
Abstract
Furosemide, a loop diuretic, is used to increase urine output in patients with acute kidney injury (AKI). It remains uncertain whether the benefits of furosemide in AKI outweigh its potential harms. We investigated if furosemide influenced oxidative stress in 30 critically ill patients with AKI by measuring changes in F2-isoprostanes (F2-IsoPs), markers of in vivo oxidative stress, in plasma and urine following intravenous furosemide. Urine F2-IsoPs were higher in sepsis (p = 0.001) and increased in proportion to urine furosemide (p = 0.001). The furosemide-induced increase in urine F2-IsoPs differed depending on AKI severity (p < 0.001) and was greatest in those with the most severe AKI. Furosemide had no effect on plasma F2-IsoPs. We demonstrate for the first time that furosemide increases renal oxidative stress in AKI and find that patients with the most severe AKI—to whom the largest doses are likely to be administered—showed the greatest increase in oxidative stress. These findings lead to the hypothesis that the common practice of administering high-dose furosemide to convert oliguric to nonoliguric AKI may induce harmful oxidative stress in the kidneys, and an adequately powered, randomized controlled trial is required to determine if clinical benefits of this dosing strategy justify its potential harms. Antioxid. Redox Signal. 26, 221–226.
Introduction
T
Furosemide is often used to increase urine output in patients with acute kidney injury (AKI), but it remains uncertain if its benefits outweigh potential harms. We demonstrate that furosemide increases renal oxidative stress in AKI and find that patients with the most severe AKI showed the greatest increase in oxidative stress. These findings have important implications for future research on the management of AKI: the clinical benefits of the common practice of administering high-dose furosemide to convert oliguric to nonoliguric AKI should be confirmed by an adequately powered, randomized controlled trial to justify inducing potentially harmful oxidative stress in the kidneys.
Furosemide, the most commonly used diuretic in AKI, inhibits Na+/K+/2Cl- cotransporters (NKCC1 and NKCC2) in the ascending loop of Henle, resulting in a natriuresis and diuresis. In addition to its diuretic effect, furosemide influences renal hemodynamics and the intrarenal oxygen supply–demand relationship (3). Protective effects of furosemide demonstrated in animal models include a reduction in the metabolic demand of medullary tubular cells due to transporter inhibition; reduced sodium transport (and therefore oxygen) requirements consequent to a reduction in glomerular filtration rate induced by renin and/or prostaglandin release; and increased renal blood flow, all of which act to improve the intrarenal oxygen supply–demand balance and decrease oxidative stress. Although total renal blood flow may be increased, furosemide has also been shown to impair intrarenal autoregulation of blood flow, resulting in a detrimental change in the medullary oxygen supply–demand balance. Human studies performed in patients with AKI have produced mixed results, with some showing benefit and others harm from the use of furosemide (3). The overall effect of furosemide on the pathophysiology of AKI in humans remains unclear—in part, due to the significant heterogeneity of published human AKI studies—but is important to define due to the frequency of its use in this setting.
F2-isoprostanes (F2-IsoPs) are derived from free radical-induced lipid peroxidation of arachidonic acid and are considered highly reliable markers of in vivo oxidative stress (6). Increased levels of F2-IsoPs predict (and may contribute to) AKI in sepsis and cardiac surgery patients (1, 9). If furosemide influences the oxygen supply–demand relationship within the kidney in either a favorable or unfavorable way, then this is likely to be detectable as a change in the level of F2-IsoPs. An increase in renal excretion of F2-IsoPs would suggest that furosemide increases renal oxidative stress and question its use in AKI, whereas a decrease in renal excretion would support the protective effect of furosemide that has been demonstrated in animal models.
Results
Thirty patients were recruited, with a median age of 58 years (interquartile range [IQR] 46–75). No patients were receiving nonsteroidal anti-inflammatory drugs, and only one patient was being treated with an angiotensin-converting enzyme inhibitor. Inspired oxygen concentration was unchanged throughout the study in 21 patients. In the nine remaining patients, the median number of stepwise changes was 1 (IQR 1–2) and the median absolute change in inspired oxygen concentration was 10% (IQR 5–15). All plasma F2-IsoP values are analyzed as concentrations (pM), and urine F2-IsoP values as mass excreted per hour (nmol/h).
Baseline F2-IsoP measurements
Across all patients, there was no correlation between baseline plasma F2-IsoP concentration and urine F2-IsoP excretion (r = 0.004, p = 0.983). After inclusion of creatinine clearance (CrCl) in the linear regression model to control for its significant effect (Δr 2 [improvement in model fit] = 0.660, p < 0.001), there was a weak nonsignificant association (semipartial correlation [sr] = 0.193, p = 0.086) between baseline plasma F2-IsoP concentration and urine F2-IsoP excretion. Baseline urine F2-IsoP excretion, but not plasma F2-IsoPs, weakly correlated with the severity of organ failure as measured by the Sequential Organ Failure Assessment (SOFA) score (urine: r = 0.453, p = 0.012; plasma: r = 0.124, p = 0.514). Neither F2-IsoP measure correlated with systemic inflammation as measured by plasma C-reactive protein concentration (p > 0.1).
In univariate analysis (Table 1), baseline urine F2-IsoP excretion decreased as AKI became more severe as defined by both Acute Kidney Injury Network (AKIN) stage (p = 0.015) and 6-h CrCl (p < 0.001). Similarly, lower baseline urine F2-IsoP excretion was associated with a subsequent requirement for continuous renal replacement therapy (p = 0.015). There was no correlation between baseline plasma F2-IsoPs and any of the factors assessed in univariate analysis, including sepsis or severity of AKI.
All values are presented as median (IQR).
AKIN, Acute Kidney Injury Network; ARDS, acute respiratory distress syndrome; BMI, body–mass index; CrCl, creatinine clearance; CRRT, continuous renal replacement therapy; F2-IsoP, F2-isoprostane; ICU, intensive care unit; IQR, interquartile range.
Effect of furosemide
The final linear mixed model analyzing urine F2-IsoP excretion included the presence of sepsis and CrCl and plasma F2-IsoPs (Table 2). Urine F2-IsoP excretion was significantly increased in the presence of sepsis (p = 0.001) and was significantly lower in patients with CrCl <20 ml/min/1.73 m2 compared with those with CrCl >20 ml/min/1.73 m2 (p < 0.001). Urine furosemide excretion increased urine F2-IsoPs (p = 0.001) and the magnitude of this increase differed depending on CrCl (p < 0.001; Fig. 1). The greatest increase with furosemide occurred in patients with CrCl <20 ml/min/1.73 m2 (p = 0.002 and 0.001 in comparison with CrCl 20–40 and >40 ml/min/1.73 m2, respectively), and the smallest increase occurred in patients with CrCl >40 ml/min/1.73 m2 (p = 0.036 in comparison with CrCl 20–40 ml/min/1.73 m2). There was no statistically significant effect of plasma F2-IsoPs on urine F2-IsoP excretion (p = 0.113).

CI, confidence interval.
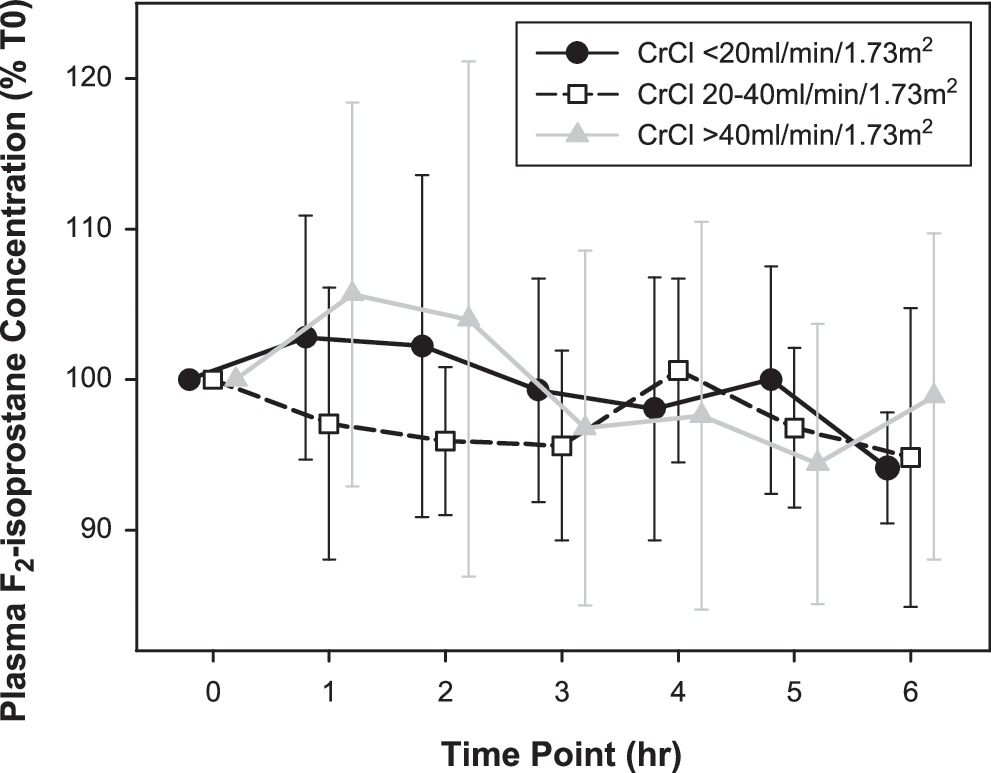
Linear mixed model analysis of plasma F2-IsoPs (Table 3 and Fig. 2) following intravenous (IV) furosemide identified a small decrease over time (p = 0.011; mean T6 concentration 96 ± 3% [standard error of the mean] of baseline). CrCl had no effect on plasma F2-IsoP concentration (p = 0.299). Inclusion of plasma furosemide concentration in the model did not change the statistical significance of included variables and resulted in an inferior model fit [χ2(1) = 4.969, p = 0.026].
Discussion
In this study of critically ill patients with AKI and no history of chronic kidney disease or recent diuretic exposure, we demonstrate for the first time that furosemide increases levels of oxidative stress in the kidney as indicated by the increased urinary excretion of F2-IsoPs. This effect was greatest in patients with the most severe AKI. There was no evidence of an increase in systemic oxidative stress following furosemide.
Urinary furosemide increased renal oxidative stress in all patients, with the greatest increase occurring in those with the most severe AKI. Although furosemide may reduce the metabolic activity of the ascending loop of Henle, an increase in renal oxidative stress could still occur through multiple other mechanisms (4). Renal oxygen demand is highly dependent on solute transport requirements. Inhibition of solute reabsorption in the loop of Henle increases distal solute delivery, shifting the transport burden to regions of the nephron that are metabolically less efficient. As furosemide also inhibits tubuloglomerular feedback, solute transport requirements will be increased by greater nephron blood flow (and therefore filtration) and renin release will be stimulated, which may further increase oxidative stress. Finally, the medullary oxygen supply–demand imbalance may be compromised by the ability of furosemide to alter the distribution of blood flow within the kidney (e.g., medullary vasoconstriction), which may exacerbate any regional hypoperfusion already present due to the AKI.
Our finding that patients with the most severe AKI had the greatest increase in renal oxidative stress following furosemide is somewhat unexpected as this group of patients has the greatest pharmacodynamic limitation (7), but could be explained by these patients having the least reserve available to deal with increased metabolic demand and changes in intrarenal blood flow. For example, tubular sodium reabsorption in AKI is known to be less efficient due to impaired transporter polarization and tight junction integrity, and these changes would likely be most pronounced in those with more severe AKI. The greater sensitivity of macula densa Na-K-Cl cotransporter 2 (NKCC2) to furosemide compared with loop of Henle (2) may also contribute. In patients with mild AKI, furosemide may reach the tubules in high enough concentration to inhibit loop of Henle sodium reabsorption to an extent sufficient to offset its effect on macula densa, resulting in no change in release of renin or tubuloglomerular feedback mediators. Conversely, in severe AKI, the lower concentration of furosemide in the tubules may only be sufficient to inhibit NKCC2 in the macula densa, increasing tubular workload through tubuloglomerular feedback inhibition and stimulating renin secretion.
This study is the largest published investigation of urine F2-IsoP excretion in patients with sepsis. We show for the first time that sepsis increases urine F2-IsoP excretion, consistent with current theories regarding the importance of systemic inflammation and oxidative stress as mediators of septic AKI. The lack of association between sepsis and plasma F2-IsoPs may be due to the unequal group sizes and small total sample size of the present study. Although retained in the final linear mixed model, the effect of plasma F2-IsoPs on urine F2-IsoPs did not reach statistical significance. A positive association would be expected as both reflect systemic oxidative stress and plasma F2-IsoPs are filtered at the glomerulus. Additionally, F2-IsoPs are physiologically active as potent vasoconstrictors and may contribute to AKI (6). A small statistically significant decrease in plasma F2-IsoPs was observed over the duration of the study; however, the clinical relevance of this finding requires further investigation as it was unrelated to plasma furosemide concentration.
The increase in renal oxidative stress following furosemide has important implications for future research on the management of AKI. An adequately powered, randomized controlled trial evaluating the common practice of administering high-dose furosemide to patients with AKI is required to justify inducing potentially harmful renal oxidative stress as furosemide has not been shown to improve patient-centered outcomes. This is especially important for patients with severe AKI in whom we found the greatest increase in urine F2-IsoPs per unit urine furosemide excretion as high doses of furosemide are often required to induce diuresis as a result of pharmacodynamic and pharmacokinetic limitations to the diuretic effect of furosemide in severe AKI, implying that to achieve an effective diuresis, higher urine furosemide concentrations may be required (7). The increase in renal oxidative stress following furosemide cautions against using furosemide in an attempt to improve or prevent progression of AKI and is consistent with AKI clinical practice guideline recommendations (5). In view of the relationship between urine furosemide and renal oxidative stress, furosemide infusions may be preferable to repeated bolus doses as they have been shown to achieve greater diuresis with lower total furosemide excretion (3), although this requires further investigation.
A number of limitations of the present study must be acknowledged. Although an increase in urine F2-IsoP excretion was observed and indicates an increase in renal oxidative stress, this study was not designed to assess the effects of this oxidative stress on renal or patient-centered outcomes. However, F2-IsoPs are potent renal vasoconstrictors (6) and levels correlate with the development of AKI in sepsis and cardiac surgery patients (1, 9). The absence of a control group in this study precludes comment on whether the increase in urine F2-IsoPs is a normal or pathological response to furosemide.
In summary, we have demonstrated the effect of a single IV dose of furosemide to increase renal oxidative stress in patients with AKI. This effect was greatest in patients with the most severe AKI to whom the largest doses of furosemide are often administered. Clinicians must be cognizant of these findings when treating patients with AKI, although further investigation into patient-centered outcomes associated with the increase in oxidative stress is warranted.
Notes
This study was performed as a substudy of a recently published investigation into furosemide pharmacokinetics/pharmacodynamics in AKI (7). Briefly, 30 critically ill patients with AKI according to the AKIN criteria (5) were prospectively recruited to an observational study investigating the pharmacokinetics and pharmacodynamics of furosemide in AKI. Patients with pre-existing chronic kidney disease and patients currently receiving dialysis were excluded. A single dose (chosen by the treating intensivist) of furosemide was administered as an IV bolus, and the diuretic response (hourly urine output) measured using an indwelling urinary catheter. Blood and urine samples were collected immediately before furosemide and hourly over the subsequent 6 h (T1–6). Six-hour CrCl was measured (using T6 plasma creatinine concentration and the urine produced between T0 [immediately before furosemide] and T6) as a marker of renal function. A full description of study methodology, including detailed inclusion/exclusion criteria, has been reported (7). Ethics approval was obtained from the Royal Perth Hospital Human Research Ethics Committee (EC 2011/130), and written informed consent obtained from all patients or their next of kin.
Laboratory methods and specimen collection
All blood samples were collected from arterial catheters. Urine samples were aspirated from the proximal access port of the indwelling urinary catheter. For measurement of plasma electrolytes, urea, creatinine, and C-reactive protein, blood was collected into specimen tubes containing lithium heparin and analyzed within 1 h (PathWest Laboratory, Royal Perth Hospital). Urine creatinine concentration was measured (at T0 and T6) within 1 h of collection (PathWest Laboratory, Royal Perth Hospital). Arterial blood for measurement of furosemide concentration was collected into specimen tubes containing lithium heparin. For measurement of plasma F2-IsoPs, arterial blood was collected into cold (4°C) specimen tubes containing ethylenediaminetetraacetic acid (EDTA), reduced glutathione, and butylated hydroxytoluene. Samples were centrifuged (3600 × g for 8 min at 4°C), and the plasma stored at −80°C. Urine samples were stored at −80°C until time of analysis. We used high-performance liquid chromatography–mass spectrometry with a Kinetex XB-C18 column (Phenomenex) on a Shimadzu Nexera X2 system coupled to a Shimadzu LCMS-8030 triple quadrupole mass spectrometer (Shimadzu, Victoria, Australia) to measure furosemide concentrations in the urine and plasma specimens. Plasma and urinary F2-IsoP concentrations were measured by gas chromatography–mass spectrometry (Agilent 6890 N gas chromatograph coupled to an Agilent 5975B mass spectrometer; Agilent) using electron capture negative ionization and selective ion monitoring, as previously described (8).
Statistical analysis
Correlation between baseline plasma and urine F2-IsoPs was assessed using Pearson's correlation coefficient after log transformation to a normal distribution. As visual inspection of the scatterplot suggested an effect of CrCl, this was tested using linear regression of plasma on urine F2-IsoPs with block-wise forced entry of CrCl, followed by the interaction term “creatinine clearance X plasma F2-IsoP.” This interaction term was not statistically significant and therefore the semipartial correlation was calculated between urine and plasma F2-IsoPs controlling for CrCl. The correlation between urine/plasma F2-IsoPs and C-reactive protein/SOFA score was calculated using Pearson's correlation coefficient after log transformation to a normal distribution. Univariate analysis of baseline variables was performed using Mann–Whitney U and Kruskal–Wallis tests.
Linear mixed model analysis was used to identify factors influencing plasma and urine F2-IsoPs (concentration and total excreted, respectively) over time. Separate models were constructed for each site of measurement, and variables were log transformed to a normal distribution before analysis if required. Variables included in the initial (full) models were selected based on theoretical plausibility, and backward variable selection (using Bayesian Information Criterion) was performed to produce the reduced (parsimonious) models presented in this article. Urine output was not included in the linear mixed model to avoid colinearity due to the strong correlation with urine furosemide excretion and CrCl. Plasma furosemide concentrations were included in the linear mixed model only as a sensitivity analysis as they were not significantly different between the study patients in our furosemide pharmacokinetics/pharmacodynamics analysis (7). Model specification allowed for random effects of intercept, CrCl, and subject, as well as appropriate repeated measures covariance structures. Measured CrCl values were categorized as <20 ml/min/1.73 m2, 20–40 ml/min/1.73 m2, and >40 ml/min/1.73 m2 for ease of interpretation. We did not normalize urine F2-IsoP excretion to urine volume or urine creatinine concentration as the former is influenced by furosemide's diuretic effect and the latter may change at a different rate or to a different extent than F2-IsoPs in the setting of AKI.
Footnotes
Acknowledgments
The authors would like to thank Ms. Jenny Chamberlain for her advice on the data collection process, Ms. Linda Gregory for her assistance with the laboratory resources, and Dr. Steven Wallis for measuring the plasma and urine furosemide concentrations for this study. This study was funded by an Australian and New Zealand College of Anaesthetists project grant (13/004) and the New Independent Researcher Infrastructure Support (NIRIS) award from the Western Australia Department of Health (2014). K.M.H. and T.B.C. are funded by Raine Medical Research Foundation and Western Australia Department of Health through the Raine Clinical Research Fellowships. J.A.R. and T.A.M. are supported by National Health and Medical Research Council of Australia Fellowships.