
Abstract
Aims:
A kidney-brain interaction has been described in acute kidney injury, but the mechanisms are uncertain. Since we recently described a reno-cerebral reflex, we tested the hypothesis that renal ischemia-reperfusion injury (IRI) activates a sympathetic reflex that interlinks the renal and cerebral renin-angiotensin axis to promote oxidative stress and progression of the injury.
Results:
Bilateral ischemia-reperfusion activated the intrarenal and cerebral, but not the circulating, renin-angiotensin system (RAS), increased sympathetic activity in the kidney and the cerebral sympathetic regulatory regions, and induced brain inflammation and kidney injury. Selective renal afferent denervation with capsaicin or renal denervation significantly attenuated IRI-induced activation of central RAS and brain inflammation. Central blockade of RAS or oxidative stress by intracerebroventricular (ICV) losartan or tempol reduced the renal ischemic injury score by 65% or 58%, respectively, and selective renal afferent denervation or reduction of sympathetic tone by ICV clonidine decreased the score by 42% or 52%, respectively (all p < 0.05). Ischemia-reperfusion-induced renal damage and dysfunction persisted after controlling blood pressure with hydralazine.
Innovation:
This study uncovered a novel reflex pathway between ischemic kidney and the brain that sustains renal oxidative stress and local RAS activation to promote ongoing renal damage.
Conclusions:
These data suggest that the renal and cerebral renin-angiotensin axes are interlinked by a reno-cerebral sympathetic reflex that is activated by ischemia-reperfusion, which contributes to ischemia-reperfusion-induced brain inflammation and worsening of the acute renal injury. Antioxid. Redox Signal. 27, 415–432.
Introduction
A
A kidney-brain interaction has been described in acute kidney injury, but the mechanisms are uncertain. This study demonstrates that renal ischemia-reperfusion injury activates a sympathetic reflex that interlinks the renal and cerebral renin-angiotensin system (RAS) axis to promote oxidative stress and progression of the kidney injury. This identifies a novel mechanism underlying reno-cerebral interaction in response to renal ischemia-reperfusion, and therefore could lead to new interventional approaches, such as the use of RAS inhibitors, sympatholytic agents, or renal nerve ablation.
Renal angiotensinogen (AGT) level is rate limiting for Ang II generation (10, 30). Urinary AGT has been proposed as a marker of the activity of the intrarenal renin-angiotensin system (RAS) (27, 29, 51) and a biomarker for AKI (1, 53), which suggests that the intrarenal RAS may modulate the severity of AKI. Indeed, pretreatment with RAS inhibitors mitigates renal ischemia-reperfusion injury (IRI) in several experimental models (13, 37, 49). Importantly, most organs possess a local RAS that is regulated independently and is somewhat compartmentalized from the circulation (10, 46). Our recent study reported activation of the local RAS not only in the damaged kidney but also in the brain in a high-salt model of CKD (6). The link between the renal and cerebral RAS axes was provided by a reflex activation of renal afferent sympathetic nerves by salt that increased cerebral and renal ROS (15) and promoted progression of CKD via activation of efferent sympathetic nerves (6). Thus, there can be a self-sustaining enhancement of the brain and the intrarenal RAS through neuronal pathways. Oxidative stress mediated much of the effect of the activated RAS to enhance damage in the kidney and to activate the brain sympathetic outflow (41). Interestingly, overactivation of the renal sympathetic nervous system (17, 33, 45) and severe intrarenal oxidative stress (23, 25) have been reported in IRI-induced AKI. Renal Nox4 expression is increased by hypoxia (12), suggesting the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in ischemic AKI.
In the present study, we used a mouse bilateral renal IRI model to test the hypothesis that the renal and cerebral RAS, which we have termed the “reno-cerebral RAS axis,” interact via changes in renal afferent and efferent sympathetic nerve activity to contribute to the renal and cerebral oxidative stress and to the progression of ischemic AKI.
Results
Time course of blood pressure, heart rate, and humoral parameters after IRI and effect of interventions
The mean arterial pressure (MAP) and heart rate increased over 1 to 3 days after IRI, accompanied by evidence of sympathetic activation (increase in serum norepinephrine), oxidative stress (elevation of serum 8-iso-prostaglandin F2α), and inflammation (increase in serum keratinocyte-derived cytokine [KC]) (Table 1). These parameters returned to baseline by 1 week, except for serum 8-iso-prostaglandin F2α that remained elevated, although it markedly reduced from the peak at 1 day. Plasma renin activity and Ang II level did not change significantly during the course of the study.
Data are expressed as mean ± SD (Each experiment has been replicated three times. For each time, each study group consists of six mice.).
p < 0.05 versus sham mice at the same time point.
8-iso-PGF2α, 8-iso-Prostaglandin2α; Ang II, angiotensin II; IRI, ischemia-reperfusion injury; KC, keratinocyte-derived cytokine; MAP, mean arterial pressure; SD, standard deviation.
The MAP and heart rate decreased after blockade of RAS (intragastric [IG] or intracerebroventricular [ICV] losartan [Los]), blockade of sympathetic outflow (ICV clonidine [Clo]) or afferent signal (renal arterial capsaicin), and treatment with anti-oxidant (ICV tempol [Tem]), accompanied by a parallel reduction in serum 8-iso-prostaglandin F2α and KC (Table 2). Importantly, IG hydralazine (Hyd) reduced mean blood pressure comparably but it did not change 8-iso-prostaglandin F2α and KC. No interventions significantly changed plasma Ang II.
Data are expressed as mean ± SD (Each experiment has been replicated three times. For each time, each study group consists of six mice.).
p < 0.05 versus ischemic mice given vehicle.
aCSF, artificial cerebrospinal fluid; Clo, clonidine; Hyd, hydralazine; ICV, intracerebroventricular; IG, intragastric; Los, losartan; Tem, tempol.
Renal IRI activated renal RAS, sympathetic activity, and oxidative stress
Bilateral renal clamping or bilateral nephrectomy in mice increased serum creatinine (24 h postoperation: sham versus 45 min of IRI versus bilateral nephrectomy: 0.58 ± 0.12 versus 2.0 ± 0.61 versus 2.6 ± 0.43 mg/dl; p < 0.05) and decreased glomerular filtration rate, which was accompanied by marked renal histological injury in the corticomedullary junction and outer medulla (Supplementary Fig. S1; Supplementary Data are available online at
IRI led to a transient increase in renal cortical and medullar expression of AGT protein that peaked at 1 day (Fig. 1A, C). However, there were opposite changes in AGT mRNA at this time after IRI (Supplementary Fig. S2B). This suggests that the increased renal Ang II was not generated from local synthesized AGT but rather from AGT that derived from the systemic circulation and was filtered through a permeable glomerular membrane. The upregulation of the intrarenal RAS after IRI was accompanied by increased renal tissue Ang II levels (Fig. 1A, B) and AGT excretion (Fig. 1D). There were strong correlations between albumin excretion and AGT excretion (r = 0.86, p < 0.05), and between AGT excretion and renal tissue AGT expression (r = 0.88, p < 0.05, Supplementary Fig. S1G, H). This contrasts with an unchanged renal renin expression that was confined to the juxtaglomerular apparatus and an unchanged plasma renin activity and plasma Ang II after IRI (Supplementary Fig. S2A, Table 1). This indicated that the regulation of intrarenal RAS was distinct from the systemic RAS.

Renal IRI was associated with increased renal norepinephrine level and upregulated expression of the NADPH oxidase subunits Nox2 and Nox4 (Fig. 1E, F).
There was no difference in renal RAS, sympathetic activity, oxidative stress, and inflammation among sham groups at different time points postoperation (data not shown); thus, only the data at 1 day were shown.
Renal tubular RAS was upregulated in mice with renal IRI
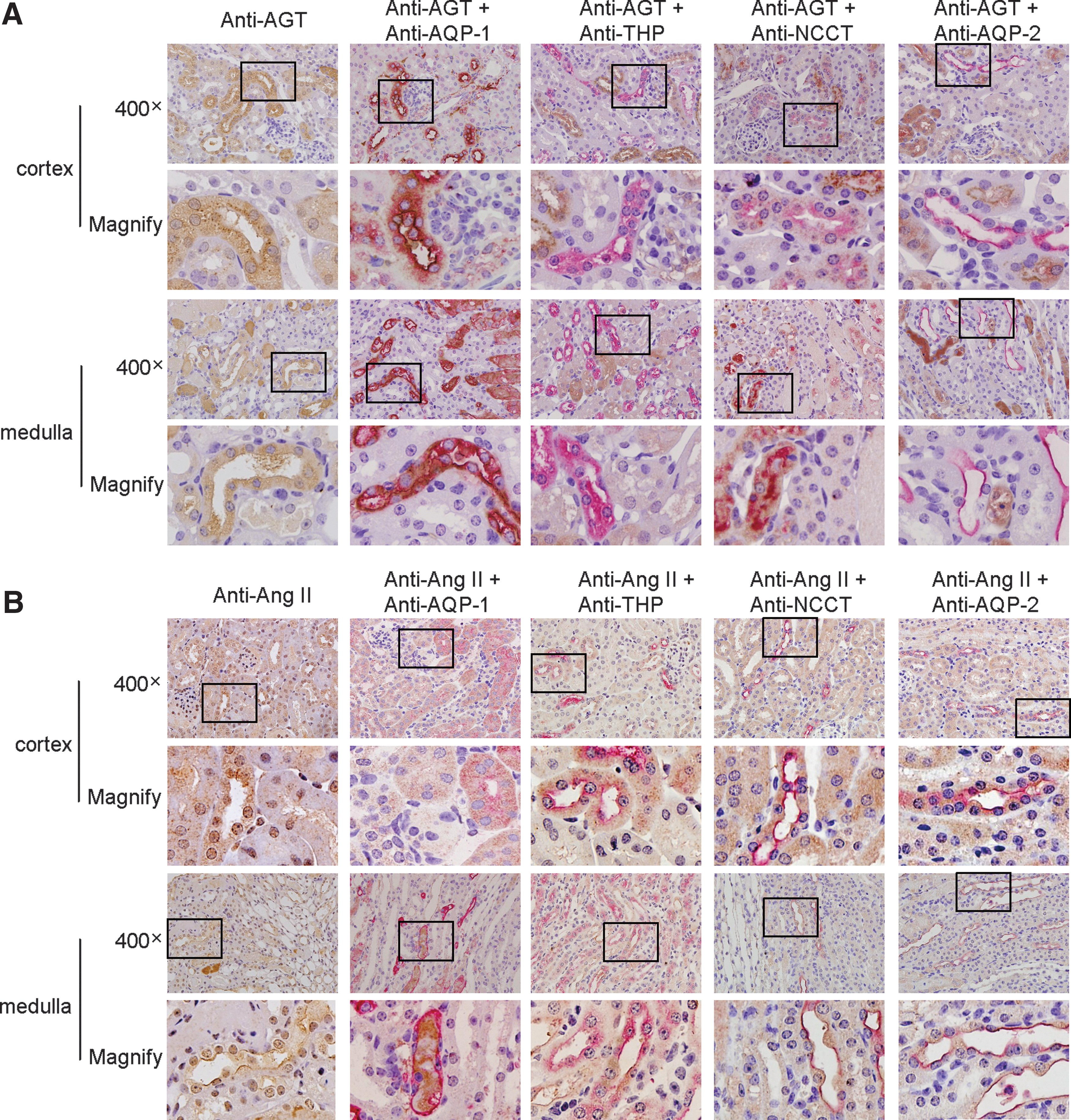
Overexpression of AGT in ischemic kidneys was localized mainly in the cortex in proximal tubular cells and in the medulla in the descending thin limb of Henle's loop (Fig. 2A). The expression of Ang II (Fig. 2B) was apparent mainly in the renal medulla in the descending thin limb and ascending thick limbs of Henle's loop, distal convoluted tubules, and collecting ducts.
Brain RAS, tyrosine hydroxylase, Noxs, and inflammation were increased in mice with renal IRI
Activation of neuron has been reported in AKI in the stress-sensitive regions (paraventricular nucleus, cingulate cortex, piriform cortex, Hippo), the regions involving regulation of salt and water balance (subfornical organ [SFO]), and area involving autonomic regulations (lateral hypothalamic area) (40).
The expression of AGT and Ang II was upregulated by IRI in all these regions (Fig. 3, Supplementary Fig. S2E–H). We, therefore, chose SFO and Hippo CA3 as the representative study regions. IRI significantly increased the numbers of AGT- and Ang II-expressing cells in SFO and Hippo CA3 and upregulated AGT mRNA and Ang II levels after IRI (Fig. 3B). The mRNA level of brain renin was detectable in SFO and Hippo, but it did not change after IRI (Supplementary Fig. S2C). Bilateral nephrectomy also increased AGT modestly in the brain, but to a much less extent (Supplementary Fig. S3A).

Using double immunofluorescence staining with antibodies recognizing the neuron-specific enolase (NSE) or glial fibrillary acidic protein (GFAP), we found that IRI-induced expression of AGT and Ang II was located mainly in neurons in SFO and Hippo CA3 (Fig. 3C), with a less degree in glial cells in the edge area of SFO and the surrounding area of Hippo CA3 (Supplementary Fig. S4). IRI-induced expression of AGT and Ang II was also observed in glial cells in the corpus callosum and cerebral cortex (Supplementary Fig. S4).
Tyrosine hydroxylase (TH) is the rate-limiting enzyme for cerebral norepinephrine synthesis. Its expression was upregulated after IRI in activated neurons (c-fos-positive) in the rostral ventrolateral medulla (RVLM), which is the gateway for activation of the central sympathetic nervous system (Fig. 4A). Upregulated Nox2 and Nox4 followed a similar time course (Fig. 4B).

IRI induced cerebral inflammation, as evidenced by an increase in GFAP-positive cells (astrocytes) in the SFO, Hippo CA3, corpus callosum, and cerebral cortex. The increase in GFAP-positive cells was mainly located in the edge area of SFO and the surrounding area of Hippo CA3 (Fig. 4C). IRI-induced cerebral inflammation was further demonstrated by increased levels of cytokines such as KC and granulocyte-colony stimulating factor in these regions (Supplementary Fig. S5A). There was only modest cerebral inflammation after bilateral nephrectomy (Supplementary Fig. S3B).
IRI-induced cerebral inflammation and TH expression were prevented by blockade of central AT1 receptors or prevention of oxidative stress
ICV administration of Los, at ∼1.3% of the dose of IG Los, inhibited the expression of TH and Noxs (Fig. 5) in the brain, whereas ICV Clo was ineffective (Fig. 5).
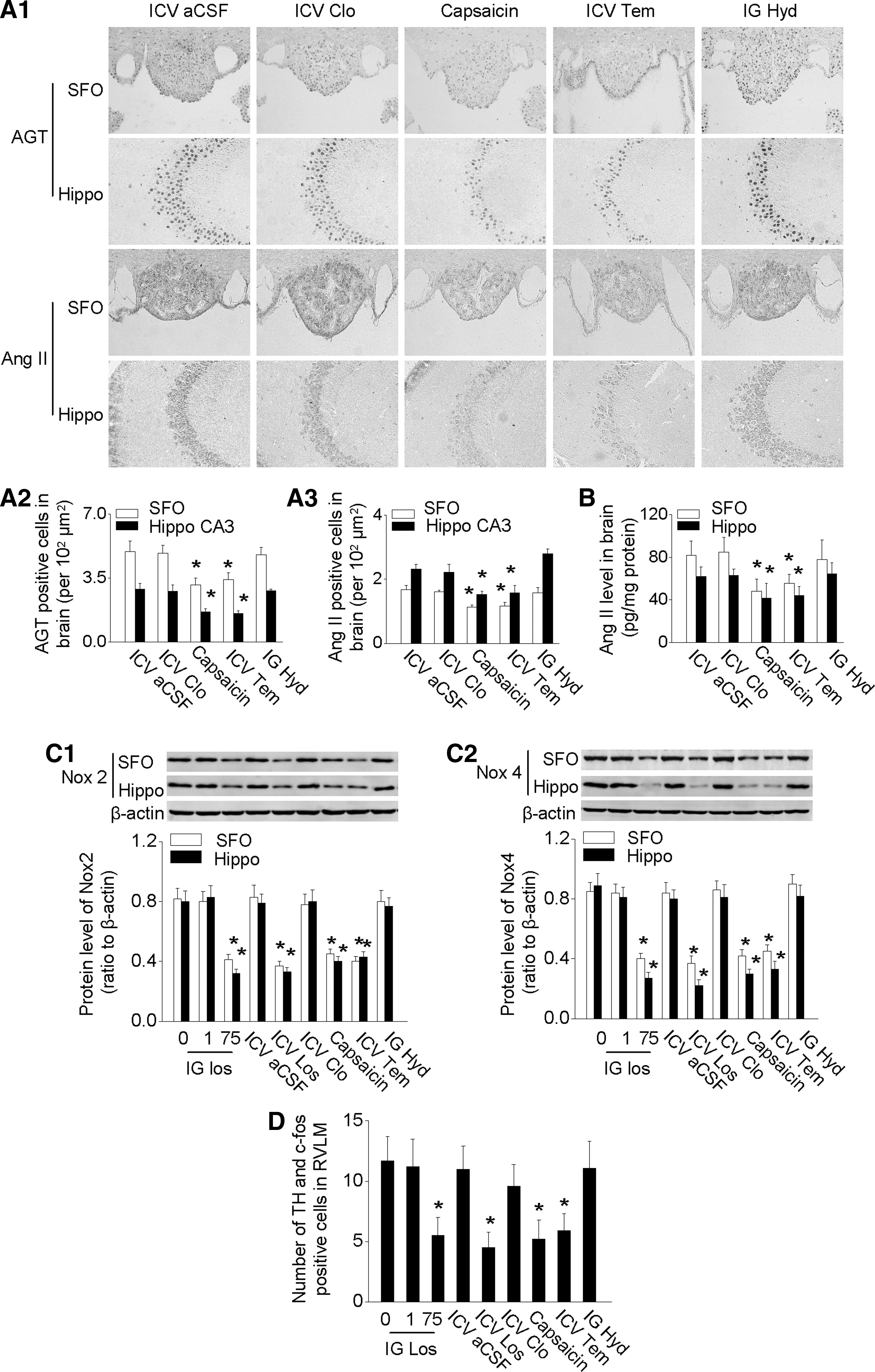
Remarkably, selective blockade of renal afferent signals by capsaicin or renal denervation prevented the IRI-induced upregulation of the brain RAS, TH, and Noxs (Fig. 5 and Supplementary Fig. S6). Brain inflammation, as indicated by GFAP, KC, and granulocyte-colony stimulating factor, was decreased by blockade of cerebral RAS, or blockade of renal afferent signals, or treatment with anti-oxidant Tem (Fig. 6 and Supplementary Fig. S5B).

The activation of the central RAS, sympathetic nervous system, oxidative stress, or inflammation persisted after controlling blood pressure with Hyd (Figs. 5 and 6).
IRI-induced renal RAS activation was prevented by blockade of RAS, reno-cerebral reflex, or oxidative stress
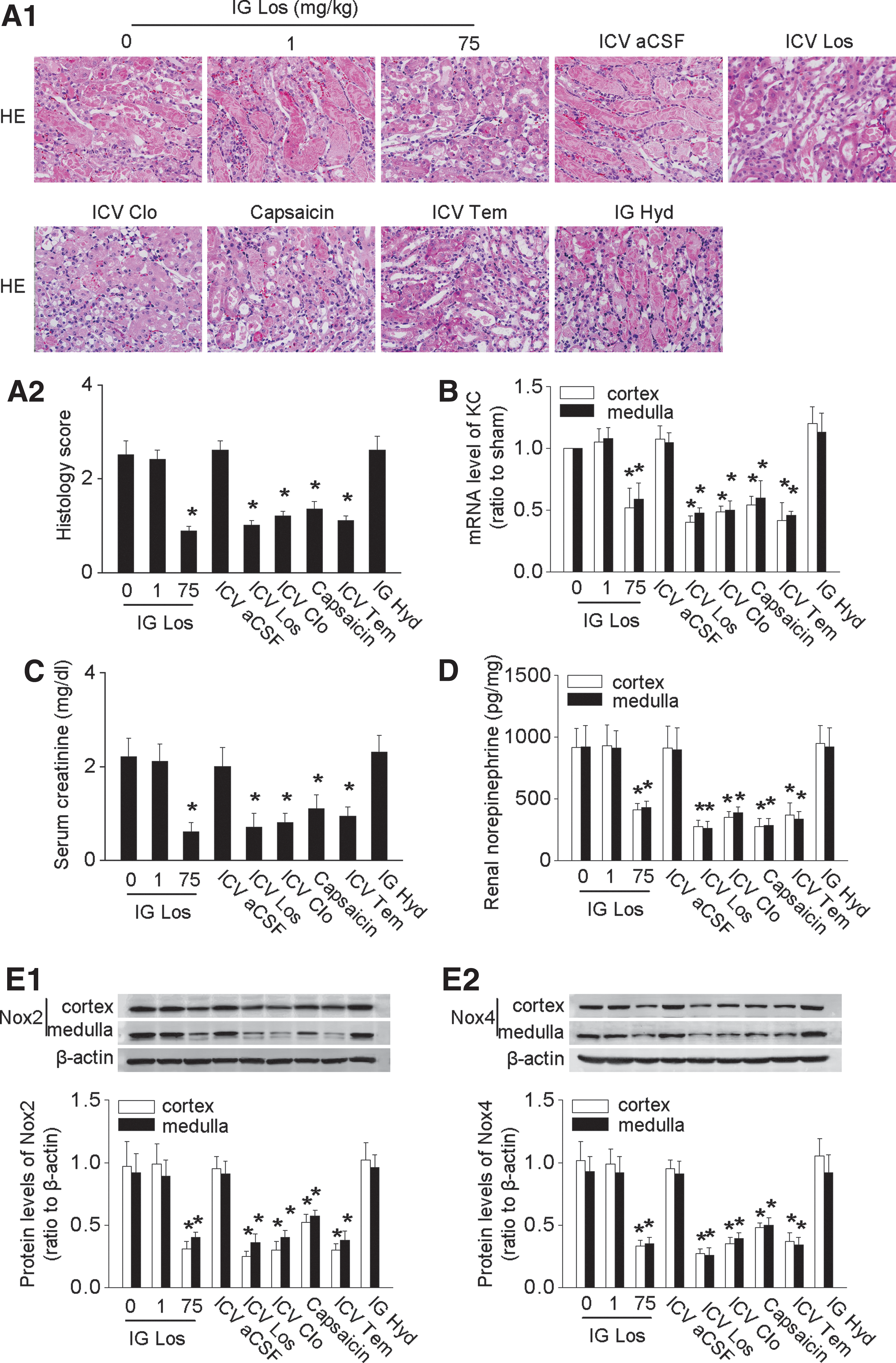
ICV administration of Los reduced IRI-induced overexpression of the intrarenal RAS (Fig. 7A, B), decreased concentration of Ang II in renal homogenates (Fig. 7C), and reduced urinary AGT excretion (Fig. 7D). Remarkably, blockade of renal afferent sympathetic activity by capsaicin or renal denervation, or efferent signals by ICV Clo, or anti-central oxidative stress by ICV Tem all alleviated the IRI-induced activation of the intrarenal RAS (Fig. 7 and Supplementary Fig. S6). Thus, renal nerves and central oxidative stress activated the intrarenal RAS after IRI, and this persisted after lowering the blood pressure with Hyd.

Renal injury and dysfunction after IRI were attenuated by blockade of RAS, sympathetic activity, or oxidative stress
ICV administration of Los decreased the renal tissue injury score (Fig. 8A) and renal KC level (Fig. 8B), reduced serum creatinine (Fig. 8C), decreased the renal norepinephrine concentration (Fig. 8D), and downregulated the renal Nox2 and Nox4 expression (Fig. 8E). Blockade of sympathetic traffic by ICV Clo or renal denervation, or renal afferent nerve output with capsaicin, or anti-oxidative stress with ICV Tem all reduced renal injury and dysfunction in mice with ischemic AKI (Fig. 8 and Supplementary Fig. S6) whereas the reduction of blood pressure with Hyd was ineffective (Fig. 8). Overall, renal injury score in this model was increased 2.2-fold compared with sham mice, but ICV Tem, Los, or Clo reduced this by 58%, 65%, or 52%, respectively, and renal denervation or renal deafferentaion by 49% or 42%, suggesting robust, but not exclusive, roles for the brain and the renal nerves in the renal dysfunction in this ischemic AKI model.
Discussion
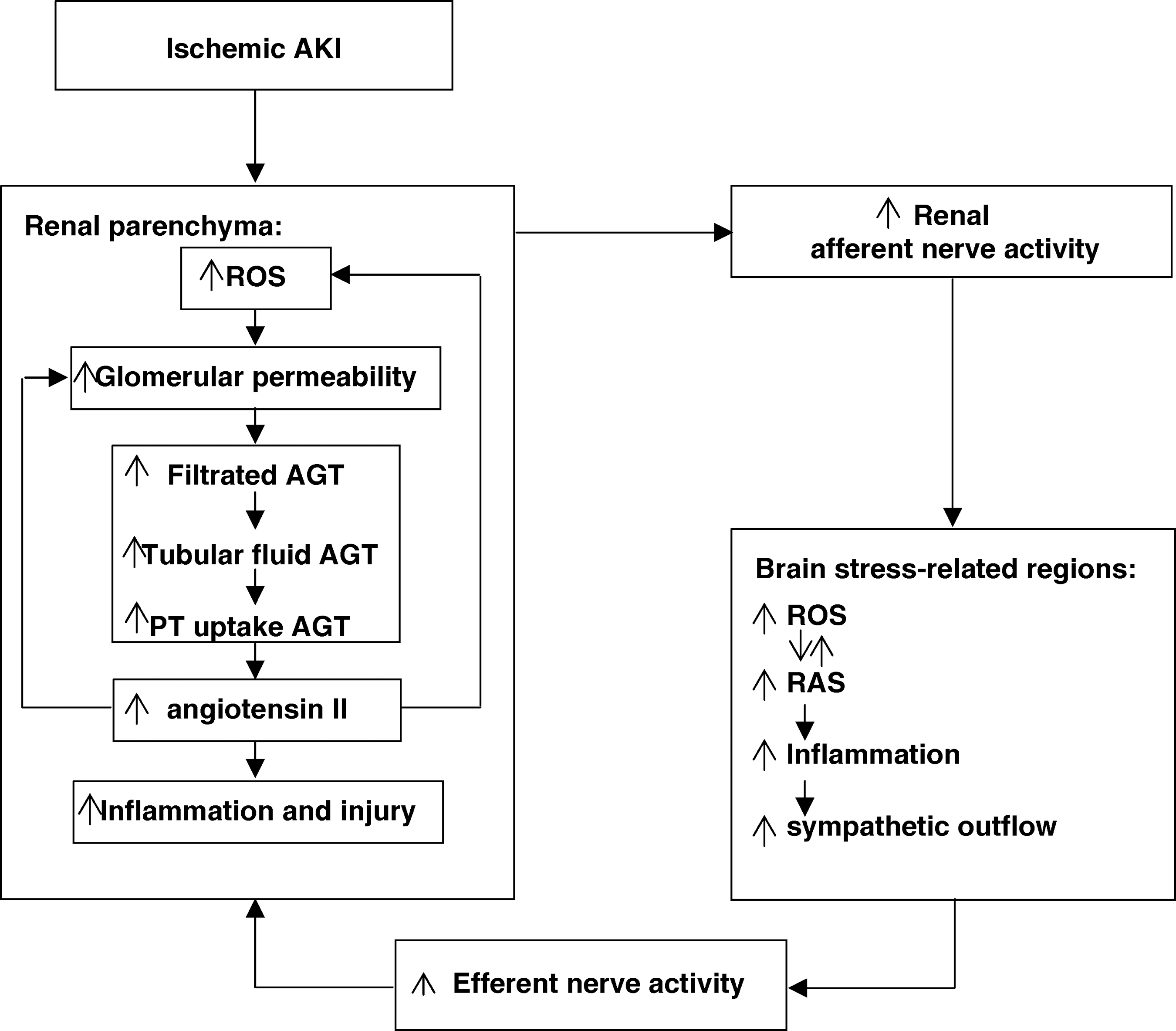
The present study has uncovered a new reflex pathway between damaged kidneys and the brain that sustains renal oxidative stress and renal RAS activation to promote ongoing renal damage. Renal IRI co-activated the intrarenal and cerebral RAS and oxidative stress, independent of systemic Ang II or blood pressure, via a robust intercommunication provided by renal afferent and efferent sympathetic nerves. Reflex activation of this reno-cerebral RAS and oxidative stress axis contributed to the brain inflammation and to worsening of the ischemic renal damage in ischemic AKI. To our knowledge, this is the first study demonstrating that interorgan crosstalk between kidney and brain may occur directly through a neuronal pathway in ischemic AKI (illustrated in Fig. 9).
Activation of intrarenal RAS is a well-established pathway for progression of CKD (4, 10), but its role in the pathophysiology of AKI remains poorly understood. Renal AGT and Ang II were upregulated in the IRI model, suggesting an over-activation of intrarenal RAS. Overexpression of AGT was most prominent in the renal cortex in proximal tubules, whereas Ang II was most prominent in the renal medulla in distal tubules. Enhancement of the intrarenal RAS components was regulated independently from circulating RAS, since plasma renin activity and circulating Ang II remained unchanged during IRI, and although leading to systemic hypertension, was independent of blood pressure since it persisted after prevention of hypertension with Hyd. This extends previous reports of increased Ang II in renal homogenates after IRI (2, 11, 31).
Whether the increases in urinary AGT in AKI reflect increased glomerular passage (36, 43) or intrarenal AGT formation (28, 30) is currently unknown. In a transgenic mouse model of podocyte-selective injury, increased renal Ang II content and markedly increased tubular and urinary AGT were attributed to increased glomerular passage of circulating AGT (36). Urinary albumin is generally believed to be plasma derived. We confirm that the levels of urinary albumin and AGT are strongly correlated, suggesting that much of the urinary AGT can be derived from the plasma. Indeed, the finding that renal tissue AGT protein expression increased approximately fivefold 1 day after IRI, yet renal tissue AGT mRNA expression fell by approximately eightfold implies that much of the increased renal AGT was not locally synthesized but rather derived by filtration from the systemic circulation.
Since activation of the intrarenal RAS was upregulated strongly in ischemic AKI, it became important to understand its cause. Previous studies have shown that central Ang II activates sympathetic tone (5, 55). We found likewise that activation of intrarenal RAS after IRI was mediated by overexpression of the brain RAS that, in turn, activated the sympathetic nervous system. Mice with ischemic AKI exhibited an increased generation of cerebral TH, a rate-limiting enzyme for norepinephrine synthesis, thereby indicating an increased central sympathetic drive. These mice also had increased renal levels of norepinephrine, indicating an enhanced renal efferent sympathetic nerve activity. This central sympathoexcitation is secondary to oxidative stress, since centrally administration of Ang II potently activates ROS in cardiovascular neurons (60). Indeed, brain sympathoexcitation during ischemic AKI was clearly dependent on the central RAS and ROS, because their blockade with ICV Los or Tem prevented the activation of intrarenal RAS and the increase in renal norepinephrine, and attenuated renal injury and dysfunction induced by IRI. In turn, the changes in renal structure and function were dependent on increased central sympathetic outflow, since its blockade with ICV Clo suppressed renal RAS activation and injury after IRI. Because ICV Clo did not affect the brain RAS activity, we conclude that brain RAS activation is upstream from central sympathetic outflow, thereby linking activation of the brain RAS and ROS with exacerbation of renal injury. Similarly, blockade of brain RAS or sympathetic outflow in salt-loaded CKD rats attenuates the renal damage (6).
After IRI, the mRNA expression of renin in the SFO increased ∼30% (not a significant change), whereas AGT expression in the SFO increased by ∼80% and Ang II expression increased by ∼90%. These figures suggest that the increased levels of Ang II in the SFO related primarily to an increase in AGT. The protein expression for AGT in the SFO increased by ∼60%, which was quite comparable to the increase of ∼80% in the local levels of AGT mRNA. This suggests that the brain AGT was synthesized locally. There were rather similar changes in the Hippo. Moreover, IRI reduced circulating Ang II by about 15%. Therefore, we consider our data compatible with the increased levels of Ang II in the brain after IRI originating from increased local expression of AGT but cannot rule out a contribution from circulating Ang II.
Interestingly, we found that capsaicin treatment (14) to interrupt renal afferent nerves or renal denervation inhibited the activation of the brain-kidney RAS axis and alleviated the renal injury, implying an essential role for renal afferent sympathetic nerves in the central sympathoexcitation and consequent renal injury. This extends previous reports in rats of activation of renal afferent nerves by ROS during neurogenic hypertension (39, 56), Moreover interruption of afferent renal nerves inhibits the activation of the brain-kidney RAS axis in salt-loaded CKD rats (6).
Blockade of the cerebral RAS/ROS attenuated IRI-induced renal damage by ∼60%, suggesting a robust but not exclusive effect of RAS/ROS activation. Other pathways might be also involved in kidney damage after IRI. A pathway for distant brain inflammation in the presence of AKI might be uremic solute retention, which, in turn, triggers a whole cascade of central proinflammatory reactions (35). Consistent with this view, our study also confirmed the presence of systemic inflammation after IRI. Furthermore, bilateral nephrectomy also induced cerebral RAS activation and inflammation, although to a much less degree. However, uremia was not a dominant cause, since brain RAS activation and inflammation occurred at 3 h of IRI when azotemia was not evident.
An unexpected finding was that renal deafferentation with capsaicin, or central blockade of Ang II type 1 receptor with ICV Los or prevention of central oxidative stress with ICV Tem, or prevention of sympathetic activation with ICV Clo all prevented the increase in mean blood pressure and systemic parameters of oxidative stress and inflammation after IRI. This suggests that a more widespread hypertensive, oxidative, and inflammatory stimulus originated from the postischemia kidney that was mediated via the renal afferent nerves, the central RAS, and the efferent sympathetic nerves. Although the focus of this study was on renal damage, the profound effects on the blood pressure, heart rate and these systemic parameters suggest that this pathway likely extends to other organs in the body. This is important, since AKI increases morbidity and mortality beyond what can be ascribed to the effect of angiotensin or uremia. Indeed, the ∼250-fold increase in serum 8-iso-prostaglandin F2α may be sufficient to activate systemic thromboxane-prostanoid receptors that are themselves implicated in hypertension, renal vasoconstriction, oxidative stress, thrombosis (47, 48), and increased mortality from shock (44).
In conclusion, this study demonstrates that ischemia-reperfusion induces AKI in a mouse model, at least in part, by activation of a reno-cerebral RAS axis that is interlinked by renal afferent and efferent sympathetic nerves. This identifies a novel mechanism underlying reno-cerebral interaction in response to renal ischemia-reperfusion, and, therefore, could lead to new interventional approaches, such as the use of RAS inhibitors, sympatholytic agents, or even renal nerve ablation.
Materials and Methods
Animals
Six-week-old male C57BL/6J mice (Nanfang Hospital Animal Experiment Center, Guangzhou, China) were maintained in a pathogen-free facility under controlled temperature (24°C ± 2°C) and humidity (55% ± 5%), with a 12-h light/dark cycle. All animal experiments were approved by the Animal Ethics Committee of Nanfang Hospital.
Study design
Protocol 1
Renal IRI, bilateral nephrectomy, or sham operation (sham) was performed at 7 weeks of age as previously described (35). Animals were hydrated with warm saline on a heating pad (40°C), keeping body temperatures constantly at 37°C until full recovery from anesthesia. An atraumatic vascular clamp was placed on both renal pedicles for 45 min to induce IRI. Sham-operated animals underwent the identical procedure but without placement of the vascular clamps. Bilateral nephrectomy animals underwent similar procedures, except that both renal pedicles were ligated and the kidneys were removed. The success of the AKI model was confirmed by increased serum creatinine concentration and renal histological damage after 24 h.
Protocol 2
Mice were assigned randomly to 10 groups and matched for body weight (n = 6 in each group). All except group 7 received the following treatments at 48, 24, and 1.5 h before IRI: (a) IG vehicle (phosphate-buffered saline, pH 7.4) (group 1) or Los (Sigma Chemical, Saint Louis, MO) at 1 mg/kg (group 2), or 75 mg/kg (group 3); (b) ICV injection of vehicle (artificial cerebrospinal fluid [aCSF]) (group 4) or Los at 1 mg/kg in 3 μl of aCSF (20) (group 5); (c) ICV Clo (Sigma Chemical) at 10 μg/kg in 3 μl of aCSF as previously described (32) (group 6); (d) Renal deafferentation with capsaicin (Sigma Chemical) treatment was performed 10 min before ischemia-reperfusion as previously described (14) (group 7); (e) Renal denervation was performed 5 min before IRI as previously described (17) (group 8); (f) ICV Tem (Sigma Chemical) at 400 μmol/kg in 3 μl of aCSF (20) (group 9); and (g) IG Hyd (Sigma Chemical) at 15 mg/kg (group 10). Animals were subjected to renal IRI and were sacrificed 24 h after ischemia for sample harvesting.
Treatment procedure
ICV administration of drug
The doses of ICV Los, Clo, or Tem were chosen from preliminary studies in which an intravenous injection of these doses had no detectable effect on Ang II-induced increase in blood pressure or renal norepinephrine levels. The accuracy of the ICV injection was confirmed by using the tracer Evans blue. The dose of IG Los was chosen to achieve similar improvement of renal injury to those given ICV Los. The dose of IG Hyd was chosen to reduce the blood pressure similarly to mice given ICV Los.
Selective renal deafferentation
Capsaicin-mediated selective renal afferent denervation was performed according to a previous report (14). Briefly, the renal vessels were exposed by gently dissecting the surrounding fat and wrapped with a small piece of gauze that was soaked in a capsaicin solution (33 mM in 5% ethanol, 5% tween 80, and 90% normal saline) for 15 min. The efficacy of capsaicin treatment was confirmed by reduced expression of calcitonin gene-related peptide, which is an afferent nerve-specific marker (Supplementary Fig. S5C) (14).
Renal denervation
Renal denervation was performed as previously described (17). Briefly, the renal vessels were exposed by gently dissecting the surrounding fat, and renal denervation was performed by surgically stripping the renal arteries and veins of adventitia, cutting all visible renal nerve bundles under a dissection microscope. Renal vessels were painted with 10% phenol to ensure the destruction of any remaining nerves. Effectiveness of renal denervation was demonstrated by a reduction of renal norepinephrine content to less than 5% of the control.
Measurement of renal function and mean arterial pressure
Serum creatinine concentration was measured as a marker of renal function by using the Quantichrom Creatinine assay kit (BioAssay Systems, Hayward, CA). Serum potassium and sodium concentrations were measured with an automated chemistry analyzer (AU480; Beckman Coulter). Urinary albumin excretion was measured with an ELISA kit (IMTEC Diagnostics). Glomerular filtration rate was determined by inulin clearance as previously described (22).
Mean blood pressure was measured in conscious rats via a catheter in the carotid artery by telemetry using TA11PA-C10 probes (Data Sciences International, St. Paul, MN), which were implanted 1 week before the renal IRI or sham operation as previously described (18).
Tissue injury analysis
Evaluation of renal injury
The kidneys were dissected and processed for hematoxylin-eosin staining as previously described (52). Morphologic damage at the corticomedullary junction and outer medullary area after IRI was assessed by two experienced blinded renal pathologists using a grading scale of 0 to 5, as described (52).
Evaluation of tissue inflammation
Whole brain was dissected and cut into thick coronal slices (2 mm) with a mouse brain slicer matrix (Zivic Instruments, Pittsburgh, PA). Slices were postfixed, embedded in paraffin, and cut into 5 μm serial thick sections. The stress-related regions, including SFO, Hippo CA3, corpus callosum, and cerebral cortex, were identified under a microscope and immunohistochemical staining was performed by using an anti-GFAP (Millipore, Temecula, CA) antibody to determine the expression of GFAP. The integrated optical density/area of the positive staining for GFAP was assessed in three sections from each part of the brain by two blinded pathologists using an Image-Pro Plus software (version 6.0, Media Cybernetics, Rockville, MD) (21).
The expression of KC and granulocyte-colony stimulating factor in homogenates of brain or kidney was determined by a real-time polymerase chain reaction (PCR) as previously described (57, 58).
RAS expression and activity
Immunohistochemistry analysis
To evaluate the expression of intrarenal RAS, 5-μm-thick sections were processed (7) by using rabbit anti-mouse AGT (1:200; IBL, Japan), rabbit anti-Ang II (1:800; Peninsula Laboratories, San Carlos, CA), and goat anti-mouse renin (1:50; Santa Cruz, Dallas, TA) antibodies. RAS expression in the renal cortex and medulla was quantitated as described (50), and intrarenal renin expression was assessed as previously described (42).
The cerebral expression of AGT and Ang II was quantitated with immunohistochemical staining as described in our previous study (6) by using the antibodies mentioned earlier.
Localization of tissue RAS
For localization of intrarenal RAS, 5-μm paraffin-embedded kidney sections were double stained by using first primary antibodies against AGT (1:200; IBL), or Ang II (1:800; Peninsula Laboratories), in separate sections, and second primary antibodies against the markers of tubular epithelial cells: anti-aquaporin 1 (AQP-1, a marker of proximal tubular epithelial cells and the descending thin limb of Henle's loop; 1:100; Abcam, UK); anti-Tamm-Horsfall protein (a marker of ascending thick limbs of Henle's loop; 1:100, Santa Cruz); anti-thiazide-sensitive NaCl cotransporter (NCCT, a marker of distal convoluted tubular epithelial cells; 1:500; Millipore); and anti-aquaporin 2 (AQP-2, a marker of collecting duct epithelial cells; 1:100; Novus Biologicals, Littleton, CO) antibodies in separate sections, separately.
Cerebral localization of AGT and Ang II was determined by double staining using the anti-AGT or anti-Ang II antibody mentioned earlier as the first primary antibody, and anti-NSE (Millipore) or anti-GFAP (Millipore) antibodies as the second primary antibody (50).
Western blot and real-time PCR
The levels of AGT in renal homogenates were determined as previously described (8) by using rabbit anti-mouse AGT (IBL) and rabbit anti-β-actin (Cell Signaling Technology, Danvers, MA) antibodies. AGT and renin mRNA expression were determined by real-time PCR as previously described (59).
ELISA
AGT levels in urine collected via cystocentesis were assessed by an ELISA kit (IBL) according to the manufacturer's instructions.
Radioimmunoassay
Ang II concentrations in plasma and renal homogenates and plasma renin activity were determined by the radioimmunoassay according to the manufacturer's instructions (Beijing North Institute of Biological Technology, China). To prevent angiotensin from degradation, blood samples were collected in ice-cooled tubes containing a cocktail of protease inhibitors (0.01 mM p-hydroxylmercuribenzoate, 1.5 mM 1,10-phenanthroline, 0.01 mM PMSF, 0.05 mM pepstatin A, and 10 mM EDTA). Renal and brain tissue was homogenized on ice with 0.045 M HCl in ethanol containing 0.9 mM p-hydroxylmercuribenzoate, 131.5 mM 1,10-phenanthroline, 0.9 mM PMSF, 1.75 mM pepstatin A, 1.1 mM EDTA, and 0.0043% protease-free BSA (11).
Evaluation of sympathetic activity
Norepinephrine concentrations
Concentrations of norepinephrine in serum and homogenates of renal tissue were assessed with an ELISA kit (Demeditec Diagnostics, Germany) according to the manufacturer's protocol.
Expression of TH in brain
Five-micrometer-thick brain stem sections were double stained with rabbit anti-mouse TH (Millipore) and rat anti-mouse c-fos (Santa Cruz) antibodies as previously described (54). The number of neurons labeled by TH and c-fos in the RVLM was quantified as previously described (54).
Evaluation of oxidative stress and inflammation
Systemic oxidative stress and inflammation
Serum concentrations of 8-iso-prostaglandin F2α and KC, markers of systemic oxidative stress and inflammation, were quantified by the ELISA kits (Enzo Life Sciences, NY; Qiagen, Valencia, CA).
Expression of Noxs in kidney and brain
The expression of NADPH oxidase subunits Nox2 and Nox4 in renal and brain homogenates was determined by Western blot using anti-Nox2 (Abcam) or Nox4 antibody (Santa Cruz) (8).
Statistic analyses
All data are expressed as mean ± SD of at least three independent experiments. Continuous variables among groups were compared by using one-way analysis of variance, followed by LSD test. An unpaired t-test was used to compare the parameters between IRI and sham mice or between animals treated with IRI and bilateral nephrectomy at the same time point. Histology scores were analyzed by nonparametric testing. Statistical analyses were conducted with SPSS 17.0 for Windows (SPSS, Inc., Chicago, IL). A value of p < 0.05 was considered statistically significant.
Footnotes
Acknowledgments
This study was supported by the National Key Technology Support Program of China (2013BAI09B06 and 2015BAI2B07 to F.F.H.), the State Key Program of National Natural Science Foundation of China (81430016 to F.F.H.), the Major International (Regional) Joint Research Project of National Natural Science Foundation of China (81620108003 to F.F.H.), the Foundation for Innovation Research Groups of the National Natural Science Foundation of China (81521003 to Y.L.), the National Natural Science Foundation of China (81570619 to W.C., 81270825 and 31201751 to A.L.), the Major Scientific and Technological Planning Project of Guangzhou (15020010 to F.F.H.), NIH (HL-68686; DK-049870; DK-036079 to C.S.W.), and funds from the George E. Schreiner Chair of Nephrology and the Georgetown University Hypertension, Kidney and Vascular Research Center (to C.S.W.).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.