
Abstract
Aims:
Acquired hearing loss is a worldwide epidemic that affects all ages. It is multifactorial in etiology with poorly characterized molecular mechanisms. Mitochondria are critical components in hearing. Here, we aimed to identify the mechanisms of mitochondria-dependent hearing loss using Fus1 KO mice, our novel model of mitochondrial dysfunction/oxidative stress.
Results:
Using auditory brainstem responses (ABRs), we characterized the Fus1 KO mouse as a novel, clinically relevant model of age-related hearing loss (ARHL) of metabolic etiology. We demonstrated early decline of the endocochlear potential (EP) that may occur due to severe mitochondrial and vascular pathologies in the Fus1 KO cochlear stria vascularis. We showed that pathological alterations in antioxidant (AO) and nutrient and energy sensing pathways (mTOR and PTEN/AKT) occur in cochleae of young Fus1 KO mice before major hearing loss. Importantly, short-term AO treatment corrected pathological molecular changes, while longer AO treatment restored EP, improved ABR parameters, restored mitochondrial structure, and delayed the development of hearing loss in the aging mouse.
Innovation:
Currently, no molecular mechanisms linked to metabolic ARHL have been identified. We established pathological and molecular mechanisms that link the disease to mitochondrial dysfunction and oxidative stress.
Conclusion:
Since chronic mitochondrial dysfunction is common in many patients, it could lead to developing hearing loss that can be alleviated/rescued by AO treatment. Our study creates a framework for clinical trials and introduces the Fus1 KO model as a powerful platform for developing novel therapeutic strategies to prevent/delay hearing loss associated with mitochondrial dysfunction. Antioxid. Redox Signal. 27, 489–509.
Introduction
A
Age-related hearing loss is a serious health problem with few efficient therapies due to its multifactorial etiology and poorly characterized molecular mechanisms. We present evidence that mitochondrial dysfunction/oxidative stress, often associated with inflammatory or mitochondrial diseases, result in a special type of progressive hearing loss manifested by mitochondrial and vascular degeneration in the cochlear stria vascularis, a decrease in endocochlear potential, and early alterations in aging-associated molecular pathways in the inner ear. Remarkably, antioxidant supplementation corrects these electrophysiological, mitochondrial, and molecular pathologies resulting in a significant delay in hearing decline and suggesting approaches to alleviate this type of hearing loss.
Unrecognized or untreated hearing loss results in widespread effects from difficulties in communicating, and affects productivity and quality of life. It may lead to social isolation, depression, and cognitive decline (2). Uncovering and detailed characterization of the mechanisms of premature hearing decline would provide a foundation for tailored prevention or treatment plans based on “individual broken systems.”
One of the critical components of hearing function is mitochondrial health. The mitochondrion is a versatile energy-producing organelle that regulates vital cellular processes, such as metabolism, calcium and redox signaling, and apoptosis. Mitochondrial involvement in auditory function was recognized by hearing impairment in many patients with mitochondrial diseases and via identification of mutations in mitochondrial DNA in patients with maternally inherited deafness (12). Mitochondrial dysfunction-mediated hearing impairment can be inborn or progressive when induced by aminoglycosides, sound exposure, or aging.
Mitochondria contain more than 1500 proteins, but only 14 are encoded by mitochondrial DNA while the rest are nuclear encoded (33). Therefore, one would expect that abnormal activities/loss of many of these mitochondria-residing proteins could affect hearing. However, the significance of nuclear-encoded mitochondrial proteins in hearing is poorly understood with only several hearing loss-linked mutations characterized in humans (73, 17).
In mice, the most extensively studied model is the so-called mtDNA-mutator mouse bearing a mutation in the mitochondrial polymerase gene (Polg) (69) that exhibits a significant elevation of auditory brainstem response (ABR) thresholds at low to middle frequencies by 9–10 months of age (63). Two other mouse models that lack the antioxidant (AO) enzymes, Gpx1 or Sod1, show enhanced susceptibility to noise-induced hearing loss (45). Scavenging of mitochondrial reactive oxygen species (ROS) by targeting an AO protein catalase to mitochondria in MCAT transgenic mice alleviates hearing loss in aging MCAT mice compared to age-matched WT mice (58, 59).
Recently, our group characterized a transgenic mouse strain that robustly overexpresses the mitochondrial methyltransferase TFB1 M (Tg-mtTFB1 mice) and exhibits progressive hearing loss most likely of strial etiology; however, no obvious signs of strial atrophy were detected. Hearing loss in this model was rescued by genetically reducing AMPK α1 activity (38). While these few models provide strong evidence that mitochondria and mitochondria-generated ROS play essential roles in progressive hearing loss, additional mouse models are required to characterize the spectrum of auditory system pathologies inflicted by mitochondrial dysfunction-induced oxidative stress and inflammation.
Our early studies showed that mitochondrial dysfunction and oxidative stress are the primary pathogenic mechanisms in Fus1-deficient cells and mice. In our KO model (22), loss of mitochondrial protein Fus1 profoundly affects several mitochondrial parameters, such as ROS production, calcium uptake, mitochondrial membrane potential (MMP), respiratory reserve, and mitochondrial fusion (70, 71). As we demonstrated, the perturbed mitochondrial homeostasis/oxidative stress in Fus1 KO mice affect steady-state mitochondrial activities and severely compromise stress response on exposure to asbestos, radiation, infection, or immune stimulation (20, 70, 71, 76). Therefore, we hypothesized that Fus1 KO mice may experience either an inborn hearing defect or develop early progressive age-related hearing loss (ARHL) and that this pathology may be alleviated by improving the redox state in the cochlea.
Here, we established that Fus1 KO mice suffer from the least characterized type of ARHL of strial origin, making this model a valuable tool for studying mitochondrial/oxidative mechanisms of age-related hearing decline. Using this model, we establish and characterize in detail the phenotypic manifestations of premature hearing loss of strial etiology based on Fus1 loss-mediated mitochondrial dysfunction, and identify the target cells and tissues in the inner ear that are most vulnerable to such dysfunction. We also identify molecular pathways responsible for early hearing loss, and explore an AO approach to slow down hearing loss and development of molecular defects in cochlear tissues associated with mitochondrial dysfunction/oxidative stress.
Results
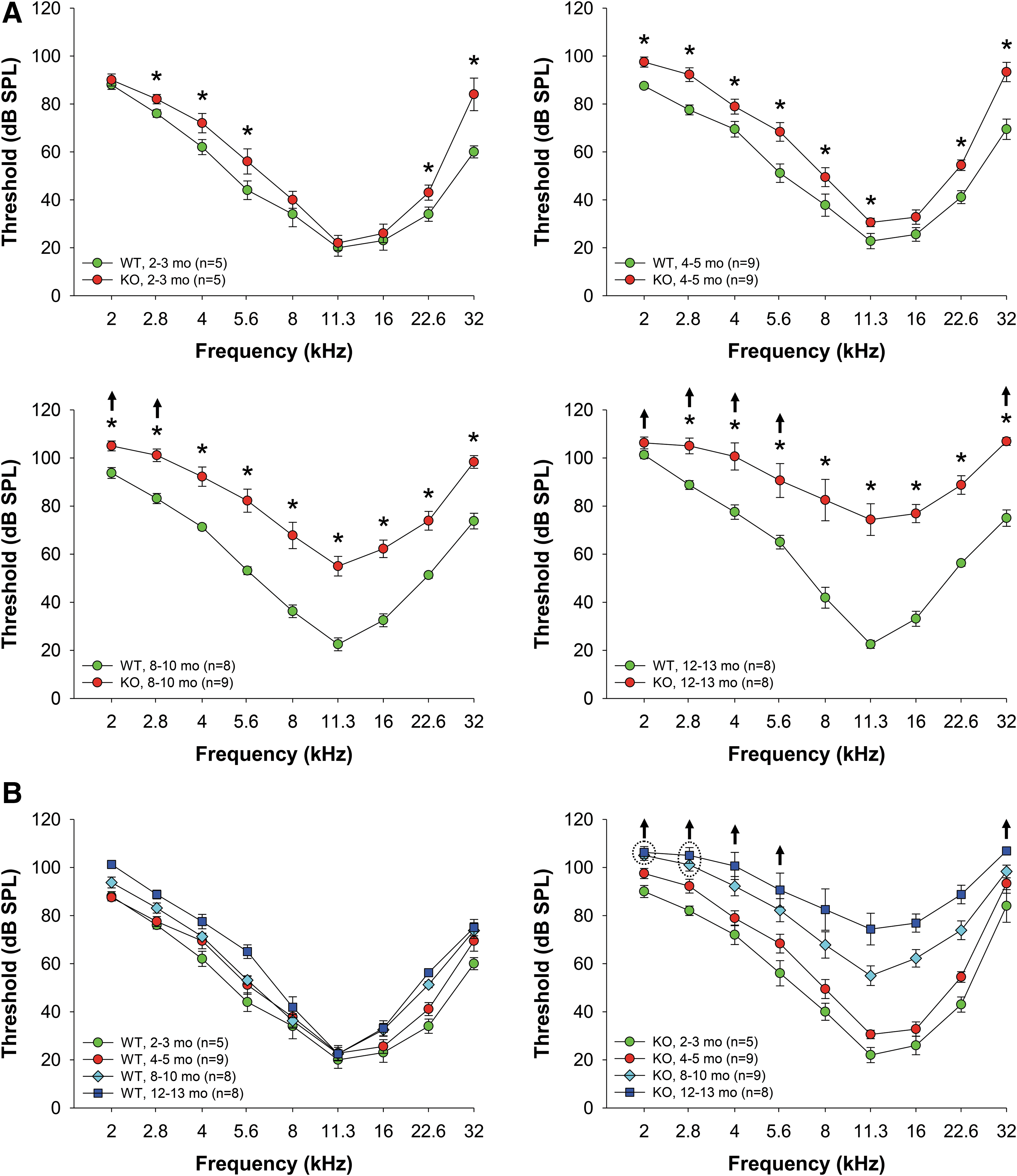
Fus1 KO mice show early-onset progressive hearing loss
Our earlier studies showed that Fus1 KO cells and tissues are characterized with high levels of ROS and insufficient AO machinery (70, 71, 77). Considering the detrimental effects of oxidative stress on hearing (19), we hypothesized that Fus1 KO mice may have progressive hearing loss. We used ABRs to measure hearing levels in Fus1 WT and KO mice of different ages. We found that while young 2-month-old KO mice had similar hearing thresholds to WT mice at middle frequencies, and slightly elevated thresholds at low and high frequencies (Fig. 1A), 4–5-month-old KO mice had moderately elevated thresholds over the 2–32 kHz frequency range, and 8–10-month-old KO mice had substantial threshold elevations of up to 50 dB across all frequencies. Hearing decline continued further in 12–13-month-old KO mice (Fig. 1A). Age-matched WT mice over the same time period showed no significant changes in thresholds at middle frequencies and a slight threshold elevation at high and low frequencies (Fig. 1B). Hearing decline in WT mice was slow and even 15–18-month-old WT mice had much better hearing than 8-month-old KO mice. Thus, systemic loss of Fus1 affects hearing at all frequencies starting from 4 to 5 months of age and results in rapid progression of hearing loss thereafter.
Young Fus1 KO mice have robust ABR wave I amplitude that rapidly diminishes with age
Amplitudes and latencies of the initial four ABR waves were analyzed. These waves are the response to 3 ms pips that represent sequential temporal events along the auditory ascending pathway. The amplitude of the waves reflects the number of activated neurons and synchrony of firing, while latency, the elapsed time from sound delivery, reflects the timing of synaptic transmission and nerve conduction. Amplitude and latency analysis of the ABR waves provides information on the integrity of auditory periphery and brainstem pathways. In humans, aging substantially reduces the amplitudes of all principal ABR waves producing significant latency shifts in waves I and III (25).
We compared ABR wave latencies and amplitudes in cohorts of 2- and 8–10-month-old WT and KO mice (Fig. 2). In young WT and KO mice, wave I latencies were similar at the majority of sound levels (Fig. 2B). Strikingly, in 8–10-month-old Fus1 KO mice, wave I latencies were profoundly delayed with the largest difference at 65–70 dB SPL (sound pressure level) (Fig. 2A).
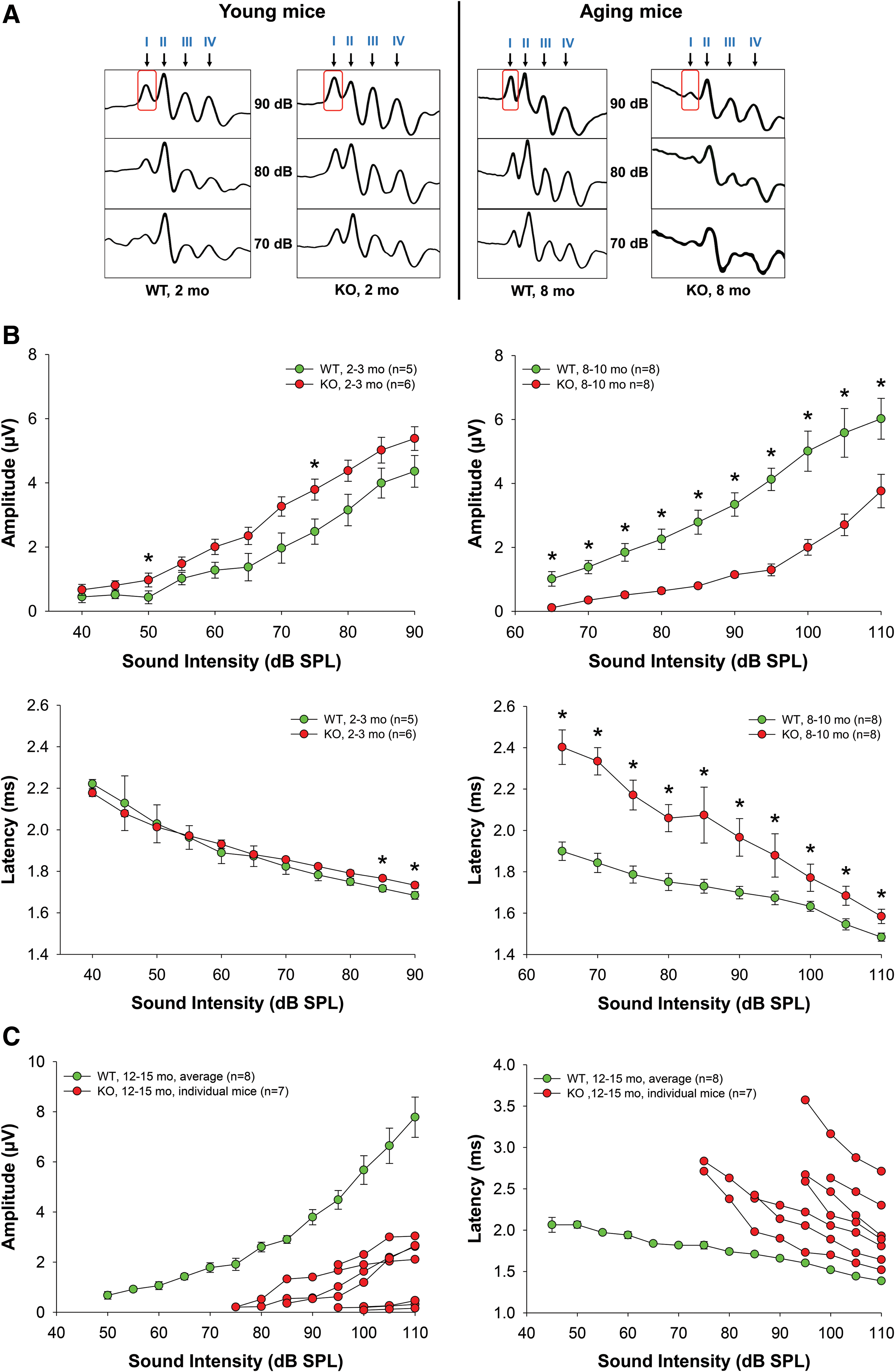
Interestingly, in young KO mice, wave I amplitude tended to be higher than in WT mice at all analyzed sound intensities and was significantly higher at 60–80 dB SPL (Fig. 2B). However, with age, wave I amplitude became profoundly lower in Fus1 KO mice (Fig. 2A, B), consistent with the correlation between hearing loss and decrease in wave I amplitude in humans (25). Figure 2C shows further deterioration of wave I parameters along with profound interindividual heterogeneity reflecting variations in the onset of ARHL in 12–13-month-old KO mice. Note that age-matched WT mice still show normal wave I parameters and low individual heterogeneity.
Latencies and amplitudes of waves II-IV in Fus1 KO mice were similar to wave I patterns (Supplementary Fig. S1; Supplementary Data are available online at
Thus, analysis of the adult Fus1 KO ABR waveforms reveals profound early changes in the integrity of auditory periphery and brainstem pathways similar to the changes that occur in aged people. These data are consistent with premature aging of the auditory system in Fus1 KO mice.
Signs of chronic inflammation in the inner ear of aging Fus1 KO mice
In our early studies, we established that Fus1 loss results in augmented/skewed innate and adaptive inflammatory responses, suggesting a preactivated state of the immune cells in KO mice (20, 70). Thus, we hypothesized that Fus1 KO mice may have low-grade chronic inflammation in cochlear tissues and surroundings that could affect auditory performance.
Indeed, hematoxylin and eosin (H&E) staining of bone marrow (BM) cells in the temporal bone surrounding the cochlea showed that in older KO mice, the BM is more hypercellular and myeloid predominant than in age-matched WT mice (Fig. 3A, lower panel). This type of BM histology is often observed in patients with low-grade chronic inflammation that results in an increased systemic level of inflammatory cytokines (68). Staining of BM with Iba-1, a microglia/macrophage-specific calcium-binding protein upregulated during macrophage activation, confirmed this finding (Fig. 3B, upper panel). BM from temporal bones of young WT and KO mice was phenotypically indistinguishable, suggesting that development of this pathology is age dependent. Staining of cochlear tissues with Iba-1 revealed that the number of activated cochlear macrophages is also higher in KO mice supporting the hypothesis of chronic inflammatory processes in the aging Fus1 KO cochlea (Fig. 3B, lower panel).

Aging KO mice with severe hearing loss have slight or no changes in auditory sensory and neuronal cell counts but showed reduced number of inner hair cell synapses and type IV fibrocytes in the cochlea
We investigated morphological changes of three major cochlear cell types that could have major effects on hearing: outer hair cells (OHCs), inner hair cells (IHCs), and spiral ganglion neurons (SGNs). Hair cells are the sensory receptors of the mammalian auditory system located within the organ of Corti. The spiral ganglion is a group of nerve cells that receive inputs from hair cells and serve to transfer these signals to auditory brainstem nuclei. Damage to any of these cells results in decreased hearing sensitivity, and because regeneration is absent, this damage is permanent (15, 42, 60).
For IHC and OHC counts, we stained the organ of Corti dissected from the cochleae of 11-month-old mice using the nuclear dye DAPI. Nuclei with uniform size, shape, and staining intensity are characteristic of healthy inner and OHCs (Fig. 3C). Despite the considerable hearing loss in KO mice, both WT and KO mice had mostly intact inner and OHCs, and, in general, a well-preserved cochlear structure. SGN count in H&E-stained cochlear sections showed only a marginal, although statistically significant, decrease in the number of SGNs in the KO mouse cochlea (Fig. 3D). It is worth noting, however, that this slight decrease in the number of SGNs could not be responsible for the severe age-related elevation of hearing thresholds and delay in the absolute wave latencies in Fus1 KO mice.
SGNs are connected to auditory hair cells via multiple synapses (65). Recent data have suggested IHC synaptic loss as an important mechanism of hearing loss after auditory damage and aging (28). We immunostained cochlear tissue sections with antibodies to CtBP2/Ribeye, a principal component of the presynaptic ribbon, to compare the numbers of IHC synapses in aging cochleae (60) with young animals that have 14–18 synapses/cell (Fig. 3E). We found an age-related decrease in all three cochlear turns with a more prominent loss in the apical turn in both WT and KO mice. However, the synaptic loss was significantly greater in KO mice (Fig. 3E).
Another histopathological change that was observed in 11-month-old KO mice but not in WT mice was the loss of a small spatially distinct class of fibrocytes (type IV) within the inferior region of the spiral ligament (SL) (Fig. 3F), which is vulnerable to sound exposure and aging. However, the impact of this type of pathology alone on hearing is inconclusive since the presence or absence of type IV fibrocytes does not completely correlate with the degree of hearing threshold shift (74, 27).
EM analysis points to the stria vascularis as the primary target tissue most vulnerable to mitochondrial damage and oxidative stress in aging Fus1 KO mice
Aging (senescence) is characterized by progressive accumulation of macromolecular damage, thought to be due to continuous minor oxidative stress associated with mitochondrial respiration. Earlier we showed that Fus1-deficient cells and tissues experience severe oxidative stress (70, 71, 76, 77). Thus, we hypothesized that accumulation of oxidative damage to macromolecules followed by loss of their function is the underlying cause of premature hearing loss in Fus1 KO mice. We performed transmission electron microscopy (TEM) analysis to identify cochlear cells dependent on Fus1 and mitochondrial activities. Cochleae were isolated from 12-month-old WT and KO mice, the age when KO mice have almost no hearing, while WT mice still maintain a robust auditory response (Fig. 1). We performed comparative analysis of four types of cells/tissues that are known to play major roles in ARHL.
Outer hair cells
In KO OHCs, we did not observe significant deviation from the WT morphology or innervation (data not shown). Also, no difference in mitochondrial morphology was observed between WT and KO OHCs (Fig. 4A).
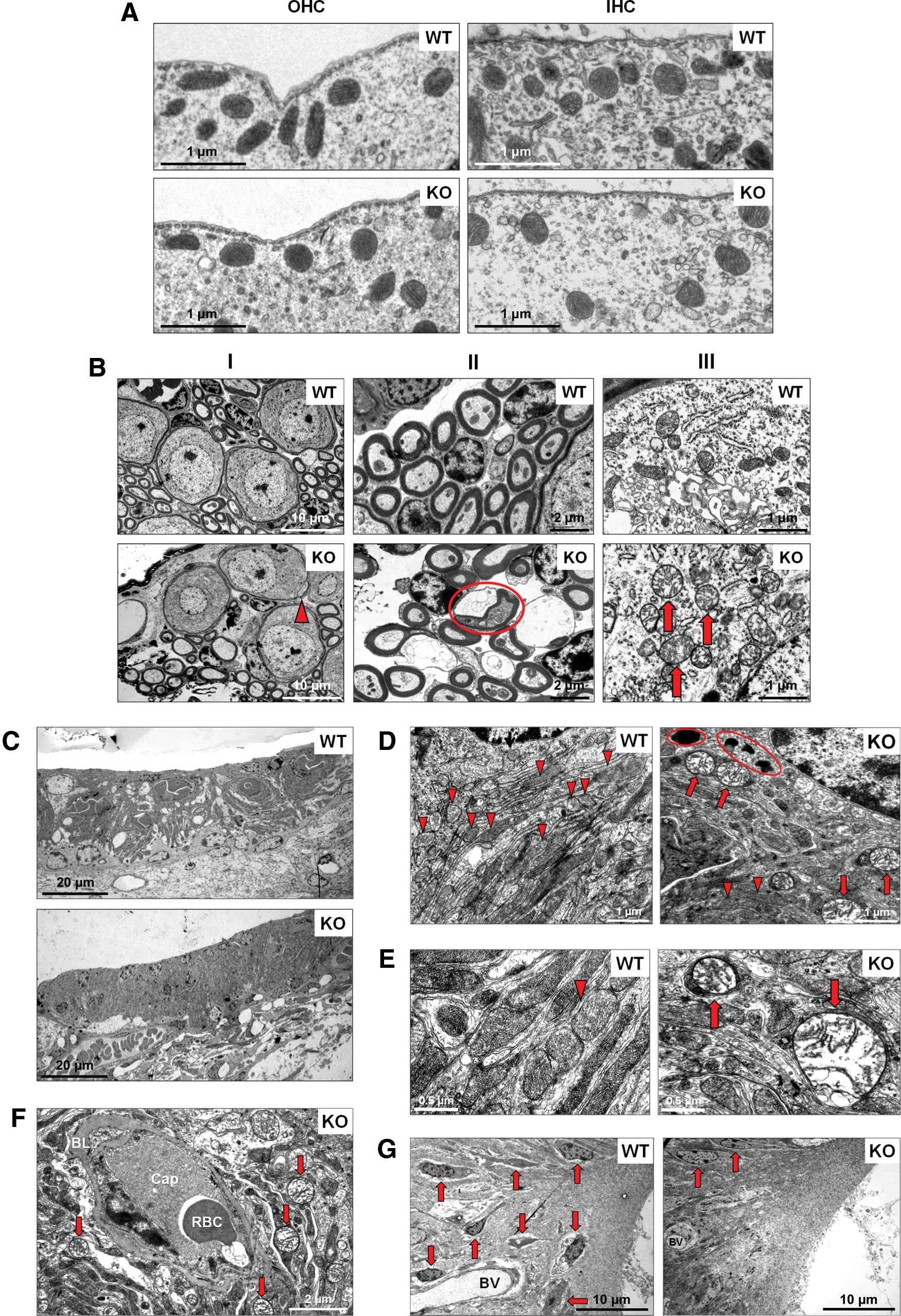
Inner hair cells
The general morphology of WT and KO IHCs was similar (data not shown). We also observed no difference in the number and structure of mitochondria between WT and KO IHCs (Fig. 4A).
Spiral ganglion neurons
In KO SGNs, we observed modest pathological alterations such as occasional myelin sheath splits (Fig. 4B-I) and areas of nerve fiber degeneration (Fig. 4B-II). At higher magnification, the differences in the morphological structure and sizes of WT and KO mitochondria were easily discernable. Mitochondria in KO SGNs were considerably larger in size and were mostly round in shape. Many of them had electron-lucent content and partially or fully disrupted cristae architecture indicative of mitochondrial pathology/degeneration (Fig. 4B-III).
Stria vascularis and SL
The SV has a dense capillary network and an exceptionally high metabolic rate, the highest of all cochlear tissues (34). The main role of the SV is to produce endolymph, the fluid within the scala media, and generate the endocochlear potential (EP) via activation of Na+-K+-ATPase pumps for ion balance maintenance. As this process requires a continuous energy supply, the SV has an extensive capillary network and numerous mitochondria inside marginal cell (MC) processes that interdigitate with intermediate cells (IC) (46). The SL is a dense layer of vascular connective tissue that participates in EP generation (46). The EP enhances the sensitivity of hair cells and, thus, is essential for audition.
TEM analysis showed no abnormality in the SV of aging WT mice while multiple signs of deterioration were observed in the KO SV and SL. First, MC of the SV contained multiple lipofuscin-like dark inclusions (Fig. 4D) that usually correlate positively with oxidative stress and mitochondrial damage (67). Second, the majority of mitochondria in MCs showed different degrees of pathological changes such as swelling, rounded shape, electron-lucent cytoplasm, as well as partially or entirely disrupted cristae architecture. Many mitochondria were drastically enlarged forming so-called giant mitochondria indicative of defective fission (Fig. 4D). Third, large mitochondria in KO SV were not surrounded by double-layered membrane, characteristic of autophagosomes, and therefore, not targeted for degradation via autophagy-like mitochondria in WT SV (Fig. 4E). Fourth, the SV appeared devoid of MC/IC interdigitations, possibly indicating a reduction in IC number, which has been shown to adversely affect EP production (24).
Another layer of pathological change was observed in the SV capillary network. Many capillaries in Fus1KO SV were occluded, filled with electron-dense material and dying blood cells (Fig. 4F). Neither endothelial nor pericapillary cells of KO vessels contained functional mitochondria (Fig. 4F), while WT mitochondria were easily discernable in cells of the capillary wall (data not shown). Moreover, while WT capillaries in SV were surrounded by numerous morphologically normal mitochondria, the KO capillary surroundings consisted of mostly swollen dysfunctional mitochondria and vacuolar structures (Fig. 4F). This suggests that occlusion and degeneration of capillaries in KO SV occur due to capillary cell energy crisis. It is noteworthy that the basal lamina was abnormally thick in KO capillaries compared to WT mice (Fig. 4F). Interestingly, these severe pathological changes were characteristic only for the SV capillaries, while capillaries located in the spiral ganglion from the same cochlea appeared normal reflecting a lower energy demand of this cell type compared to the SV network (Supplementary Fig. S2B).
The KO SL also showed multiple signs of deterioration/degradation. For example, the type IV fibrocyte area in the SL contained only a few normal fibrocytes while most of them had already been degenerated (Fig. 4G), which is consistent with our light microscopy data. The same type IV fibrocyte area in KO mice showed degenerating vessels, while WT vessels had normal morphology (Fig. 4G).
Thus, based on TEM examination, we concluded that the SV and SL, which showed signs of severe deterioration, are most likely the primary cause of premature hearing loss in Fus1 KO mice. However, pathological changes in IHCs and SGNs may be contributing factors.
Reduced EP in young adult Fus1 KO mice
The SV and fibrocytes of the SL play major roles in the generation and maintenance of the EP via constant ion transport to the endolymphatic space of the cochlea. The EP plays a critical role in maintaining normal hearing sensitivity (4, 61). In view of our EM data showing severe pathology in the SV of KO mice (Fig. 4), we hypothesized that premature loss of hearing in Fus1 KO mice was linked to altered EP. Thus, we measured EPs in 4–5-month-old WT and KO mice (Fig. 5). Representative EP recordings from WT and KO mice are shown in Figure 5B. EP measured in WT mice averaged 101.8 ± 2.1 mV (n = 3) (Fig. 5C), which is similar to published values. In contrast, the average EP in KO mice was 59.9 ± 7.6 mV (n = 3), 41.9% lower than in WT mice, p = 0.026592 (Fig. 5C), which directly demonstrates the importance of SV integrity/functionality for establishing the high EP.

These data suggest that the major mechanism of premature ARHL in Fus1 KO mice is an inability to maintain EP due to, most likely, mitochondrial dysfunction-caused energy crisis in cells of the SV and SL.
Cochlear tissues of 3-month-old Fus1 KO mice show pathological molecular changes in aging-associated pathways, antioxidant and autophagy machineries
To determine the molecular alterations in the KO mouse cochlea that initiate progressive hearing loss, we performed immunoblot analysis focusing on aging-associated pathways. To capture the onset of pathological changes, we analyzed proteins from cochleae of 1- and 3.5-month-old WT and KO mice. Cochleae extracted from five mice per group were pooled together to obtain sufficient protein amounts.
Starting from 1 month of age, we detected potentially detrimental molecular alterations in Fus1 KO mice (Fig. 6 and Supplementary Figure S3A, B). Among proteins of AO machinery, an increase in PRDX1 and a pathological decrease in mitochondrial Sod2 levels were observed (Fig. 6A). Autophagy marker LC3-II showed a Fus1-dependent decrease pointing to an early onset of autophagy dysregulation (Fig. 6D). The most dramatic and consistent changes were found in the PTEN/AKT pathway where total PTEN levels were decreased, while phosphorylation of AKT, a substrate for PTEN phosphatase activity, was increased (Fig. 6B). The level of the mitochondrial kinase PINK1, a mitochondrial quality control protein and PTEN substrate, was also noticeably downregulated (Fig. 6A).
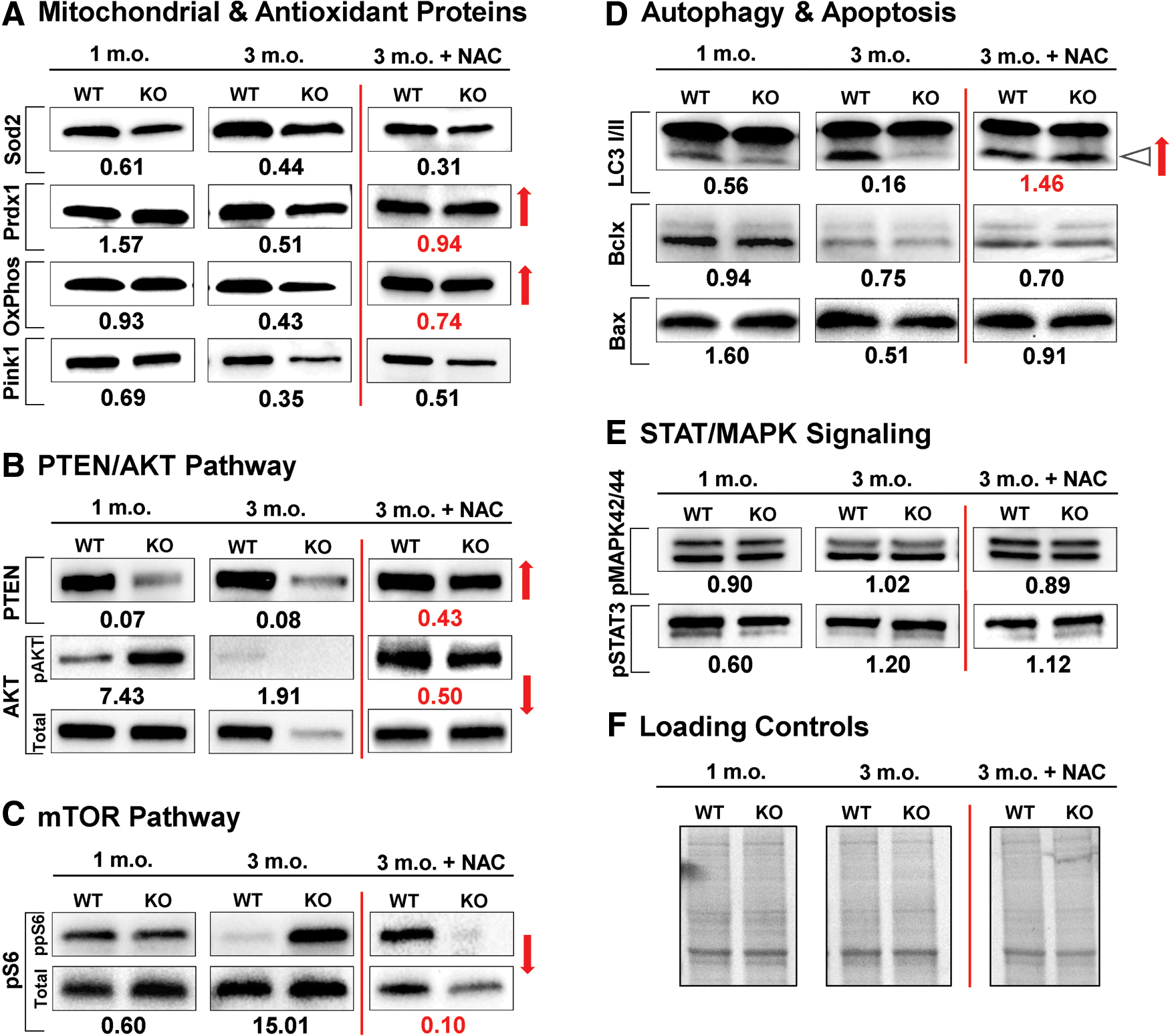
In the adult KO mouse cochlea (3.5 months), we detected escalation of molecular changes observed in younger mice as well as new critical changes (Fig. 6 and Supplementary Fig. S3A, B). The most unexpected change was a dramatic decrease/loss in total AKT levels in 3.5-month-old KO mice in contrast to 1-month-old KO mice that had increased AKT phosphorylation but no changes in total level of AKT (Fig. 6B). This suggests the existence of pathological factors other than PTEN dysregulation that affect AKT translation/degradation in adult KO cochleae. Along with dysregulation of AKT pathway, we observed an apparent mTOR activation (ppS6) in KO cochleae (Fig. 6C). It is noteworthy that mTOR activation (pS6 increase) was observed at both total and phosphorylated levels indicating that Fus1 affects different levels of protein regulation. We also found a further decrease in the levels of AO molecules and a decrease in the level of OxPhos I, a member of mitochondrial respiratory complex I (Fig. 6A). Further decrease in LC3-II levels suggested accumulation of dysfunctional mitochondria due to defective autophagy (Fig. 6D). The PTEN and Pink1 levels in both KO age groups were consistently lower than in WT mice (Fig. 6A, B).
Interestingly, some aging-associated signaling hubs, such as MAPK42/44 and STAT3 (Fig. 6E), as well as apoptotic proteins with the exception of Bax (Fig. 6D) showed no or marginal changes between KO and WT mice in young or adult mice, thus suggesting Fus1 deficiency specifically targets the PTEN/AKT and mTOR/autophagy axes.
Short-term AO supplementation alleviates/corrects pathological molecular changes in cochlear tissues of adult Fus1 KO mice
All molecular pathways analyzed in the present study are mitochondria/energy dependent. In our earlier studies and here (70, 71, 77) (Fig. 6 and Supplementary Fig. S3A, B), we showed that Fus1-deficient tissues experience chronic oxidative stress that may be detrimental for mitochondria and tissues with high-energy demand such as the cochlea. We hypothesized that relief of oxidative stress may improve mitochondrial activities, alleviate molecular pathology, and slow down hearing loss. To validate this hypothesis, 30 mM N-acetyl cysteine (NAC), an AO compound, was provided in drinking water to 3.5-month-old WT and KO mice for 30 days. Our choice of AO was substantiated by several data sets. First, NAC was one of the three AO supplements that was reported to alleviate hearing pathology in B6 mice when provided for 11 months in a study that tested the long-term effect of 17 different AO compounds (62). Second, NAC was shown to be able to penetrate the blood–brain barrier (BBB) (13), and third, it was shown to prevent mitochondria from oxidative injury, which is critical for our model of mitochondrial dysfunction (29).
To confirm that NAC relieves oxidative stress in the cochleae of KO mice, we compared the levels of AO proteins and found that PRDX1 and OxPhos I level 39 kDa were completely rescued in the cochleae of NAC-treated KO mice compared to WT mice (Fig. 6A). Remarkably, NAC treatment normalized or alleviated Fus1-dependent pathological changes in other aging-related proteins such as autophagy marker LC3-II and proteins from the nutrient (mTOR) and energy (PTEN/AKT) sensing pathways (Fig. 6, red arrows). Interestingly, not all proteins in the analysis were equally sensitive to NAC treatment. Thus, we did not observe significant NAC effects on mitochondrial proteins SOD2 or Pink1 (Fig. 6A), while levels of total and phosphorylated AKT, S6, and PTEN were drastically improved (Fig. 6B, C). This discordant response to AO intervention suggests that NAC may not efficiently penetrate mitochondria, or that in addition to oxidative stress, other pathological processes are associated with Fus1 loss in the cochlea.
Short-term antioxidant (NAC) supplementation prevents progression of hearing loss, improves ABR wave amplitudes, restores EP levels, and improves mitochondrial morphology in SV cells and SGNs in Fus1 KO mice
To determine the effect of AO treatment on progression of hearing loss in KO mice, we treated 4-month-old mice with NAC for 3 months. NAC supplementation significantly retarded hearing loss in KO mice making the ABR threshold levels of 7-month-old KO mice comparable to WT mice, while nontreated control KO mice of similar age had profound hearing loss (Fig. 7A). Moreover, NAC prevented both the reduction of wave I amplitudes (Fig. 7B) and the delay in wave I latencies (Fig. 7C), and almost restored the EP in KO mice to the level of 4–5-month-old WT mice (Fig. 5D).
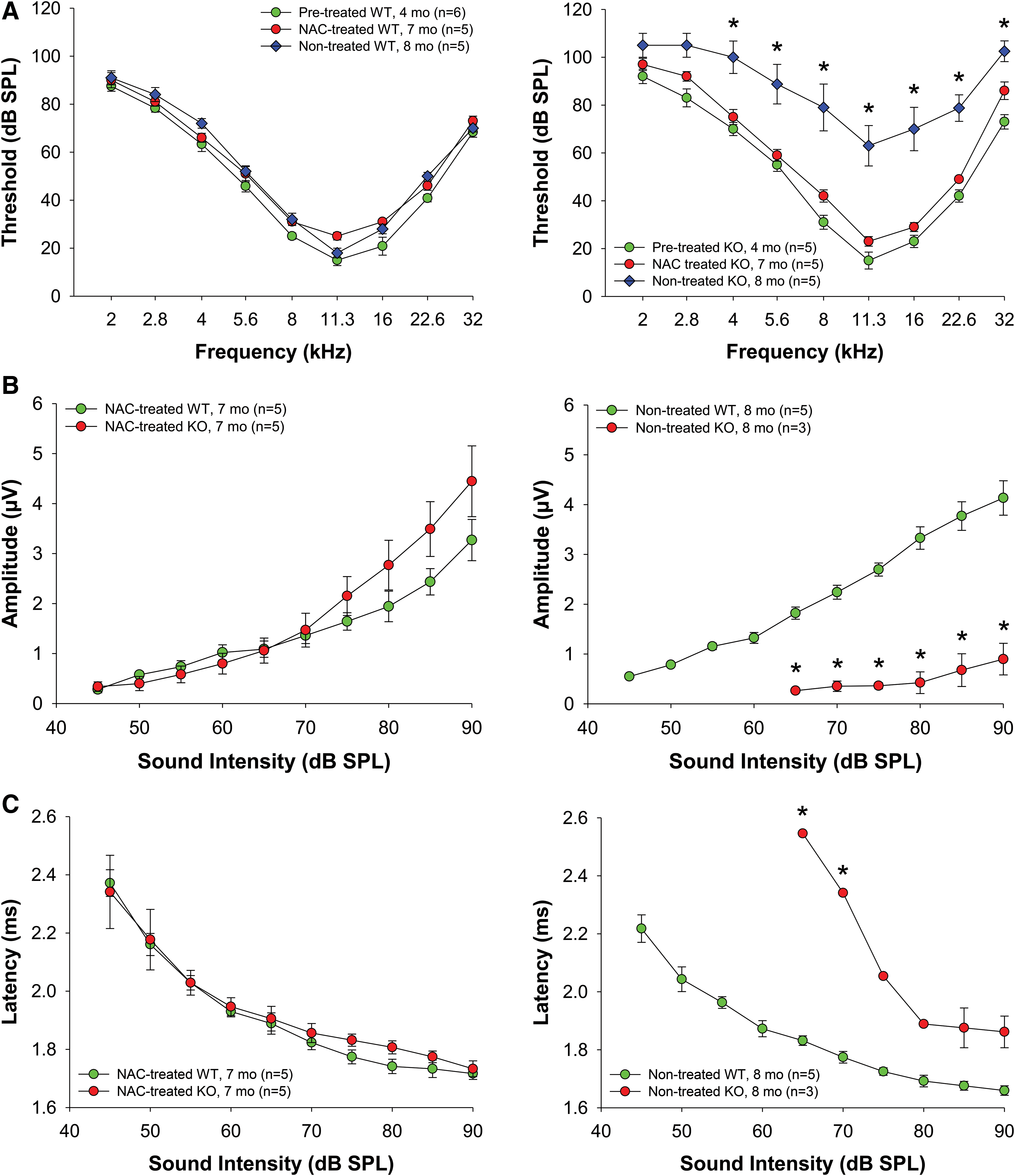
In addition, the effect of NAC at the ultrastructural level in the cochleae of KO mice was examined by TEM analysis (Fig. 8). In nontreated 5–6-month-old Fus1 KO mice, SV cells and SGNs had swollen mitochondria with disrupted cristae architecture, similar to that observed in 12-month-old KO mice, but not as severe. NAC supplementation of 4-month-old KO mice for 2 months significantly improved the morphology of mitochondria in SV cells and SGNs completely restoring cristae architecture. These results, therefore, suggest that oxidative stress is one of the major causative mechanisms of ARHL in Fus1 KO mice.

Discussion
Mitochondria present a unique and crucial set of key intracellular functions (72). The critical role of mitochondria in auditory function and ARHL began to emerge some time ago but is still understudied and underestimated (5). With regard to oxidative stress and mitochondrial function, only a few genes and loci linked to ARHL have been proposed as a result of candidate gene-based association human studies. Among them are AO enzymes superoxide dismutases (SODs) and glutathione S-transferases (GSTs), and mitochondrial uncoupling proteins (UCPs) that reduce the MMP in mammalian cells (16). Deletions/mutations in mitochondrial DNA or reduced expression of genes from mtDNA has also been associated with ARHL in humans (3, 35, 14). Animal studies largely confirmed the conclusion that oxidative stress, altered levels of AO enzymes, decreased activity of I, II, IV complexes/altered respiration, and increased mtDNA mutation rate lead to premature hearing loss (23, 37, 39, 48, 63, 69).
Types of mitochondrial dysfunction are defined by the activities of the mitochondrial protein(s) or mitochondrial hub(s) that are defective in a specific tissue. In addition to ATP production, other mitochondrial functions may define the type of dysfunction, such as maintaining calcium concentration within cellular compartments, calcium signaling that mediate responses to outside signals, regulation of the membrane potential, AO function, apoptosis, and regulation of cellular metabolism (44). It would be logical to suggest that mitochondrial impact on hearing is defined by the type of mitochondrial dysfunction. Thus, additional models of premature hearing loss linked to various mitochondrial activities are urgently needed to understand the basic mitochondrial mechanisms of ARHL and counteract them.
In the current study, we characterized a novel model of ARHL mediated by the loss of the mitochondrial protein Fus1 and determined its molecular mechanisms. In our earlier studies, we established Fus1 as one of the few regulators of mitochondrial calcium handling (71, 72). However, Fus1 KO cells were presented with dysregulation not only in calcium accumulation/distribution but also in calcium-coupled mitochondrial parameters (via so-called metabolic coupling), such as ROS production, mitochondrial potential, GSH content, mitochondrial fusion, and calcium signaling mediated via NFkB/NFAT axis. We hypothesized then that disruption of metabolic coupling in mitochondria may result in multiple cellular and systemic pathologies (71, 72).
Indeed, in this study, we provide multiple lines of evidence that Fus1 KO mice are a mechanistically novel, convenient, and clinically relevant model of premature ARHL. We found that besides early and fast progressing hearing decline (Fig. 1), young Fus1 KO animals have paradoxically increased ABR wave amplitudes (Fig. 2). Higher amplitudes normally reflect increased number of firing neurons or higher synchrony of firing. We speculate that higher ABR wave amplitudes in young KO mice may be a result of higher ROS production or altered intracellular calcium/signaling.
In our early work, we established that immune cells from young Fus1 KO mice are chronically activated, which could benefit young mice by better protecting them from acute infection, but may play a detrimental role during aging (20, 71). Here we suggest that heightened ABR responses in the young KO inform a higher metabolic demand, and may underlie the drastic drop of amplitudes and hearing later in life as we observed in aging KO mice (8–10 months) but not in age-matched WT mice (Fig. 2). We did not find that this phenotype is gender specific.
The patterns of ABR wave deterioration in adult KO mice are very similar to the data obtained from studies on aging individuals (25), suggesting that the Fus1 KO mouse is a clinically relevant model of ARHL, and therefore studying the molecular mechanisms of hearing loss in Fus1 KO mice as well as approaches for its prevention could be applicable to humans.
We ruled out death of sensory cells as a primary cause that often underlies age-related sensorineural hearing loss (75). The absence of mitochondrial pathology in OHCs and IHCs also suggests preservation of their bioenergetics levels in Fus1 KO organ of Corti. Although based on the slight but significant decrease in the number of SGNs and IHC synapses as well as on prominent mitochondrial swelling and morphological abnormalities in SGNs corrected by antioxidant treatment, we cannot exclude neural cell involvement at some stages of hearing pathology in KO mice.
However, the severe vascular and mitochondrial structural changes in the SV and SL, with reduced MC/IC interdigitations, strongly suggest that the basis of ARHL in Fus1 KO mice is of metabolic nature. The significant pathological changes of strial capillaries may cause a restrained cochlear blood flow, which can lead to mitochondria-mediated insufficiency in energy production that is needed to maintain ion pumping to the endolymph. The SV is critical for the generation and maintenance of the EP. Reduction in the EP results in a loss of electrical driving force for hair cell transduction current (4, 61). EP measurement in 4–5-month-old mice revealed a significant 41.9% reduction in KO mice (59.9 ± 7.6 mV) compared with WT mice (101.8 ± 2.1 mV) demonstrating that strial dysfunction contributes to cochlear pathology underlying progressive hearing loss in Fus1 KO mice.
Metabolic (based on strial pathology) ARHL is the least studied pathology (4, 64). No human genes that promote strial presbycusis have been identified nor is its pathophysiology well understood (45, 46). Although extensive strial degeneration coincides with hearing loss, EPs have never been measured in humans due to the absence of technology for this invasive measurement. In animal studies, until recently, only two well-studied models, Mongolian gerbils and Tyrp1(B-lt) mice (8), were known to undergo age-associated EP reduction. Recently, four other strains with modest to severe EP reduction during aging (C57BL/6-Tyr(c-2J) (50), BALB/cJ (47), CBA/CaJ, and NOD.NON-H2(nbl)/LtJ (49)) were described. So far, only one individual gene Tyrp1, which codes for tyrosinase-related protein (56), has been linked to age-related EP decline in these strains, although several candidate protein classes had been suggested such as AO machinery, ion channels, exchangers and pumps, immune-related genes, and cell repair genes (6, 46).
Therefore, our novel Fus1 KO model of mitochondrial dysfunction that affects the performance of AO machinery (76, 77), intracellular calcium distribution (71), and immune response (20, 70) may be of a great value for studying the etiology and molecular mechanisms of age-related strial degeneration.
To pinpoint the molecular basis of strial degeneration, we analyzed protein alterations in cochlear tissue of Fus1 KO mice. We used a logical candidate-based approach when choosing the proteins for analysis, including pathways and processes linked to (i) nutrient sensing and energy expenditure (PTEN/AKT and mTOR); (ii) mitochondrial activities; (iii) oxidative stress; (iv) apoptosis and autophagy. Our main quest was to identify primary Fus1-dependent molecular changes that drive other molecular and pathological changes later in life. Therefore, we compared protein levels and their phosphorylation/activation in the cochleae of 1- and 3.5-month-old animals. The most consistent and early alterations were observed in the PTEN/AKT pathway as well as the proteins of AO machinery, allowing us to consider them as the primary molecular events. Phosphatase and tensin homolog (PTEN) is involved in numerous important systemic functions, including anti-aging activities. It was shown to positively regulate longevity (51). Recently, a new metabolic role of PTEN as a positive regulator of energy expenditure through increased oxidative phosphorylation was revealed (52). ROS have been shown to oxidize the active site cysteine on PTEN (Cys124), resulting in inactivation of PTEN and perpetual activation of the AKT pathway (30). Moreover, mitochondrial ROS specifically were shown to inhibit PTEN and activate AKT (9), which is consistent with the molecular events we observed in young Fus1 KO mice.
Based on the ROS scavenging effects in Fus1 KO mice that resulted in rescue of PTEN/AKT pathway, we believe that consistently decreased PTEN levels and activation of AKT in the Fus1 KO cochlea are due to pathologically high levels of ROS. However, the exact nature of PTEN decrease in Fus1 KO cochleae is yet to be confirmed since PTEN is regulated by multiple mechanisms, including but not limited to phosphorylation, oxidation, binding to different partners, and transcriptional regulation (31). Considering the importance of PTEN in aging and hearing, there is no doubt that PTEN regulation is one of the key molecular mechanisms of Fus1 action in protection from premature hearing loss. It is worth noting that in our earlier work we showed that Fus1 is also involved in PTEN regulation in lungs during bacteria-induced inflammatory response (20).
Another important alteration we observed in 3.5-month-old KO mice is the decreased levels of OxPhos 39 kDa, a subunit of mitochondrial respiratory Complex I, a component of the oxidative phosphorylation chain. This decrease was rescued by AO treatment. Low levels of Complex I indicate low ATP levels in KO cochlear tissue since ATP is produced during oxidative phosphorylation. ATP deficiency may lead to an energy crisis and strial malfunction.
In line with PTEN inactivation/pAKT activation in young KO mice, in adult mice we observed activation of the mTOR pathway measured by ribosomal protein S6 phosphorylation, a downstream target of mTOR. MTOR lies at the heart of a nutrient-sensing signaling network that controls cellular metabolism and, thus, aging (21). mTOR activation has been linked to aging in multiple studies [(18), and reviewed in (53)]. It was suggested that aberrant mTOR activation in aging is a response to mitochondrial stress in cells (41), which is consistent with the processes that may take place in the Fus1-deficient cochlea.
Interestingly, in 1- and 3.5-month-old KO cochleae, we observed negative age-related dynamics in accumulation of the autophagy marker LC3-II. Autophagy is an evolutionarily conserved catabolic process that targets proteins and organelles to lysosomes for degradation, followed by recycling of free amino acids and ATP for biosynthesis (68). Autophagy mediates cytoprotective effects (40). During autophagy a cytosolic form of LC3 (LC3-I) is converted via conjugation to phosphatidylethanolamine to LC3-II, which is recruited to autophagosomes. High levels of LC3-II reflect a higher autophagy rate and vice versa. In our study, based on LC3-II and Pink1 decrease and accumulation of large swollen mitochondria, we concluded that Fus1 KO cochlear tissues are deficient in autophagy/mitophagy. Moreover, in line with deficient autophagy, we registered activation of mTOR in 3.5-month-old KO mice, which has been shown to suppress autophagy and, thus, promote aging [(43) and references therein]. Normalization of LC3-II levels in Fus1 KO cochleae after AO treatment confirms the link of oxidative stress and autophagy.
Finally, in line with all the data pointing to deficient autophagy/mitophagy in KO cochleae, we showed downregulation of PINK1 (PTEN-induced putative kinase 1) protein in 1- and 3-month-old KO mice. The mitochondrial protein PINK1 is a PTEN-induced putative kinase regulating mitochondrial quality control via activation of mitophagy (36). It is a critical protector from age-related diseases linked to dysfunctional mitochondria (7, 54). In the Fus1 KO setting, decreased levels of PINK1 are in line with decreased levels of its upstream regulator PTEN (Fig. 6). In terms of long-term consequences for tissue integrity, PINK1 deficiency could be detrimental for aging Fus1 KO tissues as it leads to inability to withstand oxidative and environmental stress due to defective mitochondrial quality control resulting in accumulation of dysfunctional mitochondria and deterioration of tissues due to energy crisis as we observed in the SV of aging 8–11-month-old Fus1 KO mice.
Apoptotic markers that we used to monitor cell death in young Fus1 KO cochlear tissues showed little difference in expression between KO and WT mice (Fig. 6D, Bcl-X, and Bax), suggesting that deficiency in autophagy may result in other types of cell death in the cochlea (55).
It was also interesting to find, in terms of Fus1 biology, that crucial signaling hubs such as STAT3 (11) and MAPK42/44 (66) showed no major perturbations, suggesting that the antiaging effect of Fus1 is promoted specifically through PTEN/AKT/mTOR pathways partially mediated through control of oxidative stress.
Simple supplementation of the AO NAC to Fus1 KO mice for 1 month increased the level of AO enzymes, normalized PTEN/AKT and mTOR pathways, and protected mitochondria from dysfunction and degradation. Based on our early studies of Fus1 protein biology (70 –72), we suggest that normalization of ROS levels in Fus1-deficient tissues helps in recovering mitochondrial integrity, MMP and mitochondrial calcium transport across the membrane and, thus, improve mitochondrial activities, including ATP production and Ca2+ distribution, dynamics, and calcium signaling. These changes in different mitochondrial processes could explain the multitude of effects of NAC-mediated ROS scavenging in the Fus1 KO cochlea.
Importantly, NAC supplementation for 3 months produced several highly beneficial changes in hearing of Fus1 KO mice, including prevention of age-related hearing decline, normalization of ABR peak amplitudes, and rescue of the EP. WT mice did not show significant auditory changes in response to NAC treatment at this age, most likely due to the absence of degenerative processes in auditory system at the analyzed age. Short-term NAC treatment also significantly improved the morphology of mitochondria in cells of the SV and SGNs. These results strongly suggest that oxidative stress is one of the major pathogenic factors in ARHL in Fus1 KO mice. It is likely that more potent AOs that specifically target mitochondrial ROS and have better efficacy would produce even more robust rescuing effects in Fus1 KO mice.
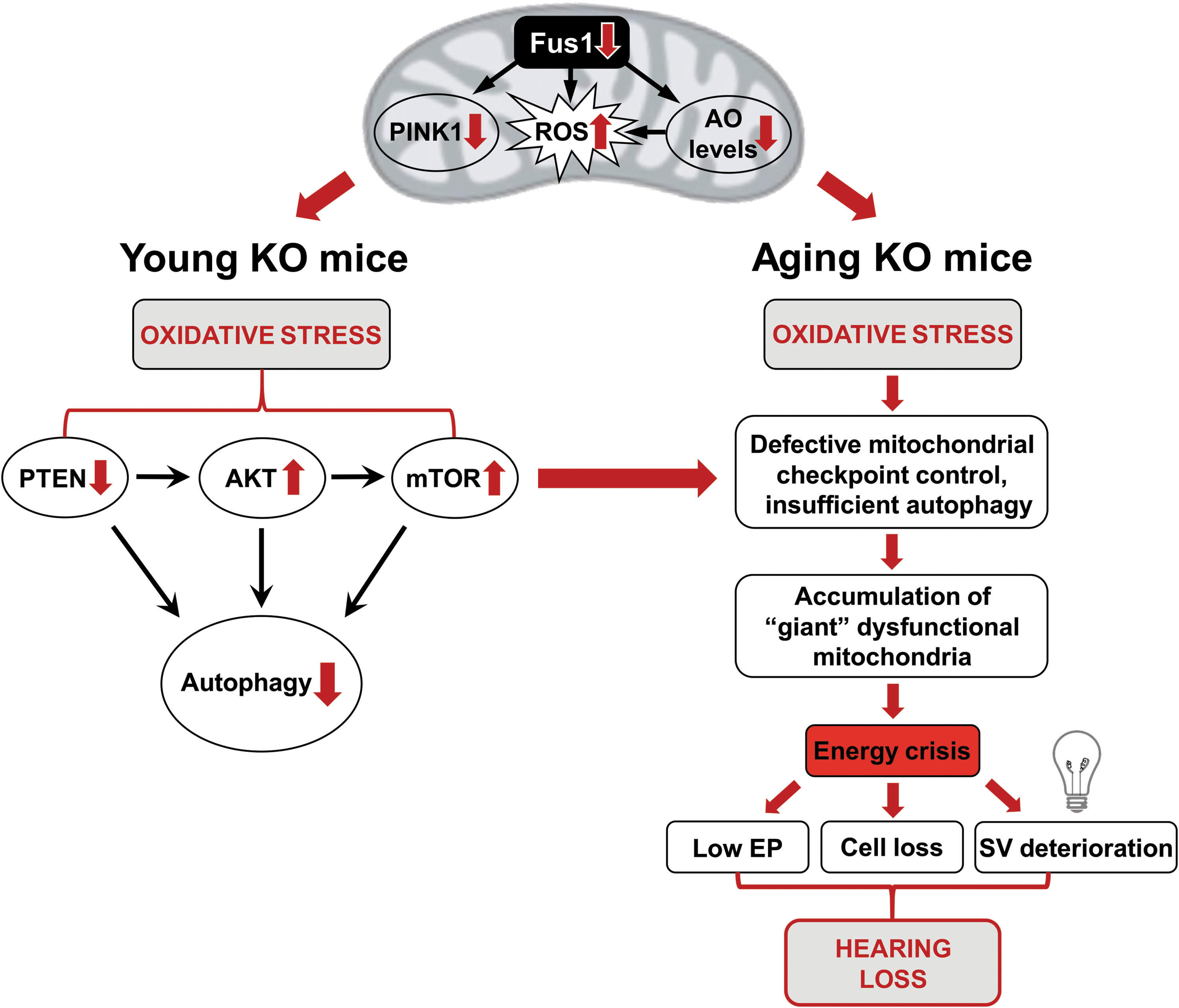
A schematic illustration of our data presented in Figure 9 suggests how loss of Fus1 results in premature hearing loss that begins with molecular changes in the cochleae of very young Fus1 KO mice. Our data suggest that efficient targeting of mitochondrial dysfunction-mediated premature hearing loss may require combinatorial approaches that will include AO, anti-inflammatory, anti-TOR, calcium balancing, and other therapies.
Materials and Methods
Fus1 KO mouse model
Fus1 KO mice generated by Dr. A Ivanova (22) were backcrossed to 129sv background in the laboratory of Dr. S Anderson (NCI-Frederick). All animal experiments were performed according to a protocol approved by the Yale University Institutional Animal Care and Use Committee (IACUC) and the animals were cared for according to the recommendations in the “Guide for the Care and Use of Laboratory Animals” (National Institutes of Health). The mice were fed with a standard diet. housed in standard cages (five per cage), and maintained in a 12-h light–12-h dark cycle. They had ad libitum access to drinking water and normal diet throughout the experiment. Mice of both genders were used in the study.
NAC supplementation
In all experiments described (30 and 90 days of treatment), N-acetyl cysteine (NAC) (Cat# A7250; Sigma-Aldrich, Inc.) was dissolved in drinking water (30 mM) and supplied ad libitum to the mice. For the first 2 days, mice were provided with 15 mM NAC water to acclimate them to the taste and then it was replaced with 30 mM NAC for the remainder of the experiment. NAC water was the only source of drinking water during the experiments. NAC water was changed twice a week.
Auditory brainstem responses
ABR represents the activity of the auditory nerve and the central auditory pathways in response to sounds. ABR measurements were carried out within a sound attenuating booth (Industrial Acoustics Corp.). Mice were first anesthetized with chloral hydrate (480 mg/kg i.p) and then placed onto a heating pad to maintain body temperature at 37°C. Subdermal needle electrodes (Rochester Electro-Medical, Inc.) were placed at the vertex (active, noninverting), the infra-auricular mastoid region (reference, inverting), and the neck region (ground). The acoustic stimuli for ABR were produced and the responses recorded using a customized TDT3 system (Tucker-Davis Technologies, Inc.) controlled by BioSig (TDT), digital signal processing software. Differentially recorded scalp potentials were bandpass filtered between 0.05 and 3 kHz over a 15-ms epoch. A total of 400 trials were averaged for each waveform for each stimulus condition. ABRs were elicited with digitally generated (SigGen; TDT, Inc.) pure tone pips presented free field via a speaker (TDT, Inc.; Part FF1 2021) positioned 10 cm from the vertex. Symmetrically shaped tone bursts were 3 ms long (1 ms raised cosine on/off ramps and 1 ms plateau) and were delivered at a rate of approximately 20 per second. Stimuli were presented at frequencies between 2 and 32 kHz and in 5 dB decrements of sound intensity from 90 dB SPL (or 110 dB SPL if thresholds exceeded 90 dB SPL). The ABR threshold was defined as the lowest intensity (to the nearest 5 dB) capable of evoking a reproducible, visually detectable response.
Amplitudes (μV) and latencies (ms) of the initial four ABR peaks (waves I, II, III, and IV) were then determined at 16 kHz. The most sensitive frequency range of hearing in mice is 11.3–22.6 kHz, and 16 kHz is half octave in-between, so was therefore chosen for analysis. The analysis was carried out offline in BioSig on traces with visible peaks by setting cursors at the maxima and minima (trough) of the peaks. Latency was determined as the time from the onset of the stimulus to the peak, while amplitude was measured by taking the mean of the ΔV of the upward and downward slopes of the peak.
EP measurement
EP was measured in nontreated and NAC-treated WT and Fus1 KO mice. The method of EP measurement is illustrated in Figure 5A. Mice were deeply anesthetized with sodium pentobarbital (45 mg/kg, i.p.) and placed onto a heating pad to maintain body temperature at 37°C. Animals were then secured onto a stereotaxic mouse head holding adaptor (MA-6 N; Narishige) mounted onto a ball-and-socket stage (Model M-RN-50; Newport Corporation) and a magnetic base (Model 100; Newport Corporation). A silver–silver chloride reference electrode was placed under the skin of the chest. A tracheotomy was first performed and then the round window of the cochlea was exposed via a ventral approach by opening the auditory bulla of the temporal bone. A microelectrode (5–15 MΩ 1B150F-4; World Precision Instruments, Sarasota, FL) filled with 3 M KCl was positioned in the round window using a micromanipulator with a pulse motor driving unit (PF5-1; Narishige). An Axon 200A patch clamp amplifier was used for current clamp recording with an Axon Digidata 1321A and jClamp software version 22.8.4 (Scisoft, Inc.). When the microelectrode was inserted into the perilymph of the scala tympani, the voltage was balanced to 0 mV and then the microelectrode was advanced in 5 μm steps through the basilar membrane and into the endolymph of the scala media to measure the EP. For confirmation of the EP recording, the microelectrode was withdrawn back into the scala tympani, advanced again into the scala media, and continued further into the scala vestibuli, and then retracted back again into the scala tympani. After the EP measurement, the animals were euthanized with an overdose of sodium pentobarbital.
Confocal immunofluorescence
Cochleae were extracted from the temporal bones of 11-month-old WT and Fus1 KO mice, perfused, fixed for 1 h in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS), then washed, and stored in PBS at +4°C. The bony capsule, lateral wall, and modiolus were removed under a dissection microscope using fine forceps. The cochleae were incubated with 5% goat serum in 0.1% Triton/PBS for 30 min at room temperature followed by incubation overnight with mouse monoclonal CTBP2 primary antibody (Cat# 612044; BD Biosciences, Inc.) diluted 1:500 in 2% goat serum/antibody diluent (Cat # S202230-2; Dako, Inc.). The next day, the cochleae were washed thrice in 0.1% Triton X-100/PBS followed by incubation with the secondary antibody Alexa Fluor 568-conjugated rabbit (Cat # A-11011; Thermo Fisher Scientific, Inc.) at room temperature for 1 h. Mounting on glass slide was done in the DAPI mounting solution (Cat# H-1200; Vector Laboratories, Inc.). During mounting, SLs were dissected away to allow good exposure of the organ of Corti, and the cochleae were separated into three parts, the apical turn (corresponding to frequencies between 200–1.5 kHz), middle turn (1.5–16 kHz), and basal turn (>16 kHz). DAPI-labeled IHCs, OHCs, and IHC synaptic ribbons were visualized with a Zeiss LSM410 laser confocal microscope (Carl Zeiss Microscopy). Sensory hair cell loss was quantitatively evaluated by counting missing hair cells in the apical, middle, and basal turns of the cochlea. Images were collected and analyzed with a ZEN camera and ZEN software. CTBP2-positive synaptic ribbons were counted per IHC (3–5 fields per region, 10–12 IHC per field) and then averaged/region.
H&E staining and immunohistology
Cochleae were fixed in 4% PFA in PBS for 12–48 h, washed in PBS, decalcified in 0.1 M EDTA for 3–4 days, dehydrated through an ethanol gradient, and then embedded in paraffin. Sections of 5 μm thick in the midmodiolar, vertical plane were stained with H&E, then analyzed using a Nikon Eclipse 80i Microscope (Nikon Instruments, Inc.). The SGN density (cells in 3–5 areas were counted and averaged) in Rosenthal's canal of the basal turn was counted. Immunostaining with rabbit anti-Iba1 antibodies (Wako Chemicals, Inc.) on thin sections was performed using automatic Omni Multimer detection. Statistical analysis was performed using the Student's t-test to compare KO genotype and WT groups.
Cochlear protein lysate preparation and Western blot analysis
Both cochleae from each mouse were harvested, and one was immersed in 4% PFA in 0.1 M phosphate buffer (PB; fixative solution; pH 7.4) and used for immunohistochemical staining. The contralateral cochlea was placed in lysis buffer (RIPA plus protease inhibitor cocktail), perfused with the lysis buffer through the oval and round windows, snap-frozen, and kept at −80°C until required for lysate preparation. For protein lysate preparation, five cochleae from five WT or KO animals were pooled into an Eppendorf tube, 100 μL of lysis buffer was added to the tube, and the cochlear tissues were homogenized using a plastic mini-pestle. Lysates were sonicated and cleared via 15 min of centrifugation at 14,000 rpm in a microcentrifuge (Eppendorf). The supernatants were then transferred to a fresh tube. Western blot analysis of cochlear tissues was performed as described in our previous articles (76, 77).
Antibodies for Western blot analysis
The following antibodies were used in the study: rabbit anti-Sod2 (Cat# ab13533; Abcam), rabbit anti-PRDX1 (Cat# HPA007730; Sigma-Aldrich, Inc.), mouse anti-OxPhos 39 kDa (Cat# 45-8199; Thermo Fisher Scientific, Inc.), and mouse anti-Pink1 (Cat# NBP2-36488; Novus Biologicals). The following antibodies were bought from Cell Signaling Technology, Inc.: rabbit anti-AKT total and pAKT (Cat## 9272 and 3787), mouse anti-pS6 total (Cat# 2317), rabbit anti-ppS6 (Cat# 2221), rabbit anti-pSTAT3 (Cat# 9131), rabbit anti-pMAPK42/44 (Cat# 4695), rabbit anti-Bax (Cat# 5023), mouse anti-BclX (Cat# 2764), and mouse anti-PTEN (Cat# 9552).
TEM analysis
Cochleae from 5-6 month-old and 12-month-old WT and KO mice were extracted and deboned as described above and fixed in 2.5% glutaraldehyde and 2% PFA in 0.1 M sodium cacodylate buffer pH 7.4 for 1 h. After fixation, the cochleae were rinsed in cacodylate buffer, then postfixed in 1% osmium tetroxide in cacodylate, and en bloc stained in 2% aqueous uranyl acetate for a further hour each. The samples were well rinsed, followed by dehydration in an ethanol series, infiltrated with epoxy resin Embed 812 (Electron Microscopy Sciences), and baked overnight at 60°C. Hardened blocks were cut using a Leica UltraCut UC7. Sections (60 nm) were collected on formvar/carbon-coated grids and contrast stained using 2% uranyl acetate and lead citrate. The sample sections were viewed using an FEI Tecnai Biotwin Transmission Electron Microscope (FEI, Hillsboro, OR) at 80Kv. Images were taken using MORADA CCD and iTEM (Olympus) software.
Footnotes
Acknowledgments
This work was supported by grants R21 DC014357-02 to A.I. and R01 000273 and 008130 to J.S., all from NIDCD, National Institutes of Health, USA and OSHE grant to L.S. I apologize to those colleagues whose work I could not cite owing to space limitations. The authors of this article declare no conflicts of interest or competing financial interests.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.