
Abstract
Aims:
Anemia of inflammation (AI), the second prevalent anemia, is associated with worse prognosis and increased mortality in numerous chronic diseases. We recently reported that the gasotransmitter hydrogen sulfide (H2S) suppressed the inflammatory activation of signal transducer and activator of transcription 3 (STAT3) and hepcidin, the critical mediators of AI. Adenosine 5′-monophosphate-activated protein kinase (AMPK) is a novel inflammatory regulator and might be activated by H2S. In this study, we determined whether AMPK played a role in H2S-mediated anti-inflammatory response in AI and evaluated the therapeutic potential of AMPK against AI by pharmacological and clinical approaches.
Results:
We showed that AMPK mediated the inhibition of STAT3, hepcidin, and AI by H2S during inflammation. Moreover, pharmacological and genetic activation of AMPK ameliorated hepcidin production, corrected iron dysregulation, and relieved hypoferremia and anemia in both acute and chronic inflammation models in mice. Mechanistic studies indicated that AMPK suppressed STAT3/hepcidin activation by promoting proteasome-mediated Janus kinase 2 (JAK2) degradation, which was dependent on the intact function of suppressor of cytokine signaling 1 (SOCS1) and increased interactions between SOCS1 and JAK2. Most importantly, the AMPK activator metformin was associated with decreased serum hepcidin content and anemia morbidity in Chinese type 2 diabetes mellitus patients.
Innovation:
This is the first study to demonstrate the inhibition of inflammatory hepcidin and AI by AMPK-induced JAK2 degradation. Our work uncovered AMPK as a novel therapeutic target, and metformin as a potential therapy against AI.
Conclusion:
The present work demonstrated that AMPK mediated the therapeutic effects of H2S and relieved AI by promoting SOCS1-mediated JAK2 degradation. Antioxid. Redox Signal. 27, 251–268.
Introduction
A
Conventional remedies for anemia of inflammation (AI) have raised concerns regarding safety and effectiveness, calling for alternative therapies. Using both in vitro and in vivo models, we demonstrated that AMPK relieved iron dysregulation and anemic symptoms in AI mice by promoting Janus kinase 2 (JAK2) degradation and, subsequently, inhibiting hepatic signal transducer and activator of transcription 3/hepcidin activation. Moreover, metformin, a typical AMPK activator, was associated with lower serum hepcidin and reduced anemia morbidity in type 2 diabetes mellitus patients. These findings reveal AMPK as a novel therapeutic target, and metformin as a potential drug against AI.
AI is characterized by disturbance of iron homeostasis and dysfunction of erythroid system (47), in which iron dysregulation is one of the hallmarks. Suppressed dietary absorption and increased retention of iron within the reticuloendothelial system limit iron availability for erythroid progenitor cells, leading to restricted erythropoiesis (18). Therefore, restoration of the iron balance is proposed as an effective approach in AI treatment.
Hepcidin, a liver-derived iron-regulating peptide, is a well-established critical player in the development of AI (17). During inflammation, hepcidin is induced by interleukin-6 (IL-6)-mediated activation of the Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) signaling pathway. Hepcidin reduces intestinal iron absorption and promotes the diversion of circulating iron to the storage sites by promoting the degradation of ferroportin, the main cellular iron exporter (54, 75). Increased urinary hepcidin excretion is observed in AI patients (49), whereas inhibition of hepcidin by Il-6 or Stat3 knockout in mice protects against AI (35, 60). These findings suggest that inhibition of hepcidin in inflammation may be an effective strategy to resolve iron disorders and AI.
Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) is a conserved metabolic sensor that monitors intracellular AMP/adenosine triphosphate (ATP) ratio (27, 39). Due to its vital role in energy regulation, AMPK was extensively studied in metabolic disorders. Importantly, metformin, a potent AMPK activator, is widely prescribed as first-line therapy in type 2 diabetes mellitus (T2DM) (20). In addition, recent studies reported that AMPK might also act as a novel suppressor of inflammation (52). Notwithstanding these findings, there remains a knowledge gap on AMPK/metformin, hepcidin, and AI.
Hydrogen sulfide (H2S), the third bio-active gasotransmitter after nitric oxide and carbon monoxide, is produced endogenously and plays a prominent role in inflammatory responses (37). We recently demonstrated that H2S inhibited hepcidin expression and hypoferremia in an acute lipopolysaccharide (LPS) model (76), as previous studies showed that H2S activated the AMPK signaling pathway in the central nervous system (45). These findings prompted us to assess the role of AMPK in H2S-mediated inhibition of hepcidin and AI by using pharmacological and clinical approaches with the ultimate goal of evaluating the therapeutic potential of AMPK activation in AI.
In the present study, we found that AMPK contributed to the inhibition of hepcidin and alleviation of AI by H2S by promoting suppressor of cytokine signaling 1 (SOCS1)-mediated JAK2 degradation. Moreover, in in vivo mouse models and by retrospective analysis of serum samples from T2DM patients, we identified AMPK as a novel therapeutic target and metformin as a potential remedy against AI. The graphic abstract is presented in Figure 1.
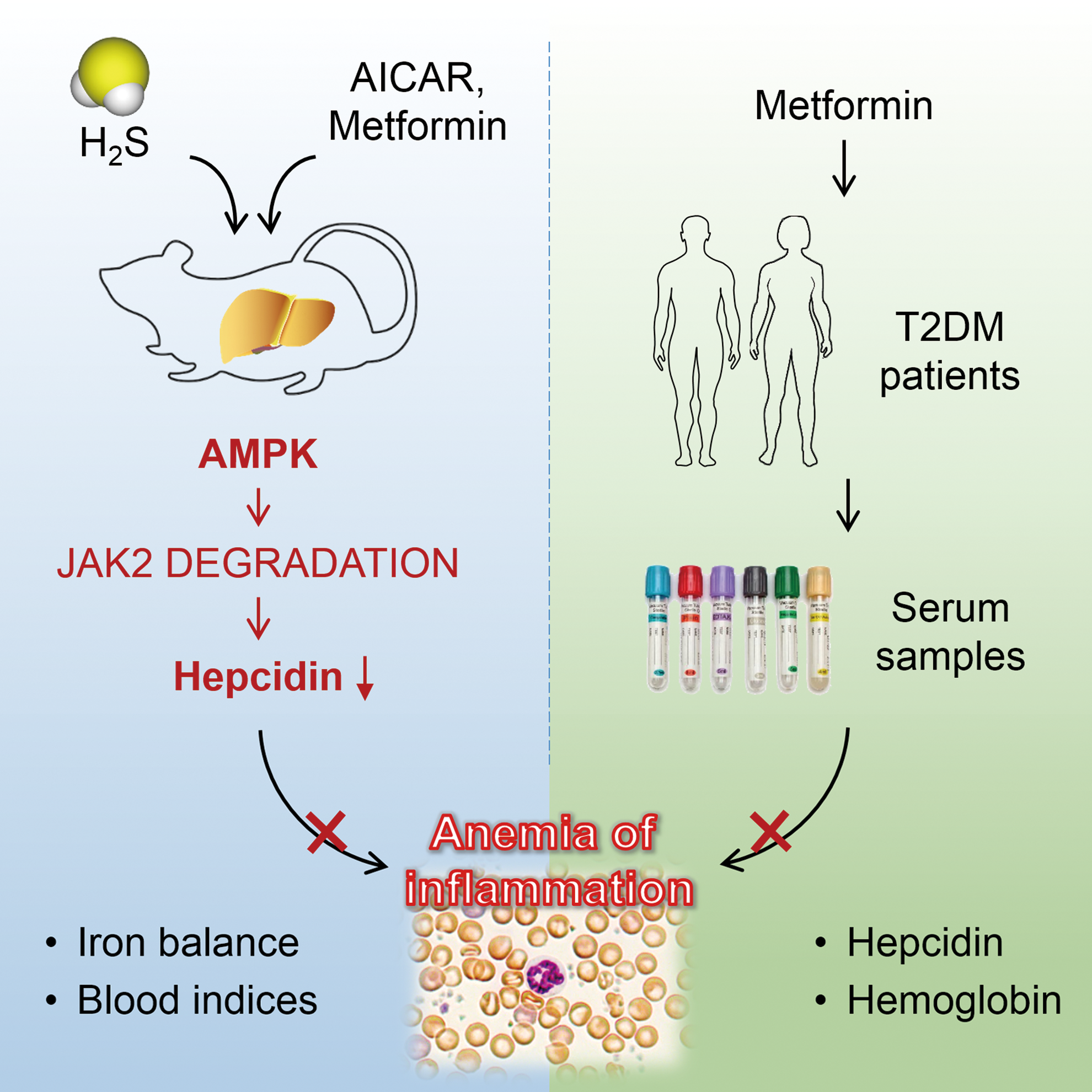
Results
H2S-mediated inhibition of STAT3 and hepcidin is dependent on AMPK activation
Based on our recent finding that H2S suppressed the inflammatory activation of STAT3 and hepcidin, we sought to identify H2S-mediated intracellular signal transduction pathways in primary mouse hepatocytes pretreated with sodium hydrosulfide (NaHS), an exogenous H2S donor, followed by IL-6 treatment using an antibody array. As illustrated in Figure 2A, H2S led to a significant increase in AMPK phosphorylation while inhibiting the activation of many transcriptional factors controlled by IL-6 included in the array.

To investigate the role of AMPK in our system, we pretreated HepG2 hepatoma cells (Fig. 2B and Supplementary Fig. S1A; Supplementary Data are available online at
We next assessed the involvement of AMPK by gene silencing and pharmacological inhibition by using Huh7 cells that exhibited a higher response than HepG2 cells in the luciferase activity assay. As shown in Figure 2G, H and Supplementary Figure S1E, knockdown of AMPK by two specific siRNAs successfully abolished the suppression of STAT3 and hepcidin by H2S. As hepcidin was previously shown to be suppressed by compound C, the classic AMPK inhibitor, by suppressing the bone morphogenetic protein pathway in an AMPK-independent manner (53), instead we introduced a lentiviral vector expressing dominant negative AMPK (DN-AMPK) and demonstrated the abrogation of H2S-mediated STAT3 and hepcidin inhibition (Fig. 2I, J and Supplementary Fig. S1F). These data suggested that AMPK contributed to the inhibition of inflammatory STAT3 and hepcidin by H2S.
AMPK mediated the treatment effects of H2S on AI
Next, we examined the effects of H2S on AI and the interplay between AMPK and H2S. Constitutively active AMPK (CA-AMPK), DN-AMPK, or empty lentiviral vectors were i.v. injected to C57BL/6 mice, at 2 × 107 lentiviral particles/mouse. One week later, mice were treated with weekly subcutaneous injections of turpentine (100 μl/20 g weight) for 4 weeks to induce a chronic non-infectious model of AI. Starting in the second week of turpentine treatment, NaHS (6 mg/kg) was i.p. administered twice a week, according to a previous report (38). Mice were sacrificed after anesthesia 1 week after the final injection of turpentine. The workflow of the chronic AI model is presented in Figure 3A.
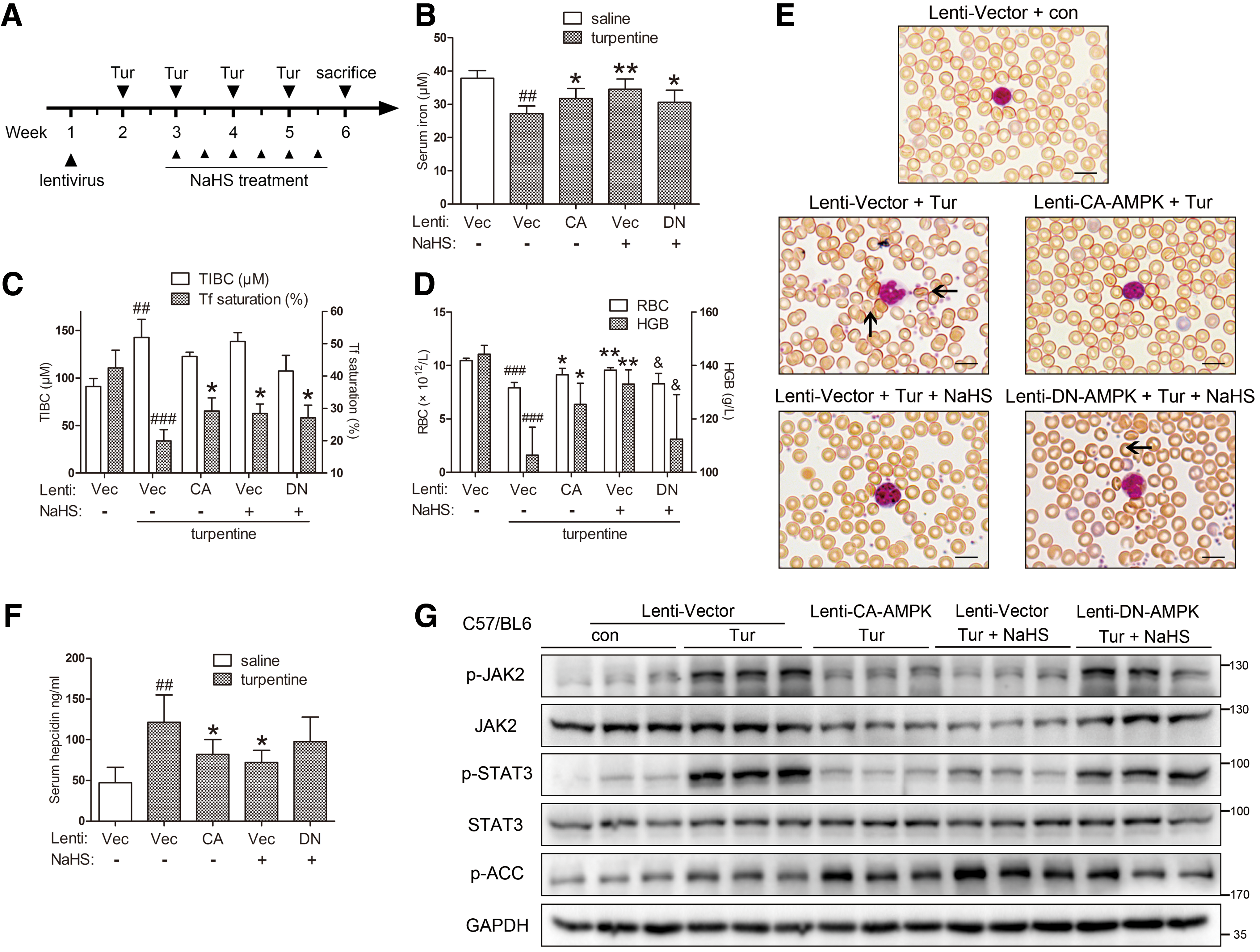
Serum iron levels, as well as transferrin (Tf) saturation, were restored in mice treated by NaHS and those infected with CA-AMPK, though no significant change was observed with DN-AMPK (Fig. 3B, C). To better evaluate the parameters of circulating erythrocytes, a complete blood count was conducted. As demonstrated in Supplementary Table S1, the chronic AI model using turpentine shared the normocytic normochromic nature observed in clinical AI (34, 56). Both NaHS treatment and CA-AMPK infection successfully improved erythrocyte numbers and hemoglobin content (Fig. 3D). Moreover, the effects of NaHS were reduced in DN-AMPK-infected mice. These results were confirmed by blood smear analysis with Wright-Giemsa staining, which demonstrated reduced erythrocyte membrane irregularity in NaHS-treated and CA-AMPK-infected mice (Fig. 3E). Immunoblots and enzyme-linked immunosorbent assay (ELISA) showed that CA-AMPK infection and NaHS treatment markedly ameliorated JAK2/STAT3 signaling and downstream hepcidin production, as opposed to DN-AMPK (Fig. 3F, G). Intriguingly, total protein levels of JAK2, the critical signal transducer within the IL-6/STAT3 pathway, were also decreased. These data not only demonstrated that H2S relieved AI in an AMPK-dependent manner but also suggested that direct AMPK activation might elicit similar therapeutic effects.
AMPK activation ameliorates IL-6-induced hepcidin levels both in vitro and in vivo
We next examined the effects of pharmacological and genetic activation of AMPK on inflammatory hepcidin. As a potent AMPK activator, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR, 0.5/2 mM) (73) dramatically suppressed hepcidin promoter activity and the transcriptional function of STAT3 as assessed by a dual luciferase reporter assay, in parallel with reduced phosphorylation and nuclear translocation of STAT3 (Fig. 4A–C). Similar results were obtained with the CA-AMPK construct (Fig. 4D and Supplementary Fig. S2A).

Our hypothesis was then tested in vivo. C57BL/6 mice were pretreated with one of the two AMPK activators, AICAR (i.p., 250 & 500 mg/kg) (80) and metformin (i.g., 250 mg/kg) (22), 1 h before a 3-h challenge with IL-6 (i.p., 3 μg/20 g weight). The dose of metformin used here was converted from a human dose of 1500 mg/75 kg by following a translation formula previously described (57). The mice were sacrificed as stated earlier (54). As illustrated in Figure 4E and Supplementary Figure S2B, both AICAR and metformin suppressed hepatic hepcidin mRNA (Hamp) and serum hepcidin levels evoked by IL-6. As expected, serum iron levels were restored and splenic iron content was reduced in AICAR- and metformin-treated mice (Fig. 4F, G). Immunoblot assays indicated that both AICAR and metformin decreased hepatic phospho-STAT3 (p-STAT3) levels induced by IL-6 (Fig. 4H and Supplementary Fig. S2C–E). Taken together, these results suggested that AMPK activation ameliorated inflammatory STAT3 and hepcidin activation both in vitro and in vivo.
AMPK activators improve turpentine-induced AI in vivo
To support our hypothesis that AMPK was a therapeutic target against AI, the effects of AICAR (i.p., 500 mg/kg) and metformin (i.g., 250 mg/kg) were evaluated in turpentine-treated C57BL/6 mice as described in Figure 5A.

Both AMPK activators successfully increased serum iron levels and transferrin saturation that were compromised in turpentine-treated mice (Fig. 5B, C). Consistently, hematological indices and erythrocyte morphology were also improved (Fig. 5D, E). In addition, inflammatory splenomegaly induced by turpentine was relieved by AMPK activation (Fig. 5F). In agreement with Perl's Prussian blue staining, splenic iron accumulation was ameliorated as evidenced by non-heme iron analysis (Fig. 5G, H). Similar to that observed in the acute IL-6 model, serum hepcidin was reduced and hepatic JAK2/STAT3 was suppressed (Fig. 5I, J and Supplementary Fig. S3). Collectively, these results indicated that both AICAR and metformin might have therapeutic potential against AI.
H2S and AMPK suppress inflammatory STAT3 and hepcidin by promoting proteasome-mediated JAK2 degradation
Janus family of tyrosine kinases is activated on ligand binding to associated cytokine receptors, and it modulates the phosphorylation of related signaling molecules (36). Among these kinases, JAK1 and JAK2 play key roles in the IL-6/STAT3 pathway (11, 46). Based on our observation of JAK2 regulation by AMPK in Figures 3G, 4H, and 5J, we sought to examine whether JAK2 contributed to the inhibition of STAT3 and hepcidin.
As demonstrated in Figure 6A and Supplementary Figure S4A, JAK1 and JAK2 phosphorylation levels were markedly suppressed by AICAR treatment. However, contrary to JAK2, there was minimal phosphorylation of JAK1 in response to IL-6 challenge, suggesting that JAK2 was the predominant JAK involved in signal transduction in this experimental paradigm. In addition, as was observed in vivo, AICAR decreased the protein levels of JAK2 but not JAK1 in mouse primary hepatocytes treated by IL-6. Moreover, lactate dehydrogenase (LDH) assay ruled out the potential possibility of contribution of AICAR-induced cytotoxicity to decreased JAK2 protein levels (Supplementary Fig. S5A). The involvement of JAK2 in STAT3 and hepcidin activation was further confirmed with specific JAK2 inhibitors (AG490, WP1066) and JAK2 overexpression (Fig. 6B and Supplementary Fig. S5B–D). These findings were further corroborated in experiments using CA-AMPK construct (Fig. 6C and Supplementary Fig. S4B) and in the absence of IL-6 (Supplementary Fig. S5E). In agreement with AMPK, NaHS exerted a similar effect on JAK2 (Fig. 6D and Supplementary Fig. S4C). These data showed that H2S and AMPK inhibited STAT3/hepcidin by suppressing JAK2.
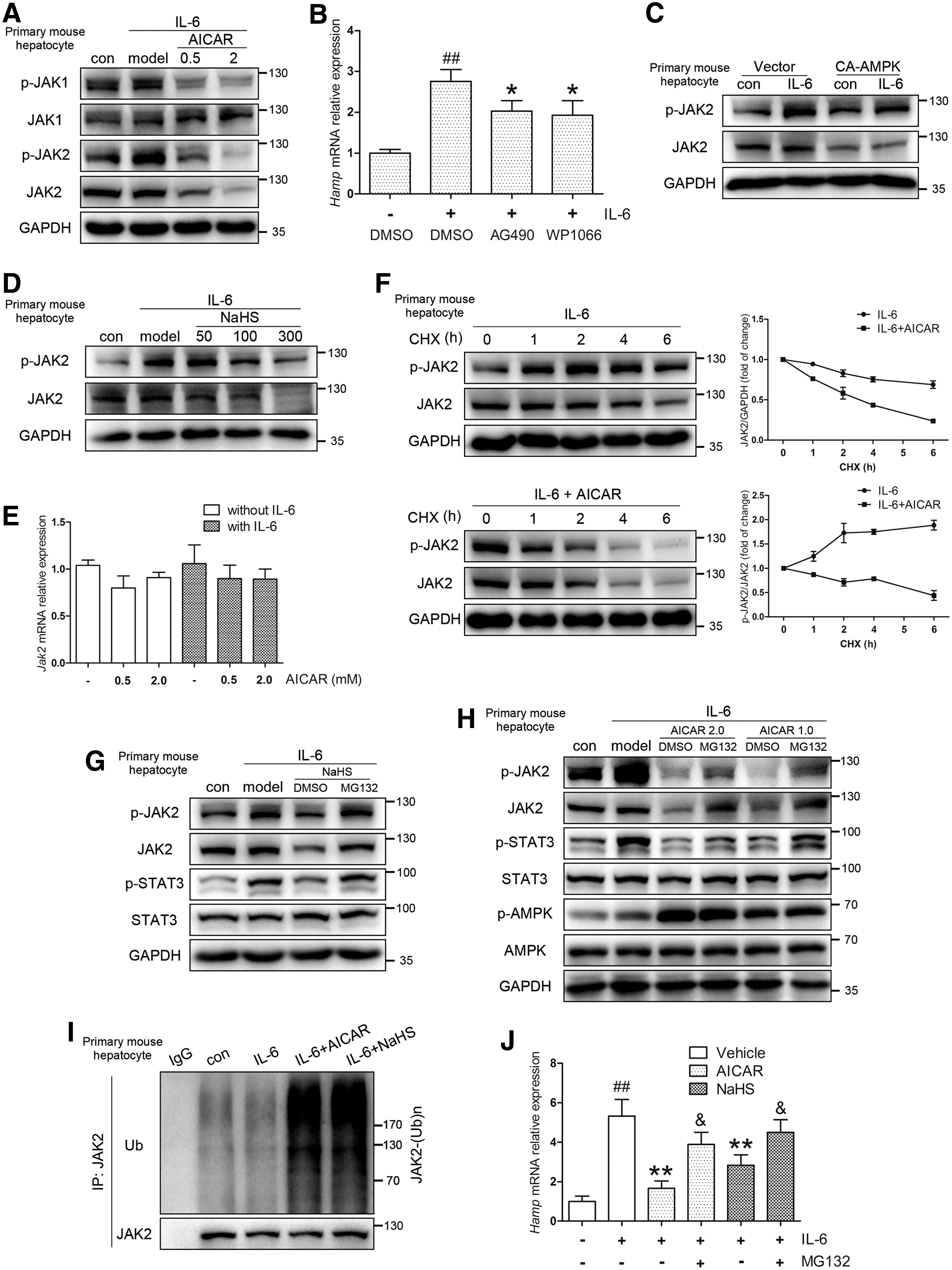
To further explore the regulation of JAK2 by AMPK, we focused on the mechanisms within the decrease of JAK2 protein levels. As shown in Figure 6E, Jak2 mRNA levels were not significantly decreased by AICAR, independent of IL-6. Considering the rapid changes observed in JAK2 protein following AICAR application (1–3 h), these results indicated that the reduction of JAK2 was probably achieved at the posttranscriptional level. Indeed, cycloheximide (CHX) chase assay revealed that JAK2 degradation was promoted in mouse primary hepatocytes treated with AICAR, with a shift of its half-life from more than 6 h to about 3 h (Fig. 6F). The inhibition of STAT3 with JAK2 abrogation was confirmed in a time-course experiment of mouse primary hepatocytes that were pretreated with AICAR for 3 h followed by IL-6 treatment (Supplementary Fig. S5F).
These data prompted us to ask which pathway of protein degradation is involved. By applying proteasome inhibitor MG132, we found that the effects of AICAR, NaHS, and CA-AMPK were diminished, as opposed to ectopically expressed ubiquitin (Fig. 6G, H, Supplementary Figs. S4D, E and S5G, H). These observations were supported by in vivo JAK2 ubiquitylation assay and hepcidin levels in mouse primary hepatocytes (Fig. 6I, J). Taken together, our data indicated that both AMPK activation and H2S reduced inflammatory hepcidin levels by promoting proteasome-mediated JAK2 degradation.
SOCS1 mediates the JAK2 degradation induced by AMPK activation
As a member of the SOCS family, SOCS1 plays a prominent role in the negative regulation of cytokine signal transduction (30). With the help of a conserved carboxy-terminal motif, SOCS box, SOCS1 interacts with elongin B/C and acts as an E3 ligase in the degradation of JAK2 (78). As demonstrated in Figure 7A and B, silencing of Socs1 reduced JAK2 ubiquitylation levels and prolonged JAK2 half-life in AICAR-treated mouse primary hepatocytes. As expected, suppression of the JAK2/STAT3 pathway was also reversed with Socs1 silencing (Fig. 7C and Supplementary Fig. S6A). Co-immunoprecipitation assay indicated that AICAR and NaHS significantly promoted JAK2-SOCS1 interactions (Fig. 7D, E and Supplementary Fig. S6B, C). These observations were further supported by transfection of constructs bearing wild-type SOCS1 and a truncated fragment with SOCS box deleted (SOCS1ΔBox, Fig. 7F, G and Supplementary Figs. S6D–F and S7).
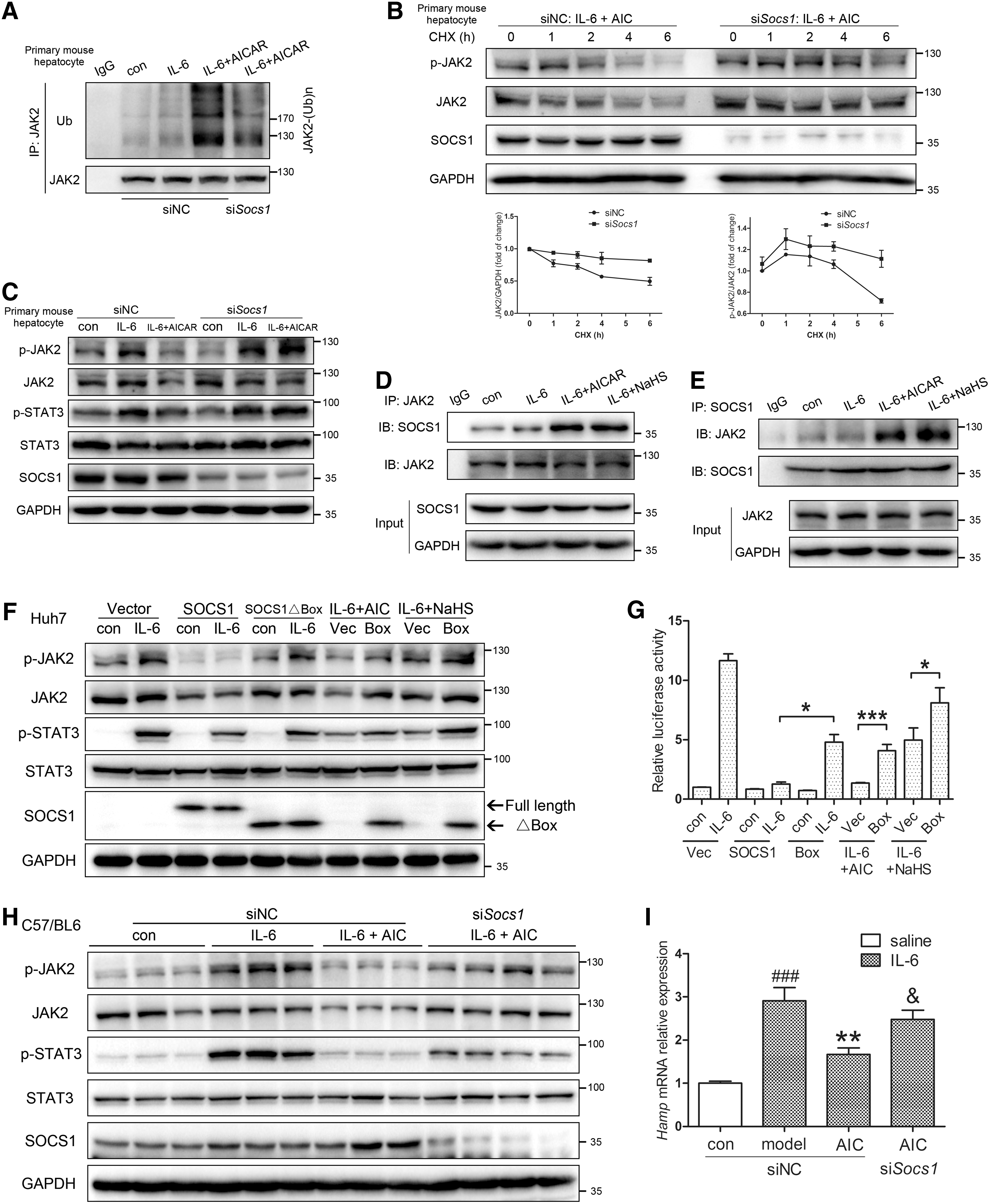
We next determined the potential in vivo role of SOCS1 by hydrodynamic transfection of Socs1 siRNA in C57BL/6 mice. Knockdown of Socs1 abrogated the AMPK-mediated inhibition of JAK2/STAT3 and hepatic hepcidin (Fig. 7H, I and Supplementary Fig. S6G). In conclusion, our data suggested that SOCS1 mediated the JAK2 degradation and hepcidin inhibition induced by AMPK activation.
Metformin treatment is associated with lower serum hepcidin levels and reduced anemia morbidity in patients with T2DM
Metformin is a first-line medication for T2DM, which is closely associated with chronic inflammation (72), as evidenced by elevated IL-6 content, and increased anemia risk (67). To assess whether metformin elicits similar clinical effects on anemia and hepcidin, we retrospectively analyzed the hematological indices of 137 T2DM patients, from whom 83 serum samples were collected. The remaining patients were classified by gender and metformin therapy.
No significant difference was observed in baseline characteristics of the entire cohort (Fig. 8A). In addition, baseline characteristics except for the mean age of female patients were similar among patients with serum samples (Supplementary Fig. S8A).

Importantly, as demonstrated in Figure 8B, metformin therapy was associated with decreased serum hepcidin in male patients. Accordingly, erythrocyte number and hemoglobin content were significantly higher in male patients taking metformin (Fig. 8C, D). An increasing trend was also observed in hematocrit of the male patients on metformin, and in erythrocyte number and hemoglobin content of the female patients taking metformin (Supplementary Fig. S8B–E). Moreover, an insignificant increase in the serum H2S content was observed in male patients taking metformin (Fig. 8E).
We next examined the association of anemia with metformin therapy and serum hepcidin. As shown in Figure 8F, among all patients, anemia was significantly lower in the metformin group than in the non-metformin group (odds ratio = 0.081, 95% confidence interval = 0.010–0.639; p = 0.004). Similarly, the rate of anemia was significantly lower in male patients treated with metformin (p = 0.010) and it was borderline significantly higher in male patients with high serum hepcidin levels (p = 0.050). The odds ratio and 95% confidence interval could not be calculated, as no male patient had anemia in the metformin and hepcidin groups. Overall, our results indicated that metformin was, at least for men, associated with reduced serum hepcidin content and anemia prevalence in Chinese T2DM patients.
Discussion
In the present study, we demonstrated that AMPK mediated the treatment effects of H2S on inflammatory hepcidin and AI. In addition, we elucidated the therapeutic potential of AMPK in the resolution of AI and the restoration of iron balance, and we demonstrated that AMPK induced JAK2 degradation via the ubiquitin-proteasome pathway (Fig. 8G). These results provided new insights into the anti-inflammatory roles of AMPK activators, including metformin. Notably, evaluation of clinical samples showed that metformin treatment was associated with decreased serum hepcidin levels and anemia in T2DM patients. On the whole, our work uncovered AMPK as a novel therapeutic target, and metformin as a potential drug against AI.
Conventional remedies for AI include blood transfusion, iron supplementation, and erythropoietic stimulating agents (ESAs). However, blood transfusion is associated with a high risk of multi-organ failure and immunomodulation, such as allergic reaction, infection transmission, and immunosuppression (69). Conversely, in addition to well-known issues such as limited oral absorption and potential cardiovascular events (10), iron therapy was shown to be associated with increased infection risk in hemodialysis patients (65). Although the beneficial effects of ESAs are well documented, a meta-analysis examining 13,933 patients with cancer in 53 trials demonstrating an association of ESA use with increased mortality and worse overall survival raises concerns about their use (3). These studies highlight the urgent need for alternative therapies and illustrate the significance and implications of our findings in clinical management of AI.
AI is a complex disease arising from a confluence of inflammatory responses by multiple organs; therefore, a variety of infectious and non-infectious mouse models have been established to date. Compared with the currently used infectious models, non/pseudo-infectious AI models achieve improved reproducibility and are safer for both experimenters and other experimental animals. LPS and recombinant IL-6 are commonly used to induce inflammatory hepcidin and hypoferremia not only in animal models but also in human trials (29, 48); however, there still remains a gap between acute inflammation insult and AI. Turpentine has been extensively studied in inflammatory animal models for decades. A number of studies previously used turpentine to induce normocytic normochromic AI (34, 50). We previously utilized LPS to induce hepcidin levels. In the present study, both IL-6 and turpentine were used to investigate the therapeutic potential of AMPK by several methods. Our results demonstrated that AMPK not only reversed acute inflammatory hepcidin and hypoferremia but also improved AI symptoms in a chronic model.
A large number of studies explored the role of H2S in diseases of the cardiovascular, central nervous systems as well as in inflammation (38). We herein reported that H2S successfully improved AI symptoms and corrected iron disturbance in animal models via AMPK activation. In line with our observations, other groups also reported the activation of AMPK by H2S in cerebral cortex (45) and microglial cells (81). However, few studies sought to assess the role of AMPK in the H2S-mediated anti-inflammatory processes. Zhou et al. (81) suggested that AMPK was responsible for the suppression of neuroinflammation by H2S. In accordance with their findings, our data indicated that AMPK contributed to the suppression of hepatic inflammation by H2S both in vitro and in vivo, suggesting multiple physiological functions of AMPK.
As shown in Figure 2, the inhibition of hepcidin by H2S was not completely abolished by AMPK siRNA or DN-AMPK, which might be attributed to limited knockdown or inhibition efficiency and potential compensation by alternative H2S-mediated signaling pathways (38). We recently demonstrated that H2S inhibited STAT3 phosphorylation by activating Sirtuin 1 (SIRT1) (65). Since AMPK was reported to regulate SIRT1 activity in skeletal muscle (6), we tested whether a similar pattern was also in the present model. As shown in Supplementary Figure S9, the suppression of STAT3 and hepcidin activation by AICAR was not rescued by EX-527, a specific SIRT1 inhibitor. These data indicated that AMPK suppressed STAT3/hepcidin in an SIRT1-independent manner, at least in the current cell model, and that H2S might activate AMPK and SIRT1 by distinct mechanisms.
AMPK is a metabolic sensor that is highly conserved from yeast to mammals. As a heterotrimeric complex consisted of three subunits, AMPK is activated by phosphorylation at Thr172, which leads to stimulation of glucose utilization and inhibition of gluconeogenesis; therefore, AMPK is regarded as an ideal therapeutic target for T2DM. In addition to its role in energy homeostasis, AMPK is also involved in cancer, autophagy, and inflammation (15, 44). By suppressing the mammalian target of rapamycin signaling and ACC activity, AMPK and metformin impair protein and fatty acid synthesis, and, consequently, inhibit proliferation of breast cancer cells (13). Similar signaling patterns were also observed in induction of autophagy by AMPK (31, 59).
Previous studies investigating the crosstalk between inflammation and AMPK suggested that IL-6, a typical inflammatory cytokine, activated AMPK in a tissue-specific manner. Specifically, IL-6 increased AMPK phosphorylation in both skeletal muscle and adipose tissue, whereas exercise-mediated AMPK activation was diminished in Il-6 knockout mice (28). However, in the same study, the authors found that hepatic AMPK activation was IL-6 independent. In agreement with these earlier observations, we did not find any significant change in AMPK phosphorylation during IL-6 and turpentine; however, we observed a decrease in AMPK kinase activity.
On the contrary, the mechanisms underlying the anti-inflammatory effects of AMPK are not clear. According to a recent review by O'Neill (52), AMPK-mediated pseudo-starvation leads to increased recruitment of anti-inflammatory M2 macrophages and regulatory T cells. In the present study, one of the most notable findings was AMPK-induced JAK2 degradation via the ubiquitin-proteasome pathway. By promoting the interaction between JAK2 and SOCS1, as an endogenous suppressor of cytokine signaling, AMPK blocked IL-6/STAT3 signaling, thus ameliorating downstream inflammatory response. A similar strategy was used in the development of antitumor drugs, such as the targeted degradation of androgen receptor (74) and the cellular bromodomain protein BRD4 (42). Conversely, JAK family members are attractive pharmacological targets based on their roles in immune-driven diseases. Several JAK inhibitors, such as tofacitinib and ruxolitinib, have been approved for treatment of rheumatoid arthritis and myelofibrosis, respectively. Our findings might provide new perspectives and therapeutic targets for AI.
Previous studies indicated diabetes as an independent risk factor for anemia (14, 23), whereas anemia was shown as a risk factor for cardiovascular disease and all-cause mortality in diabetes (70). It is generally accepted that kidney failure contributes to anemia in diabetic patients. However, Grossman et al. reported that anemia was more common even in diabetic patients with normal renal function compared with nondiabetic subjects (10.8% vs. 2.7%, p < 0.001) (19). In addition, anemia may develop before renal impairment (12) and has a two- to threefold higher prevalence rate (23%) in diabetic patients than in subjects in the general population with comparable renal impairment and iron stores (67). These observations suggest additional causes for diabetic anemia.
Indeed, chronic inflammation, indicated by elevated serum IL-6, is intimately involved in the pathogenesis of T2DM (55). Furthermore, the presence of AI is evidenced by both iron dysregulation (25) and suppressed erythropoiesis despite normal or increased erythropoietin (9). Accordingly, several groups reported increased hepcidin levels in T2DM patients (1, 24), whereas others claim conflicting results (61). In the present work, T2DM patients with renal impairment were excluded to rule out the potential confounding effect of renal anemia. Consequently, our analysis determined that the metformin-treated group had lower serum hepcidin levels and anemia prevalence, supporting our hypothesis that hepcidin was a mediator in diabetic anemia. Future investigation is necessary to further elucidate this outcome.
Metformin, a biguanide antidiabetic drug, has been applied clinically for more than half a century. Recent studies revealed that metformin's beneficial effects extended beyond diabetes. Several groups reported that metformin was related to a reduced risk of cancer in diabetic patients (40). Other reports suggested that metformin retarded aging in Caenorhabditis elegans (4) and improved lifespan in mice (43). However, the relationship between metformin and anemia is not clear. In the only large study published to date, although anemia prevalence was higher in patients taking metformin, the results were debatable due to the lack of proper controls and statistically significant difference (2).
Conversely, few studies explored the relationship between metformin and hepcidin, with conflicting results. Here in the present study, we demonstrated for the first time that metformin was associated with reduced serum hepcidin levels in Chinese male T2DM patients. What is more, the inverse correlation of serum hemoglobin with cardiovascular outcomes (70), and cardiac and renal benefits and improved quality of life with correction of hemoglobin levels are well established (58, 63). Our results showing the association of metformin with increased hemoglobin in Chinese T2DM patients, therefore, warrant further investigation into the role of hemoglobin and hepcidin in the improved clinical outcomes by metformin in diabetic patients. It is also worth noting that anemia morbidity is decreased in the metformin group, suggesting metformin as a potential therapeutic agent for anemia not only in diabetes but also in other inflammatory diseases.
Materials and Methods
Reagents and antibodies
NaHS, turpentine, metformin, and 1,10-phenanthroline monohydrate were purchased from Sigma-Aldrich. AICAR, AG490, WP1066, and MG132 were obtained from Selleck Chemicals. CHX was purchased from Melonepharma.
Recombinant human and murine IL-6 were obtained from Peprotech. Antibodies to total JAK2 (catalog 3230), phospho-ACC (catalog 3661), total (catalog 9139) and phospho-STAT3 (Tyr705, catalog 9145), and total (catalog 5832) and phospho-AMPK (Thr172, catalog 2535) were purchased from Cell Signaling Technology. Antibody to phospho-JAK2 (Tyr221, catalog BS4837) was acquired from Bioworld Technology. Antibodies to ubiquitin (catalog 10201-2-AP), HA-tag (catalog 66006-1-Ig), and GAPDH (catalog 60004-1-Ig) were obtained from Proteintech Group. Antibodies to SOCS1 (catalog RLT4362), total (catalog RLP0154) and phospho-JAK1 (catalog RLP2424) were purchased from Ruiyingbio. In addition, antibodies to CSE (catalog sc-374249) and CBS (catalog sc-67154) were obtained from Santa Cruz Biotechnology.
Animals and induction of chronic turpentine model
All animal experimental protocols complied with the Animal Management Rules of the Ministry of Health of the People's Republic of China, and they were approved by the Animal Care Committee of Fudan University.
Eight-week-old male C57BL/6 mice were purchased from Sippr-bk Experimental Animal Center. Mice were housed in specific pathogen-free rooms at 25°C and maintained under a 12-h/12-h light/dark cycle with ad libitum access to food and water.
For the turpentine model, C57BL/6 mice were subcutaneously injected with turpentine (100 μl/20 g weight) once a week for 4 weeks to induce chronic AI as described earlier (50). Compound treatment was performed twice a week, starting from the second injection of turpentine. Mice were sacrificed 1 week after the last injection, after anesthesia with pentobarbital sodium.
Cell culture and isolation of mouse primary hepatocytes
Two human hepatoma cell lines, Huh7 (Japanese Collection of Research Bioresources) and HepG2 (American Type Culture Collection), were cultured in high-glucose DMEM that was supplemented with 10% fetal bovine serum (ExCell Bio) and 1% penicillin and streptomycin (Gibco).
Mouse primary hepatocytes were isolated from 8-week-old C57BL/6 mice by using a modified two-step collagenase perfusion protocol as previously described (76).
Antibody array
Antibody arrays were conducted with the PathScan Intracellular Signaling Array Kit (#7744; Cell Signaling Technology) by following the manufacturer's instructions. The array slides were visualized and analyzed with the LICOR Odyssey Sa Infrared Imaging System. The intensity of each array was balanced by internal negative controls and then normalized to internal positive controls. The MeV software (
Plasmid, lentivirus, and siRNA preparation
Human CBS lentiviral vector was purchased from Obio Tech. pcDNA3.1 CSE construct was from Dr. Yaqi Shen as previously described (62). Human JAK2 overexpression plasmid was kindly provided by Dr. Shenmeng Gao (77). HA-ubiquitin construct was donated by Dr. Qunying Lei (41). Murine CA-AMPKα2 (1–312, T172D) construct was kindly provided by Dr. Xiaolong Liu (7); dominant negative AMPKα2 was obtained by K45R mutation of the wide type as previously described (79). The 2 AMPK plasmids were then subcloned to lentiviral vectors. SOCS1 construct and the SOCS Box deleted form were kindly provided by Dr. Gerardo Ferbeyre (5). All plasmids were purified by using an EndoFree Plasmid Maxiprep Kit (Biotool). Transient transfection was performed by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Lentivirus packaging was carried out with the psPAX2/VSVG system and HEK 293T cells by using FuGENE HD transfection reagent (Promega Biotech). For in vivo application, the lentivirus was concentrated with ultra-centrifugation to reach a titer of 5–10 × 108 TU/ml.
For silencing experiments in vitro, Huh7 cells and mouse primary hepatocytes were transfected with siRNA (siAMPK-1: 5′-GAGGAGAGCUAUUUGAUUATT-3′; siAMPK-2: 5′-GCGUGUACGAAGGAAGAAU-3′; siSocs1: 5′-CTACCT GAGTTCCTTCCCCTT-3′) by using Lipofectamine 2000, as previously described (76). For murine RNAi in vivo, 2′OMe-modified siRNA of Socs1 was rapidly injected into mouse tail vein (50 μg in 1 ml normal saline). Analysis was performed 24 h later.
AMPK kinase assay
After the indicated treatment, mouse primary hepatocytes were lysed in immunoprecipitation buffer and immunoprecipitated with AMPK antibody. The precipitates were combined with 0.2 mM SAMS peptide (HMRSAMSGLHLVKRR; Biotool). The kinase reaction was conducted at 37°C for 30 min by using ADP-Glo™ Kinase Assay system (Promega).
Immunofluorescence staining
Immunofluorescence staining was conducted as previously described (76). Briefly, mouse primary hepatocytes were fixed by 4% formaldehyde and treated with cold methanol for 10 min at −20°C. The cells were incubated with specific primary antibodies overnight after blocking for 1 h. The slides were observed by confocal laser scanning microscopy (Zeiss LSM 710).
RNA isolation and real-time qRT-PCR
RNA extraction and real-time qRT-PCR were carried out as previously described (76) with Bio-Rad CFX Connect Real-Time PCR System and primers as follows: human β-ACTIN forward 5′-AGGATGCAGAAGGAGATCACTG-3′, reverse 5′-GGGTGTAACGCAACTAAGTCATAG-3′; human HAMP forward 5′-CTGCAACCCCAGGACAGAG-3′, reverse 5′-GGAATAAATAAGGAAGGGAGGGG-3′; murine β-actin forward 5′-TGTTACCAACTGGGACGACA-3′, reverse 5′-GGTGTTGAAGGTCTCAAA-3′; murine Hamp forward 5′-AGAGCTGCAGCCTTTGCAC-3′, reverse 5′-GAAGATGCAGATGGGGAAGT-3′; murine Il-6 forward 5′-TGTGCAATGGCAATTCTGAT-3′, reverse 5′-CCAGA GGAAATTTTCAATAGGC-3′; murine Jak2 forward 5′-TTGTGGTATTACGCCTGTGTATC-3′, reverse 5′-ATGC CTGGTTGACTCGTCTAT-3′; specificity of all PCR products was confirmed by melting curve analysis.
Immunoprecipitation and immunoblot analysis
Immunoprecipitation and immunoblot were performed as previously described (76). In brief, cells were lysed in IP-specific buffer (20 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, pH 7.5) and immunoprecipitated with antibody and Protein A/G Plus-Agarose (Santa Cruz Biotechnology). SDS-PAGE and transferring procedure were carried out with the standard Western blot system (Bio-Rad).
In vivo ubiquitylation assay
In vivo ubiquitylation assay was performed as previously described (26). Briefly, mouse primary hepatocytes were first lysed in 1% SDS buffer (50 mM Tris, 0.5 mM EDTA, 1 mM dithiothreitol, pH 7.5) with MG132 and boiled for 10 min. The lysates were 10-fold diluted in Tris-HCl buffer before analysis of ubiquitylation levels by immunoprecipitation and Western blot.
Dual-luciferase reporter assay
Dual-luciferase reporter assay was conducted by using the pGL3 vector containing human hepcidin promoter (−1000/+71) and the pGL4.47 [luc2P/SIE/Hygro] vector (Promega Biotech). Relative luciferase activities were determined with the Dual Luciferase System (Promega Biotech) as previously described (76).
LDH assay
The LDH leakage assay was conducted as described by the manufacturers (Beyotime). The LDH leakage (%) was calculated by using the following equation: [(A − C)/(P − C)] × 100%, where A is the absorbance of the tested samples, C is the medium control, and P is the positive control (Triton X-100).
Serum iron parameters, hepcidin, and IL-6 analysis
Mouse serum iron levels and total iron-binding capacity (TIBC) were determined with a colorimetric method (Jiancheng Bioengineering Institute). Transferrin saturation was calculated as serum iron/TIBC × 100%.
Serum hepcidin and IL-6 were quantified by ELISA following protocols described by the manufacturers (both murine and human hepcidin kit from Uscn) (66, 68) (IL-6 kit from Boatman).
Tissue non-heme iron analysis
Tissue non-heme iron was measured by 1,10-phenanthroline monohydrate method as described in our previous work (76). Generally, mouse spleen samples were digested in acid solution after being dried at 65°C. The supernatant was collected and mixed with 1,10-phenanthroline monohydrate. The absorbance at 510 nm was measured for determination of Fe (II) concentration.
Perl's Prussian blue staining of paraffin-embedded sections
Perl's Prussian blue staining of mouse spleen paraffin sections was conducted as described and captured with the Zeiss Axio Scope A1 system (76).
Hematological analysis and Wright-Giemsa staining
After anesthesia with pentobarbital sodium, whole blood samples were obtained from the retro-orbital sinus and collected in tubes that were pretreated with EDTA. Complete blood count was performed with SYSMEX XT-2000i automatic analyzer.
Peripheral blood smears were carried out by using 3 μl fresh whole blood that was treated with EDTA, and prepared for Wright-Giemsa staining (Yeasen Biotech). The slides were visualized with the Zeiss Axio Scope A1 system.
Collection and analysis of serum samples from type 2 diabetic patients
Serum samples that were frozen between September 2015 and March 2016 were collected from the hospital tissue bank, and the results of complete blood count from T2DM were acquired from the Department of Endocrinology, Huashan Hospital, Fudan University, Shanghai, China. Written informed consent was obtained from all patients before inclusion. All procedures related were approved by the Ethics Committee of Huashan Hospital, Fudan University, and in accordance with the Declaration of Helsinki. Anemia was determined by the reference hemoglobin values in Huashan Hospital: <115 g/L for women and <130 g/L for men.
Exclusion criteria include infections during the previous week, tumor-related diseases, chronic kidney diseases, auto-immune diseases, history of organ transplantation and on anti-rejection drugs, and participation in clinical trials during the past 6 months. Patients taking metformin for less than 3 months were also excluded.
Measurement of serum H2S levels
The H2S levels of serum samples from T2DM patients were measured as previously described (33) with minor revision. Briefly, 30 μl serum samples were mixed with 10 μl Tris-HCl buffer (100 mM, pH 8.5) and 70 μl monobromobimane reagent. After vortexing for 1 h in the dark, the samples were centrifuged at 13,000 g, 4°C for 20 min after the addition of 10 μl 20% formic acid. The supernatant was subjected to reverse-phase HPLC for H2S determination.
Statistics
Data are expressed as the mean ± SEM. Statistical analysis of continuous variables was performed with t-test, Mann–Whitney U test, or one-way ANOVA followed by Turkey's test. For counting variables, chi-square or Fisher's exact probability test was conducted. A two-tailed p < 0.05 was considered statistically significant.
Footnotes
Acknowledgments
The authors would like to thank the following individuals for providing plasmids as stated in the methods: Dr. Shenmeng Gao from the First Affiliated Hospital of Wenzhou Medical College, Dr. Qunying Lei and Dr. Yaqi Shen from Fudan University, Dr. Xiaolong Liu from Shanghai Institutes for Biological Sciences, and Dr. Gerardo Ferbeyre from Université de Montréal. This work was supported by grants from the National Natural Science Foundation of China (No. 81330080; 81573421; 81402917) and the Shanghai Committee of Science and Technology of China (No. 14JC1401100).
Authors' Contributions
M. W. and H. X. designed and conducted the experiments. M. W. and W. T. analyzed the data and wrote the article. Y. L., Z. Z., and L. F. collected clinical samples. L. M. and X. W. performed animal experiments. B. T. analyzed serum H2S levels. H. X. and Y.Z.Z. reviewed and edited the article.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.