
Abstract
Aims:
The glutathione (GSH), thioredoxin (Trx), and Nrf2 systems represent a major defense against reactive oxygen species (ROS), the cellular imbalance of which in cancer promotes growth and therapeutic resistance. This study investigated whether targeting the GSH, Trx, and Nrf2 antioxidant systems effectively eliminated head and neck cancer (HNC).
Results:
At high concentrations, auranofin, but not buthionine sulfoximine (BSO) alone, decreased the viability of HNC, whereas even at low concentrations, auranofin plus BSO synergized to kill HNC cells. Dual silencing of the genes for GCLM and TrxR1 induced GSH depletion, Trx activity inhibition, and ROS accumulation, synergistically killing HNC cells. Inhibition of the GSH and Trx systems resulted in activation of the Nrf2-antioxidant response element (ARE) pathway, which may result in suboptimal GSH and Trx inhibition where HNC is resistant. Genetic inhibition of Nrf2 and/or HO-1 or trigonelline enhanced growth suppression, ROS accumulation, and cell death from GSH and Trx inhibition. The in vivo effects of GSH, Trx, and Nrf2 system inhibition were confirmed in a mouse HNC xenograft model by achieving growth inhibition >60% compared with those of control.
Innovations:
This study is the first to show that triple inhibition of GSH, Trx, and Nrf2 pathways could be an effective method to overcome the resistance of HNC.
Conclusions:
Inhibition of the Nrf2-ARE pathway in addition to dual inhibition of the GSH and Trx antioxidant systems can effectively eliminate resistant HNC. Antioxid. Redox Signal. 27, 106–114.
Introduction
T
These results identify the effectiveness of targeting the glutathione (GSH), thioredoxin (Trx), and Nrf2 antioxidant systems to eliminate head and neck cancer (HNC). Dual blockade of the GSH and Trx systems synergized to kill HNC cells, but this may be suboptimal in some resistant HNCs because of activation of the Nrf2-antioxidant response element (ARE) pathway. Inhibition of the Nrf2-ARE pathway in addition to dual inhibition of the GSH and Trx antioxidant systems effectively eliminated resistant HNC.
Tripeptide glutathione (GSH), thioredoxin (Trx), and transcription factor nuclear factor (erythroid-derived 2)-like 2 (Nrf2) systems represent a major defense against ROS. GSH, the most abundant cellular antioxidant, is synthesized through the rate-limiting step by glutamate cysteine ligase (GCL) with a heterodimer of GCL modifier (GCLM) and catalytic (GCLC) subunits (19). The Trx system is also essential for maintaining cellular redox at a reduced status in the presence of Trx reductase (TrxR) (20). Nrf2 is a central player in regulating cellular redox homeostasis by its promoter binding to the antioxidant responsive element (ARE) (15). Cancer cells buffer cellular ROS levels by actively upregulating the multiple antioxidant pathways that contribute to tumor progression (6, 7, 24).
Recently, the impact on tumorigenesis by inhibiting the GSH and Trx antioxidant systems has been highlighted (14). Inhibition of both the GSH and the Trx pathways synergistically induces cancer cell death in vitro and in vivo and decreases resistance to anticancer therapy (2). However, targeting of Trx or GSH systems leads to robust activation of Nrf2, resulting in maintaining the cellular environment in a reduced state (4). It may therefore be postulated that inhibition of the Nrf2-ARE pathway in addition to a dual blockade of the GSH and Trx antioxidant systems would effectively eliminate cancer cells. However, the effects of combined inhibition of the GSH, Trx, and Nrf2 systems have rarely been tested in head and neck cancer (HNC) and other cancer types. This study investigated whether targeting the GSH, Trx, and Nrf2 antioxidant systems effectively eliminated HNC. In this study, triple inhibition of GSH, Trx, and Nrf2 systems appeared to be more effective than a dual blockade of GSH and Trx systems for eliminating HNC in vitro and in vivo, particularly when the HNC shows resistance.
Results
Buthionine sulfoximine and auranofin synergized to induce HNC cell death
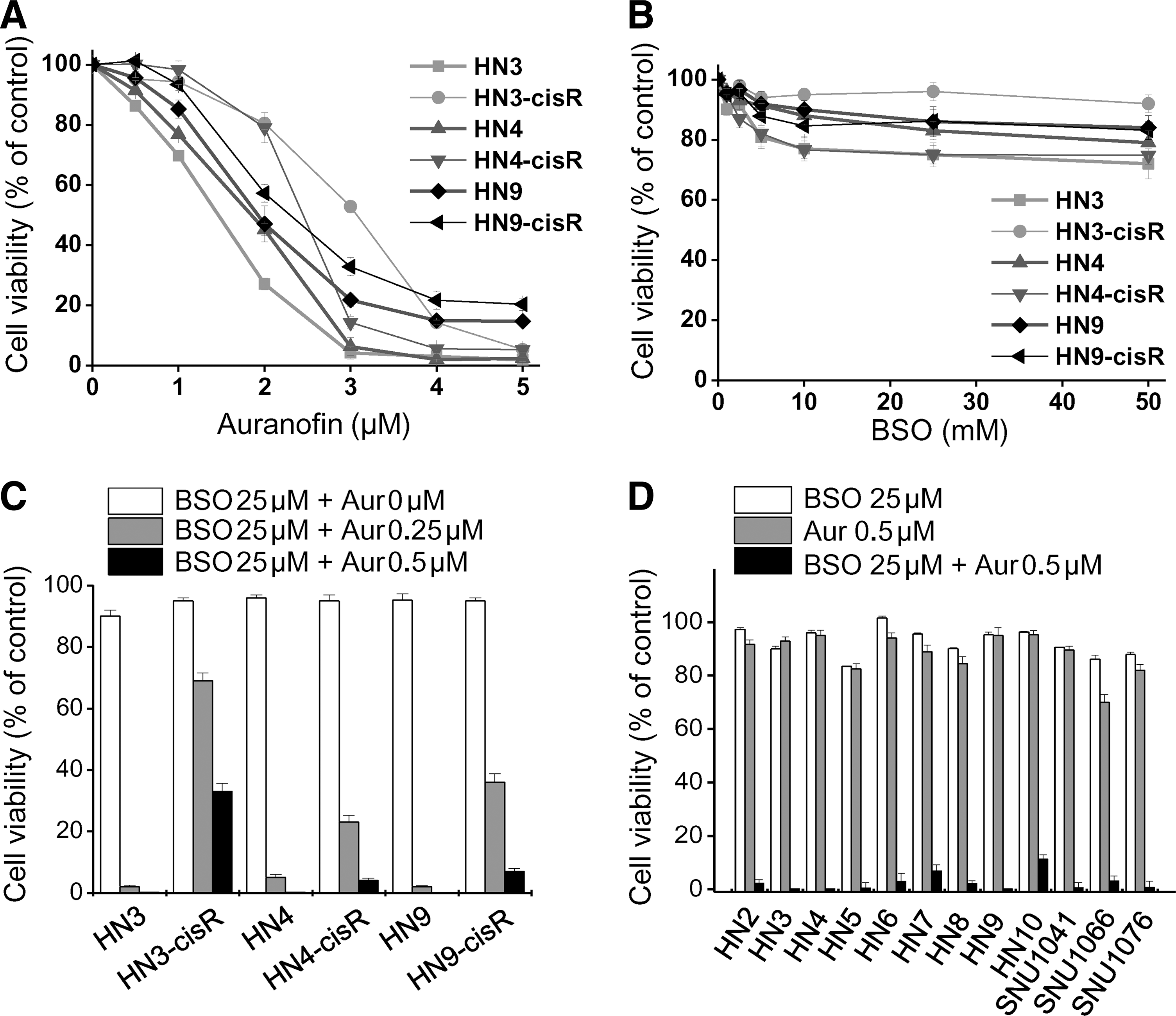
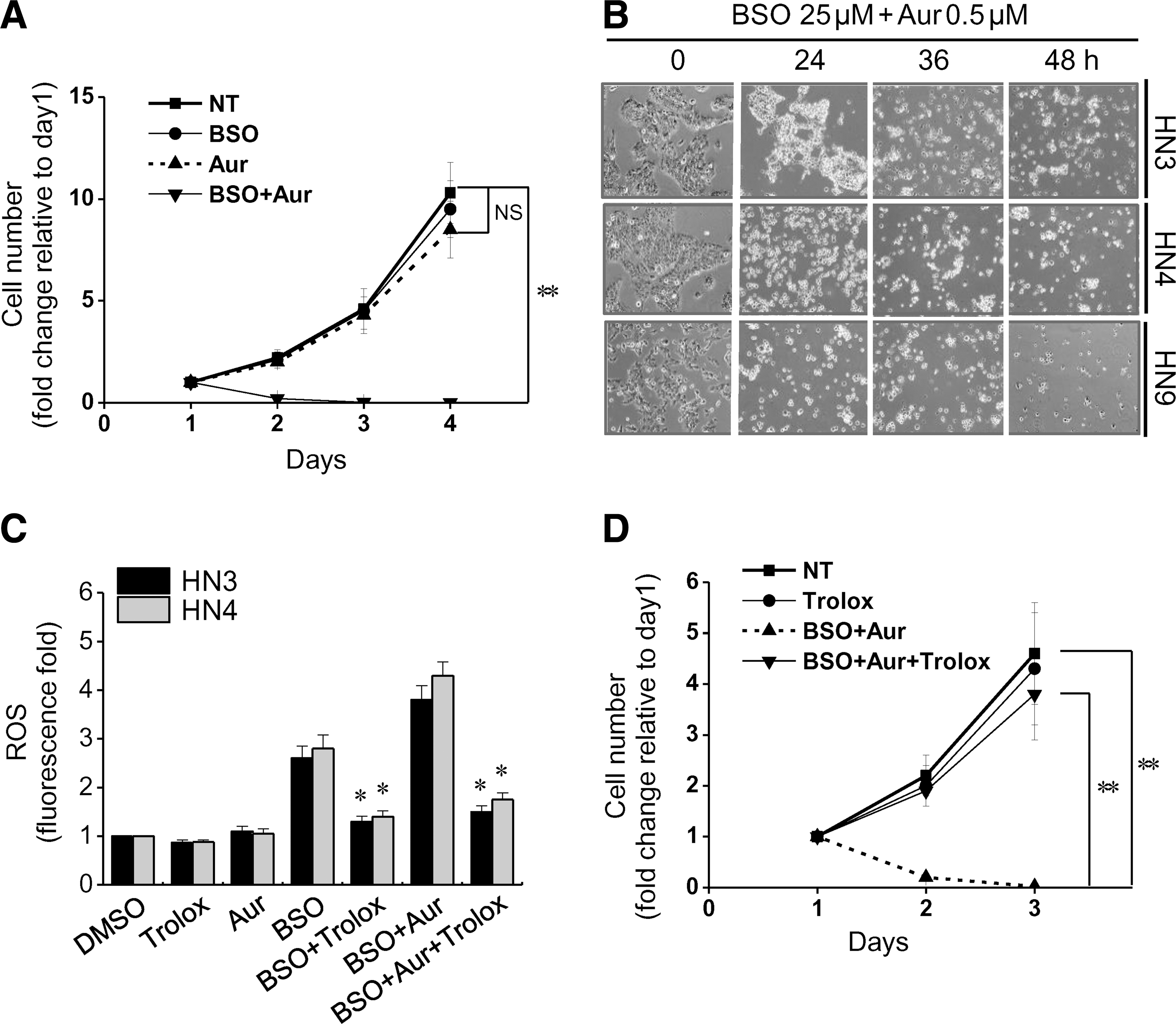
Auranofin decreased the viability of cisplatin-sensitive and -resistant HNC cells in a dose-dependent manner (Fig. 1A), whereas buthionine sulfoximine (BSO), even in high concentrations (up to 50 mM), did not induce cell viability inhibition by >50% of the control (Fig. 1B). A combination of low-dose BSO (5–25 μM) and auranofin (0.1–0.5 μM) synergistically induced the effective death of different HNC cells (combination index [CI] <1.0; Fig. 1C, D). A combination of BSO and auranofin significantly decreased the cell numbers from day 1 after the treatment (CI <1.0), although low concentrations of either of these alone did not (Fig. 2A, B). BSO, or BSO combined with auranofin, induced cellular ROS accumulation, which was abrogated by the pretreatment of trolox, an antioxidant (Fig. 2C). Trolox also prevented the inhibition of cell growth by BSO plus auranofin (CI <1.0; Fig. 2D). Therefore, BSO was acting as a sensitizer of auranofin-induced cell death. The combined treatment increased mitoSOX and decreased TMRM staining in HNC cells (data not shown).
Dual blockade of GSH and Trx systems induced HNC cell death
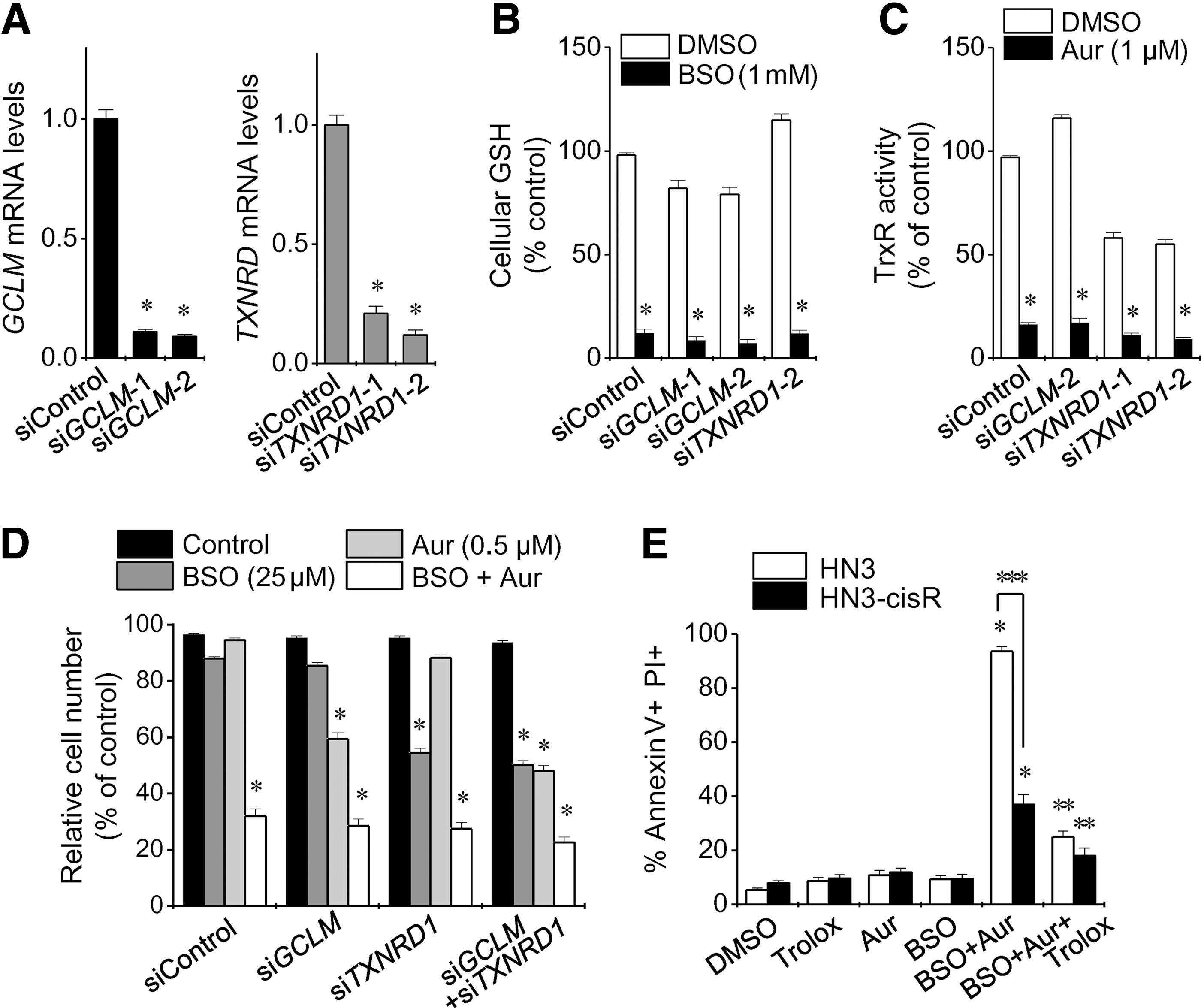
Silencing of the GCLM and TXNRD1 genes induced GSH depletion and Trx activity inhibition, respectively (Fig. 3A–C and Supplementary Fig. S1; Supplementary Data are available online at
Inhibition of GSH and Trx systems resulted in activation of the Nrf2-ARE pathway
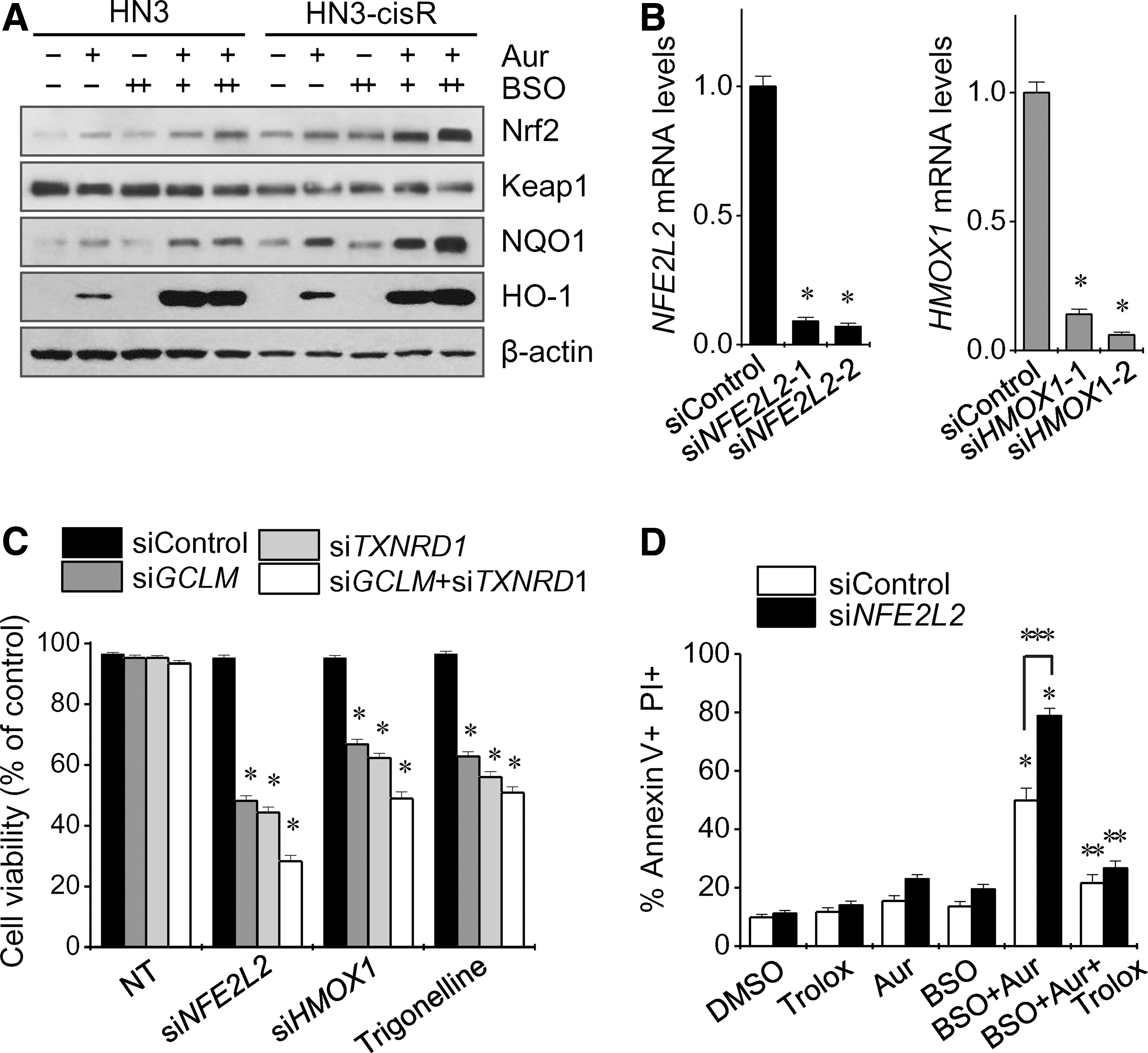
Auranofin and auranofin plus BSO increased the expression of Nrf2 and its ARE pathway, including NQO1 and HO-1 (Fig. 4A). Silencing of the NFE2 L2 or HMOX1 genes, or pharmacological inhibition of Nrf2 with trigonelline, resulted in decreased cell viability of HNC cells transfected with siGCLM and/or siTXNRD1 (Fig. 4B, C). Genetic silencing of Nrf2 and/or HO-1 enhanced the synergistic cell death from BSO plus auranofin in cisplatin-resistant HN3-cisR and HN9-cisR cells (Figs. 4D and 5A). Pharmacological inhibition of Nrf2 with trigonelline induced greater cell growth inhibition, ROS accumulation, and cell death from BSO plus auranofin than did no treatment of trigonelline or pretreatment with the antioxidant trolox (Fig. 5B–D). The increased Nrf2 levels were found in cisplatin-resistant HNC cells compared with those of their parental cisplatin-sensitive HNC cells (Supplementary Fig. S3). Nrf2 silencing prevented Nrf2 increase induced by BSO plus auranofin in a time-dependent manner (Supplementary Fig. S4). The triple combinations did not significantly suppress the growth of normal cells (Supplementary Fig. S5).

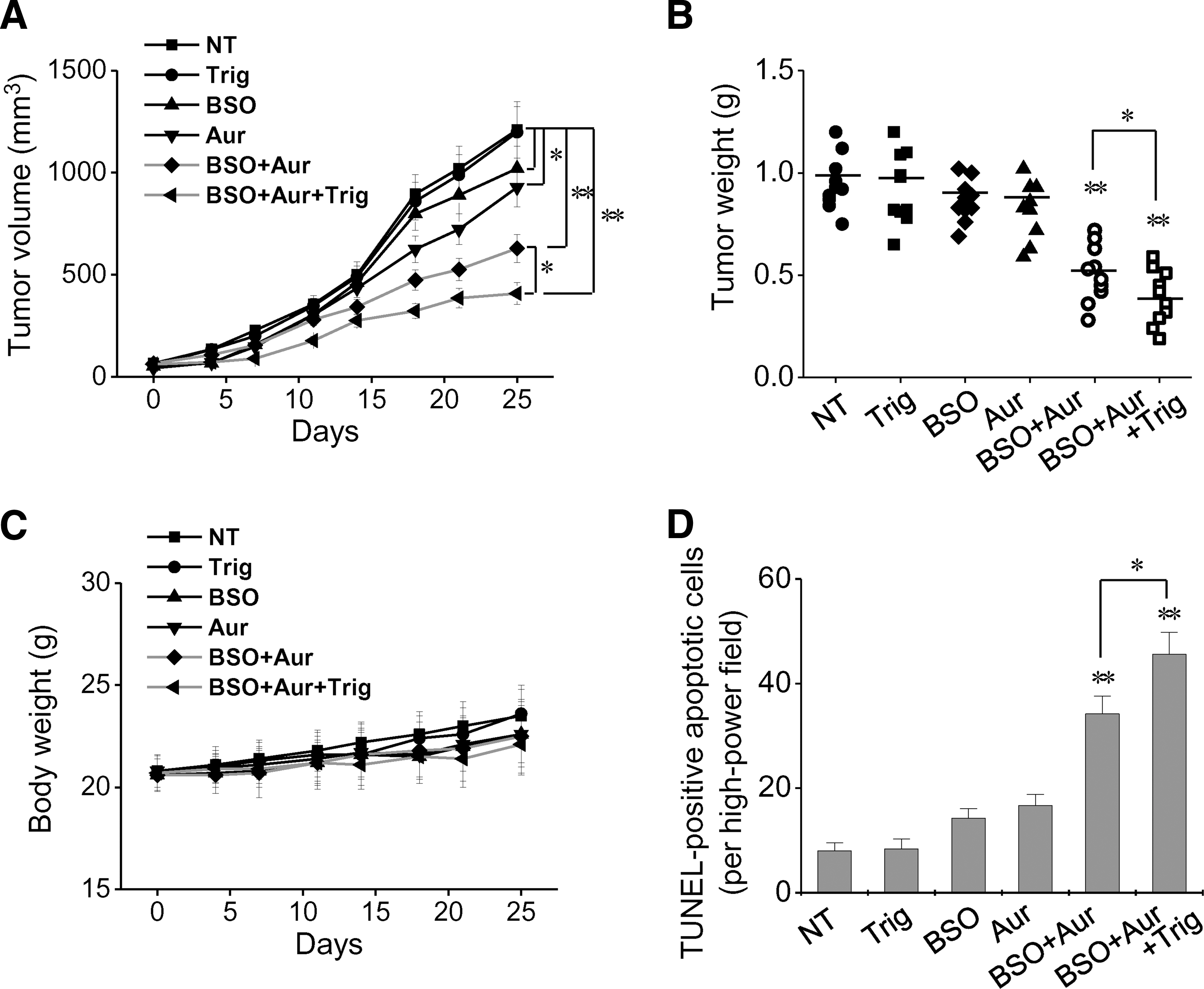
Blockade of GSH, Trx, and Nrf2 systems induced the effective inhibition of in vivo HNC growth
The pharmacological dual blockade of the GSH and Trx systems with BSO plus auranofin resulted in significant inhibition of HNC in vivo growth, which was augmented by the addition of trigonelline (Fig. 6A, B and Supplementary Fig. S6). BSO, auranofin, or trigonelline alone, or combinations of these, did not result in significant changes in body weight of the tumor-transplanted mice (Fig. 6C) or other complications of major organs (Supplementary Fig. S7). BSO plus auranofin, or these combined with trigonelline, significantly increased apoptotic cells in transplanted tumors (Fig 6D and Supplementary Fig. S8). Cellular GSH levels were significantly depleted in tumors of mice treated with BSO, BSO plus auranofin, or the combination of BSO, auranofin, and trigonelline (Supplementary Fig. S9). Therefore, BSO was acting as a sensitizer to other drugs and showing the bioavailability of in vivo tissues.
Discussion
This study examined whether targeting of the GSH, Trx, and Nrf2 antioxidant systems effectively eliminated HNC in vitro and in vivo. Pharmacological or genetic inhibition of the GSH or Trx system alone appeared not to be effective in inducing cell death, but dual inhibition, even at low concentrations, synergized to kill HNC cells. However, inhibition of the TrxR1 or GSH system activated Nrf2-ARE pathways, resulting in dual inhibition of Trx and GSH pathways being suboptimal against some cisplatin-resistant HNC. Inhibition of the Nrf2-ARE pathway in addition to GSH and Trx dual blockade resulted in the most HNC cell deaths, particularly of those that are resistant. Our study is the first to show that triple inhibition of the GSH, Trx, and Nrf2 pathways could be an effective method to overcome the resistance of HNC.
GSH is the most abundant antioxidant in the cells, and yet experiments that targeted the inhibition of GSH synthesis alone failed to eliminate the cancer cells. This may be supported by a previous finding in which clinical trials of BSO alone inhibiting GSH synthesis had little success (1). Regardless of GSH depletion, cellular ROS levels may be decreased because of activation of the Trx antioxidant pathway from increased cystine import (14). In addition, genetic silencing of TrxR1 can increase the compensatory activity of the alternative GSH antioxidant system (21). Tumors with TrxR1 genetic ablation are highly susceptible to treatment with BSO to induce GSH depletion (21). Perturbing either of the GSH and Trx redox systems is likely to affect the other because the two systems have overlapping and widespread redundancies (2). The combined inhibition of the GSH and Trx systems synergistically acts to decrease their cellular antioxidant functions, resulting in extremely increased cellular oxidative stress and, subsequently, cancer cell death (2, 11). Increased cellular oxidative stress is likely to kill cancer cells by disrupting the well-adapted endogenous antioxidant capacity of tumors resistant to exogenous stress (31). Auranofin is a gold complex that has been used to treat rheumatoid arthritis and recently become repurposed as an anticancer agent by inducing apoptosis in cancer cells via inhibition of TrxR and overproduction of cellular ROS (8, 10). In the present study, both BSO and auranofin synergistically acted to decrease the antioxidant functions of the GSH and Trx systems, resulting in cancer cell death.
The synergy of GSH and TrxR reduction has previously been well described in recent reports of lung, colon, and breast cancer, glioblastoma, lymphoma, and leukemia (9, 14, 16). Increased susceptibility of cancer cells to the dual inhibition of GSH and Trx pathways was first reported in 2009 by Simons et al. following experimental studies (26). They reported that combined inhibition of GSH and Trx metabolism enhanced sensitivity to the Akt inhibitor, perifosine, and perifosine-induced oxidative stress, resulting in clonogenic killing and GSH depletion (26). They also reported that increased GSH and Trx metabolism in HNC cells was a potential mechanism related to resistance to chemotherapy (27). The combination of BSO and auranofin synergistically induced HNC cell death and sensitized the cells to cell death induced by erlotinib, an EGFR inhibitor (27). The synergy of GSH and Trx inhibition in HNC cells was confirmed in our study. The simultaneous inhibition of GSH and Trx systems is likely to potentiate the killing of cancer cells via oxidative stress induced by other chemotherapeutic agents (9, 23). The combined blockade of GSH and Trx systems may be a promising treatment strategy for inducing cancer cell death in established cancers. However, this could be suboptimal in some cancer types that show resistance because of the activation of other important antioxidant systems.
Nrf2 is the central player regulating the expression of antioxidant molecules in cells (29). Cancer cells equipped with a sufficient amount of antioxidants together with the oncogenic transcription factor Nrf2 play a key role against environmental or intracellular stress (17). Nrf2 controls the cellular antioxidant systems responsible for GSH production (12). Growing evidence indicates that Nrf2 is frequently overexpressed in several types of cancers and is linked with the increased resistance of cancer cells to chemotherapeutic agents and poor survival outcomes in cancer patients (28, 32). Genetic or pharmacological inhibition of Nrf2 results in a marked depletion of GSH and increases chemotherapeutic cytotoxicity (28, 32). Kelch-like ECH-associated protein 1 (Keap1) constantly targets the proteasomal degradation of Nrf2; however, TrxR1 has also been suggested as a potent regulator of the Nrf2-Keap1 response system (4). In the present study, dual inhibition of the GSH and Trx systems was suboptimal in some resistant HNC cells because of activation of the Nrf2-ARE pathway. The cisplatin-resistant cancer cell lines showed less susceptibility to the combined blockade of the GSH and Trx pathways. Increased expression of Nrf2 and ARE molecules appeared to be a suboptimal result of dual inhibition. Furthermore, genetic inhibition of Nrf2 and/or HO-1 or trigonelline enhanced the growth suppression, ROS accumulation, and cell death arising from combined GSH and Trx inhibition. The inhibition of Nrf2 additional to the dual blockade of GSH and Trx systems effectively killed HNC cells (Fig. 7).
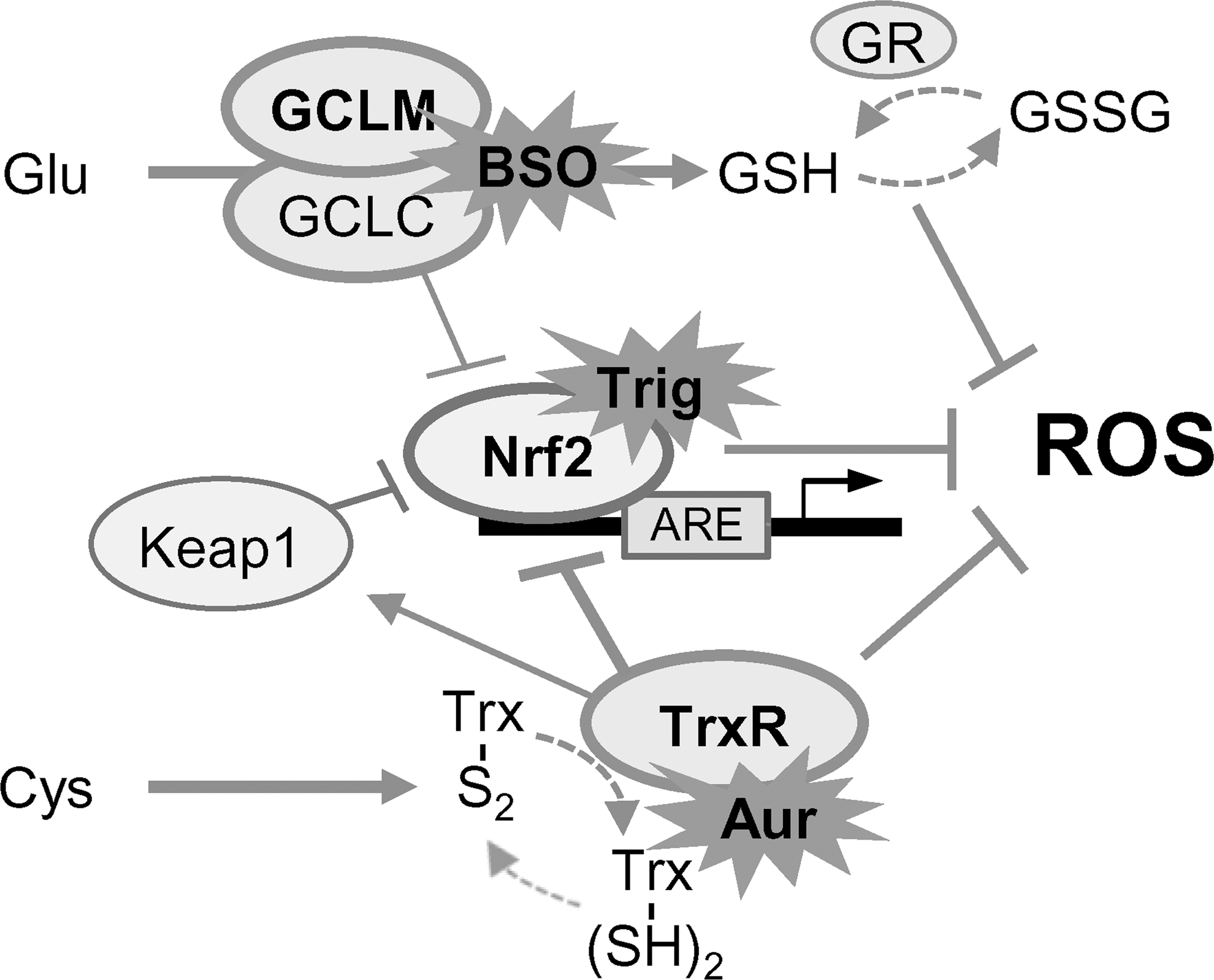
In conclusion, our study suggested that triple inhibition of GSH, Trx, and Nrf2 systems is an effective treatment strategy for eliminating HNC cells, particularly those that show resistance. The dual blockade of GSH and Trx systems appears to induce cancer cell death, but this may be suboptimal in some resistant cancer cells because of activation of the Nrf2-ARE pathway. Further preclinical and clinical investigations of triple targeting of GSH, Trx, and Nrf2 antioxidant systems should be conducted in patients with resistant cancer types to explore this promising cancer therapy.
Materials and Methods
Cell lines
The HNC cell lines used in our studies were previously established HN2 to 10 and SNU cell lines purchased from Korea Cell Line Bank (Seoul, Republic of Korea) and authenticated by STR-based DNA fingerprinting and multiplex PCR. The cells were cultured in Eagle's minimum essential medium or Roswell Park Memorial Institute (RPMI) 1640 (Thermo Fisher Scientific, Waltham, MA) with 10% fetal bovine serum at 37°C in a humidified atmosphere containing 5% CO2. Normal oral keratinocytes or fibroblasts were obtained from patients undergoing oral surgery and used in in vitro assays. In addition, three cisplatin-resistant HNC cell lines (HN3-cisR, HN4-cisR, and HN9-cisR) were used; these were developed from the parental HN3, HN4, and HN9 cells, respectively, with continuous exposure to increasing cisplatin concentrations (22). The half-maximal inhibitory concentrations (IC50) of cisplatin (Sigma-Aldrich, St. Louis, MO) in the parental (2.2–3.5 μM) and cisplatin-resistant (25.5–38.9 μM) HNC cells were determined by using cell viability assays.
Cell viability assays
Cell viability after exposure to BSO, auranofin, or trigonelline (Sigma-Aldrich) was assessed using MTT and trypan blue exclusion. The MTT assays were performed with the tetrazolium compound, MTT (3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide; Sigma-Aldrich), for 4 h, followed by a solubilization buffer for 2 h, and then absorbance was measured at 570 nm using a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA). The trypan blue exclusion was performed with 0.4% trypan blue staining and counting using a hemocytometer. A cell death assay was also performed using staining with Annexin V and propidium iodide (PI) (Sigma-Aldrich) and then counting Annexin V or PI-positive cells with flow cytometry and Cell Quest Pro software (BD Biosciences, Franklin Lakes, NJ). All assays were performed with triplicate samples and repeated three times. The CI of drug interaction was calculated using the Chou–Talalay method, which defined CI <1 as being a synergistic interaction (ComboSyn, Inc., Paramus, NJ) (5).
Glutathione synthesis, ROS production, and Trx activity measurement
A GSH colorimetric detection kit (BioVision, Inc., Milpitas, CA) was used to measure the cellular GSH levels of lysates of HNC cells exposed to different drugs for 24 h. Cellular ROS generation in a supernatant of HNC cell lysates treated differently for 24 h was measured using 2′,7′-dichlorofluorescein diacetate (DCF-DA) (cellular ROS; Enzo Life Sciences, Farmingdale, NY). The ROS levels were analyzed using an FACSCalibur flow cytometer equipped with CellQuest Pro (BD Biosciences). Trx activity was measured in HNC cells treated with different drugs for 24 h using a Trx activity fluorescent assay kit (Cayman Chemical, Ann Arbor, MI).
siRNA transfection
For the silencing of the GCLM, TXNRD1, NFE2 L2, and HMOX1 genes, cisplatin-resistant HN3- and HN9-cisR cells were seeded and transfected 24 h later with 10 nM small interfering RNA (siRNA), which targets human genes, or scrambled control siRNA (Integrated DNA Technologies, Coralville, IA). The siRNA-induced gene silencing was confirmed by Western blotting and reverse transcription–quantitative polymerase chain reaction (RT-qPCR) from 1 to 2 μg of total RNA for each sample using a SuperScript® III RT-PCR system (Thermo Fisher Scientific).
Western blotting
Cells were plated, grown with 70% confluence, and then subjected to treatment with the indicated drugs or conditioned media. Cells were lysed at 4°C in a radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Fisher Scientific). A total of 50 μg of protein was resolved by SDS-PAGE on 10%–12% gels, transferred to nitrocellulose or polyvinylidene difluoride membranes, and probed with primary and secondary antibodies. The following primary antibodies were used: Nrf2, Keap1, NQO1, and HO-1 (Abcam, Cambridge, United Kingdom). β-Actin (Sigma-Aldrich) was used as a loading control. All of the antibodies were diluted to between 1:500 and 1:5000.
In vivo mouse xenograft models
All animal study procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of our institution. Six-week-old athymic BALB/c male nude mice (nu/nu) were purchased from Central Lab Animal, Inc., (Seoul, Republic of Korea). HN3-cisR cells were injected subcutaneously into the flank of nude mice. From the day of detection of gross nodules from tumor implants, the mice were subjected to six different treatments: vehicle, BSO (450 mg/kg daily intraperitoneal injection), auranofin (2 mg/kg daily intraperitoneal injection), trigonelline (50 mg/kg daily per oral administration), BSO plus auranofin, or a combination of BSO, auranofin, and trigonelline (n = 10 each). The tumor size and body weight of each mouse were measured twice per week, and volume was calculated as (length × width2)/2. The mice were sacrificed on day 25, and the tumors were isolated and analyzed by cellular GSH measurement. Arbitrary fluorescence units were compared between the differently treated tumors. The apoptotic cells in the tumors were counted in a blind manner in 10 randomly selected high-power fields after an in situ terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay (Promega, Fitchburg, WI). Two-tailed Mann–Whitney U tests were used to compare statistical differences between the treatment groups.
Footnotes
Acknowledgments
This study was supported by a grant (no. 2015R1A2A1A15054540) from the Basic Science Research Program through the National Research Foundation of Korea (NRF), Ministry of Science, ICT, and Future Planning, and a grant (no. HI15C2920) from the Korean Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), Ministry of Health and Welfare, Seoul, Republic of Korea (J-L. Roh).
Author Disclosure Statement
The authors declare no conflicts of interest exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.