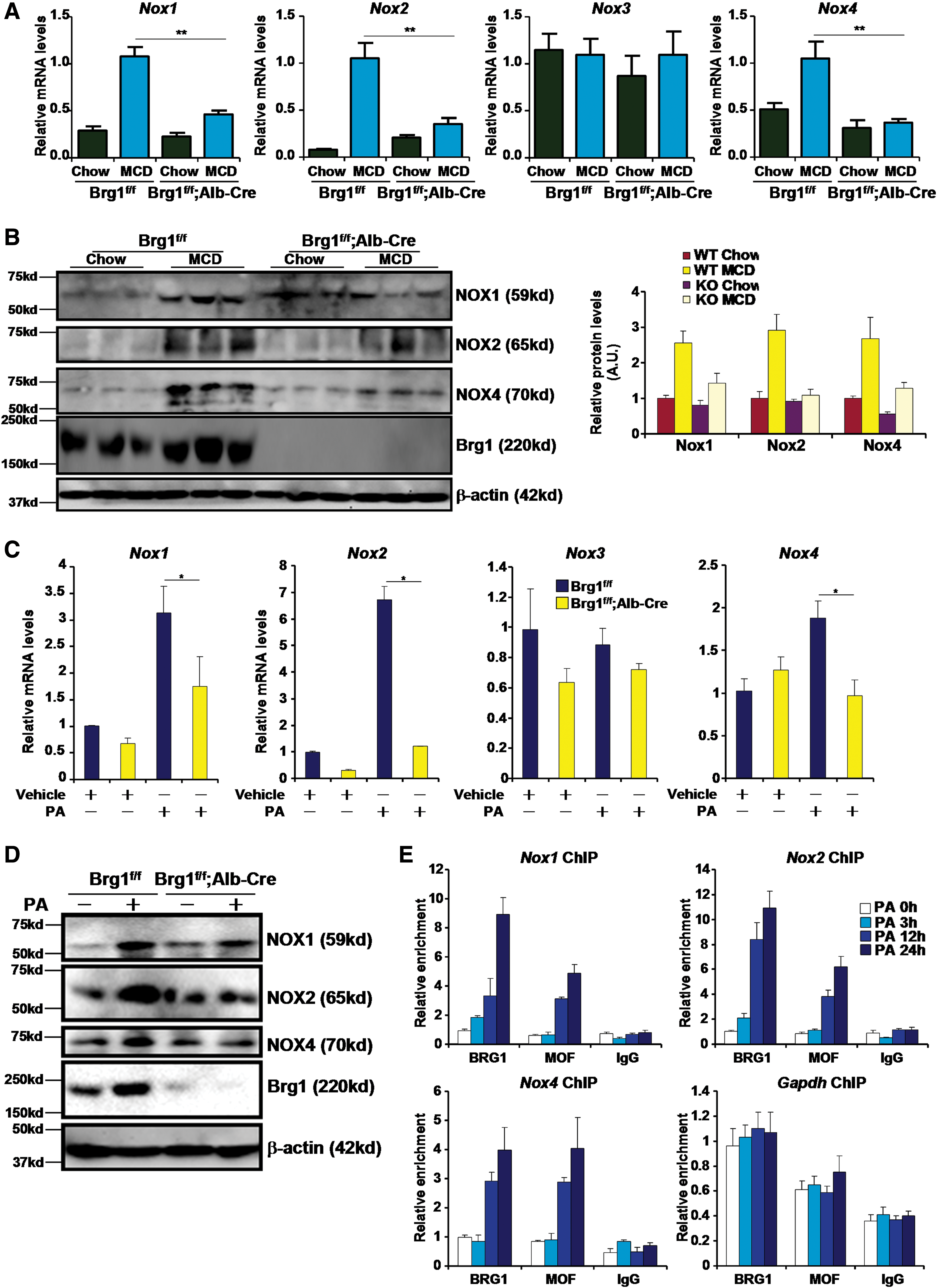
Brg1 regulates histone modifications on the NOX promoters
We have previously shown that the ability of Brg1 to regulate transcription is a function of its interaction with histone modifying enzymes (12, 17, 42, 44). Now that we observed a decrease in NOX transcription in the absence of Brg1, we hypothesized that Brg1 deficiency might render a repressive chromatin structure surrounding the NOX promoters. To test this hypothesis, we performed a series of chromatin immunoprecipitation (ChIP) experiments. In response to PA stimulation, active histone marks, including acetylated H3 (Fig. 4A), acetylated H4 (Fig. 4B), and trimethylated H3K4 (Fig. 4C), quickly accumulated on the NOX proximal promoters, but not on GAPDH promoter, in WT instead of HepcKO hepatocytes. Likewise, Brg1 was necessary for the NOX promoter-specific accumulation of active histones in the livers in mice (Supplementary Fig. S11). On further examination, we found that whereas there was varied dependence of Brg1 with regard to H3K9 (Fig. 4D and Supplementary Fig. S12A) and H3K27 (Fig. 4E and Supplementary Fig. S12B) acetylation on the NOX promoter, Brg1 was invariably indispensible for H4K16 acetylation (Fig. 4F and Supplementary Fig. S12C).
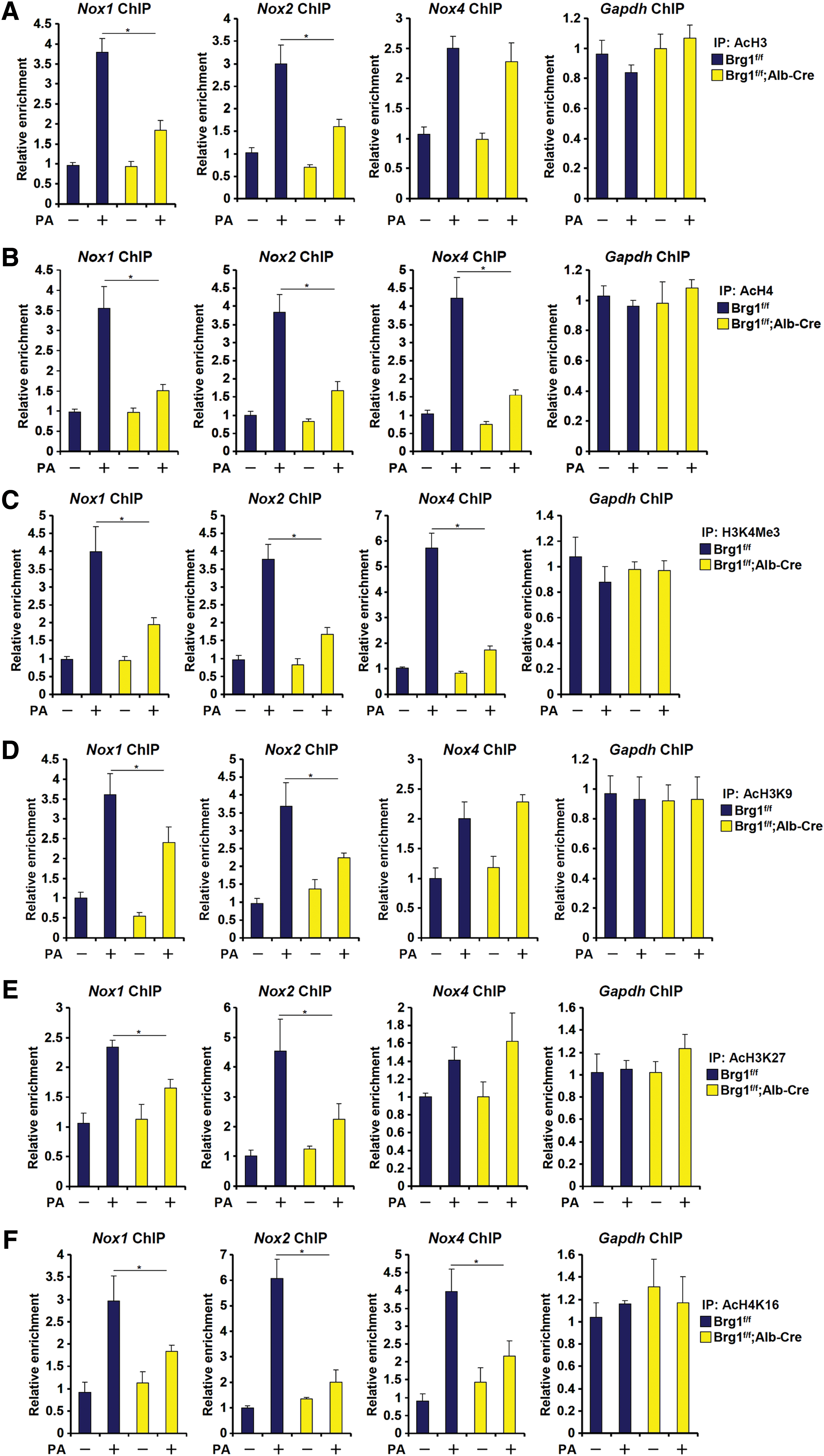
We also examined the levels of two repressive histone modifications, namely trimethylated H3K9 (H3K9Me3) and trimethylated H3K27 (H3K27Me3), in the same experimental settings. Consistent with the notion that methylation and acetylation of the same lysine residue are antagonistic to each other, we observed that PA treatment downregulated the levels of H3K9Me3 and H3K27Me3, paralleling increased levels of H3K9Ac and H3K27Ac in WT hepatocytes; Brg1 deficiency, however, significantly attenuated the loss of H3K9Me3 and H3K27Me3 on the NOX1 and NOX2, but not the NOX4, promoters (Supplementary Fig. S13). Together, these data suggest that Brg1 might regulate NOX gene transcription by influencing differential histone modifications on the NOX proximal promoters.
MOF inhibition attenuates ROS levels in hepatocytes
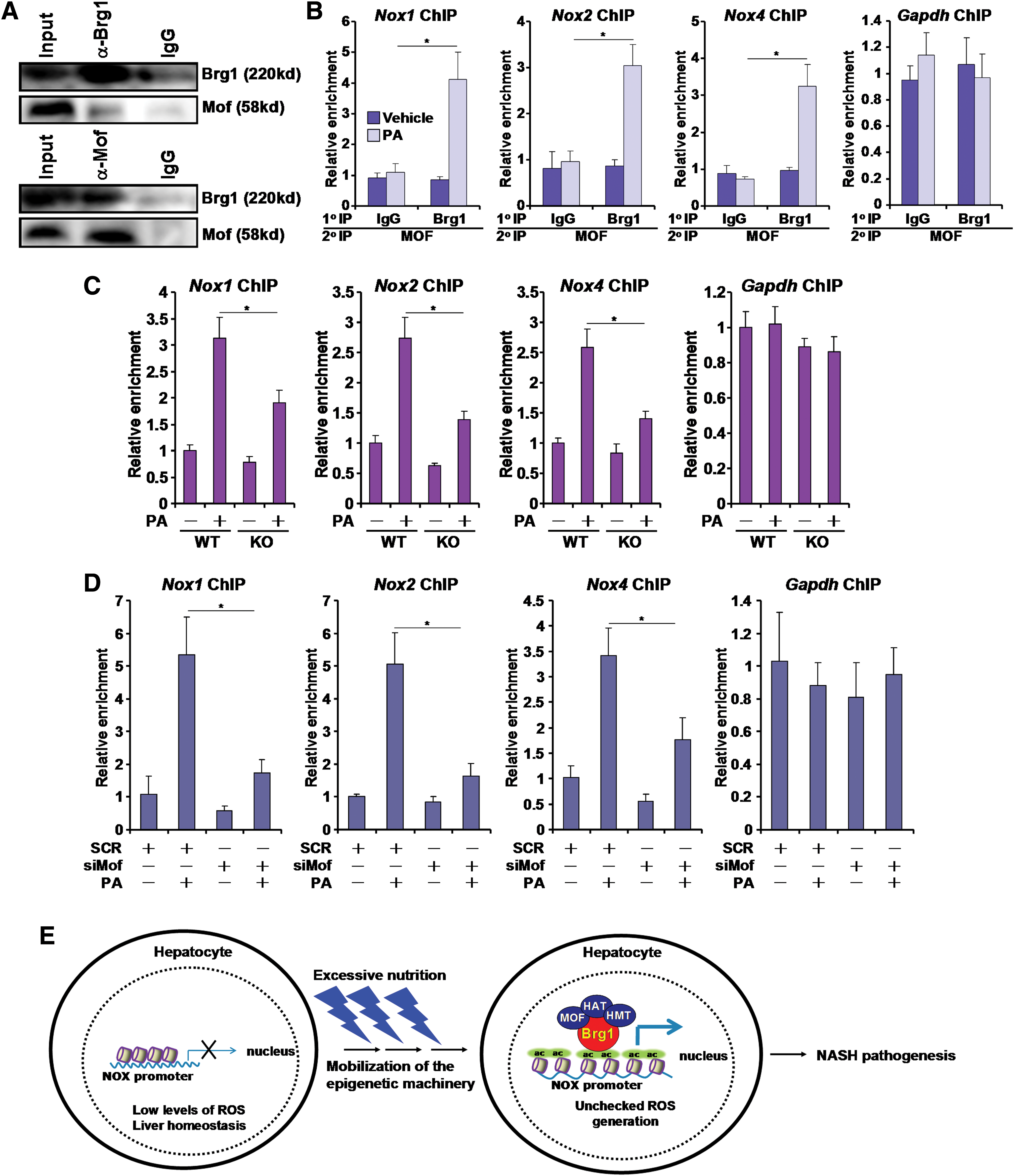
We and others have previously demonstrated that Brg1 can potentially interact with histone H3K4 methyltransferase (44), H3K27 acetyltransferase (1), and H3K9 acetyltransferase (28). Since Brg1 was uniformly required for H4K16 acetylation across all three NOX promoters and that no previous investigation had evaluated the mechanism through which Brg1 contributes to H4K16 acetylation, we decided to focus the remainder of the study on a possible interaction between Brg1 and H4K16 acetyltransferase. In eukaryotes, histone H4K16 acetylation is catalyzed by the conserved acetyltransferase MOF. We hypothesized that MOF could participate in PA-induced ROS production in hepatocytes. Indeed, MOF occupancies on the NOX promoters were upregulated in primary hepatocytes treated with PA (Fig. 4E), in the livers of MCD-fed mice (Supplementary Fig. S14), and in the livers of HFD-fed mice (Supplementary Fig. S15). In addition, overexpression of a WT form but not an enzyme-deficient form of MOF enhanced transactivation of NOX promoters by PA (Supplementary Fig. S16). MOF silencing by small interfering RNA (siRNA) abrogated the induction of NOX1, NOX2, and NOX4 mRNA (Fig. 5A) and protein (Fig. 5B) levels. In the meantime, intracellular ROS levels were also downregulated following MOF knockdown (Fig. 5C and Supplementary Fig. S17). We then used a specific MOF inhibitor MG149 to treat primary hepatocytes. Similar to MOF depletion, MG149 treatment aborted NOX induction (Fig. 5D, E) and intracellular ROS spike (Fig. 5F and Supplementary Fig. S18) in hepatocytes exposed to PA. MG149 treatment also abominated the augmentation of NOX promoter activities by PA (Supplementary Fig. S19). Taken together, these data implicate MOF in NOX transactivation and ROS production in hepatocytes.

Brg1 interacts with MOF to regulate NOX transcription
Finally, we tackled the functional synergy between Brg1 and MOF in NOX transcription. Coimmunoprecipitation assay indicated that Brg1 and MOF could form a complex (Fig. 6A). To further demonstrate a functional interplay between Brg1 and MOF in NOX transactivation, we performed Re-ChIP assay. PA treatment significantly strengthened the Brg1–MOF interaction on the NOX promoters instead of GADPH promoter in primary hepatocytes (Fig. 6B). Re-ChIP experiments also revealed a stronger Brg1–MOF interaction on the NOX promoters in the livers of MCD-fed mice (Supplementary Fig. S20) compared with the chow-fed mice. Similar observations were made in the livers of HFD-fed mice (Supplementary Fig. S21). Furthermore, coexpression of Brg1 and MOF activated NOX promoter activities better than when they were individually expressed (Supplementary Fig. S16). Finally, ChIP assays showed that Brg1 deficiency significantly attenuated the occupancies of MOF on the NOX promoters (Fig. 6C). Reciprocally, MOF depletion downregulated Brg1 recruitment to the NOX promoters (Fig. 6D), suggesting that a cross talk between Brg1 and MOF may contribute to NOX transactivation in hepatocytes.
Discussion
A key feature in the pathogenesis of NASH is imbalanced ROS generation and elimination resulting in accumulation of excessive free radicals in the liver (7). We present evidence here that a cross talk between BRG1 and MOF contributes to ROS generation in hepatocytes by epigenetically activating the transcription of NOX genes (Fig. 6E). Consistent with our previous observation that whole-body knockdown of Brg1 alleviates NASH in mice, our data as reported here indicate that hepatocyte-restricted deletion of Brg1 is sufficient to attenuate NASH. Although Brg1 is essential for organogenesis during development, mounting evidence suggests that it is dispensable for postnatal activities (9, 49). Not coincidently, mice with conditional Brg1 knockout in the myocardium after reaching adulthood, while not exhibiting any detectable phenotype under physiological conditions, are protected from mechanical stress-induced pathological hypertrophy likely because Brg1 promotes the transactivation of neonatal cardiac genes such as β-MHC (23). Furthermore, we have shown that endothelial-specific depletion of Brg1 in adult mice renders them resistant to angiotensin II-induced cardiac remodeling, the underlying mechanism of which seems to be that Brg1 is necessary for the synthesis of a prohypertrophic factor endothelin (44). Combined, these observations collectively seem to argue for a model in which postnatal Brg1 expression/activity is associated with disease-prone transcription events. On the contrary, several studies have pointed out that Brg1 may also exert protective roles during carcinogenesis in the lungs (21) and the pancreatic duct (35). Therefore, the benefits of Brg1 deletion/inhibition have to be weighed against its potential risks in the development of novel therapeutic strategies to treat NASH.
We limited our investigation on the role of Brg1 in NASH pathogenesis to the transactivation of NOX genes, which certainly is far from exhaustive since several alternative theories exist. First, we were able to confirm our previous observation (42) that Brg1 plays an essential role in hepatic inflammation and that hepatocytes represent a major source of inflammation during NASH pathogenesis (Supplementary Fig. S2). Second, several independent reports have implicated Brg1 in the regulation of cell metabolism. Salma et al., for instance, have shown that Brg1 promotes a prolipogenic transcriptional program by serving as a cofactor for C/EBPα, C/EBPβ, and/or PPARγ (36). Wang et al., on the contrary, demonstrated that Brg1 might be involved in insulin-induced lipogenesis by directly binding to the promoter region of FASN (fatty acid synthase) gene (43). Of intrigue, a metabolomic analysis revealed that Brg1 regulates, along with another SWI/SNF family member Brm, cellular metabolism by maintaining an extricate equilibrium between fatty acids, glucose, and amino acids (5). These observations suggest that reduced liver injury in HepcKO mice may be secondary to improved metabolic profile as a result of Brg1 deficiency in hepatocytes. Finally, previous investigations support a protective, rather than deleterious, role for Brg1 in safeguarding the function of mitochondria, a major source of ROS generation (10, 39). Tan et al. have shown that during heat shock response, Brg1 is recruited by heat shock factor 1 to activate the transcription of genes encoding mitochondrial chaperones and consequently contributes to the maintenance of mitochondrial membrane potential and cell survival (39). Similarly, Bultman et al. have observed that Brg1-Brm double knockout mice exhibit small and fragmented mitochondria with reduced mitochondrial number and size in the heart (10). They further proposed that Brg1 and Brm play intricate roles regulating the mitochondrial dynamics in cardiomyocytes by controlling the expression of genes responsible for mitochondrial fusion and fission. There is also evidence indicating that Brg1 can empower liver progenitor cells (LPCs) with increased reprogramming capacity (25). Because LPCs may contribute to liver regeneration and alleviate steatotic injury in the course of NASH pathogenesis in mice (14), Brg1 could potentially antagonize NASH pathogenesis by boosting and/or maintaining the LPC population. These lingering issues clearly warrant further investigation.
One major finding in the present investigation is that Brg1 relies on the H4K16 acetyltransferase MOF to activate NOX transcription. It is well known that Brg1 contains a bromo domain that recognizes acetylated lysines although its functional relevance has never been clearly delineated in a context-specific manner (30). We show here that while Brg1 deficiency dampens MOF binding on the NOX promoters, MOF depletion causes reduced Brg1 recruitment as well. This is in agreement with a previous report showing that a dynamic interaction between Brg1 and the H3K9 acetyltransferase p300/CBP associated factor mediates the activation of myogenin transcription during myogenic cell differentiation (28). It remains to be determined how the Brg1–MOF interplay modulates genome-wide transcriptional events in hepatocytes in the context of NASH pathogenesis. Recently, Alexander et al. have reported, using ChIP coupled with deep sequencing (ChIP-seq), that there is an enrichment of Brg1 on acetyl H3K27-demarcated (catalyzed by the acetyltransferase CBP/p300) enhancers to activate mesoderm differentiation-related genes (1). Therefore, Brg1 might be corecruited to the chromatin with a specific histone modifying enzyme to regulate transcription in a cell- and stimulus-dependent manner. Further investigation exploiting the ChIP-seq technique will reveal genome-wide colocalization of Brg1 and MOF and its implications in NASH pathogenesis.
Curiously, Brg1 deletion appears to have little effect on basal levels of NOX expression either in mice or in cultured hepatocytes. One possible explanation is that Brg1 is not present in the complex responsible for maintaining the basal transcription of NOX genes. Rather, Brg1 becomes activated and incorporated into a new complex that mediates the transactivation of NOX genes in response to excessive nutrition. We also observed that Brg1 is essential for the activation of NOX1, NOX2, and NOX4, but not NOX3 expression in hepatocytes, although the underlying mechanism is not clear at this point. Of note, Brg1 relies on sequence-specific transcription factors (TFs) to be recruited to the chromatin to regulate transcription (17, 42). Transcription of different NOX genes is mediated by different sets of TFs (6). It is thus possible that one or more TFs that are specifically required for NOX3 transactivation, but not for NOX1/2/4 transactivation, fail to interact with and recruit Brg1, which in turn contributes to the varied dependence on Brg1.
There are several limitations associated with the present study that we wish to stress. First, we only examined ROS production without touching on other pathophysiologically critical processes in NASH such as lipid metabolism, inflammation, fibrosis, and autophagy, in which Brg1 and/or MOF clearly play a regulatory role. Second, we focused on the interplay between Brg1 and MOF, whereas other epigenetic factors may be present in the same complex and participate in regulating ROS production and NASH pathogenesis. Third, although we have shown that MOF knockdown or inhibition dampens ROS production in cultured hepatocytes, it is not clear whether targeted deletion/inhibition of MOF in the liver could protect the mice from NASH pathogenesis. Finally, the relevance of our findings in NASH pathogenesis in humans remains questionable. For instance, do levels of Brg1 and/or MOF alter in patients with NASH compared with healthy individuals? If so, does this alteration correlate with ROS production and NASH severity? These limitations clearly need to be addressed in future studies.
In summary, we provide evidence here that a cross talk between two epigenetic factors, Brg1 and MOF, contributes to NOX transactivation in hepatocytes in the context of NASH. Recent advances in cancer research have provided a success story of using small-molecule compounds that can inhibit proteins with bromo domains to treat various malignancies (19). In light of our finding showing that a small-molecule MOF inhibitor could potentially dampen excessive ROS generation, it is thus possible to harness these data for the development of novel anti-NASH strategies.
Materials and Methods
Animals and histology
All animal procedures were reviewed and approved by the intramural Committee on Ethical Conduct of Animal Studies of Nanjing Medical University. Smarc4-Flox mice (20) were crossed with Alb-Cre mice (Model Animal Research Institute, Nanjing, China) to obtain hepatocyte conditional Brg1 knockout (HepcKO) mice. Six- to 8-week-old male HepcKO mice and their WT littermates were fed on an MCD diet (A02082002B; Research Diets) for four consecutive weeks. Alternatively, the mice were fed on HFD (D12492; Research Diets) for 16 weeks to induce NASH. ALT levels were measured by enzyme-linked immunosorbent assay (ELISA) as previously described (41).
Histological analyses were performed essentially as described before (16, 40). Paraffin sections were stained with H&E (Sigma) according to standard procedures. Parallel sections were stained for cell surface markers. Briefly, the sections were blocked with 10% normal goat serum for 1 h at room temperature and then incubated with anti-F4/80 (1:200; Abcam) antibodies. Staining was visualized by incubation with anti-rabbit secondary antibody (1:1000) and developed with a streptavidin–horseradish peroxidase kit (Pierce) for 20 min. Pictures were taken using an Olympus IX-70 microscope. Frozen sections were stained with Oil Red O (Sigma) according to standard procedures. Slides were visualized on a cofocal microscope (LSM 710; Zeiss). Quantifications were performed with ImageJ. NAFLD activity score (NAS) evaluation was performed as previously described by two independent pathologists in a blinded manner (26). Agreement rate between the blinded observers was 93%.
Cell culture, treatment, and transfection
Primary mouse hepatocytes were isolated and maintained as previously described (48). HepG2 cells (ATCC) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. NOX1 promoter construct, NOX4 promoter construct, MOF expression constructs, Brg1 expression construct, and Brg1 shRNA construct have been previously described (3, 11, 12, 22, 31). PA was purchased from Sigma and MG149 was obtained from Selleck. siRNA for mouse MOF gene was purchased from Dharmacon. Transient transfections were performed with Lipofectamine 2000 (for HepG2 cells) or Lipofectamine RNAiMAX (for primary hepatocytes). Briefly, siRNA (∼3 μg/p35 culture plate for RNA analyses, ∼5 μg/p60 plate for protein analyses, or ∼10 μg/p100 plate for ChIP assay) was mixed with RNAiMAX (2 μL/μg siRNA) in serum-free media for 20 min before being added dropwise to each plate.
RNA isolation and real-time PCR
RNA was extracted with the RNeasy RNA isolation kit (Qiagen). Reverse transcriptase reactions were performed as previously described using a SuperScript First-strand Synthesis System (Invitrogen) (38a). Primers are listed in the Supplementary Table S1.
Protein extraction, immunoprecipitation, and Western blot
Tissue and cell lysates were obtained as previously described (50). Antibodies were incubated with cell lysates overnight before being absorbed by protein A/G-plus agarose beads. Precipitated immune complex was released by boiling with 1 × sodium dodecyl sulfate electrophoresis sample buffer. Western blot analyses were performed with anti-NOX1, anti-NOX2, anti-NOX4 (Proteintech), anti-MOF (Bethyl Laboratory), anti-Brg1, anti-RNA polymerase II (Santa Cruz), and anti-β-actin (Sigma) antibodies.
Chromatin immunoprecipitation
ChIP and Re-ChIP assays were performed essentially as described before (42). Briefly, 100 μg formaldehyde crosslinked nuclear proteins were precipitated with anti-Brg1 (Santa Cruz), anti-MOF (Bethyl Laboratories), anti-acetyl H3 (Millipore), anti-acetyl H4 (Millipore), anti-trimethyl H3K4 (Millipore), anti-acetyl H3K9 (Millipore), anti-acetyl H3K27 (Millipore), and anti-acetyl H4K16 (Millipore). Precipitated genomic DNA was amplified by real-time PCR with primers listed in the Supplementary Table S2. Serially diluted genomic DNA extracted from normal cells/tissues was used to generate a standard curve to calculate the amount of DNA being precipitated by a particular antibody. A total of 10% of the starting material is also included as the input. Data are then normalized to the input and expressed as fold changes compared to the control group.
DHE and DCFH-DA staining
DHE and DCFH-DA (dichlorodihydrofluorescein diacetate) stainings were performed essentially as previously described (42). Frozen liver sections or primary hepatocytes were stained with DHE (10 μM) or DCFH-DA (10 μM) at 37°C for 30 min. Fluorescence was visualized by cofocal microscopy (LSM 710; Zeiss). Quantifications were performed with ImageJ.
Statistical analysis
Data are presented as mean ± standard deviation. For experiments concerning multiple groups, one-way ANOVA with post hoc Scheffe analyses was performed to evaluate the differences. The differences between two (control and experimental) groups were determined by two-sided, unpaired Student's t-test. p-Values smaller than 0.05 are considered significant.
Footnotes
Acknowledgment
This work was supported, in part, by the Natural Science Foundation of China (81725001).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.