
Abstract
Aims:
Protein succination by fumarate increases in the adipose tissue of diabetic mice and in adipocytes matured in high glucose as a result of glucotoxicity-driven mitochondrial stress. The endoplasmic reticulum (ER) oxidoreductase protein disulfide isomerase (PDI) is succinated in adipocytes that are matured in high glucose, and in this study we investigated whether succination would alter PDI oxidoreductase activity, directly linking mitochondrial stress and ER stress.
Results:
Protein succination and the ER stress marker C/EBP homologous protein (CHOP) were diminished after pharmaceutical targeting of mitochondrial stress with the chemical uncoupler niclosamide in adipocytes matured in high-glucose concentrations. PDI was succinated by fumarate on both CXXC-containing active sites, contributing to reduced enzymatic activity. Succinated PDI decreased reductase activity in adipocytes matured in high glucose, and in db/db epididymal adipose tissue, in association with increased levels of CHOP. PDI succination was increased in fumarase knockdown adipocytes, leading to reduced PDI oxidoreductase activity, increased CHOP levels, and pro-inflammatory cytokine secretion, confirming the specific role of elevated fumarate levels in contributing to ER stress. In addition, PDI succination and ER stress were decreased, and PDI reductase activity was restored when exposure to chronic high glucose was limited, highlighting the importance of calorie restriction in the improvement of adipocyte metabolic function.
Innovation:
These experiments identify PDI succination as a novel biochemical mechanism linking altered mitochondrial metabolism to ER stress in the adipocyte during diabetes.
Conclusion:
The current study demonstrates that early biochemical changes in mitochondrial metabolism have important implications for the development of adipocyte stress. Antioxid. Redox Signal. 27, 1281–1296.
Introduction
P
We examine the role of protein succination as a novel physiological initiator of prolonged endoplasmic reticulum (ER) stress. Succination of protein disulfide isomerase (PDI) active-site cysteines inhibits PDI enzymatic activity, contributing to an accumulation of unfolded proteins during diabetes. Both the chemical uncoupler niclosamide and high-glucose limitation protect against ER stress via reduction of protein succination in the adipocyte.
Hyperinsulinemia, hypoxia, and inflammation have been proposed to initiate the development of ER stress in human adipose tissue (9, 19). ER stress has also been described in 3T3-L1 adipocytes (particularly through protein kinase RNA-like ER kinase signaling) and in the adipose tissue of type 2 diabetic mice (16, 25, 31, 39). The treatment of adipocytes from obese subjects with lipopolysaccharides, high-glucose concentrations, or free fatty acids has been shown to promote ER stress in vitro (2, 21). Although all of these may be physiologically relevant contributors to ER stress, there remains limited insight on the mechanistic processes that directly cause disturbed protein folding in the adipocyte (49, 52). In this study, we identify a novel biochemical mechanism that is responsible for reduced protein folding and increased ER stress in the adipocyte during diabetes.
Protein disulfide isomerase (PDI) is the most abundant ER oxidoreductase that facilitates the formation of correctly configured substrate disulfide bonds (17, 45). PDI contains two active sites with the sequence motif -CGHC- in the a and a′ domains, and both of these play crucial roles in the reduction and oxidation of substrate disulfide bonds in the ER (3, 18, 23, 30). ER oxidoreductin 1α (Ero1α) functions in re-oxidizing PDI by using similar CXXC active sites, to sustain oxidative protein folding (57). S-glutathionylation of PDI active-site cysteines in both HL60 leukemia cells and SKOV3 ovarian cancer cells alters PDI protein structure and inhibits PDI isomerase activity in vitro, leading to stimulation of the UPR and the production of CHOP (56, 62). In addition, nitrosative stress-induced S-nitrosylation of PDI promotes UPR activation, ER stress, and eventual cell death in cerebrocortical neurons (58). These models demonstrate that the post-translational modification of PDI active-site cysteines precedes the development of ER stress.
Oxidized low-density lipoprotein (oxLDL) treatment of HMEC-1 cells augments CHOP messenger RNA (mRNA) levels and ER stress, specifically due to decreased PDI reductase activity (37). However, HMEC-1 cells that over-express native PDI are immune to the induction of ER stress with oxLDL treatment (37). Importantly, the restoration of functional ER chaperone levels is sufficient to decrease CHOP protein expression and ameliorate ER stress in the adipocyte (32, 64); therefore, it is critical to mechanistically understand how chaperone function is altered under diabetic conditions.
We had previously confirmed that the ER residents PDI and 78 kDa glucose-regulated protein (GRP78) are succinated by endogenously produced fumarate in the adipocyte under high-glucose conditions (38). Protein succination occurs when fumarate reacts non-enzymatically with cysteine thiols to produce the irreversible modification S-2(succino)cysteine (2SC) (1, 36, 38). The analysis of proteomic targets identified to date suggests that low pKa thiols that may also be susceptible to oxidation as well as those with increased solvent accessibility may be the most likely to react with endogenously produced fumarate (34, 35). Protein succination is increased in the adipose tissue of db/db mice and in 3T3-L1 adipocytes that are matured in high glucose (10–30 mM) (11, 38).
We previously demonstrated that the adenosine triphosphate (ATP):adenosine diphosphate (ADP) ratio, Nicotinamide adenine dinucleotide (NADH):NAD+ ratio, and the mitochondrial membrane potential are significantly increased in 3T3-L1 adipocytes matured in high (30 mM) glucose versus normal (5 mM) glucose concentrations (12, 53). In the metabolically challenged adipocyte, the intracellular fumarate levels are elevated due to glucotoxicity-driven mitochondrial stress and this results in the accumulation of succinated proteins (Fig. 1) (12). Considering that protein succination occurs as a direct result of mitochondrial stress in the adipocyte, and that functional PDI is critical to maintain ER proteostasis, we propose that the succination of PDI active-site thiols would inhibit its oxidoreductase activity, contributing to ER stress.
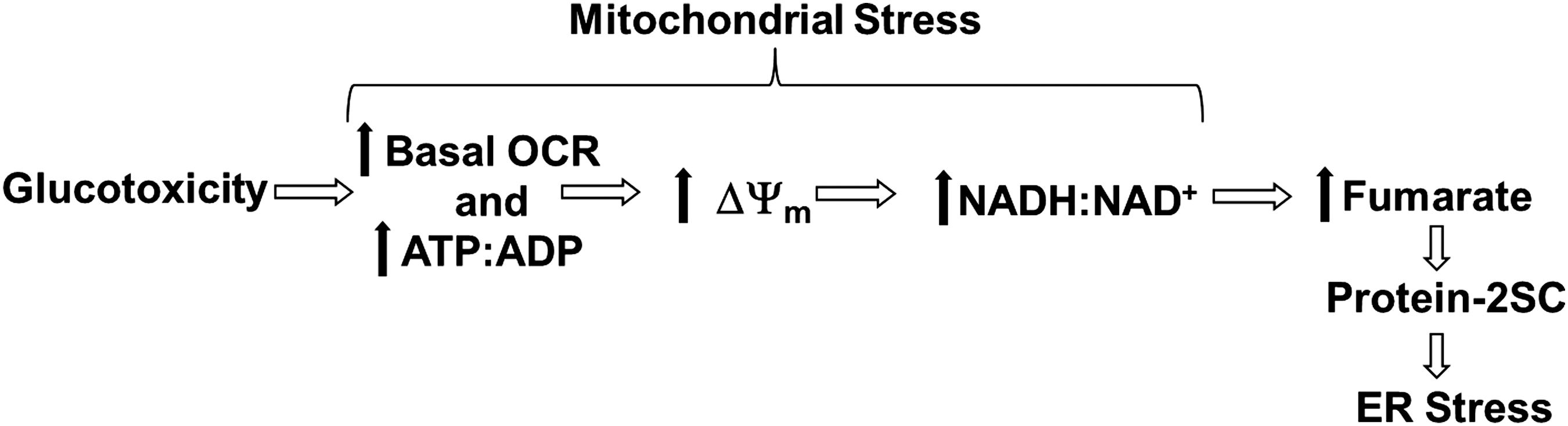
Mitochondrial uncouplers lower the inner mitochondrial membrane potential, created during glucose oxidation, by shuttling protons back into the mitochondrial matrix without generating additional ATP. Mitochondrial uncouplers are increasingly being investigated as therapeutic strategies to protect against coronary heart disease, ischemia-reperfusion injury, nonalchoholic steatohepatitis, obesity, and diabetes (26, 33, 41, 63). We had previously shown that the treatment of adipocytes matured in high glucose with mitochondrial uncouplers, 2,4-dinitrophenol (DNP) and carbonyl cyanide m-chlorophenyl hydrazone (CCCP), reduces both the intracellular fumarate concentration and the degree of protein succination (12). Although DNP and CCCP are effective in vitro, both drugs are considered toxic and are unsuitable therapeutics.
Niclosamide is a mild mitochondrial uncoupler (47, 54) that is approved by the US Food and Drug Administration as an anthelmintic drug for treating intestinal infections of tapeworms. Importantly, Tao et al. have demonstrated the beneficial outcomes of niclosamide in db/db mice after 2 months of oral administration, where niclosamide lowered both blood glucose and glycated hemoglobin levels and normalized plasma insulin levels (54). We investigated whether targeting mitochondrial stress by using niclosamide or a model of simple caloric restriction would subsequently reduce protein succination and eliminate ER stress in the adipocytes matured in high glucose. The results of this study provide a novel mechanistic link between mitochondrial stress and the development of sustained ER stress, as evidenced by increased CHOP production, in the adipocyte during diabetes.
Results
Mitochondrial stress drives ER stress in adipocytes matured in high glucose
The development of mitochondrial stress as a direct result of increased glucose oxidation (Fig. 1) is expected to increase the basal oxygen consumption rate (OCR) of the adipocyte under diabetic conditions. We matured 3T3-L1 adipocytes in glucose and insulin concentrations that we had previously shown as physiologically representative of normal versus hyperglycemic/hyperinsulinemic conditions (5 mM glucose/0.3 nM insulin vs. 30 mM glucose/3 nM insulin) (53). We observed a ∼90% increase in the basal OCR of adipocytes matured in 30 mM glucose compared with in 5 mM glucose after 2 days of maturation (Supplementary Figs. S1B and S12B; Supplementary Data are available online at
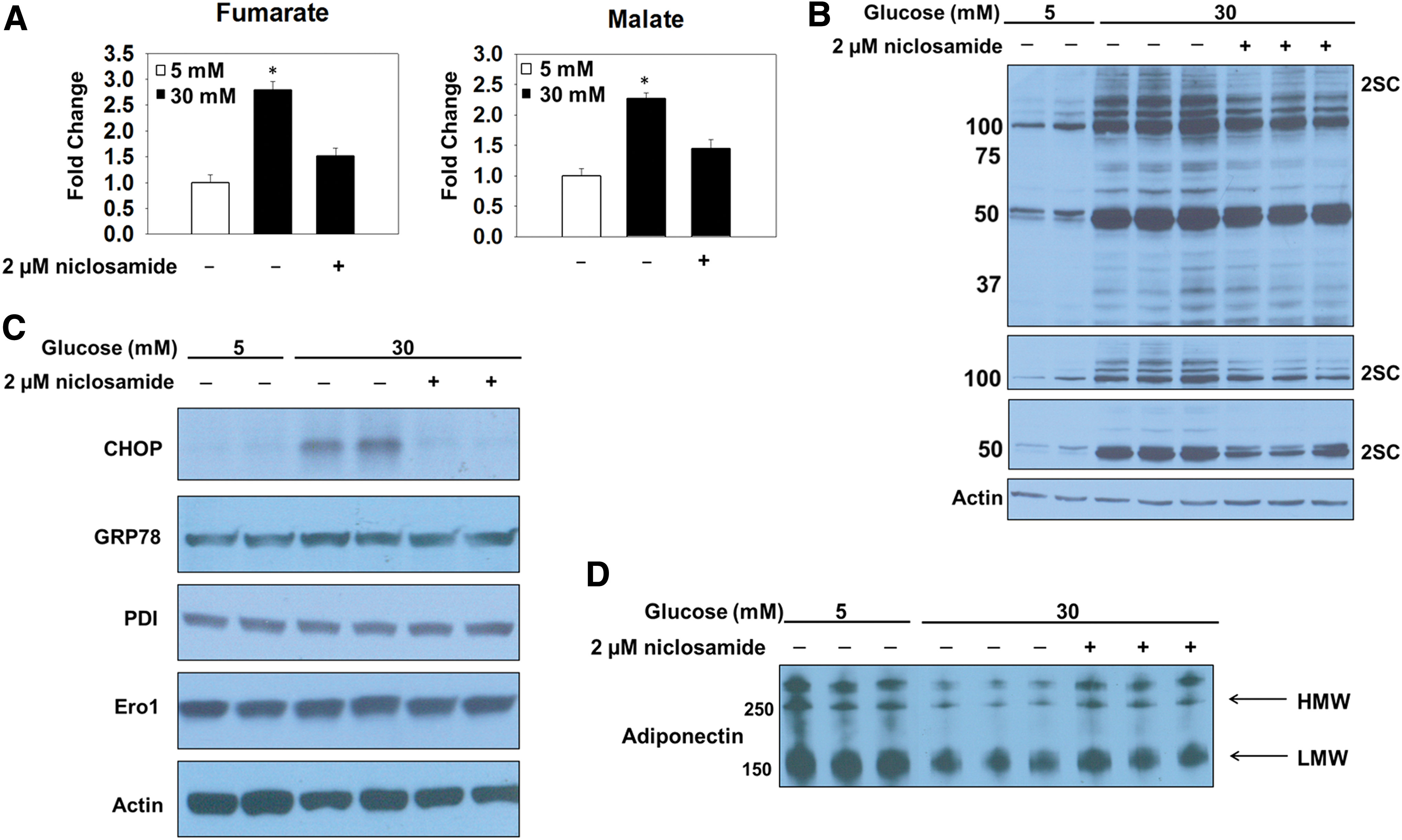
Protein succination occurs as a direct result of mitochondrial respiratory control in the metabolically challenged adipocyte (Fig. 1) (12); therefore, we hypothesized that uncoupling the inner mitochondrial membrane by using niclosamide would lower fumarate and 2SC levels. 3T3-L1 adipocytes were matured in 5 or 30 mM glucose or 30 mM glucose and treated with 2 μM niclosamide for 8 days. Metabolomic analyses substantiated the data that uncoupling the inner mitochondrial membrane, demonstrated by the acute increase in the basal OCR (Supplementary Fig. S1D) (54), prevents the accumulation of Krebs cycle intermediates in adipocytes matured in high glucose concentrations (Fig. 2A and Supplementary Fig. S1E). Immunoblotting with the anti-2SC antibody demonstrates that niclosamide treatment reduces the degree of protein succination in adipocytes matured in 30 mM glucose concentrations (Fig. 2B and Supplementary Fig. S6A). The lower panels depict shorter exposure times of the same anti-2SC IB and highlight the decreased succination of several higher-molecular-weight proteins (50–150 kDa) (Fig. 2B and Supplementary Fig. S6A). Examining the relationship between protein folding, ER stress, and glucose concentration, we assessed the levels of CHOP, Ero1, and PDI in adipocytes matured in 5 or 30 mM glucose. The levels of CHOP were pronouncedly increased in adipocytes matured in high glucose compared with normal glucose (Supplementary Figs. S1C and S12C), confirming that persistent ER stress is selectively present in adipocytes matured under hyperglycemic conditions (53). It is important to note that we did not detect differences in the protein levels of GRP78 or any components of the PERK, IRE1, or ATF6 branches of the UPR in adipocytes in normal/high glucose as the UPR is induced as a normal function of adipocyte maturation in the presence of insulin (16, 36, 53). To establish a novel link between mitochondrial stress and the sustained ER stress, we examined CHOP protein expression levels in adipocytes matured in 5, 30, or 30 mM glucose treated with 2 μM niclosamide for 8 days (Fig. 2C and Supplementary Fig. S6B). The data confirm that alleviating mitochondrial stress prevents the presence of prolonged ER stress generated by high-glucose conditions. Although the transcription of ER chaperones may be upregulated in parallel with ER stress, we did not detect any changes in the total protein levels of Ero1 or PDI between groups (Fig. 2C, Supplementary Figs. S1C, S6B, and S12C). Importantly, the alleviation of both mitochondrial and ER stress with niclosamide corresponds to improved adiponectin secretion (Fig. 2D and Supplementary Fig. S6C), confirming that mitochondrial stress and succination impair adiponectin oligomer processing in the ER (11). These data confirm that mitochondrial stress and protein succination influence the development of ER stress and reduce adiponectin secretion from the adipocyte.
Protein succination induces ER stress independent of glucose concentration
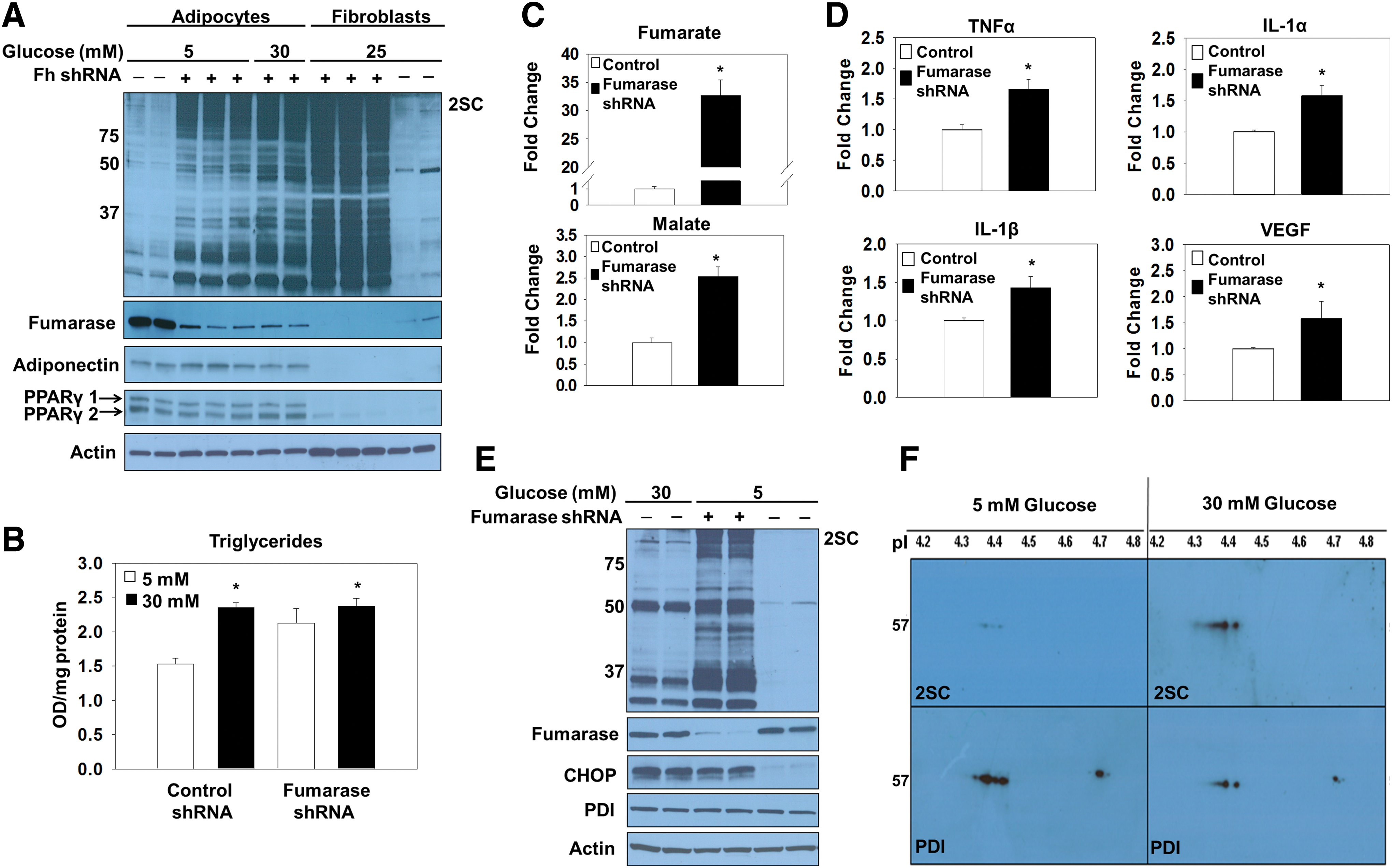
Lentiviral delivery of fumarase short hairpin ribonucleic acid (shRNA) to adipocytes matured in normal glucose was used to increase fumarate levels in the adipocyte independent of glucotoxicity, since this prevents the hydration to malate and permits fumarate accumulation. The successful transduction of both the scrambled control and fumarase knockdown lentivirus was evaluated by monitoring green fluorescent protein (GFP)-positive adipocytes 6 days post-transduction (Supplementary Fig. S2A). Adipogenesis proceeded normally in fumarase knockdown cells and was confirmed by the elevated expression of adiponectin and peroxisome proliferator-activated receptor gamma (PPARγ) isoforms (Fig. 3A and Supplementary Fig. S7A). Reduced serum adiponectin species are a hallmark of type 2 diabetes. We observed a decrease in secreted adiponectin profiles from the fumarase knockdown adipocytes versus the scrambled controls (Supplementary Figs. S2D and S13), again confirming that elevated fumarate and succination lead to reduced serum adiponectin levels (11). The quantification of the triglyceride content in control and fumarase knockdown adipocytes matured in 5 or 30 mM glucose (Fig. 3B), as well as the documented lipid accumulation by Oil Red O staining (Supplementary Fig. S2B) verified that knocking down the expression of fumarase does not interfere with adipogenesis. The ∼50% knockdown of fumarase levels observed was sufficient to increase protein succination in the adipocytes (Fig. 3A and Supplementary Fig. S7A). The pronounced escalation in protein succination in the fumarase knockdown adipocytes is directly proportional to the dramatic increase in the intracellular fumarate production. Analysis of mitochondrial metabolites from control and fumarase knockdown adipocytes matured in 5 mM glucose reveals a ∼30-fold increase in fumarate compared with controls (Fig. 3C and Supplementary Fig. S2C). Fumarase knockdown adipocytes displayed significant increases in the secretion of the pro-inflammatory cytokines tumor necrosis factor alpha (TNFα), interleukin 1 alpha (IL-1α), and interleukin 1 beta (IL-1β), as well as vascular endothelial growth factor (VEGF), versus control adipocytes (Fig. 3D and Supplementary Fig. S2E). This was similar to the significant increases in TNFα, MCP-1, and VEGF that we previously observed in adipocytes matured in 30 mM versus 5 mM glucose (53). These data suggest that elevated fumarate and protein succination may play a role in the chronic low-grade inflammatory state of adipose tissue observed during diabetes. Elevated CHOP levels were observed in both 30 mM glucose and fumarase knockdown adipocytes matured in 5 mM glucose (Fig. 3E and Supplementary Fig. S7B), confirming that protein succination alone functionally contributes to both the development of ER stress and the pro-inflammatory state of adipocytes, independent of other glucose-derived metabolic stressors. To mechanistically understand how succination might contribute to ER stress, we examined the succination of PDI in adipocytes during glucotoxicity. PDI was immunoprecipitated from adipocytes matured in 5 or 30 mM glucose and examined by two-dimensional immunoblotting to detect succination (Fig. 3F, top panels) followed by reprobing with anti-PDI (Fig. 3F, lower panels). Compared with the total levels of immunoprecipitated PDI, there was significantly more succinated PDI in adipocytes matured in high glucose. We also confirmed increased PDI succination in fumarase knockout (Fh −/−) mouse embryonic fibroblasts (MEFs), whose endogenous fumarate concentrations are ∼5 mM (55) (Supplementary Figs. S3 and S14). Overall, the results confirm that PDI is succinated in the presence of excess fumarate, potentially interfering with protein folding.
We were also interested in determining whether Ero1, the thiol-dependent protein responsible for re-oxidizing PDI active-site cysteines, is also susceptible to succination by fumarate. Since mammalian Ero1 also contains redox-sensitive cysteine thiols (20), we hypothesized that Ero1 might be a novel target of succination in the adipocyte. We incubated recombinant Ero1α (4) with increasing concentrations of fumarate in vitro to investigate whether Ero1 is susceptible to succination. One-dimensional (1D) immunoblotting with anti-2SC and anti-Ero1 antibodies confirmed that Ero1 is succinated when incubated with 5 or 25 mM fumarate (Supplementary Figs. S4A and S15A). We next immunoprecipitated Ero1 from adipocytes matured in 5 or 30 mM glucose and immunoblotted with the anti-2SC antibody. However, Ero1 succination was not detected in the adipocytes (Supplementary Figs. S4B and S15B). The blot was reprobed with an anti-Ero1 antibody to confirm the presence of Ero1 in the immunoprecipitates (Supplementary Figs. S4B and S15B). Therefore, although recombinant Ero1 could be succinated in vitro, there were no detectable increases in the endogenous succination of Ero1 in adipocytes.
Succination inhibits PDI reductase activity in vitro
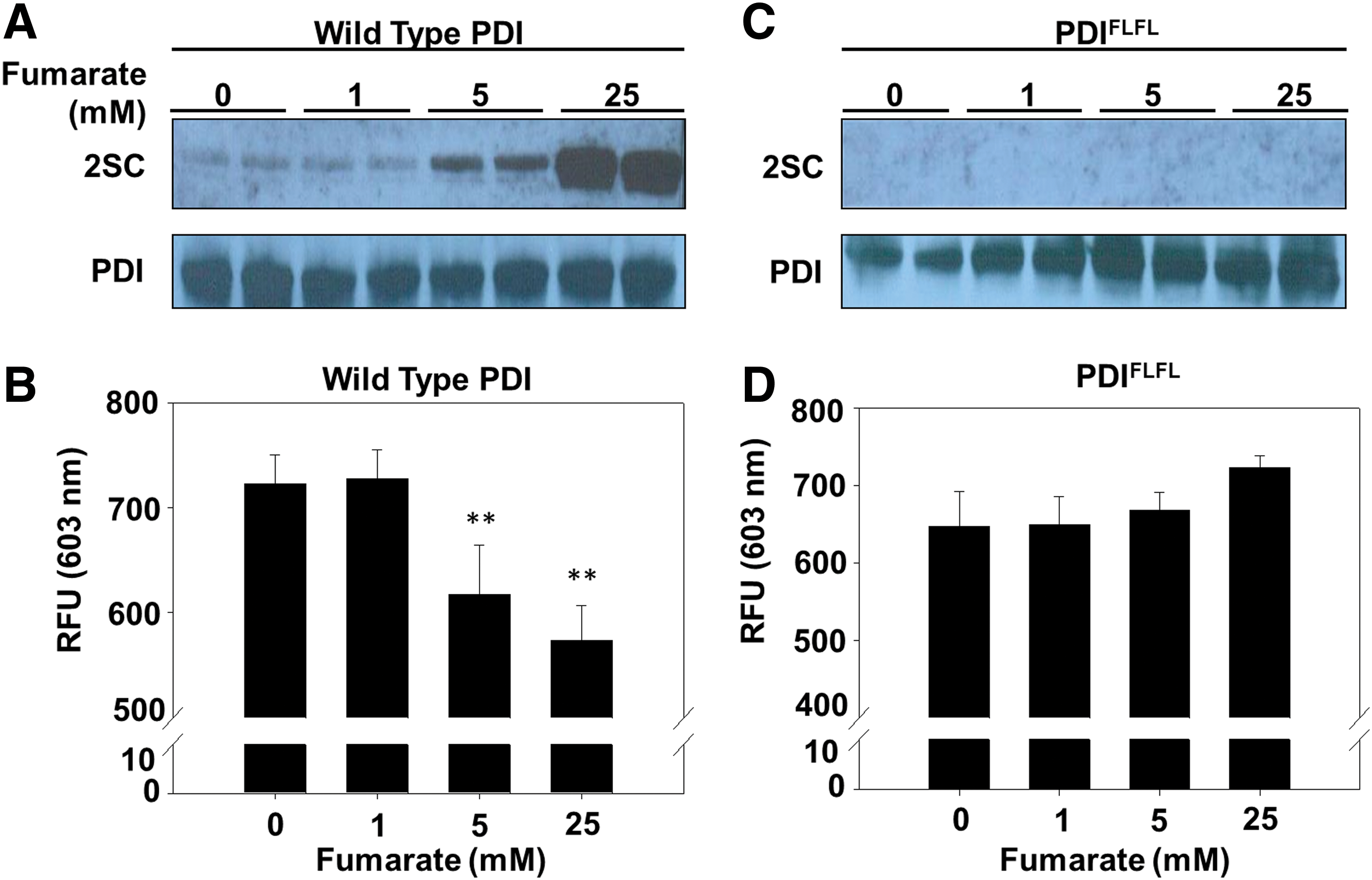
The succination of catalytically important thiols in the metabolic enzymes GAPDH and aconitase has been shown to reduce their enzymatic activity (7, 55). We hypothesized that succination at the active-site cysteines of PDI might also reduce enzymatic activity. Incubation of recombinant wild-type PDI (PDI-WT) with either 5 or 25 mM fumarate increased the PDI succination compared with the unmodified control (Fig. 4A and Supplementary Fig. S8A). Succination of PDI in vitro functionally decreased PDI reductase activity by 15% and 21% for 5 and 25 mM fumarate, respectively (n = 5, **p < 0.01) (Fig. 4A, B). In addition, we used a mutant form of PDI (PDIFLFL) to determine whether succination of PDI active-site cysteines is essential for inhibition of PDI reductase activity. The residues His and Lys, adjacent to either side of the C-terminal active-site cysteines, are mutated to Phe and Leu (H55ΔF:K57ΔL and H399ΔF:K401ΔL), respectively, in both the a and a′ domains of the mutant form of PDI (62). While remaining enzymatically functional, these simultaneous mutations are predicted to alter the active-site thiol pKa, and to prevent S-gluthionylation of PDI (62). In contrast to the data obtained with PDI-WT, the mutant PDI could not be succinated in the presence of increasing fumarate concentrations in vitro, and its reductase activity was unaffected (Fig. 4C, D and Supplementary Fig. S8B), indicating that active-site cysteine modification by fumarate is necessary for inhibition of PDI reductase activity.
We confirmed that direct succination of active-site cysteines was mechanistically responsible for the inhibition of PDI reductase activity by LC-MS/MS mass spectrometry. Recombinant PDI that had been incubated in the presence or absence of fumarate (Supplementary Figs. S5A and S15C) was alkylated with 4-vinylpyridine to produce a control pyridylethyl (PE) modification (CPE, +105.058 Da) on the remaining free thiols. The tryptic precursor ion masses for control (CPE, CPE) and succinated (CPE, C2SC, +116.011 Da) peptides were analyzed by LC-MS/MS to determine the specific site of succination. We identified the MS/MS spectrum corresponding to the succinated peptide 45YLLVEFYAPWCPEGHC2SCK59 ([M + 3H]3+: 683.9882 mass/charge ratio [m/z]) and 388KNVFVEFYAPWCPEGHC2SCK403 ([M + 3H]3+: 717.0033 m/z) in the recombinant PDI sample modified by fumarate (Supplementary Fig. S5B, D), as well as each analogous control peptide (Cys55PE, Cys58PE) (Cys399PE, Cys402PE) in the unmodified recombinant PDI sample (Supplementary Fig. S5C, E). In the case of both active-site peptides, the succination site was confirmed to be the second (C-terminal) cysteine, and it was only detected in the PDI samples incubated with fumarate in vitro.
Succination inhibits PDI reductase activity in 3T3-L1 adipocytes
Succination of PDI active-site cysteines inhibits reduction of insulin disulfide bonds in vitro (Fig. 4), whereas the assay is not specific for PDI activity in cell lysates due to the presence of other oxidoreductases. Di-Eosin-GSSG is a fluorescent, self-quenching, synthetic PDI-specific substrate (28, 43) that is frequently utilized to evaluate PDI reductase activity in protein lysates (37, 61). PDI reductase activity was decreased by ∼36% (Fig. 5A, D, n = 3, *p < 0.05) in adipocytes matured in high glucose versus normal glucose, and increased succination was detected at the molecular weight of PDI (∼57 kDa, arrow, Fig. 5B and Supplementary Fig. S9A) where the total levels of PDI remained similar between all samples (Fig. 5B and Supplementary Fig. S9A). One-dimensional immunoblotting of PDI immunoprecipitates with anti-2SC verified that succination of PDI increased in fumarase knockdown versus control adipocytes (Fig. 5C and Supplementary Fig. S9B). The evaluation of PDI reductase activity in fumarase knockdown adipocytes demonstrated specifically that succination of PDI accounted for a 15% decrease in PDI reductase activity versus controls (Fig. 5D, n = 3, *p < 0.05).

PDI was enriched from adipocytes matured in 5 or 30 mM glucose and analyzed by LC-MS/MS to confirm the endogenous sites of succination. A fragment ion (y13) representative of both control precursor ions containing active-site cysteines was detected in adipocytes matured in both 5 mM ([M + 3H]3+: 680.3434 m/z) ([M + 3H]3+: 670.6406 m/z) and 30 mM ([M + 3H]3+: 680.3387 m/z) ([M + 3H]3+: 670.6228 m/z) glucose (Fig. 5E, F; panels 1 and 3). In contrast, the mass of the representative y13 fragment ion from each succinated precursor ion containing active-site cysteines ([M + 3H]3+: 683.3942 m/z) ([M + 3H]3+: 674.3534 m/z) was only detected in adipocytes matured in 30 mM glucose (Fig. 5E, F; panel 2 vs. 4). The fragment ion profile was unique to these succinated peptides, whereas the N-terminal or C-terminal location of the succinated cysteine could not be assigned; however, modification of either cysteine is sufficient to impair PDI function as both thiols participate in the oxidoreductase mechanism of PDI. Importantly, these measurements confirm that the endogenous succination of active-site cysteines in PDI selectively occurs in adipocytes matured in high glucose.
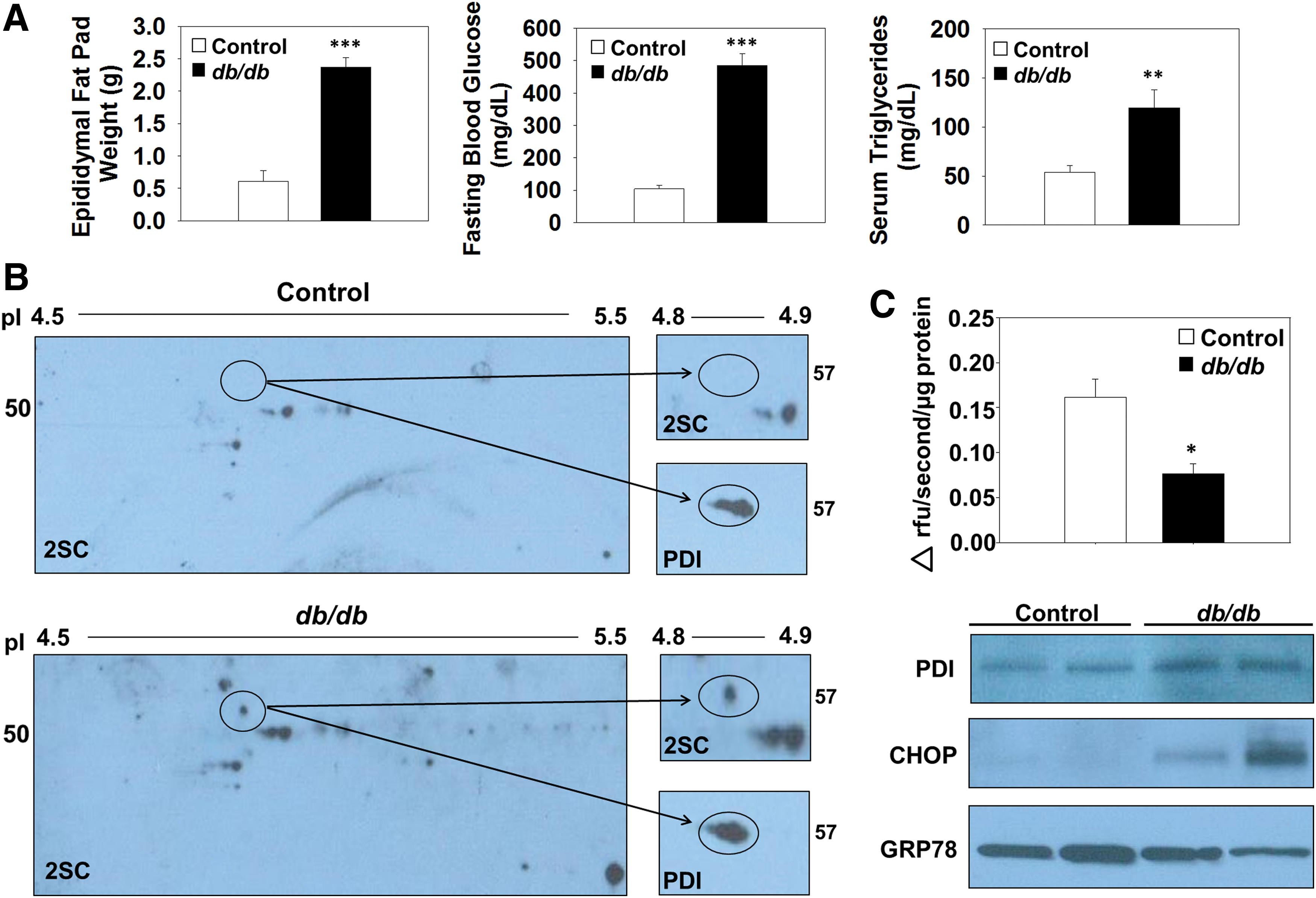
Succination inhibits PDI reductase activity in adipose tissue of db/db mice
Control and db/db mice at 15 weeks of age were used for the analysis of protein succination. The total epididymal fat pad weight, fasting blood glucose, and plasma triglyceride levels of the db/db mice were all elevated compared with age-matched control mice (Fig. 6A). To establish whether succination impacts PDI functionality in vivo, we examined PDI enzymatic activity in the epididymal adipose tissue of db/db diabetic mice. Succinated PDI is found only in the epididymal adipose tissue of db/db mice and is absent in the epididymal adipose tissue of the control mice (Fig. 6B, upper panels and Supplementary Fig. S10A). Total PDI is detectable in both the control and db/db adipose tissue samples ∼57 kDa (Fig. 6B, lower panels and Supplementary Fig. S10A). Assessment of PDI reductase activity by using Di-Eosin-GSSG in control and db/db adipose tissue demonstrated that activity was reduced by 52.6% in the diabetic mice (n = 4, *p < 0.05, Fig. 6C), whereas immunoblotting demonstrated equal amounts of PDI (Fig. 6C and Supplementary Fig. S10B). Immunoblotting with anti-GRP78 antibody verified our in vitro results that emphasize no alteration in the protein levels of GRP78 in the adipose tissue during diabetes; however, probing with anti-CHOP confirmed the existence of ER stress in the epididymal adipose tissue from db/db mice versus controls, (Fig. 6C and Supplementary Fig. S10B) (64). The succination of PDI occurs in parallel with reduced PDI reductase activity and the development of ER stress in the epididymal adipose tissue of db/db mice.
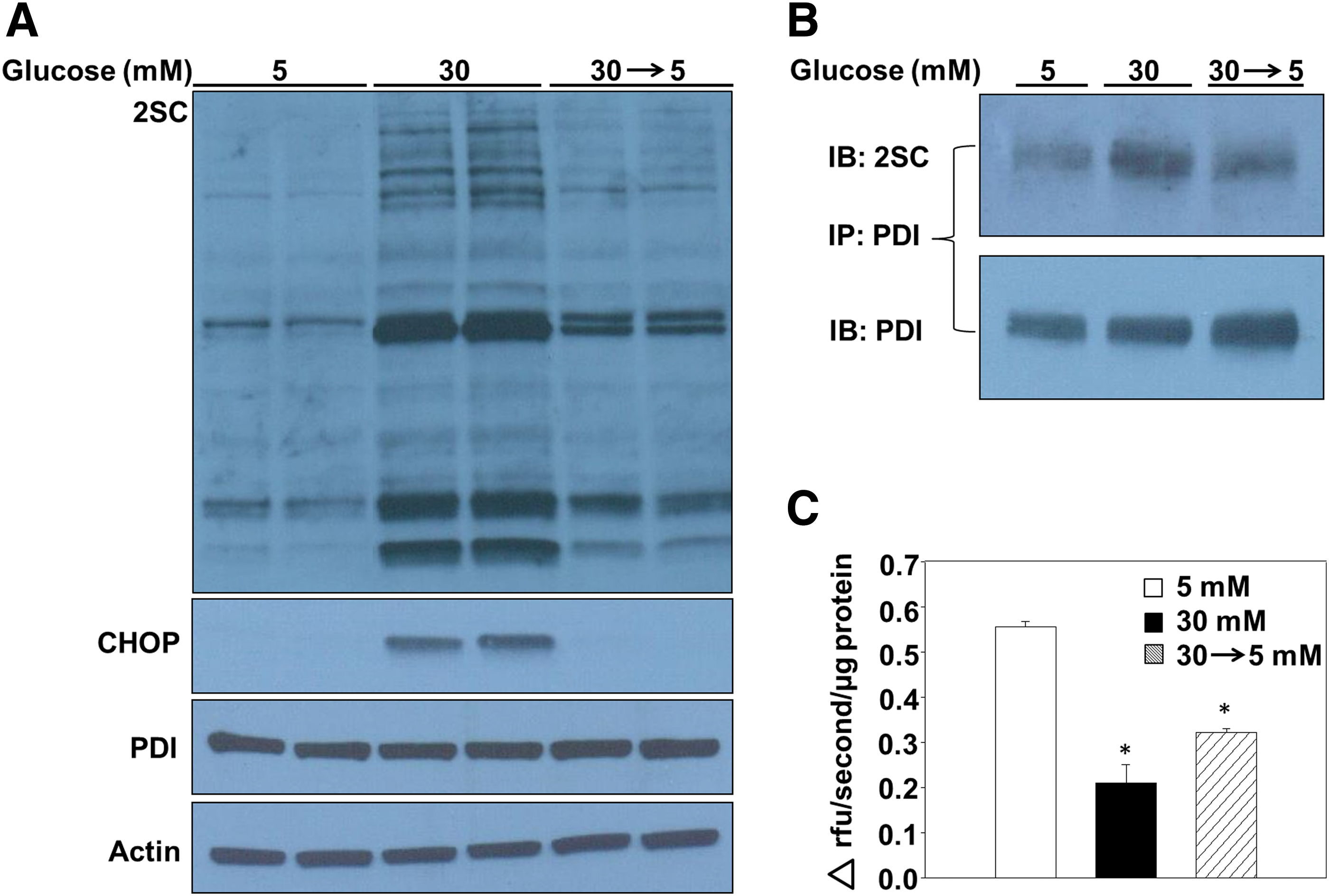
Glucose levels regulate succination of PDI in adipocytes
We have reported that decreasing glucotoxicity prevents the accumulation of succinated proteins in adipocytes (12), alleviating mitochondrial stress and permitting the turnover of succinated proteins. In the current study, we examined whether limiting chronic glucose exposure would be beneficial for recovery from ER stress in adipocytes. The accumulation of succinated proteins after 4 days of maturation (53) was prevented when the high-glucose medium was removed and replaced with normal glucose concentrations (30 → 5, Fig. 7A and Supplementary Fig. S11A). Notably, CHOP levels were also restored to those observed in normal glucose concentrations (Fig. 7A and Supplementary Fig. S11A), suggesting that limiting chronic high-glucose exposure is sufficient to ameliorate ER stress in the adipocyte. To determine the mechanism of ER stress reduction, PDI was immunoprecipitated from adipocytes matured in 5, 30, or 30 → 5 mM glucose. Immunoblotting verified that succination of PDI was the greatest in adipocytes matured in 30 mM glucose for all 8 days of maturation, whereas 4 days of recovery under normal glucose concentrations rescued the amount of succinated PDI (Fig. 7B and Supplementary Fig. S11B). PDI reductase activity was 60% lower in adipocytes matured in 30 mM glucose versus 5 mM glucose, whereas the adipocytes matured in 30 → 5 mM glucose displayed a 46% decrease in PDI reductase activity (Fig. 7C), indicating that a significant fraction of PDI functionality had been restored. The rescue of PDI reductase activity due to reduced succination of PDI contributes to an improved protein folding capacity, and to diminished levels of ER stress in the adipocyte.
Discussion
In this study, we mechanistically demonstrate that succination of PDI, as a direct result of glucotoxicity-driven mitochondrial stress, contributes to the presence of ER stress in the adipocyte. Consistent with other reports (9, 15), we document a significant increase in the protein levels of CHOP in adipocytes matured in a high-glucose medium, and notably this occurs in parallel with an increase in protein succination, suggesting a link between mitochondrial stress and ER stress. Niclosamide is a mild mitochondrial uncoupler that reduces mitochondrial stress by lowering the mitochondrial membrane potential and increasing the ADP:ATP ratio in vitro, and it significantly improves glucose homeostasis in diabetic mice (54). In Figure 2, we demonstrate two novel roles for niclosamide: Succination of proteins, including PDI, is decreased by directly reducing mitochondrial stress, and it prevents increased protein levels of CHOP, indicative of protection against ER stress in adipocytes matured under diabetic conditions. In addition, we detected a significant improvement in high-molecular-weight adiponectin secretion from adipocytes in the presence of niclosamide. Zhou et al. have described the requirement for normal levels of the ER chaperone disulfide-bond-A oxidoreductase-like protein to successfully combat ER stress in the adipocyte (64). Our initial assessment of ER stress markers demonstrated that increased CHOP was present; however, we did not observe altered PDI or Ero1 protein levels in the adipocytes matured in high glucose. Instead, our findings suggest that succination-mediated inhibition of the enzyme determines the ability of PDI to facilitate protein folding in a metabolically challenging environment.
The mitochondrial-associated ER membrane (MAM) facilitates the passage of lipids and several ions through several defined channels (5, 46). MAM enrichment, facilitating calcium transport, has been reported in hepatocytes of ob/ob- and high-fat diet-fed mice (5). Excess fumarate produced in the mitochondria is transported to the cytosol and out of the cell (29), or it may enter the ER lumen through the MAM, facilitating the succination of ER proteins (34). In this study, we have confirmed that endogenously produced fumarate increased succination of PDI in adipocytes matured in high-glucose concentrations, and that succination inhibits PDI reductase activity in these adipocytes and in the adipose tissue of diabetic db/db mice. Similar to the inhibitory effect of S-glutathionylation and S-nitrosylation on PDI enzymatic activity (56, 58), increasing PDI succination is also indirectly correlated with its reductase activity. Alongside this, we specifically confirmed that the cysteine-containing active-site peptides of PDI are endogenously succinated in adipocytes matured in a high-glucose medium. The significant reduction in PDI reductase activity contributes to disturbed protein folding by decreasing the capabilities of the major ER oxidoreductase, ultimately resulting in the pronounced CHOP levels sustained in adipocytes in vitro and in vivo.
PDI-A1 is the most abundant isoform of the PDI oxidoreductase family in the adipocyte. Despite the fact that all PDI family member (A1–A6) active-site peptides were analyzed by MS/MS mass spectrometry, the active-site peptides of PDI-A1 remained the most abundantly detected, suggesting that PDI-A1 has a major role in adipocyte ER protein folding. In addition, Muller et al. have demonstrated that increased expression of functional PDI-A1 alone is sufficient to preclude ER stress in HMEC-1 cells (37). We originally hypothesized that the N-terminal low pKa active-site Cys55 and Cys399 of PDI would be more reactive with fumarate than the higher pKa C-terminal active-site Cys58 and Cys402 (30). However, MS/MS analysis of recombinant PDI that had been succinated in vitro confirmed that the C-terminal Cys58 and Cys402 are the targets of succination. The recombinant PDI mutant used was refractory to modification by fumarate, suggesting that the pKa of at least one of the thiols is an important determinant of reactivity to succination. Importantly, mutation of the N-terminal, C-terminal, or both active-site Cys of PDI to Ser decreases PDI oxidoreductase activity and increases the accumulation of PDI-substrate intermediates (59), suggesting that modification of the active site will promote the accumulation of misfolded proteins in the ER.
High-glucose conditions facilitate not only increased metabolic stress that is derived, in part, from fumarate but also increased oxidative and lipotoxic stress. To determine the contribution of fumarate and protein succination to reduced PDI functionality, we knocked down fumarase expression and generated mature adipocytes with elevated levels of protein succination. Strikingly, the elevation in CHOP protein levels in fumarase knockdown adipocytes demonstrates that ER stress is a direct negative consequence of mitochondrial stress in adipocytes. In addition, Figure 4 suggests that half of the total decrease in PDI reductase activity in adipocytes matured in high glucose may be specifically attributed to increased succination of PDI.
Chemical chaperones, such as PBA, have been employed to combat ER stress in several rodent models of obesity and diabetes (6, 40), as continuous ER stress ultimately culminates in cell death (15). The accumulation of adipose tissue macrophages as a result of hypertrophied adipocyte death is central to the chronic low-grade inflammatory response observed in obese adipose tissue (13, 60). We have reported that greater amounts of inflammatory cytokines are released from adipocytes matured in high glucose compared with normal glucose levels in parallel with increased protein succination (53). In Figure 3, we show that increased inflammatory cytokine secretion occurs as a result of increased fumarate content and protein succination in the fumarase knockdown adipocyte. These data suggest that intra-organelle stress derived from elevated mitochondrial intermediary metabolism may be an early contributor to adipose tissue inflammation in diabetes.
The accumulation of the mitochondrial metabolite fumarate and resultant succination and inhibition of PDI activity uniquely links mitochondrial stress, a decreased protein folding capacity, and ER stress. The reduction of ER stress in obese humans using chemical chaperones has had some success (24); however, we have mechanistically demonstrated that targeting mitochondrial stress is a necessary component of successfully preventing ER stress-associated increases in CHOP. Since mitochondrial stress is a product of nutritional overload, we reduced the glucose concentration to normal levels in maturing adipocytes for several days, similar to a calorie restriction intervention, and demonstrated a significant reduction in PDI succination and an improvement in PDI reductase activity. Stanford et al. have described the importance of healthy adipose tissue for systemic glucose homeostasis by transplanting the subcutaneous white adipose tissue from exercised lean animals into sedentary animals fed a high-fat diet, improving insulin sensitivity and blood glucose levels in the sedentary animals (51). We demonstrate that a reduction in exposure to glucotoxicity limits CHOP production as a result of reduced mitochondrial stress and improved PDI reductase activity. Although specific agents that prevent protein succination in adipose tissue, such as niclosamide, may have therapeutic utility, our data clearly demonstrate that simple nutritional interventions will restore ER oxidative protein folding capacity.
In summary, we find that the active-site cysteines of PDI are succinated during glucotoxicity-induced mitochondrial stress, reducing PDI oxidoreductase activity and contributing to disturbed protein folding and adipocyte ER stress. The improvement in PDI enzymatic function and the reduction in ER stress as a direct result of reduced glucose concentration fortify the importance of emphasizing the link between healthy dietary consumption and proper adipocyte function in the management of type 2 diabetes.
Materials and Methods
Animals
Homozygous male db/db mice (C57BLKS-Leprdb) and their heterozygous littermates (Leprdb/+) were purchased at 6 weeks of age from Jackson Laboratories (Bar Harbour, ME). All mice were housed until 15 weeks of age, according to the guidelines of the University of South Carolina Institutional Animal Care and Use Committee. Fasting blood glucose measurements were performed after an overnight fast by collecting tail vein blood by using a Bayer (Whippany, NJ) Contour® blood glucose meter for control animals and the glucose oxidase assay for db/db mice. The mice were sacrificed after CO2 asphyxiation, and adipose tissues were collected and immediately frozen.
Cell culture and lentiviral transduction
3T3-L1 mouse fibroblasts were differentiated and matured as previously described (38). After differentiation (∼3 days), the adipocytes were matured in 5 mM glucose and 0.3 nM insulin or 30 mM glucose and 3.0 nM insulin for an additional 8 days. These conditions were selected as it had been observed that adipocytes cultured in normal glucose/insulin are a more appropriate control for the high-glucose/-insulin (diabetic) conditions (53). The adipocytes were treated with 2 μM niclosamide on standard medium change every 48 h. 3T3-L1 fibroblasts were incubated overnight with 150 μL of filtered conditioned medium containing Fh1 shRNA or control lentivirus. Successfully transduced fibroblasts were selected by using 1 μg/ml puromycin. The selected fibroblasts were propagated until confluent; then, they were differentiated to adipocytes and matured for 8 days in 5 or 30 mM glucose as described earlier. The accumulation of triglycerides was observed by light microscopy as the cells matured in both glucose concentrations (38, 53). Adipocytes were incubated in 1 ml of serum-free and phenol red-free medium with 5 or 30 mM glucose for 4 h. The medium was collected and frozen at −80°C for analysis of secreted adiponectin. Total cellular protein was collected after lysis in radioimmunoprecipitation assay (RIPA) buffer as previously described (38) and was quantified by the Lowry assay.
Lentiviral vector production
The lentiviral vectors were prepared by the University of South Carolina Viral Vector Facility. Lentiviral vectors were generated by using a transient transfection protocol, as previously described (22). Briefly, TRC2 Fh1 shRNA, clone-TRCN0000246831, or SHC202 MISSION TRC2 pLKO.5-puro non-mammalian shRNA control plasmids (Sigma-Aldrich, St. Louis, MO) were used to generate the lentiviral vectors. The vectors also contained a puromycin resistance gene. Fifteen micrograms of vector plasmid, 10 μg of psPAX2 packaging plasmid (No. 12260; Addgene, Cambridge, MA), 5 μg of pMD2.G envelope plasmid (No. 12259; Addgene), and 2.5 μg of pRSV-Rev plasmid (No. 12253; Addgene) were transfected into 293T cells. The filtered conditioned medium was collected and stored at −80°C until use.
Measurement of metabolites
The quantification of metabolites was performed by GC-MS at the David H. Murdock Research Institute (DHMRI, Kannapolis, NC). Metabolite extraction was performed in an adaptation of previous methods (42). Briefly, adipocyte lysates harvested in methanol from confluent 100 × 20 mm plates were washed three times with ice-cold phosphate-buffered saline (PBS) followed by the immediate addition of 20 volumes of ice-cold chloroform:methanol (2:1). The samples were vortexed and allowed to stand on ice for 10 min with intermittent vortexing before the addition of 0.2 volumes of H2O. The samples were sonicated and allowed to stand on ice for an additional 2 min, followed by centrifugation at 3220 × g for 20 min. The aqueous supernatant was transferred into a clean tube and dried under air. The extraction was repeated an additional time by adding equal parts of methanol and deionized water, centrifuging, and transferring the aqueous layer into the respective tube to dry. The protein interface for each sample was removed for quantification of protein by the Lowry assay. Before derivatization, the extracts were resuspended in ethyl acetate and transferred to GC-MS vials. The samples were dried with N2, and an internal standard (100 μM succinate-13C4; Cambridge Isotope Laboratories, Inc.) was added to each of the samples and fumarate standards. The samples and standards were derivatized with 200 μl of methylamine (20 mg/ml in pyridine; M.P. Biomedicals, Solon, OH) at 30°C for 90 min, followed by drying under N2. This was followed by the addition of 120 μl of N-methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA; Sigma, St. Louis, MO) with 1% trimethylchlorosilane (TMCS, Sigma); the mixture was incubated at 70°C for 60 min. The derivatized product was stored in a −20°C freezer for 1h. A 100 μl aliquot of the prepared product was transferred to a deactivated glass insert in a 2 ml glass vial (Agilent Technologies, Santa Clara, CA) for GC/MS analysis. An Agilent 7890A GC system, coupled to an Agilent 5975C electron ionization (EI) mass selective detector (MSD), was used to analyze the TMS-derivatized samples. A column with dimensions 30 m × 0.25 mm I.D., 0.25 μm film thickness (Restek, Bellefonte, PA) was used, and the column operation conditions were as follows: helium carrier gas at 1.0 ml/min, GC oven temperature program of 70°C (2 min), 70–100°C (30°C/min, 1 min), 100–140°C (10°C/min, 4 min), 140–188°C (4°C/min, 12 min), and 188–280°C (5 min), with a transfer line temperature of 270°C. The GC-MS was operated under a splitless mode (inlet at 250°C). The mass spectrometry was performed at an electron energy of −70 eV, and the ion source temperature was 230°C. Selected ion monitoring (SIM) was performed for fumarate, and the peak areas obtained were normalized to the added internal standard. Absolute quantitation was performed based on standard curves obtained from the normalized reference standards, and the final metabolite concentrations were normalized to the protein content of the cells.
Measurement of triglycerides and inflammatory markers
Control and fumarase knockdown adipocytes were matured for 8 days in 5 or 30 mM glucose concentrations and harvested in PBS solution for triglyceride quantification. Control and db/db serum triglyceride levels were analyzed in 15-week-old mice. The triglyceride concentration was determined by using Infinity™ Triglycerides assay kit (Thermo Fisher Scientific, Waltham, MA), according to the manufacturer's instructions. The media on scrambled control or fumarase knockdown 3T3-L1 adipocytes matured for 6 days were replaced with serum-free Dulbecco's modified Eagle's medium (DMEM) for 6 h before collection of the media. Pro-inflammatory cytokines in serum-free conditioned media were analyzed by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (Mouse Obesity ELISA; Signosis, Santa Clara, CA).
Measurement of basal respiration
3T3-L1 fibroblasts were plated in V7-microassay plates on 0.2% gelatin at 10,000 cells per well and differentiated as described earlier. XF Assay Medium (Seahorse Biosciences, North Billerica, MA), supplemented with glucose, insulin, and 1 mM sodium pyruvate, was added to replace the culture media 1 h before the analysis (53). The basal OCR (pmol/min) of adipocytes matured in 5 or 30 mM glucose for 2 days was assessed on an XF Flux Analyzer (Seahorse Biosciences). The OCR data were normalized to the total DNA content of the wells by using the CyQuant® Assay (Invitrogen, Grand Island, NY), and the data were expressed in pmoles/min/ng DNA as previously described (53).
Immunoblotting and immunoprecipitation
Protein electrophoresis and Western blotting were performed on a BioRad Criterion system as previously described (38). Polyclonal antibodies to actin (sc-1616), Ero1α (sc-365526), and PDI (sc-30932) were from Santa Cruz Biotechnology, Inc. (Dallas, TX); adiponectin (AF1119) was from R&D Systems (Minneapolis, MN). Monoclonal antibodies to PDI (MA3-019) and CHOP (MA1-250) were from Pierce (Rockford, IL). The polyclonal anti-2SC antibody was prepared as previously described (36). Ero1α and PDI were immunoprecipitated from 300 to 500 μg adipocyte cell lysates by using 5 μg of anti-PDI or anti-Ero1α, as previously described (11). The beads were re-suspended in 2 × loading buffer and boiled at 95°C for 8 min to remove the bound antibody-antigen complex before immunoblotting. To quantify secreted adiponectin before immunoblotting, the adipocytes were incubated with 1 ml of serum-free medium after 8 days of maturation for 4 h, and total adiponectin levels were quantified by using the mouse adiponectin ELISA according to the manufacturer's instructions (MRP300; R&D Systems). An equal amount of adiponectin from each sample was subjected to non-reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blotting with anti-adiponectin antibody.
Two-dimensional gel electrophoresis
Two-dimensional polyacrylamide gel electrophoresis was performed as previously described (11, 38). Briefly, 200 μg of protein from WT or Fh1−/− MEFs, control and db/db adipose tissue, or the total PDI immunoprecipitate sample were re-suspended in rehydration buffer (7 M urea, 2 M thiourea, 2% CHAPS, 1% IPG buffer, and 50 mM dithiothreitol [DTT], 3.0% DeStreak, 0.5% ampholytes) and focused on IPG strips with a pI range of 3.9–5.1 (No. 163–2024; BioRad, Richmond, CA) before SDS-PAGE in the second dimension. Western blotting was performed with anti-2SC antibody, followed by IB stripping (62.5 mM Tris, pH 6.8, 2% SDS, and 0.7% [v/v] beta mercaptoethanol) and reprobing with anti-PDI antibody.
Protein identification by mass spectrometry
For the identification of succination sites in PDI, we first incubated 2.0 μg of recombinant PDI with 100 mM fumarate for 24 h at 37°C. We then investigated the sites of endogenous succination of PDI via immunoprecipitation of PDI from adipocytes matured in 5 or 30 mM glucose for 8 days. In all cases, samples were resolved by SDS-PAGE, and the gels were stained with Coomassie Brilliant blue. After destaining, PDI was excised from the gel and incubated in the presence of 10 mM DDT, followed by alkylation by 170 mM 4-vinylpyridine and proteolytic digestion in 50 mM ammonium bicarbonate buffer with 2 pmol of Promega sequencing grade modified trypsin (Promega, Madison, WI). SIM was performed to identify select pyridylethylated and succinated cysteine-containing tryptic peptides. The digested samples were analyzed on a Dionex Ultimate 3000-LC system (Thermo Scientific, Rockford, IL) that was coupled to a Velos Pro Orbitrap mass spectrometer (Thermo Scientific). A 75-μm C18 stationary-phase LC column was used with a 60-min gradient from 2% acetonitrile in 0.1% formic acid solution (solvent A) to 70% solvent A and 30% solvent B (40% water in acetonitrile containing 0.1% formic acid). The Orbitrap was operated in the data-dependent MS/MS analysis mode and excluded all ions below 200 counts. An inclusion list of a maximum of six abundant isotopic masses ±3 amu was used to select the specific peptides for selected resonance monitoring and for determination of the site of modification. To further identify specific succinated sites, multiple reaction monitoring was used to monitor select pyridylethylated and succinated tryptic peptide masses of interest (generated using Protein Calculator software) for CID (collision-induced dissociation)-MS/MS analysis. The data-dependent and CID-MS/MS data were analyzed by using Proteome Discover 1.4 software with SEQUEST search engine against the uniprot_database October 2014 (Mus musculus 52,474 proteins). The CID-MS/MS data were sequenced manually by using Thermo Xcalibur 2.2 software to confirm the modified peptides. The variable modifications of methionine oxidation, proline hydroxylation, cysteine pyridylethylation (CPE, +105.058 Da), or cysteine succination by fumarate (C2SC, +116.011 Da) were considered in each data search.
PDI reductase activity assays
Recombinant PDI-WT and a mutant form of PDI (PDIFLFL) were generously provided by Dr. Danyelle Townsend (Medical University of South Carolina, Charleston, SC). 13.85 μg of recombinant PDI-WT or PDIFLFL was incubated in 0.1 M potassium phosphate buffer (pH 7) with 2 mM ethylenediaminetetra acetic acid (EDTA) and 0, 1, 5, or 25 mM fumarate. PDI reductase activity was monitored according to the PROTEOSTAT® PDI assay kit instructions (Enzo Life Sciences, Inc., Farmingdale, NY). Di-Eosin-GSSG was generously provided by Dr. Bulent Mutus, University of Windsor (Ontario, Canada) (43). Six micrograms of protein lysate from 3T3-L1 adipocytes or control and db/db adipose tissue was incubated with 1.06 μM Di-Eosin-GSSG, 5 μM DTT, and 100 μM diethylenetriaminepenta acetic acid (DTPA) in 0.1 M potassium phosphate buffer, pH 7. Fluorescence was measured kinetically (30 s intervals) on a TECAN Safire2 microplate reader at λex = 510 nm and λem = 550 nm.
Oil Red O staining
0.5 g of Oil Red O was thoroughly mixed in 100 ml isopropanol and diluted 6:4 in distilled water. The dye was allowed to stand for 10 min and filtered through Whatman No. 1 filter paper. One hundred percent of confluent 3T3-L1 fibroblasts and differentiated fumarase knockdown adipocytes were fixed for 1 h in 4% formaldehyde. The cells were washed with PBS, stained with the Oil Red O dye for 1 h, and washed with water. The lipid droplets were visualized by brightfield microscopy, and the images were obtained with the Invitrogen™ EVOS™ FL Auto Cell Imaging System (Thermo Fisher Scientific).
Detection of Ero1 succination
The Ero1α immunoprecipitate samples from 3T3-L1 adipocytes were immunoblotted with anti-2SC and anti-Ero1α antibodies as described earlier. Recombinant Ero1α isolated from Escherichia coli strain BL21(DE3) was generously provided by Dr. Kazutaka Araki (Kyoto Sangyo University) (4); 2.77 μg of protein was incubated in 0.1 M potassium phosphate buffer with 2 mM EDTA, 100 μM DTPA, and 0, 1, 5, or 25 mM fumarate at 37°C for 24 h before SDS-PAGE and immunoblotting with anti-2SC and anti-Ero1 antibodies.
Statistical analysis
The comparison of metabolite and inflammatory marker differences and the initial rate of reaction corresponding to PDI reductase activity was analyzed in Sigmaplot 11, by using the student t-test (n = 3, *p < 0.05, **p < 0.01, ***p < 0.001). PDI reductase activity in vitro was analyzed by using a one-way analysis of variance (ANOVA; n = 5, **p < 0.01).
Footnotes
Acknowledgments
The authors are grateful to Professor John Baynes, USC School of Medicine (Columbia, SC), for helpful discussion. They would like to thank Dr. Kazutaka Araki, Kyoto University (Kyoto, Japan), for generating and providing the recombinant ERo1α. This work was supported by grants from the American Diabetes Association (1-11-JF-13) and the National Institutes of Health (F31DK108559, R56DK105087, R37DK19971, P20GM109091, and R01NS92938).
Authors' Contributions
A.M.M. designed and performed experiments, analyzed data, contributed to the discussion, and wrote the article; M.D.W., S.L.M., and R.M.T. performed experiments and analyzed data; G.G.P. analyzed data and contributed to the discussion; A.F. and B.M. produced the Di-Eosin-GSSG; J.A. generated the WT and Fh1−/− MEFs; B.K. produced the lentiviral vectors; D.M.T. produced the control and mutant recombinant human PDI, contributed to the discussion, and edited the article; and N.F. designed and performed experiments, analyzed the data, and edited the article before submission. No potential conflicts of interest relevant to this article were reported.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.