
Abstract
Aims:
Recent studies have shown that cigarette smoke (CS)-induced oxidative stress impairs autophagy, resulting in aggresome-formation that correlates with severity of chronic obstructive pulmonary disease (COPD)-emphysema, although the specific step in autophagy pathway that is impaired is unknown. Hence, in this study, we aimed to evaluate the role of master autophagy transcription factor EB (TFEB) in CS-induced COPD-emphysema pathogenesis.
Results:
We first observed that TFEB accumulates in perinuclear spaces as aggresome-bodies in COPD lung tissues of tobacco smokers and severe emphysema subjects, compared with non-emphysema or nonsmoker controls. Next, Beas2b cells and C57BL/6 mice were exposed to either cigarette smoke extract (CSE) or subchronic-CS (sc-CS), followed by treatment with potent TFEB-inducing drug, gemfibrozil (GEM, or fisetin as an alternate), to experimentally verify the role of TFEB in COPD. Our in vitro results indicate that GEM/fisetin-mediated TFEB induction significantly (p < 0.05) decreases CSE-induced autophagy-impairment (Ub/LC3B reporter and autophagy flux assay) and resulting aggresome-formation (Ub/p62 coexpression/accumulation; immunoblotting and staining) by controlling reactive oxygen species (ROS) activity. Intriguingly, we observed that CS induces TFEB accumulation in the insoluble protein fractions of Beas2b cells, which shows a partial rescue with GEM treatment. Moreover, TFEB knockdown induces oxidative stress, autophagy-impairment, and senescence, which can all be mitigated by GEM-mediated TFEB induction. Finally, in vivo studies were used to verify that CS-induced autophagy-impairment (increased Ub, p62, and valosin-containing protein in the insoluble protein fractions of lung/cell lysates), inflammation (interleukin-6 [IL-6] levels in bronchoalveolar lavage fluid and iNOS expression in lung sections), apoptosis (caspase-3/7), and resulting emphysema (hematoxylin and eosin [H&E]) can be controlled by GEM-mediated TFEB induction (p < 0.05).
Innovation:
CS exposure impairs autophagy in COPD-emphysema by inducing perinuclear localization of master autophagy regulator, TFEB, to aggresome-bodies.
Conclusion:
TFEB-inducing drug(s) can control CS-induced TFEB/autophagy-impairment and COPD-emphysema pathogenesis. Antioxid. Redox Signal. 27, 150–167.
Introduction
E
Cigarette smoke-induced autophagy-impairment mediates chronic obstructive pulmonary disease (COPD)–emphysema progression, but underlying mechanisms are unclear. Our study identifies cigarette smoke-induced accumulation of transcription factor EB (TFEB) in perinuclear aggresome-bodies as a novel mechanism for TFEB/autophagy-impairment. Moreover, our data demonstrate the potential of prognosis-based intervention strategy for COPD-emphysema.
The search for transcription factors that govern the expression of key autophagy-related genes led to identification of transcription factor EB (TFEB), a master regulator of important genes that govern lysosomal biogenesis and autophagy (35, 48, 49). The activation of TFEB is regulated by mammalian target of rapamycin protein kinase complex (mTORC1), which under normal cellular environment, phosphorylates TFEB at the lysosomal surface leading to binding of YWHA/14-3-3 proteins and cytosolic retention of TFEB (28, 33, 45, 50). Under conditions of starvation or alteration of lysosomal functions, TFEB translocates to the nucleus, where it enhances transcription of numerous genes involved in autophagy and lysosomal biogenesis (33, 39, 50). Several reports highlight the crucial role of TFEB in age-related neurodegenerative diseases, such as Parkinson's, Huntington's, and Alzheimer's, all of which involve autophagy-impairment and accumulation of disordered or aggregated proteins (34, 56, 60). Moreover, genetic or pharmacological induction of TFEB has been reported to benefit in several of these pathological conditions caused by autophagy-impairment, which includes lysosomal storage disorders, Alzheimer's disease (AD), and α1-antitrypsin (α1-AT) deficiency. (18, 26, 43, 44, 53).
Hence, based on our recent studies identifying tobacco smoke exposure and/or aging-mediated autophagy-impairment and resulting aggresome-formation as a prognostic indicator of COPD-emphysema severity (5, 37, 51, 55, 58), we wanted to evaluate the pharmacological effects of TFEB-mediated autophagy induction in CS-induced COPD-emphysema (using human/murine lung tissues) to develop a prognosis-based intervention strategy. We first tested the therapeutic potential of a known TFEB inducer, gemfibrozil (GEM), which is an FDA-approved fibrate drug used for lowering serum lipid levels (22, 46), in controlling CS-induced autophagy-impairment and the resulting alveolar tissue damage (emphysema). GEM treatment is known to not only induce TFEB protein levels and activity in brain cells (46) but also initiates anti-inflammatory and antioxidant responses (46). Hence, apart from its well-known application in controlling hyperlipidemia in coronary heart disease (2, 54), treatment with GEM has proven beneficial in several other pathological conditions, for example, rheumatoid arthritis (12, 46), diabetes (6, 46), and murine experimental autoimmune encephalomyelitis (13, 46). Moreover, it is important to note here that the prominent comorbidities of COPD are coronary heart disease and metabolic syndrome (7), for which GEM is already shown to be clinically beneficial (46, 54). We verified the GEM results with an alternate TFEB-inducing flavonoid drug, fisetin (FIS), which is available over the counter as a dietary antioxidant for brain health.
Thus, we evaluated the impact of GEM (or FIS) treatment on CS-induced oxidative stress, autophagy-impairment, and/or resulting alveolar damage (emphysema) to validate the role of TFEB in COPD-emphysema pathogenesis.
Results
Perinuclear accumulation of TFEB protein in human lungs correlates with severity of COPD-emphysema
We have recently observed that CS or aging induces autophagy-impairment and resulting aggresome-formation that correlates with the severity of COPD-emphysema lung disease, which can serve as a prognostic biomarker (55, 58). Thus, we found that underlying autophagy-impairment is a common mechanism for CS/age-related chronic obstructive lung disease initiation and progression. To identify autophagy mechanism impaired by CS exposure, we wanted to verify the role of the master autophagy regulator, TFEB, in the pathogenesis of COPD-emphysema in murine and human lungs. Hence, we first analyzed changes in expression and localization of TFEB protein in longitudinal lung tissue sections of COPD-emphysema subjects (GOLD 0 to GOLD I–IV) and found that nuclear localization of TFEB (TFEB activation) decreases with severity of emphysema, while its perinuclear localization (in aggresome-bodies) increases with emphysema severity (from GOLD 0 to GOLD IV; Fig. 1A, D, p < 0.05, r = −0.95; white arrows: nuclear TFEB localization and red arrows: perinuclear TFEB accumulation). We observed a significant increase in perinuclear TFEB localization in severe (GOLD III) and very severe (GOLD IV) emphysema subjects compared with non-emphysema (GOLD 0) controls as well as mild or moderate emphysema (GOLD I–II). Furthermore, we demonstrate that smokers (GOLD I–IV) show a significant increase in perinuclear localization of TFEB compared with nonsmokers (Fig. 1A–C, GOLD 0 or GOLD I–IV, p < 0.05). In addition, smokers with greater disease severity (GOLD I–IV) show a comparatively higher increase in TFEB accumulation in aggresome-bodies compared with GOLD 0 subjects (Fig. 1C). Moreover, we verified that TFEB protein accumulates in the insoluble protein fractions (aggresomes) isolated from the lung tissues of moderate and severe COPD-emphysema subjects (GOLD II and III) compared with non-emphysema controls (GOLD 0) (Fig. 1E, F, and Supplementary Fig. S1; Supplementary Data are available online at

Cigarette smoke-induced TFEB accumulation in aggresome-bodies impairs autophagy in COPD-emphysema
Next, we verified our observation that TFEB protein accumulates in the perinuclear spaces as aggresome-bodies using an aggresome-specific dye. Thus, we analyzed the changes in colocalization of TFEB protein with an aggresome dye (PROTEOSTAT® Aggresome detection kit) in longitudinal lung tissue sections of COPD-emphysema subjects (GOLD I–IV) and observed that TFEB-aggresome colocalization (yellow) increases with emphysema severity in COPD subjects (GOLD I–IV) compared with non-emphysema (GOLD 0) controls (Fig. 2A, C; red arrows and insets, r = −0.88). We also found that smokers (GOLD 0 to GOLD IV) have higher TFEB-aggresome colocalization compared with nonsmoker COPD-emphysema or control subjects (Fig. 2B, p < 0.05). The Hoechst (Blue) dye was used to stain the nuclei to visualize the tissue area used for immunostaining (Supplementary Fig. S2). This suggests that CS induces accumulation of TFEB into aggresome-bodies that results in TFEB/autophagy-impairment in COPD-emphysema subjects (4, 55). We also verified this hypothesis by autophagy flux assay (colocalization of green fluorescent protein [GFP] and red fluorescent protein [RFP], LC3B-yellow puncta bodies, red arrows) and observed that CSE-impaired autophagy can be controlled by GEM or FIS-mediated TFEB induction (Fig. 2D, E, p < 0.05). The data suggest the significant role for TFEB in regulating CS-induced autophagy-impairment that required further analysis of TFEB induction strategies in controlling CS-induced oxidative stress, autophagy-impairment, and resulting emphysema.
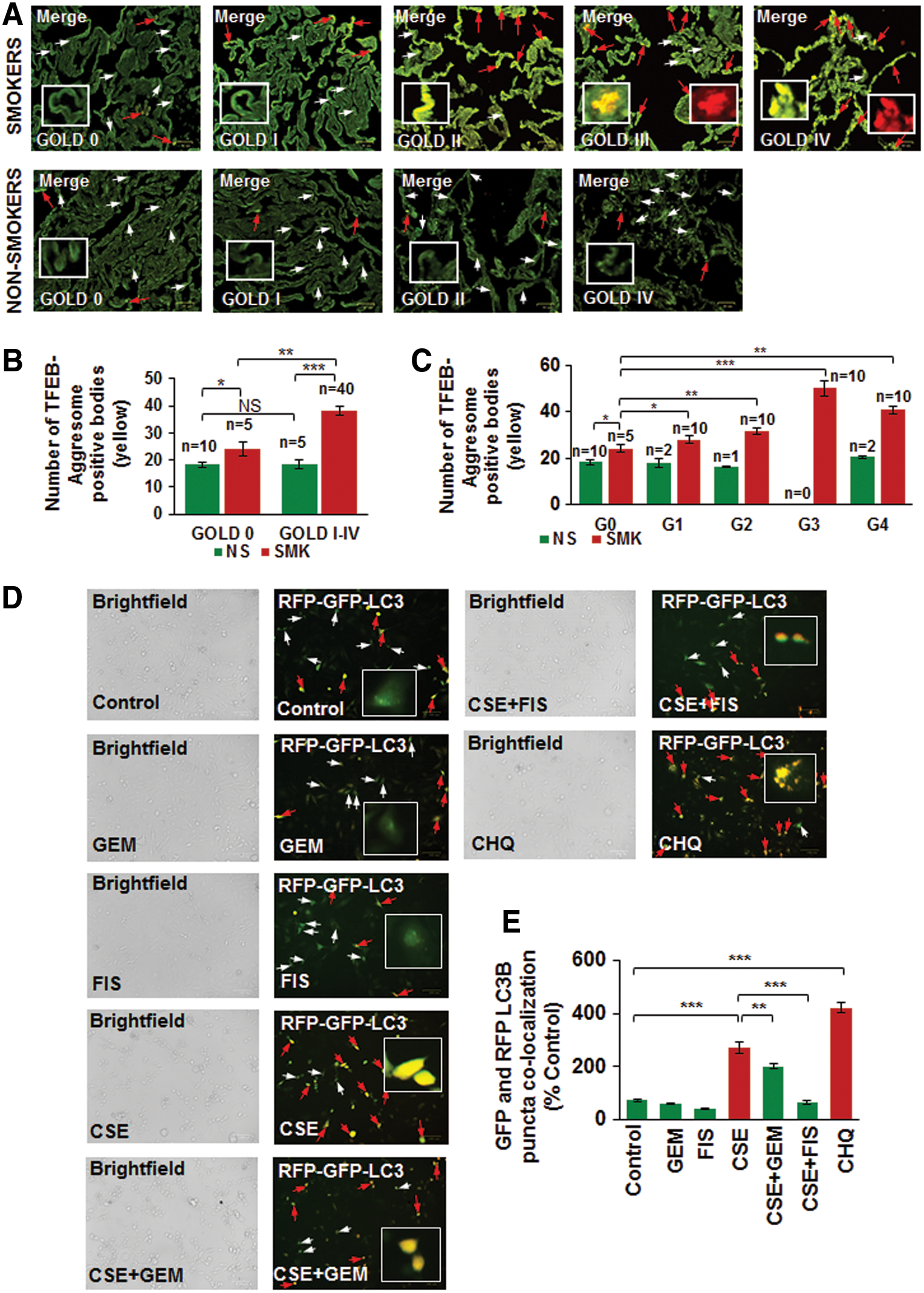
Pharmacological TFEB inducers restore CSE-mediated autophagy-impairment and inhibit aggresome-formation
Several studies have identified the therapeutic benefit of TFEB induction in age-related pathological conditions that involve aggregation of damaged/misfolded proteins (11, 14, 15). We have earlier shown that CS and aging also induce aggregation of misfolded proteins (as aggresome-bodies) due to proteostasis/autophagy-impairment in murine/human lung (58), thus we evaluated here the pharmacological potential of TFEB induction in rescuing CS-induced aggresome-formation and resulting emphysema. First, we compared GEM, a known TFEB inducer (18), with previously tested autophagy inducer drugs (5, 51, 55, 58) and found that GEM treatment significantly (p < 0.01) elevates TFEB protein expression levels in Beas2b cells, compared with cysteamine (CYS) and carbamazepine (CBZ) (Fig. 3A, B, and Supplementary Fig. S3). Next, we found that GEM/FIS-mediated TFEB induction ameliorates CSE-induced autophagy-impairment (Ub-RFP and LC3B-GFP colocalization; aggresome-formation) in human bronchial epithelial cells (Beas2b) (Fig. 3C–F, p < 0.05). Thus, GEM/FIS-mediated TFEB induction is capable of controlling CSE-induced autophagy-impairment and resulting aggresome-formation and/or emphysema progression.
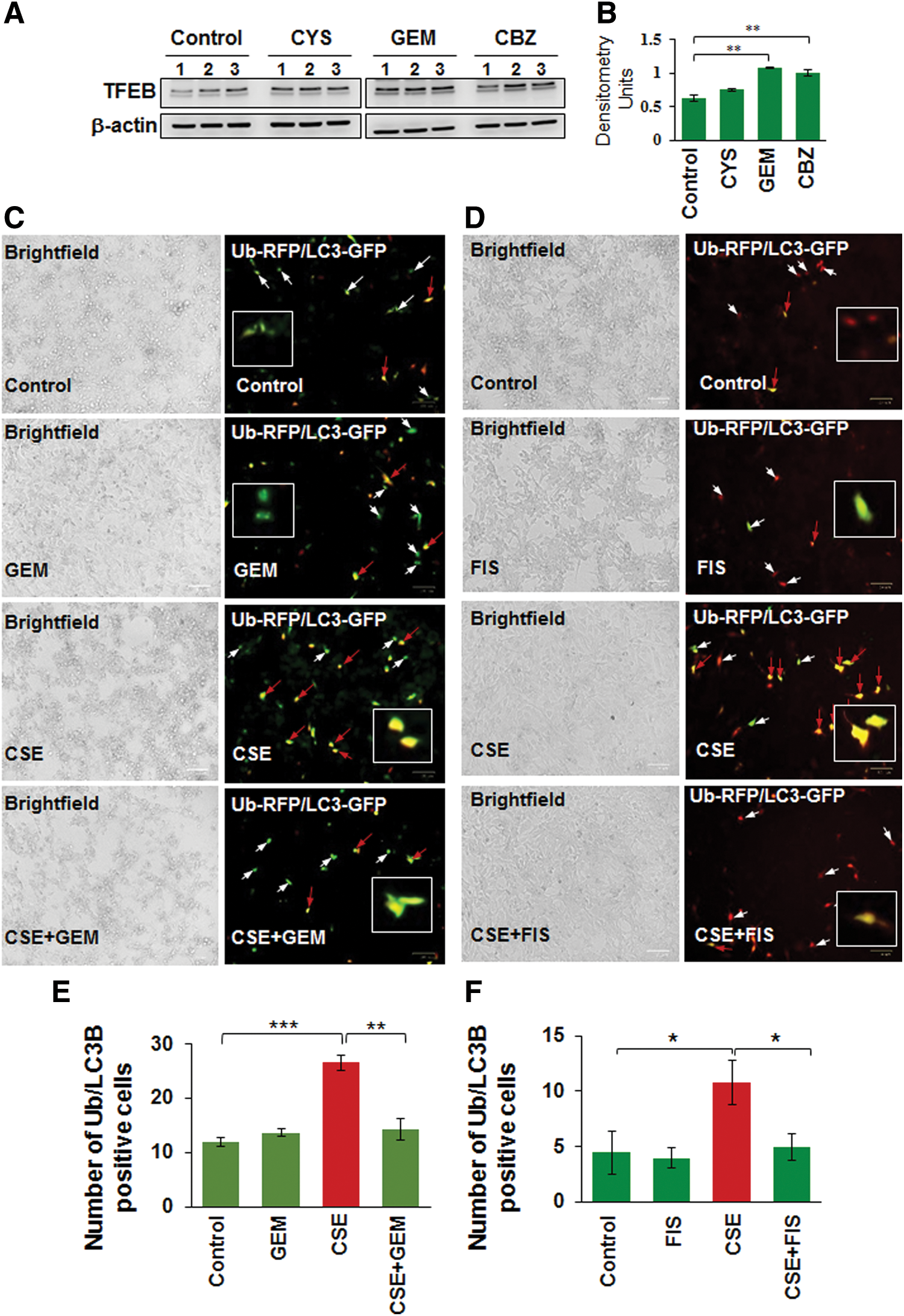
We demonstrate here that CSE-induced ubiquitin (Ub) and/or p62/sequestosome 1 accumulation in the insoluble protein fractions as aggresome-bodies can be controlled by using GEM-mediated TFEB induction (Fig. 4A, B). Furthermore, we demonstrate that the CSE-induced accumulation of ubiquitinated proteins in the insoluble protein fractions of Beas2b cells is significantly (GEM, p < 0.05) reduced by GEM/FIS treatment (Fig. 4C, D, and Supplementary Figs. S4, S5). Moreover, CSE treatment also induced the accumulation of valosin-containing protein (VCP), p62, and/or the master autophagy regulator, TFEB, in the insoluble protein fractions, which can be controlled by GEM/FIS-mediated autophagy induction (Fig. 4C, D, and Supplementary Figs. S4, S5). This suggests the critical role of TFEB in CSE-induced autophagy-impairment as GEM/FIS-mediated TFEB-autophagy induction can control CSE-induced aggresome-formation. The data also verify our findings in COPD subjects (Figs. 1 and 2) and suggest a novel mechanistic role of TFEB in CS-induced COPD-emphysema pathogenesis.

Master autophagy regulator, TFEB, controls CSE-induced oxidative stress, aggresome-formation, cellular viability, and senescence
To further verify the role of TFEB in CS-induced COPD-emphysema pathogenesis, we modulated TFEB expression in Beas2b cells, followed by functional assays. Our data shows that CSE-induced significant increase in ROS activity is controlled by TFEB induction, using GEM/FIS (Fig. 5A, p < 0.05). Moreover, TFEB induction by GEM/FIS also rescues the CSE-mediated decrease in cell viability, as shown by the MTS assay (Fig. 5B, p < 0.05). We and others have recently observed that tobacco smoke/e-cigarette vapor (eCV) exposure leads to cellular senescence (1, 51) via ROS activation that accelerates lung aging and COPD-emphysema pathogenesis (36). We demonstrate here that CSE-mediated increase in number of senescent cells is significantly diminished by treatment with TFEB-inducing drugs, GEM/FIS (Fig. 5C, D, p < 0.05). The present data confirm the importance of TFEB-mediated functional autophagy in regulating CS-induced cellular senescence.

Next, we utilized TFEB knockdown (using TFEB shRNA) in Beas2b cells and observed that even partial inhibition of TFEB protein expression induces significant accumulation of ubiquitinated proteins in the insoluble protein fraction (aggresomes, similar to CSE treatment) that could be restored by treatment with TFEB-inducing drug, GEM (Fig. 5E, F, and Supplementary Fig. S6, p < 0.05). We also observed that TFEB inhibition leads to increased ROS activity and senescence in Beas2b cells, which can be controlled by GEM-mediated TFEB induction (Fig. 5G–I). Thus, our data provide substantial proof-of-concept evidence supporting the crucial role of TFEB in CS-mediated oxidative stress, autophagy-impairment, and aggresome-formation involved in COPD-emphysema pathogenesis.
GEM alleviates CS-mediated autophagy-impairment in murine lungs by rescuing TFEB from aggresome-bodies
Recent studies from our group demonstrate that tobacco smoke/nicotine vapor exposure leads to autophagy-impairment and resulting aggresome-formation in the murine and/or human lungs (5, 55, 58), although the exact mechanism of autophagy-impairment is unknown. Hence, we utilized a similar in vivo approach as previously described (58) to evaluate the effect of GEM-mediated master autophagy regulator induction on CS-induced aggresome-formation. Our data shows that subchronic-CS (sc-CS)-induced accumulation of ubiquitinated proteins (Ub, aggresomes), p62 (impaired autophagy marker), and valosin-containing protein (VCP, a component of the retrograde translocation complex) in the insoluble protein fractions diminished by GEM treatment (Fig. 6A, B, and Supplementary Figs. S7, S8). Moreover, we report a novel observation that sc-CS exposure leads to increased accumulation of TFEB (master autophagy regulator) protein in the insoluble protein fractions as aggresome-bodies. This aggregation may plausibly render TFEB protein unavailable for performing its normal cellular functions (transcriptional regulation of crucial autophagy genes), thus leading to autophagy-impairment.
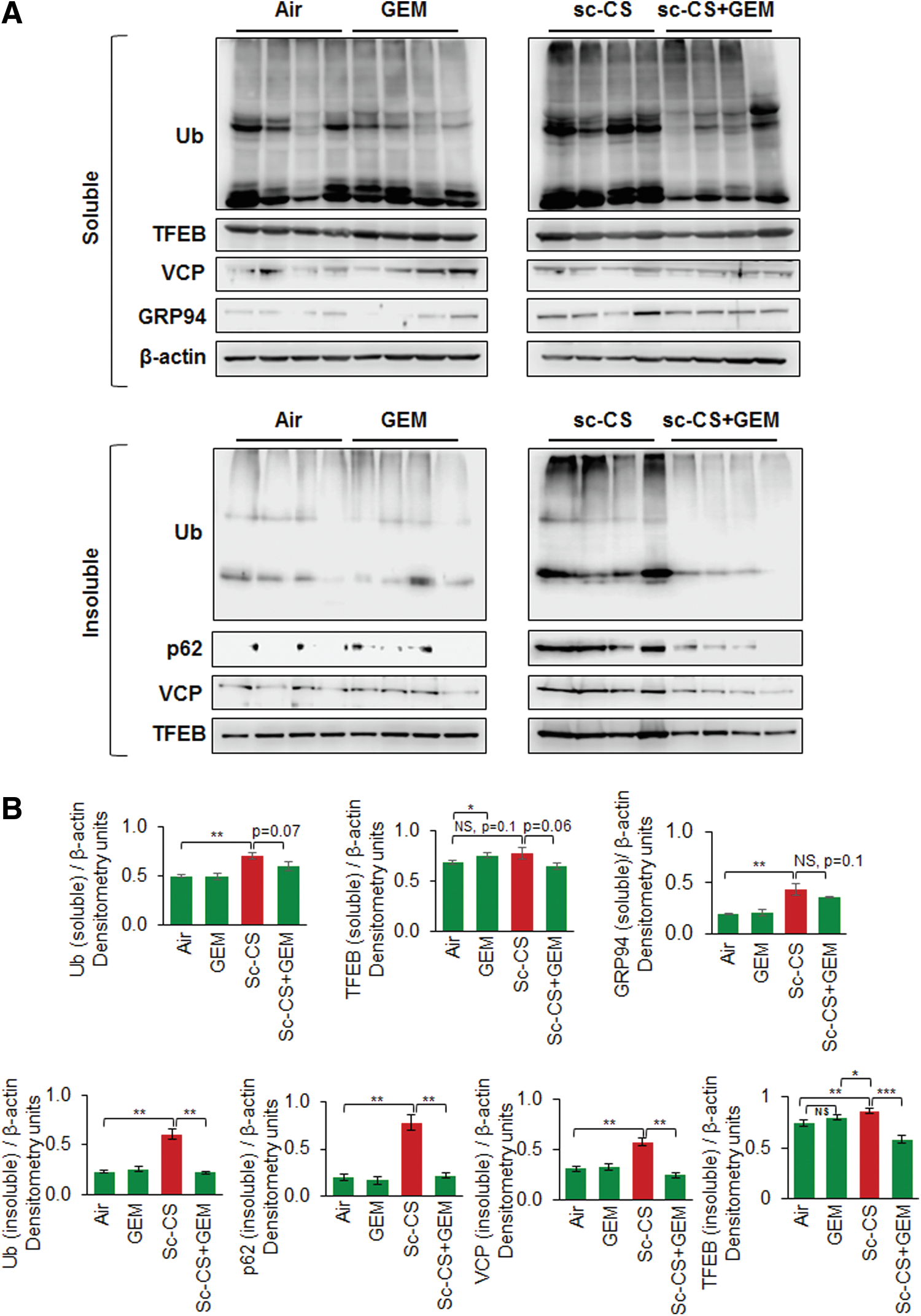
Apart from the impairment of autophagy, CS-induced oxidative stress triggers the unfolded protein response (UPR), which regulates the transcriptional program toward either cell survival or apoptosis, depending on the strength and duration of the oxidative insult (23). Sustained accumulation of misfolded or damaged proteins in the lungs due to perturbed autophagic clearance mechanisms leads to unabated activation of UPR in COPD subjects (23), promoting COPD progression (23). GRP94 is an ER-resident molecular chaperone (ER stress protein) that plays a central role in induction of the UPR (10, 59). Upon sc-CS exposure, we observed a modest induction of GRP94 protein levels (Fig. 6A, soluble fraction) compared with aggresome/impaired autophagy markers (Fig. 6B, Ub, p62, VCP, insoluble protein fractions), suggesting that long-term CS exposure induces significant accumulation of proteins in aggresome-bodies instead of ER. Overall, our data identify CS-induced accumulation of TFEB in aggresome-bodies as a novel mechanism for autophagy-impairment in murine lungs that is rescued by GEM treatment.
GEM-mediated TFEB activation rescues sc-CS-induced inflammation and autophagy-impairment
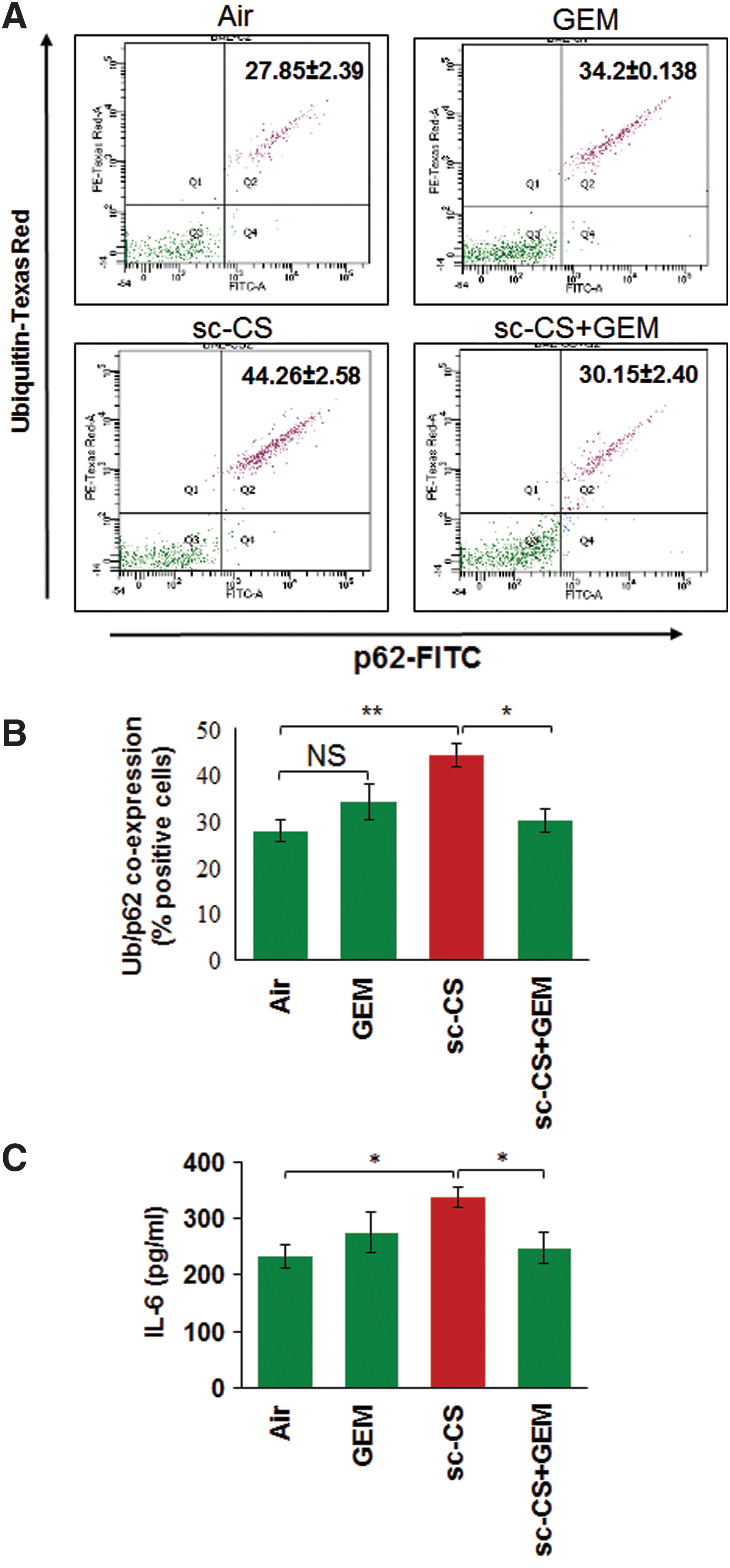
The coexpression/localization of ubiquitinated proteins (Ub) and p62 (impaired autophagy marker) is a marker of autophagy-impairment (5, 51, 58) that can be used to quantify changes in number of aggresome-bodies. Thus, to further ascertain the efficacy of GEM-mediated TFEB activation in controlling sc-CS-induced aggresome-formation, we performed flow cytometry analysis of bronchoalveolar lavage fluid (BALF) cells and show that GEM-mediated TFEB induction significantly (p < 0.05) rescues sc-CS-induced Ub and p62 (impaired autophagy marker) coexpression, indicating restoration of autophagy (Fig. 7A, B). TFEB induction by GEM also has the potential to alter CS-induced inflammatory cell phenotypes (Th1 to Th2 switching) by initiating anti-inflammatory mechanisms such as reduction of IL-6 and other inflammatory cytokines (46). Similarly, we demonstrate here that GEM-mediated TFEB induction significantly (p < 0.05) reduces sc-CS-induced IL-6 levels (Fig. 7C). Since elevated IL-6 levels are associated with CS-induced emphysema (4), reduction in sc-CS-induced IL-6 levels supports our notion that GEM-mediated TFEB induction is a potent therapeutic strategy for controlling COPD-emphysema pathogenesis.
GEM-mediated TFEB/autophagy activation rescues sc-CS-induced alveolar apoptosis and emphysema
In this study, we verified that sc-CS-mediated colocalization of ubiquitinated proteins (Ub) and p62 (impaired autophagy marker) in the murine lung sections (Fig. 8A, merge panel, white arrows) is alleviated by GEM treatment (Fig. 8B). The high-resolution confocal images from these lung sections show a clear increase in perinuclear accumulation of Ub-p62 (yellow) in sc-CS-exposed mice that is reduced by GEM treatment (Fig. 8C). Moreover, we also validate our human lung tissue (COPD subjects) and cell line (Beas2b) data and report that CS exposure induces a significant increase in perinuclear localization of TFEB in aggresome-bodies (Fig. 8D, F, yellow arrows), which was rescued by treatment with its therapeutic inducer, GEM. Finally, GEM-mediated TFEB activation significantly decreases the CS-induced alveolar airspace enlargement (mean alveolar diameter, Lm, Fig. 8E, G) and caspase-3/7 activity (lung cell apoptosis; Fig. 8H), validating that our strategy of TFEB induction has a potential to therapeutically control the progression of COPD-emphysema. Overall, our data suggest that CS induced perinuclear accumulation of TFEB as a novel mechanism for CS-mediated autophagy-impairment and pharmacological induction of TFEB by GEM as a strategy to compensate for nonfunctional aggresome-trapped TFEB protein by inducing TFEB/autophagy for controlling inflammatory-oxidative stress, apoptosis, and resulting COPD-emphysema.

Discussion
Tobacco smoke/nicotine vapor exposure induces inflammatory-oxidative stress via autophagy-impairment-mediated aggresome-formation (5, 51, 58) that is central to controlling the initiation and progression of tobacco smoke-induced obstructive lung pathology (55). Although we and others have successfully tested the efficacy of pharmacological autophagy induction in both in vitro and in vivo models of smoke-induced COPD-emphysema (55, 58), there is a gap in the understanding of the precise mechanism of tobacco smoke-induced autophagy-impairment. The activation of ROS by free radicals and pro-oxidative components of tobacco is obviously a major factor that may hamper cellular homeostasis by causing autophagy/proteostasis impairment (31), although we found that both eCV and nicotine can also induce ROS-mediated autophagy-impairment (5, 51) similar to tobacco smoke. Studies from other groups initially suggested that CS induces autophagy in COPD-emphysema (8, 9, 25, 27), although it was impaired in other lung diseases, which is discussed in detail in the recent review articles (32, 38). Subsequent studies have clarified that although acute CS exposure activates autophagy, repeated chronic exposures lead to autophagy flux impairment. The previous studies used LC3B as a marker to report CS-related autophagy changes, but subsequent studies utilized autophagy flux markers (p62 and ubiquitin accumulation in autophagosomes) and reporters (Premo™ Autophagy Tandem Sensor RFP-GFP-LC3B) to demonstrate that CS-induced autophagy-impairment accelerates COPD-emphysema progression (58). Thus, in this study, we wanted to further investigate the mechanism of CS-ROS-mediated autophagy-impairment.
We first evaluated the effect of CS exposure on transcription factor EB (TFEB; a key master regulator, which controls lysosomal biogenesis and expression of autophagy genes) expression and localization (33, 49) based on our observation in human COPD subject lungs. We initially found that TFEB protein localizes in the perinuclear region as aggresome-bodies in severe COPD-emphysema subjects (Fig. 1A), suggesting that aggresome sequestration of TFEB protein could be a potential mechanism of tobacco smoke-induced autophagy-impairment in COPD. In support of this finding, we demonstrate that significant increase in TFEB-aggresome colocalization statistically correlates with the severity of emphysema and smoking history in COPD-emphysema subjects (Fig. 2A, B). Thus, we used GEM (18) or FIS (26)-mediated TFEB/autophagy induction as an experimental tool with our well-established models of CS-induced COPD-emphysema progression to evaluate its functional role. Using both our in vitro (Beas2b cells) and murine experimental models of COPD-emphysema (CS-exposed mice), we show that CS exposure leads to accumulation of TFEB protein in the perinuclear aggresome-bodies that can be rescued by treatment with GEM-mediated TFEB activation (Figs. 4, 6, and 8D). Moreover, we also demonstrate that TFEB induction by GEM/FIS treatment can control CS-induced inflammation (Fig. 7C), oxidative stress (ROS activation; Fig. 5B), autophagy-impairment, and resulting aggresome-bodies (Figs. 2 –6), thus providing a proof-of-concept evidence that TFEB-mediated autophagy induction could rescue COPD-emphysema pathophysiology (Fig. 9).

Previous studies highlight the critical role of TFEB in the regulation and expression of autophagy genes and its mechanisms are well described (33, 39). Briefly, in normal physiological conditions, TFEB is retained in the cytoplasm by mammalian target of rapamycin (mTORC1)- or mitogen-activated protein kinase 1 (ERK2)- mediated phosphorylation, while in nutrient-deprived or lysosomal stress conditions, TFEB is dephosphorylated by the phosphatase calcineurin, which then promotes nuclear translocation of TFEB, where it regulates the transcription of numerous lysosomal biogenesis and autophagy genes. TFEB directly binds to the coordinated lysosomal expression and regulation (CLEAR) motif (lysosomal biogenesis) and also the promoter regions of various autophagy genes to induce target gene expression (39). The aberrant expression/activity of TFEB is evident in several pathological conditions, especially in age-related degenerative or lysosomal storage disorders, where TFEB impairment leads to accumulation of misfolded or damaged proteins in aggresome-bodies (34, 47). A recent study shows that genetic and chemical activation of TFEB clears the α-synuclein aggregates, which are associated with neurodegenerative diseases such as Parkinson's (24). These α-synuclein aggregates are formed by accumulation of misfolded α-synuclein due to impairment of protein degradation machinery that includes the ubiquitin–proteasome system and autophagy (24). Thus, TFEB-mediated autophagy induction is proposed as a strategy to clear α-synuclein protein aggregation (15, 24). Similarly, in AD, the neuronal accumulation of phosphorylated tau plays an important pathological role (26), and strategies to induce clearance of tau in neurons are developed as a therapeutic strategy to circumvent AD pathology (44, 52). Briefly, a recent report shows the potential of FIS, an organic flavonoid compound, in enhancing clearance of aggregated tau protein via TFEB-mediated autophagy induction (26). In the context of lung diseases, pharmacological or genetic activation of TFEB, the master transcriptional activator of the autophagolysosomal system, is shown to be beneficial in rescuing the spontaneous pulmonary fibrosis developed in the PiZ murine model, wherein accumulation of misfolded α1-antitrypsin Z (ATZ) leads to lung proteinopathy (21). These promising findings led us to test whether pharmacological TFEB induction could rescue CS-induced autophagy-impairment and the ensuing aggresome pathologies initiating COPD-emphysema.
We first chose to use GEM as a pharmacological TFEB inducer based on previous reports (18) and its known antioxidant and anti-inflammatory properties, which may provide additional therapeutic advantage, as we have recently observed with autophagy-inducing antioxidant drug, CYS (5, 51, 58). Additionally, GEM is an FDA-approved lipid-lowering drug and is shown to be clinically beneficial in coronary heart disease and metabolic syndrome (46), two prominent comorbidities of COPD (7), thus warranting its evaluation in CS-induced chronic COPD-emphysema. Our present data demonstrate that GEM-mediated TFEB induction can alleviate CS-induced inflammatory-oxidative stress (IL-6 levels, ROS activation), autophagy-impairment, aggresome-formation (Ub-p62 coexpression/localization), apoptosis (caspase-3/7 activity), and resulting alveolar airspace enlargement (Lm, H&E staining). Mechanistically, our novel data shows that CS triggers accumulation of TFEB in perinuclear aggresome-bodies, thus rendering it unavailable to perform its normal function as a master autophagy activator. It is obvious that similar to other misfolded (ubiquitinated) proteins, CS modulates TFEB protein folding that promotes its accumulation into aggresome-bodies. Thus, we used GEM treatment to induce both the expression of TFEB protein and autophagy that will also allow rescue of additional TFEB protein from aggresome-bodies.
As discussed above, TFEB-inducing strategies have been successfully used to rescue misfolded α-synuclein and tau proteins in neurological diseases (24, 26). In addition, GEM was recently shown to upregulate the expression of tripeptidyl peptidase (TPP1) in brain cells via the PPARα/RXRα pathway in late infantile neuronal lipofuscinosis, a disease caused by accumulation of mutant TPP1 protein (17). Thus, we concluded that GEM treatment similarly rescues misfolded TFEB (and other ubiquitinated proteins) from aggresomes by similar mechanisms, thereby restoring TFEB expression and transcriptional activity for normal functional autophagy response. To further validate the role of TFEB in COPD-emphysema pathogenesis, we utilized an alternative TFEB/autophagy inducer, FIS. In addition to TFEB-mediated autophagy induction (26), FIS is a potent antioxidant activator of the Nrf2 pathway (26, 29) that can alleviate CS-ROS-mediated protein misfolding and aggregation as aggresome-bodies. As a proof of concept, fisetin-mediated TFEB/Nrf2 activation shows more significant inhibition of CSE-induced ROS activity and cellular senescence (Fig. 5B–D) compared with GEM treatment. Thus, FIS is a novel therapeutic agent to potentially rescue CS-mediated oxidative stress and protein aggregation by activating antioxidant response (via Nrf2) and autophagy (via TFEB). We also verified the role of TFEB in regulating CS-induced mechanisms involved in COPD-emphysema by using TFEB shRNA. Our data shows that TFEB inhibition induces ROS activity, ubiquitinated protein accumulation (autophagy-impairment), and cellular senescence (Fig. 5E–H) similar to CS/CSE exposure (Figs. 4C, 5B–D, and 6A). Moreover, these functional effects of TFEB knockdown were rescued by GEM-mediated TFEB induction.
In conclusion, we identify CS-induced accumulation of TFEB in aggresome-bodies as a specific novel mechanism for CS-mediated autophagy-impairment and resulting aggresome-formation and emphysema progression. Moreover, we propose that TFEB-positive aggresome-bodies can serve as prognostic biomarker for tobacco smoke-induced COPD-emphysema that can allow early detection and treatment of chronic obstructive lung diseases with proposed prognosis-based intervention strategy employing TFEB/autophagy-inducing drugs.
Materials and Methods
Human subject samples
Our study protocol was approved by the Institutional Review Board (IRB), Central Michigan University, and Johns Hopkins University as not a human subject research, under exemption no. 4, as subject's lung function data and clinical parameters were obtained from NHLBI Lung Tissue Research Consortium (LTRC, NIH) (4, 58) without disclosing any of the subject's identifiers or demographic information. The clinical severity, sample size, and classification of COPD subjects utilized GOLD stage classification as defined by Global Initiative for Chronic Obstructive Lung Diseases (GOLD). The sample size included lung samples from GOLD 0, n = 15 (smokers: 5 and nonsmokers: 10); GOLD I, n = 12 (smokers: 10, nonsmokers: 2); GOLD II, n = 11 (smokers: 10, nonsmokers: 1); GOLD III, n = 10 (smokers: 10, nonsmokers: 0), and GOLD IV, n = 12 (smokers: 10, nonsmokers: 2) COPD-emphysema subjects. The smokers, who were mostly ex-smokers (as expected for COPD subjects), also included few current smokers; GOLD 0—0, GOLD I—1, GOLD II—2, GOLD III—1, and GOLD IV—0. We have previously reported the demographic information of the non-emphysema and COPD-emphysema subjects used in this study (4). Briefly, non-emphysema or COPD subjects had no other underlying condition (or α1-antitrypsin deficiency), other than emphysema for COPD subjects, although we had one patient in each COPD group (GOLD I–IV) who had his/her first-degree blood relative suffering from chronic bronchitis.
Murine experiments
All animal experiments were performed as per the guidelines of CMU Institutional Animal Care and Use Committee (IACUC). C57BL/6 mice (8 weeks, n = 4) were separated into four experimental groups: (1) room air, (2) GEM, (3) sc-CS, and (4) sc-CS+GEM. The room air or side-stream CS exposures were performed for 2 months, as per our previously described protocol (58). CS was generated by burning 3R4F (0.73 mg nicotine per cigarette) research-grade cigarettes (Tobacco Research Institute, University of Kentucky, Lexington, KY) and mice were exposed to direct whole-body smoke exposure (3 h/day, followed by another 2 h in the same chamber without burning more cigarettes) using TE2 smoking machine (Teague Enterprises). This methodology resulted in an average total particulate matter of 150 mg/m3. The mice were either treated with GEM (40 mg/kg body weight; Sigma) or equal volume PEG vehicle control by intraperitoneal (i.p.) injection on alternate days for 10 days (5 doses) before the termination of experiment. Mice were sacrificed in accordance with our IACUC-approved protocols, and BALF and lung tissues were collected for further analysis by flow cytometry, immunoblotting, and immunostaining.
Cell culture, autophagy flux/reporter, and ROS assays
The human bronchial epithelial cell line, Beas2b, was used in the study as an in vitro model for cigarette smoke extract (CSE) exposure. Standard cell culture conditions were utilized as recently described (5). Briefly, cells were maintained at 37°C, 5% CO2 atmosphere in DMEM/F12 media with 10% fetal growth serum (RMBIO) and 1% PSA (penicillin, streptomycin, and amphotericin; Invitrogen). The CSE was prepared by burning two to three 3R4F (0.73 mg nicotine per cigarette) research-grade cigarettes (Tobacco Research Institute, University of Kentucky, Lexington, KY) and bubbling the smoke directly into the serum-free DMEM/F12 media (20 ml). An OD (320 nm) of 0.74 was considered as 100% CSE and CSE final concentration was adjusted by using the cell culture media. For autophagy reporter assay, cells were transiently cotransfected with ubiquitin-RFP and LC3B-GFP plasmids using Lipofectamine® 2000 (24 h; Invitrogen) and treated with vehicle control (PEG), CSE (5%), and/or GEM (10 μM) for 12 h. The effect of CSE and the TFEB-inducing drugs on functional autophagy was quantified using the Premo Autophagy Tandem Sensor RFP-GFP-LC3B assay kit (Molecular Probes), as recently described (51). Briefly, Beas2b cells were incubated with BacMam reagent for 16 h, followed by treatment with CSE (5%), GEM (10 μM), and/or fisetin (FIS, 25 μM; Indofine Chemical Company) for 12 h. Images for both experiments were captured using the ZOE ™ Fluorescent Cell Imager (Bio-Rad) as we recently described (5, 58). The changes in ROS levels were quantified using the CM-DCFDA ROS indicator dye (Invitrogen) as per the protocol reported recently (5, 51). Briefly, cells were treated as above with CSE (5%), FIS (25 μM), and/or GEM (10 μM) for 12 h and dye was added for 30 min, followed by fluorescence measurement as described before (5). In parallel set of experiments, the effect of TFEB knockdown (using human TFEB-Mission™ shRNA; Sigma and Lipofectamine 2000 transfection reagent; Invitrogen) and/or GEM treatment on ROS activation was quantified using the chloromethyl derivative of 2′,7′-dichlorodihydrofluorescein diacetate (CM-DCFDA) ROS indicator dye.
Immunoblotting, flow cytometry, and ELISA
We used our previously described (5, 58) immunoblotting method to quantify changes in ubiquitinated proteins (Ub), p62 (aggresome marker), VCP (UPR/autophagy mediator), TFEB (master autophagy regulator), GRP94 (ER stress marker), and β-actin in soluble and/or insoluble protein fractions of murine lung or Beas2b cell protein lysates. All antibodies were procured from Santa Cruz Biotechnology (scbt), except β-actin, which was from Sigma. To verify immunoblotting data, flow cytometry analysis was used to quantify the percentage of Ub-p62+ cells (either in human Beas2b or murine BALF cells), using BD FACS Aria and FACS Diva software as we described recently (5, 58). Finally, downstream effects were quantified by measuring changes in levels of IL-6 cytokine in BALF samples using a mouse-IL-6 ELISA kit (eBiosciences), following the manufacturer's instructions as previously described (51, 58).
Immunofluorescence microscopy and lung morphometry
Paraffin-embedded longitudinal lung tissue sections (5 μM) were prepared from all four experimental murine groups (Control vehicle PEG, GEM, sc-CS, and sc-CS+GEM). The lung sections were deparaffinized and immunostained using our previously described protocol (58). Briefly, primary antibodies (1 μg/ml) against ubiquitin (mouse monoclonal, scbt), p62 (rabbit polyclonal, scbt), or TFEB (rabbit polyclonal, scbt), followed by secondary antibodies (1 μg/ml) anti-rabbit CFL488 and/or anti-mouse-Texas Red (Santa Cruz), were used to localize changes in protein expression and/or co/localization. The Hoechst dye was used to stain the nuclei for identifying nuclear/perinuclear localization and expression. Images were captured by either the ZOE Florescent Cell Imager (Bio-Rad) or the Nikon Eclipse TI confocal inverted laser scanning microscope and NIS Elements software using a 60 × /1.42 numerical aperture (NA) oil immersion objective. The sections were also stained with H&E to assess the inflammatory state and morphometric changes. Briefly, changes in mean alveolar diameter, Lm, were quantified by bright-field microscopy as we recently described in detail (58).
We also used paraffin-embedded longitudinal lung sections (5 μM) from normal/non-emphysema and COPD-emphysema subjects for quantifying changes in TFEB expression and localization. Briefly, lung sections from normal control subjects (GOLD 0) and each COPD-emphysema GOLD stage (GOLD I–IV) were immunostained, using a TFEB (1 μg/ml) primary antibody and anti-rabbit CFL-488 (1 μg/ml) secondary antibody. Nuclei were stained using the Hoechst dye, followed by microscopy analysis under ZOE Fluorescent Cell Imager and confocal microscopy, as described above. The PROTEOSTAT Aggresome Detection kit (Enzo Life Sciences) was used to perform the co-immunostaining with TFEB.
Cell proliferation, senescence, and caspase-3/7 assays
Beas2b cells were exposed to CSE (5%) and/or GEM/FIS at the indicated doses for 12 h, and cell proliferation was quantified using the standard MTT/MTS assay kit (Promega) following the manufacturer's protocol, as we recently described (5). The Senescence Cells Histochemical Staining Kit (Sigma) was used to quantify changes in number of senescent cells in the different treatment groups as previously described (5, 51). The caspase-3/7 activity was also quantified in murine lung tissue lysates using the Caspase Glo™ assay kit (Promega) following the manufacturer's protocol.
Statistical analysis
Data are shown as mean ± SEM (or SD, as indicated) of each experimental group. A two-tailed unpaired Student's t-test was performed to determine the significance between the data sets and a p-value of less than 0.05 was considered a significant change. In addition, Pearson's correlation analysis was used to determine the correlation between the two data sets and the correlation coefficient is shown as “r” value. For immunoblotting data, densitometry was used to quantify changes using the ImageJ software (NIH).
Footnotes
Acknowledgments
The authors would like to thank the NHLBI Lung Tissue Research Consortium (LTRC, NIH) for providing human lung tissue sections and Philip Oshel, Director of the Microscopy Core Facility, Central Michigan University, for help with the confocal microscopy experiments. The authors also thank April Ilacqua, FACS student technician, for assistance during the flow cytometry experiments.
Authors' Contributions
Conception and design: N.V.; analysis and interpretation: M.B. and N.V.; experimental contributions: M.B., N.V., N.P., D.S., and K.W.; and drafting of manuscript and editing: M.B. and N.V.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.