
Abstract
Aims:
Hyperlipidemia-induced oxidative stress is considered to be one of the main pathogenic factors that contribute to pancreatic beta cell dysfunction in the development of type 2 diabetes (T2D). Sirtuin 3 (Sirt3) is abundantly expressed in the mitochondria as an NAD+-dependent deacetylase and regulates mitochondrial adaptive responses to oxidative stress. We examined the antioxidant defense mechanism of Sirt3 in pancreatic beta cells in the context of hyperlipidemia.
Results:
Chronic high-fat diet (HFD) feeding caused elevated oxidative stress accompanied by reduced Sirt3 expression in the pancreatic beta cells of wild-type mice. Primary pancreatic islets of Sirt3 knockout (KO) mice and murine pancreatic MIN6 cells with downregulated Sirt3 expression showed increased superoxide dismutase 2 (SOD2) acetylation and reduced glucose-stimulated insulin secretion and glucose-stimulated adenosine triphosphate (ATP) generation. Moreover, Sirt3 deficiency sensitized the pancreatic islets and MIN6 cells to palmitate- and H2O2-induced beta cell dysfunction linked with aggravated c-Jun N-terminal kinase phosphorylation and cleaved caspase-3 expression. These negative effects were reversed by antioxidant chemical treatment or restoration of Sirt3 in KO islets. Finally, overexpression of Sirt3 in MIN6 cells partially rescued palmitate-induced reactive oxygen species generation, pancreatic and duodenal homeobox-1 (Pdx-1) nucleo-cytoplasmic translocation, and beta cell dysfunction.
Innovation:
We present that Sirt3 expression protected pancreatic beta cells from lipotoxicity by antagonizing oxidative stress-induced cell damage.
Conclusion:
These results suggest that Sirt3 may be a target for amelioration of beta cell dysfunction due to obesity and T2D. Antioxid. Redox Signal. 27, 962–976.
Introduction
P
Our study demonstrated the role of Sirt3 in the regulation of pancreatic beta cell function. The key novel finding is that Sirt3 deficiency accelerated high-fat diet-induced impairment of glucose metabolism through increased oxidative stress in beta cells. Overexpression of Sirt3 protected beta cells from lipotoxicity-induced dysfunction through ameliorated oxidative stress. These findings provide new insights into the mechanisms of the protective role of Sirt3 in pancreatic beta cell function with potential therapeutic implications in type 2 diabetes.
Sirtuin 3 (Sirt3) is an NAD+-dependent deacetylase within the mitochondria. It is abundantly expressed and actively participates in the metabolism of multiple tissues such as liver, adipocytes, and pancreatic beta cells (4, 8, 36, 37). Overexpression of Sirt3 ameliorated palmitate-induced oxidative stress and inflammation in the proximal tubular cells, while Sirt3-mediated superoxide dismutase 2 (SOD2) deacetylation and activation protected against oxidative stress in mouse liver during caloric restriction (23, 31). However, the antioxidative effects of Sirt3 may be tissue specific. In liver and muscle-specific Sirt3 knockout (KO) mice fed a high-fat diet (HFD), despite hyperacetylation of mitochondrial proteins, glucose homeostasis was not affected (12). In contrast, in chow diet-fed mice, global ablation of Sirt3 caused protein hyperacetylation with over 50% loss of adenosine triphosphate (ATP) generation in liver and other tissues (2). In these systematic Sirt3 KO mice, obesity and impaired glucose tolerance (IGT) rapidly developed upon HFD feeding (14). These results suggested that Sirt3 in tissues or organs other than muscle and liver could be involved in glucose homeostasis. In this light, reduced Sirt3 protein expression has been reported in islets isolated from T2D patients, while in experimental studies, Sirt3 overexpression in pancreatic islets ameliorated palmitate-induced beta cell dysfunction (8, 22), although the underlying mechanism requires further elucidation.
Based on these premises, our current study extended previous reports on the effects of lipotoxicity on Sirt3 expression in beta cell and the role of Sir3 on beta cell function. We hypothesized that Sirt3 deficiency may induce pancreatic dysfunction due to oxidative stress under a hyperlipidemic environment, through mitochondrial dysfunction and activation of the JNK pathway. To test this hypothesis, we examined the effects of HFD in pancreatic islets of wild-type (WT) and Sirt3 KO mice as well as that of H2O2- and palmitate-induced oxidative stress on insulin secretion in in vitro MIN6 cell-based models.
Results
Sirt3 deficiency accelerates HFD-induced impairment of glucose homeostasis
As Sirt3 expression is decreased in the islets of T2D patients, we first examined whether Sirt3 would be reduced in mouse islets under hyperlipidemia-induced pancreatic beta cell dysfunction. Adult male mice were treated with HFD and islets were isolated for Western blot analysis. As shown in Figure 1A and B, 2 weeks of HFD feeding did not affect Sirt3 expression, but by week 8, Sirt3 expression was significantly reduced in pancreatic islets, suggesting that prolonged feeding with HFD is able to reduce Sirt3 expression in islets.
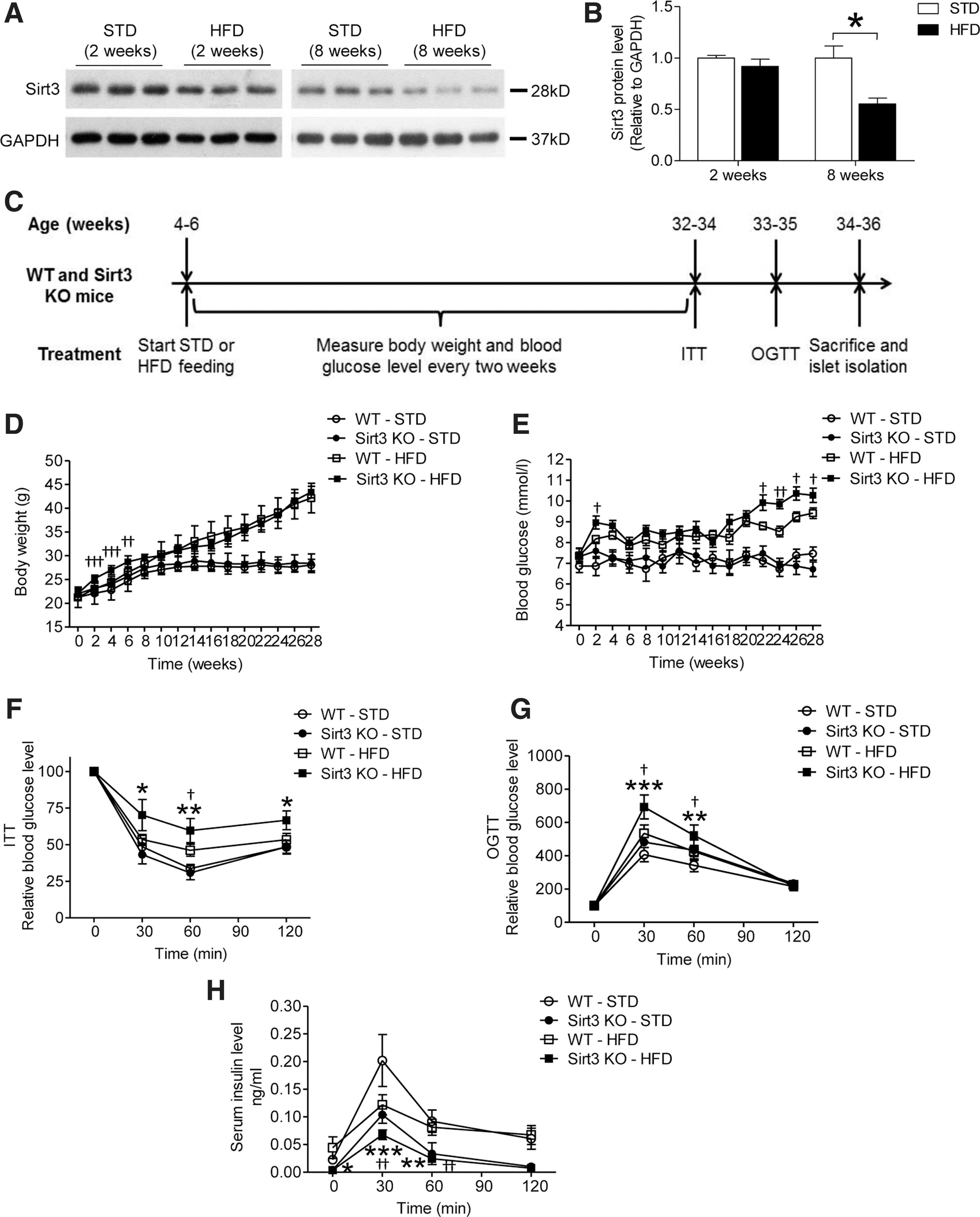
Then, we utilized the Sirt3 KO mouse model to determine the effects of Sirt3 on glucose homeostasis. We measured glucose tolerance and insulin sensitivity in WT and Sirt3 KO mice fed a standard diet (STD). Consistent with previous reports (14), parameters were not affected in Sirt3-deficient mice (data not shown) fed an STD. Then, we challenged WT and Sirt3 KO mice with HFD for 28 weeks starting from 4 to 6 weeks of age (Fig. 1C). There was a significant elevation in the body weight of Sirt3 KO mice fed an HFD immediately after the experiment started. However, the body weight was similar in HFD-fed WT and Sirt3 KO mice 6 weeks later (Fig. 1D). There was marked weight gain in both WT and Sirt3 KO mice with HFD (Fig. 1D) with blood glucose levels higher than the STD-fed mice (Fig. 1E). By week 22, the glucose levels were higher in the Sirt3 KO than WT mice fed an HFD (Fig. 1E). Insulin sensitivity and glucose tolerance were impaired in WT and Sirt3 KO mice, but to a greater extent in the Sirt3 KO mice (Fig. 1F, G). During oral glucose tolerance test (OGTT), there was marked reduction in glucose-induced serum insulin in HFD-fed Sirt3 KO mice than the other groups (Fig. 1H), suggesting Sirt3 might play a protective role in glucose homeostasis.
Sirt3 deficiency accelerates HFD-induced oxidative stress and beta cell apoptosis
Oxidative stress generated from obesity and/or diabetic conditions can cause pancreatic beta cell dysfunction. Since Sirt3 regulates oxidative stress, we investigated whether ablation of Sirt3 could impair pancreatic beta cell function during HFD feeding via inducing oxidative stress. After 28 weeks of HFD, we isolated islets from the mice for beta cell functional study. We first found that Sirt3 expression levels in WT islets after 28 weeks of HFD were reduced by fourfold compared with those from mice fed an STD (Fig. 2G, H). HFD feeding impaired insulin secretion in islets of WT mice when islet Sirt3 expression was reduced. This impairment was accentuated in islets from Sirt3 KO mice (Fig. 2A). Then, we employed immunohistochemistry of 3-nitrotyrosine to evaluate the oxidative stress level in islets. We found that positive staining with 3-nitrotyrosine in WT islets was only detected in HFD-fed, but not STD-fed, mice. In contrast, positive staining of 3-nitrotyrosine was observed in Sirt3 KO islets from both STD- and HFD-fed mice. Stronger signals were observed in islets isolated from Sirt3 KO mice fed an HFD (Fig. 2B, C). It was reported that oxidative stress induces Pdx-1 nucleo-cytosol translocation, which thereby impairs beta cell function. Results from immune staining with Pdx-1 in pancreatic islets demonstrated that HFD feeding increased cytosolic Pdx-1 levels in both WT and Sirt3 KO islets with a higher magnitude observed in Sirt3 KO islets (Fig. 2D). With STD feeding, positive staining of TUNEL was nearly undetectable in WT or Sirt3 KO islets (Fig. 2E and Supplementary Fig. S1; Supplementary Data are available online at
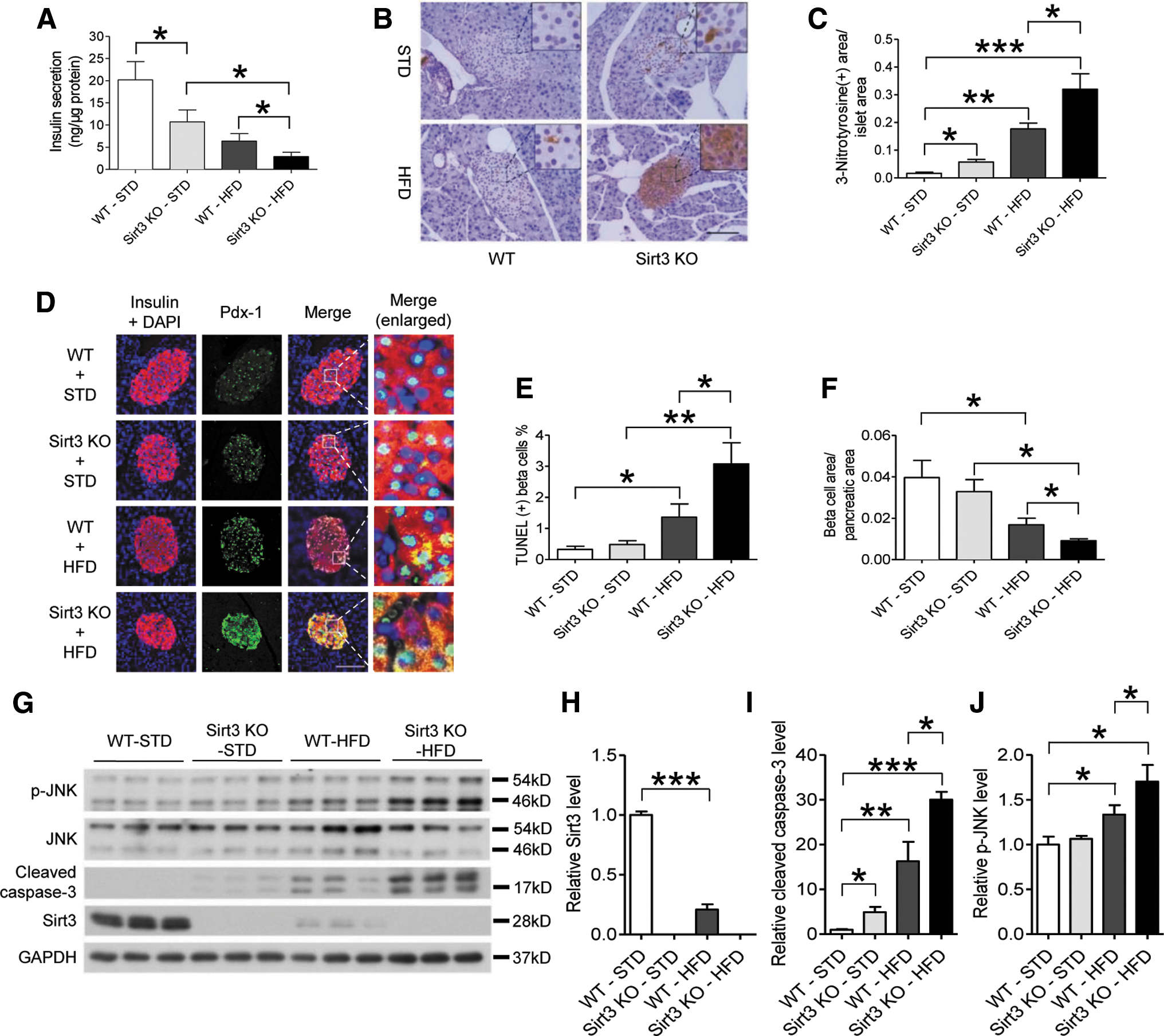
Consistent with the TUNEL staining results, cleaved caspase-3, as a surrogate marker for apoptosis, was elevated in WT islets with HFD diet. Both STD and HFD feeding enhanced expression levels of cleaved caspase-3 in Sirt3 KO islets. With HFD, there was a twofold increase of cleaved caspase-3 level in Sirt3 KO islets compared with WT islets (Fig. 2G, I). Moreover, JNK phosphorylation, a marker of oxidative stress, was increased in WT islets after HFD, which was further increased in the Sirt3 KO islets (Fig. 2G, J). These results suggested that Sirt3 expression in beta cells plays a protective role in regulating apoptosis and related factors, such as cleaved caspase-3 and JNK phosphorylation, after prolonged treatment with HFD.
Reduction of Sirt3 expression in pancreatic beta cells after palmitate or H2O2 treatment impairs beta cell function
To investigate the mechanism of how Sirt3 functions in beta cells, we employed isolated islets from mouse and the MIN6 beta cell line to evaluate the effects of Sirt3 during treatment with the saturated fatty acid palmitate or with H2O2. We first found that both palmitate (Fig. 3A) and H2O2 treatments (Fig. 3B) reduced Sirt3 protein expression in mouse islets. In the MIN6 beta cell line, both treatments decreased Sirt3 expression in a dose- and time-dependent manner (Fig. 3C–E). However, Sirt3 expression was not affected by treatment with the unsaturated fatty acid, sodium oleate (Supplementary Fig. S2A, B), suggesting that lipotoxicity caused by saturated fatty acid and oxidative stress might reduce Sirt3 expression in beta cells. In addition, palmitate and H2O2 treatments also significantly induced MIN6 cell apoptosis (Supplementary Fig. S3A, B). We also observed that mitochondrial mass was decreased in MIN6 cells after palmitate or H2O2 treatment (Supplementary Fig. S4A, C). Furthermore, a similar trend for the degree of attenuation of Sirt3 protein expression and reduction of mitochondrial mass was observed (Supplementary Fig. S4B, D), indicating that Sirt3 expression level might be closely related to the mitochondrial mass in beta cells.

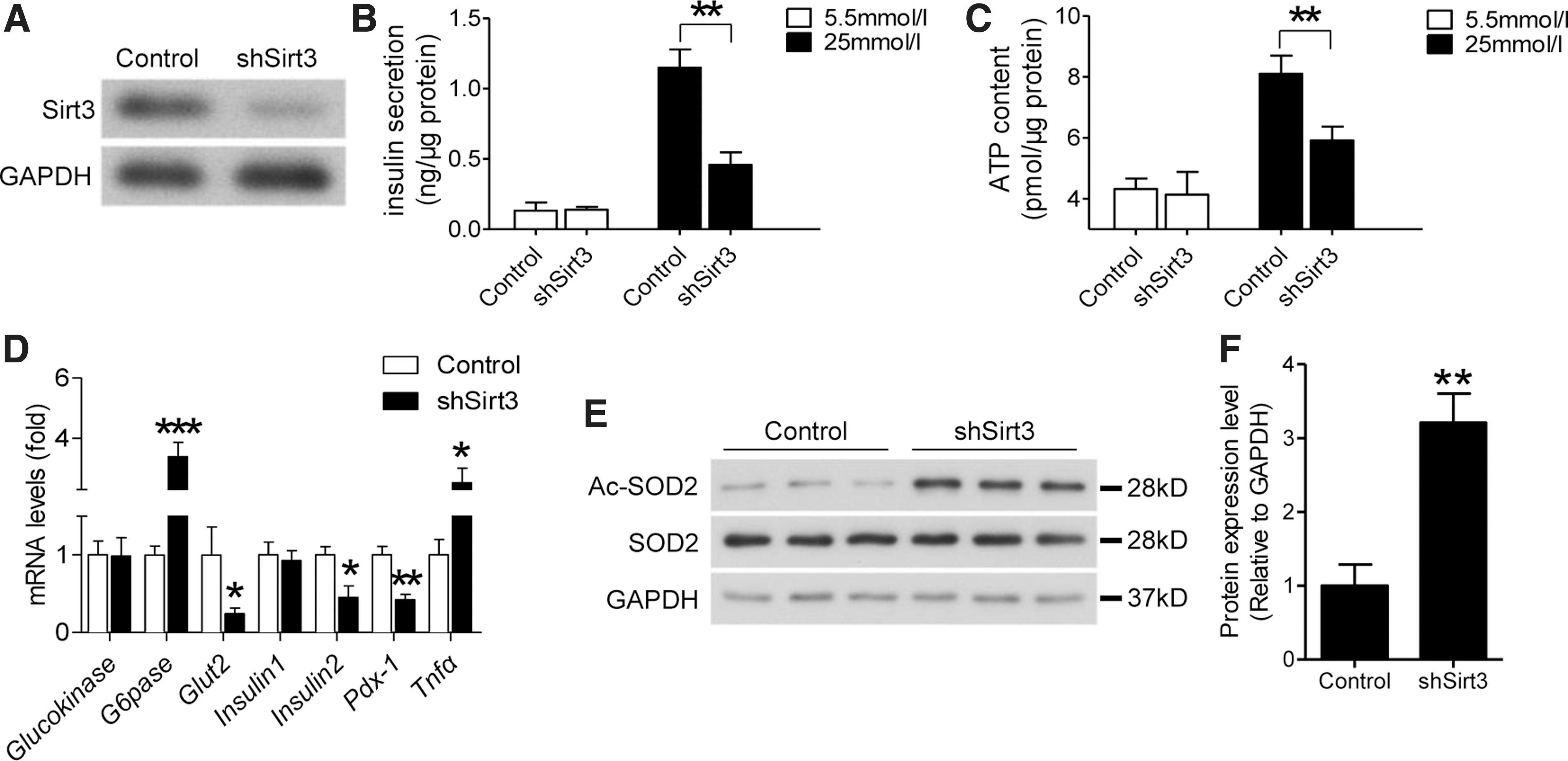
We then examined the effects of Sirt3 deficiency on beta cells by knocking down Sirt3 in MIN6 cells through shRNA lentivirus infection (Fig. 4A). This manipulation decreased GSIS (Fig. 4B), which was correlated with attenuated glucose-stimulated ATP generation (Fig. 4C). Knocking down Sirt3 expression in MIN6 cells also affected mRNA levels of marker genes related to beta cell function (Fig. 4D). Among these key markers, glucokinase mRNA level was not affected and G6pase level was increased, while expression levels of Glut2, Insulin2, and Pdx-1 were downregulated. Knocking down Sirt3 expression in MIN6 cells also increased expression of inflammatory cytokine, Tnfα (Fig. 4D). SOD2, a well-known antioxidant enzyme, was reported as one of the direct downstream targets of Sirt3, so we then evaluated the acetylation level of SOD2 in Sirt3-deficient MIN6 cells. As expected, knockdown of Sirt3 significantly increased SOD2 acetylation, which indicates that SOD2 activity was inhibited (Fig. 4E, F). These results suggested that reduction of Sirt3 expression reduced GSIS due to altering expression of genes related to beta cell function and impairing SOD2 activity.
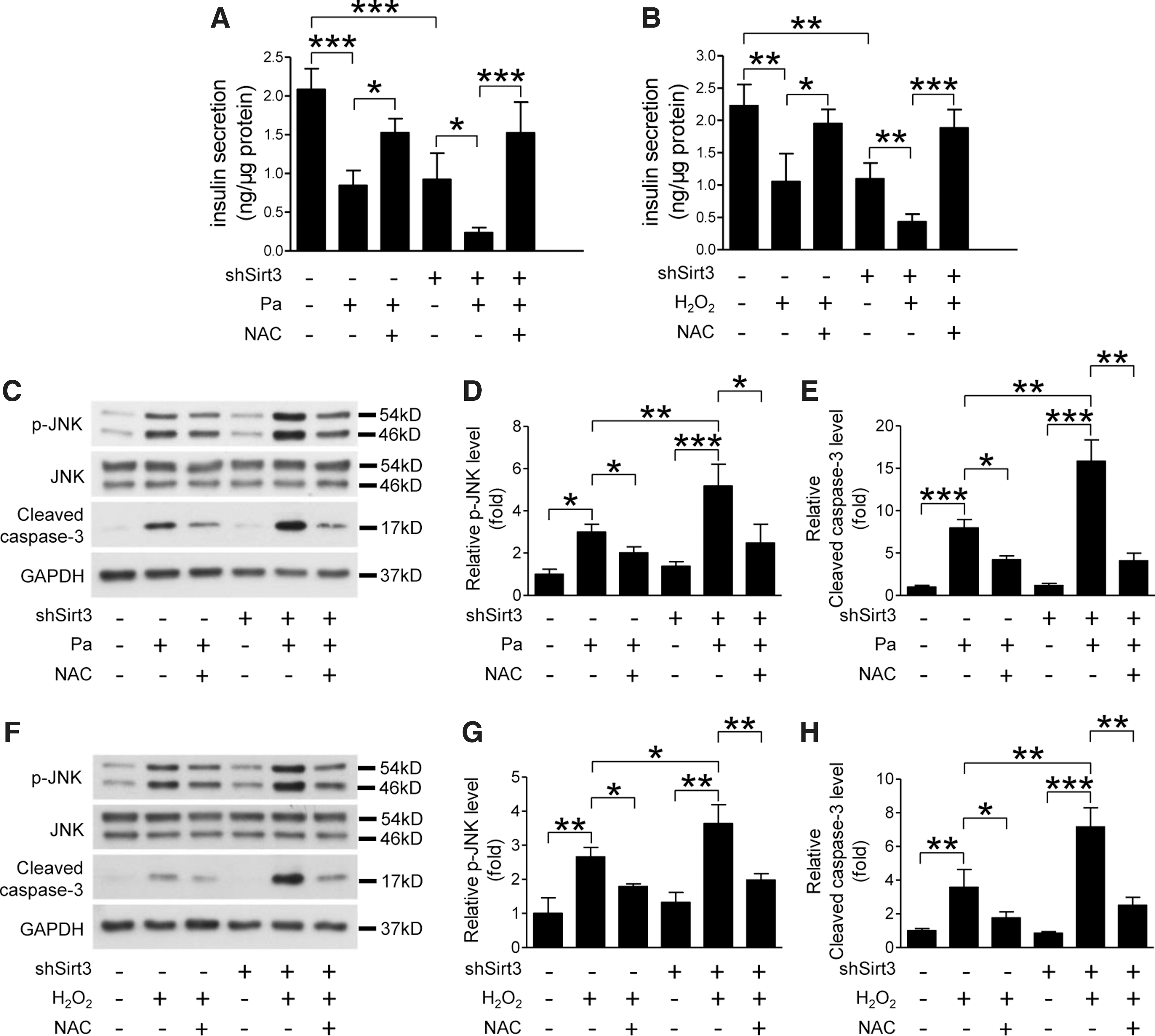
To further clarify the relationship between Sirt3 function and oxidative stress, we challenged the Sirt3 knockdown MIN6 cells with palmitate or H2O2 in combination with or without N-acetyl-cysteine (NAC), an antioxidizing agent. In control MIN6 cells, palmitate- or H2O2-induced impairment of insulin secretion was partially restored by NAC treatment. Reduced Sirt3 expression enhanced palmitate- or H2O2-induced beta cell dysfunction, which was also ameliorated by antioxidant treatment with NAC (Fig. 5A, B). JNK phosphorylation is a marker of oxidative stress, which was increased in Sirt3 KO islets with HFD feeding (Fig. 2G). Both palmitate and H2O2 treatments increased JNK phosphorylation and cleaved caspase-3 levels in MIN6 cells, which were accentuated by knockdown of Sirt3 and attenuated by NAC treatment (Fig. 5C–H).
Previous reports demonstrated that JNK activation caused by oxidative stress promotes Pdx-1 translocation from the nucleus to the cytosol, which thereby impairs beta cell function. We evaluated the Pdx-1 translocation in MIN6 cells after palmitate treatment with or without JNK inhibition. Consistent with previous reports, Pdx-1 translocation caused by palmitate treatment was suppressed by JNK inhibition (Supplementary Fig. S5A, B). Then, we further observed aggravated Pdx-1 translocation in Sirt3 knockdown MIN6 cells after palmitate or H2O2 treatment. However, this phenomenon was restored by antioxidant treatment with NAC (Supplementary Fig. S6A, B), indicating that Sirt3 plays a protective role in maintaining normal beta cell function.
Restoring Sirt3 expression in Sirt3 KO islets improves pancreatic beta cell function
To confirm the protective role of Sirt3 in beta cell function, we next infected islets isolated from 8- to 12-week-old Sirt3 KO and WT mice with Ad-Sirt3 and restoration of Sirt3 expression in isolated islets was confirmed by Western blot (Fig. 6A). In Sirt3 KO islets, GSIS and ATP production was decreased compared with WT islets. This reduction was ameliorated when Sirt3 expression was restored in KO islets (Fig. 6B, C). Similar to MIN6 cells, ablation of Sirt3 in islets also affected mRNA expression of genes related to beta cell function (Fig. 6D). In these Sirt3 KO islets, G6pase mRNA level was suppressed, while the expression level of Glut2 was increased after restoration of Sirt3 (Fig. 6D). Sirt3 deficiency-induced impairment of Pdx-1 and Insulin2 mRNA levels and stimulation of Tnfα level were not affected by Sirt3 restoration (Fig. 6D). Restoration of Sirt3 expression also increased SOD2 acetylation, which was also downregulated in Sirt3 KO islets (Fig. 6E).
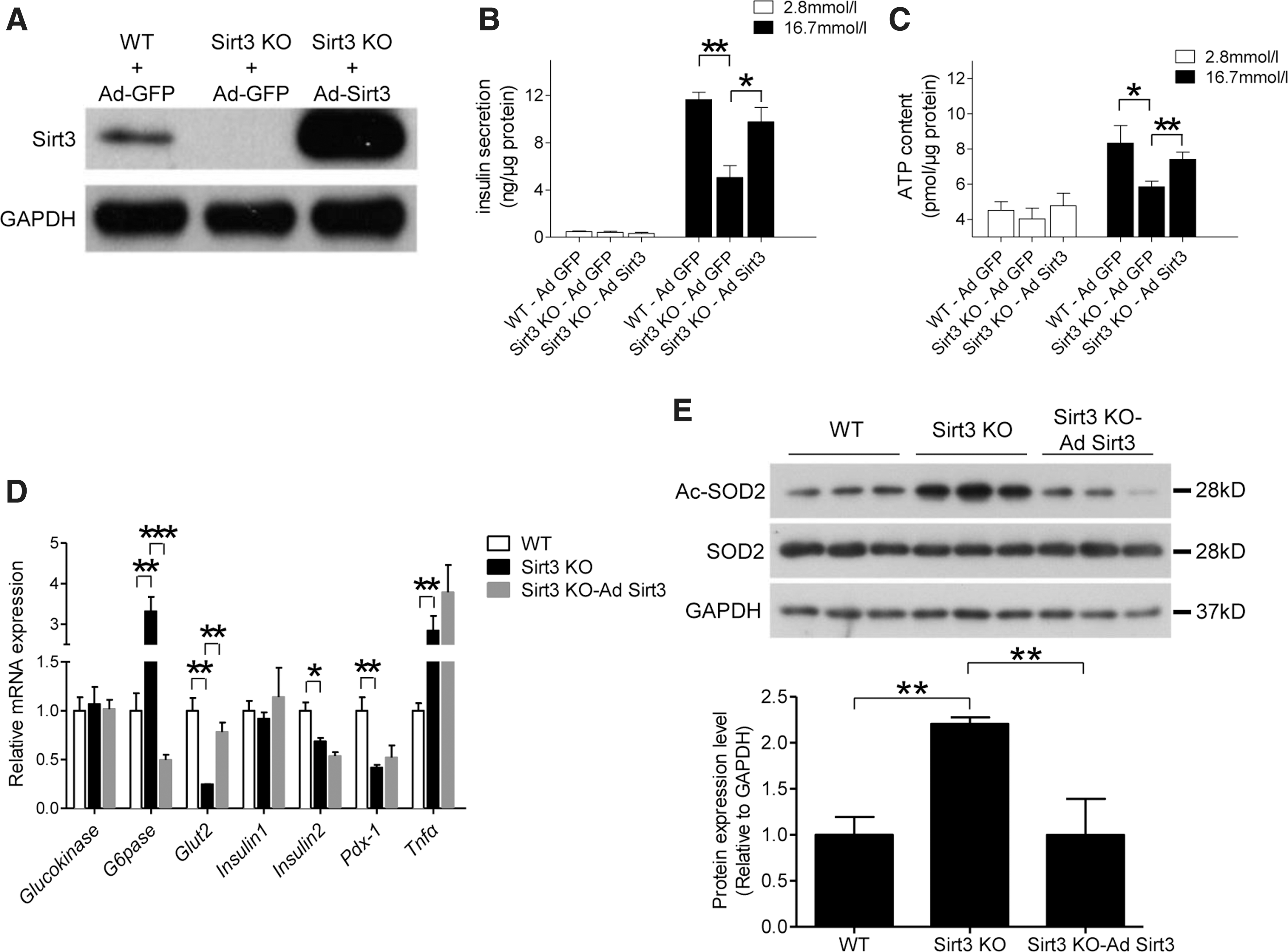
We then treated primary mouse islets with Ad-Sirt3 infection or NAC together with palmitate or H2O2. Palmitate or H2O2 treatment reduced GSIS in WT islets and this impairment was accentuated in Sirt3 KO islets. However, decreased insulin secretion was restored by Sirt3 expression or antioxidant treatment with NAC in both WT and Sirt3 KO islets (Fig. 7A, B). Similarly, JNK phosphorylation levels were increased by palmitate and H2O2 treatments in WT islets. This elevation was further enhanced in Sirt3 KO islets, but this was suppressed with restoration of Sirt3 expression or NAC treatment (Fig. 7C–H). These results confirmed that Sirt3 expression might protect beta cells by suppressing oxidative stress.

Overexpression of Sirt3 protects beta cells from palmitate-induced impairment
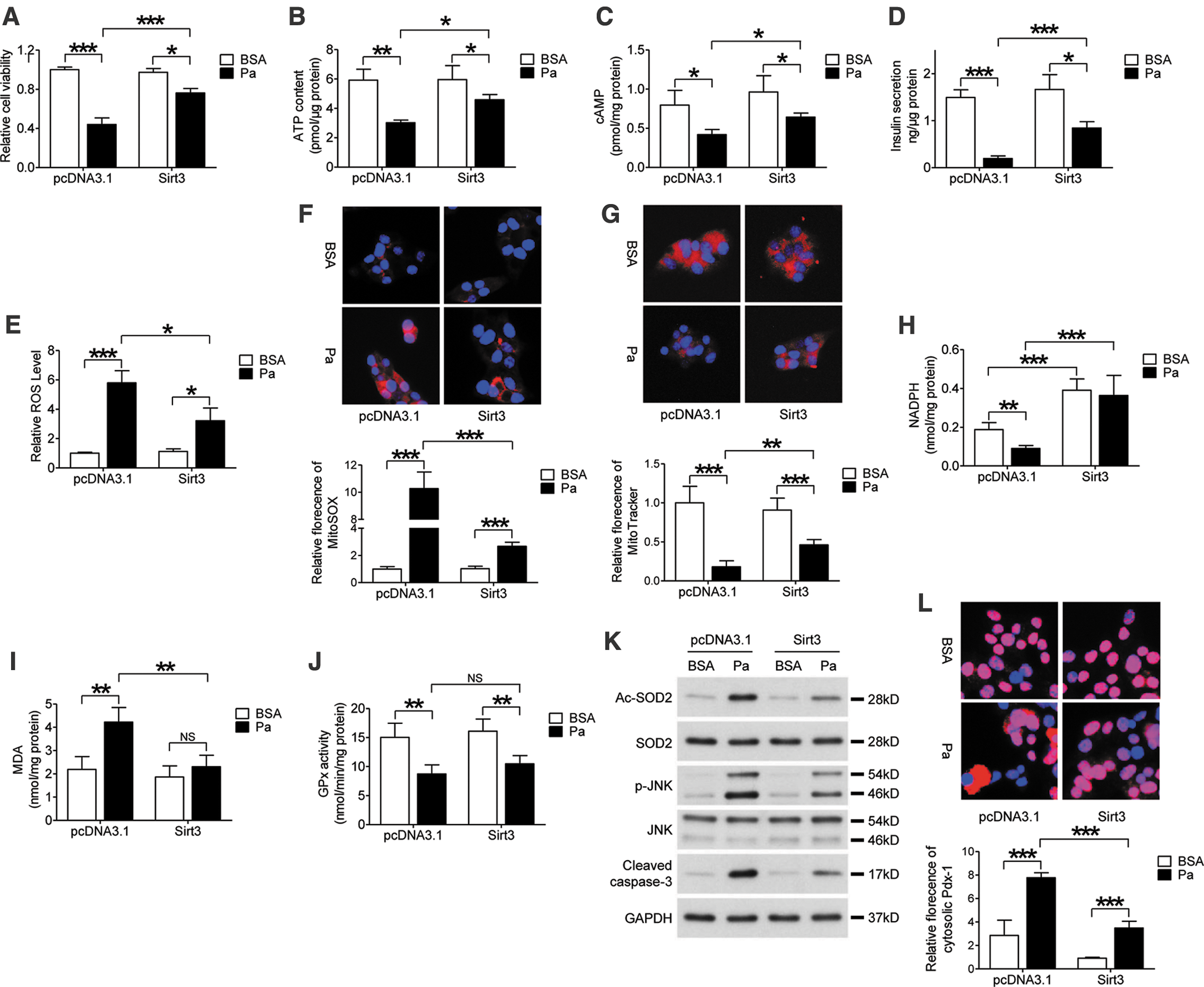
Since hyperlipidemia impaired Sirt3 expression in beta cells, we sought to investigate the therapeutic effect of Sirt3 in palmitate-treated MIN6 cells. Sirt3 overexpression plasmid or control empty vector pcDNA3.1+ was transfected in MIN6 cells. The stable Sirt3 overexpression cell line was screened by the antibiotic G418 and overexpression of Sirt3 was confirmed by Western blot (Supplementary Fig. S7A). To identify the localization of overexpressed Sirt3, a FLAG tag was ligated to the 3′ end of Sirt3 DNA fragment. Double staining of FLAG and MitoTracker suggested that overexpressed Sirt3 is localized inside the mitochondria (Supplementary Fig. S7B).
MTT results indicated that palmitate-induced suppression of cell viability was improved with Sirt3 overexpression (Fig. 8I). In the beta cell functional study, overexpression of Sirt3 improved glucose-induced ATP generation (Fig. 8J) and cAMP levels (Fig. 8K) when compared with the control group after palmitate treatment. Consistently, the reduction of GSIS in MIN6 cells after palmitate treatment was partially rescued by overexpression of Sirt3 (Fig. 8L). In addition, excessive ROS generation caused by palmitate treatment was also ameliorated by Sirt3 (Fig. 8D). In addition, increased superoxide generation and decreased mitochondrial mass caused by palmitate were both improved after Sirt3 overexpression (Fig. 8E, F). Palmitate treatment significantly impaired NADPH, but increased malondialdehyde (MDA) level in MIN6 cells (Fig. 8A, B). Overexpression of Sirt3 improved NADPH levels in both control and palmitate-treated cells. Besides, Sirt3 overexpression also inhibited MDA content in the presence of palmitate (Fig. 8A, B). However, palmitate-induced suppression of glutathione peroxidase (GPx) activity was not affected by Sirt3 overexpression (Fig. 8C). Furthermore, palmitate-stimulated SOD2 acetylation, JNK phosphorylation, and cleaved caspase-3 expression were partially decreased in Sirt3-overexpressed cells (Fig. 8G). As expected, palmitate treatment-induced Pdx-1 translocation was also suppressed by Sirt3 overexpression (Fig. 8H). These results suggested that restoration of Sirt3 expression is able to protect beta cells from damage caused by hyperlipidemia and oxidative stress.
Discussion
Insufficient insulin release from pancreatic beta cells is a key feature of T2D (1). Diet-induced obesity in mice is a well-described T2D model with impaired insulin secretion (30). In the current study, we investigated the protective role of Sirt3 with antioxidant capacity in the regulation of beta cell function. In this animal study, we demonstrated that Sirt3 deficiency promoted HFD-induced oxidative stress and pancreatic beta cell dysfunction. Similar to previous findings (14), Sirt3 KO mice treated with STD did not exhibit a glucose-related phenotype, but under metabolic stress, such as prolonged HFD feeding, there was metabolic decompensation with reduced GSIS and glucose intolerance. Using extracted islets and the MIN6 cell line exposed to palmitate or H2O2, we demonstrated the effects of Sirt3 on beta cell apoptosis, oxidative stress, Pdx-1 translocation, reduced ATP production, and dysregulation of gene expression related to beta cell function. These effects were partially rescued by restoring Sirt3 expression. Taken together, these findings suggested that Sirt3 expression played a key role in maintaining beta cell function, especially under hyperlipidemic condition, in part, mediated by ameliorating oxidative stress and might be a potential therapeutic target.
When mice are challenged with nutrition overload, Sirt3 acts as a main regulator of fatty acid oxidation in the liver and skeletal muscle through deacetylation of a series of key enzymes to protect the mice from developing severe insulin resistance (14, 21, 25). Our data showed that HFD slightly increased oxidative stress levels in islets of WT mice, but these were dramatically increased with Sirt3 deficiency. Other researchers have reported that oxidative levels increased in peripheral tissues (14, 41) of Sirt3 KO mice fed an HFD for 13 weeks (14). Aging is associated with cellular senescence, which may cause beta cell apoptosis and dysfunction (10, 24). Since Sirt3 is involved in the regulation of the aging process and oxidative stress (6, 28), we examined whether HFD might worsen age-related beta cell dysfunction. In our experiments, we subjected the mice to 28 weeks of STD and HFD and found that cleaved caspase-3 level, a marker of apoptosis, was increased in Sirt3 KO islets fed an STD in contrast to the WT mice, supporting the protective effects of Sirt3 in age-related changes in the islets.
In this study, we measured the JNK phosphorylation level as a marker of oxidative stress in pancreatic beta cells (5, 9). Ablation of Sirt3 promoted palmitate or H2O2-induced JNK phosphorylation in primary islets and cell-based experiments. Overexpression of Sirt3 in MIN6 cells suppressed palmitate-induced JNK phosphorylation, SOD2 acetylation, MDA expression, ROS generation, and Pdx-1 translocation from nucleus to cytoplasm. Consistent with previous reports from studies in HEK293 cells (39), overexpression of Sirt3 elevated NADPH levels in MIN6 cells, which was inhibited by palmitate treatment. However, palmitate-induced suppression of GPx activity was not affected by Sirt3 overexpression, suggesting that GPx activation may not be involved in Sirt3-mediated regulation of beta cell function. ROS generation and JNK phosphorylation were not affected in Sirt3-deficient MIN6 cells or islets under physiological conditions despite the reduced SOD2 activity and impaired GSIS in these cells, suggesting that JNK might be important for beta cell function only under metabolic stress conditions. In addition, compensatory pathways could be activated in Sirt3-deficient beta cells to offset the negative effects of suppressed SOD2 activity and maintain normal cellular oxidative stress level under physiological condition. Decreased Pdx-1 mRNA expression in Sirt3-deficient beta cells might contribute to impaired GSIS in addition to the oxidative pathway (18). Moreover, NAC treatment fully reverses H2O2-induced beta cell dysfunction, but only partially rescues palmitate-induced impairment. These findings imply that some other pathways in addition to oxidative stress are involved in Sirt3 deficiency-induced beta cell dysfunction, such as the activated peroxisome-proliferator-activated receptor gamma co-activator-1alpha (PGC-1α) pathway (42). Elevated G6pase and reduced Glut2 mRNA levels were observed in Sirt3-deficient beta cells, suggesting that ablation of Sirt3 increased glycolysis and impaired glucose sensitivity (16). These negative effects were reversed by Sirt3 expression in KO islets with normalization of mRNA levels. Taken together, some of these inconsistent results might be due to activation of other pathways in Sirt3-deficient beta cells to offset the negative effects of suppressed SOD2 activity to maintain normal cellular redox level under physiological condition. As such, a more discovery-oriented approach, for example, using multiomic analysis, will be needed to explain the complexity of these interacting pathways.
A recent study suggested that 24-h treatment with 0.5 mM palmitate did not affect Sirt3 expression in NIT1 cells, but overexpression of Sirt3 partially ameliorated palmitate-induced beta cell apoptosis and dysfunction (22). In this experiment, we increased the time and concentration exposure to palmitate and found reduced expression of Sirt3 protein in MIN6 cells, which was not seen with oleate treatment. Consistent with previous reports (22), we also detected ameliorated endoplasmic reticulum stress marker CCAAT/-enhancer-binding protein homologous protein (CHOP) expression (Supplementary Fig. S8) and partially improved beta cell function in palmitate-treated MIN6 cells after Sirt3 overexpression. These results suggested that different stressors for lipotoxicity might elicit a heterogeneous response, which might be partially linked by Sirt3 as a key, but not the only, antioxidant protein.
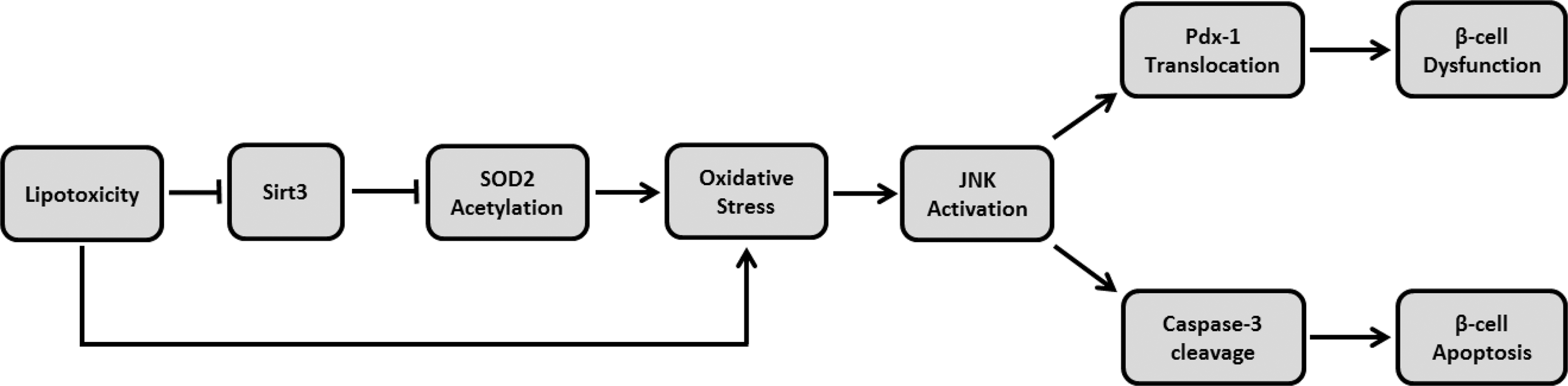
In animal and cell-based models, prolonged exposure to HFD feeding, H2O2, or palmitate reduced Sirt3 expression, resulting in reduced beta cell function and survival through activation of oxidative stress pathways, as evidenced by JNK activation (Fig. 9). These negative changes were, in part, reversed by overexpression of Sirt3 in pancreatic beta cells. These findings suggested that Sirt3 may be a therapeutic target for preservation and restoration of beta cell function due to metabolic stress such as obesity and hyperlipidemia. However, further investigations on human beta cells are still necessary to confirm the clinical relevance of these findings.
Materials and Methods
Cell culture and treatment
The MIN6 pancreatic beta cell line (passages 20–30, kindly provided by Prof. Guang Ning, School of Medicine, Shanghai Jiao-Tong University, Shanghai, China) was cultured in standard medium as previously described (20). Palmitate was prepared and used to treat MIN6 cells or islets using published methodology (20). Cells were starved in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) without fetal bovine serum (FBS) overnight, followed by treatment with H2O2 (Sigma-Aldrich, St. Louis, MO) in full medium using different concentrations and for different time periods. NAC and SP600125 (both from Sigma-Aldrich) were used to treat cells in a final concentration of 10 mM and 20 μM separately.
Plasmid, adenovirus, stable cell line establishment, and shRNA lentivirus
To construct the Sirt3 overexpression plasmid, mouse cDNA encoding Sirt3 was amplified using the following primers: forward (F) 5′-GGGGTACCATGGCGCTTGACC CTCTAGG-3′ and reverse (R) 5′-CGGAATTCTTATCTG TCCTGTCCATCCA-3′. Then, the amplified fragment was subcloned into pcDNA3.1+ vector between KpnI and EcoRI restriction sites for the following sequencing analysis. Sirt3 overexpression plasmid or empty vector was transfected into MIN6 cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Stable Sirt3 overexpression cell lines were screened for 3 weeks in the full culture medium containing 0.5 mg/ml G418 (Sigma-Aldrich). The cells resistant to G418, an antibiotic, were isolated and cultured for the following treatments.
The following primers were used to construct FLAG tag ligated Sirt3 overexpression plasmid (Sirt3-FLAG): forward 5′-GGGGTACCATGGCGCTTGACCCTCTAGG-3′ and reverse 5′-CGGAATTCTTACTTGTCGTCATCGTCTTTG TAGTCTCTGTCCTGT-3′. Previously constructed pcDNA3.1-Sirt3 plasmid was used as the template for polymerase chain reaction (PCR) amplification.
To construct Sirt3 overexpression adenovirus (Ad-Sirt3), Sirt3 DNA fragment was amplified from pcDNA3.1-Sirt3. pENTR-Sirt3 shuttle plasmid was generated by insertion of Sirt3 fragment into pENTR directional TOPO vector (Invitrogen) according to the manufacturer's instructions. After being recombined to pAd/CMV/V5-DEST Gateway vector (Invitrogen), Ad-Sirt3 was linearized by PacI digestion and transfected into 293A cells for virus production and amplification. Adenovirus overexpressing green fluorescent protein (Ad-GFP) was used as control. The titration of the virus was measured by Adeno-X Rapid Titer Kit (Clontech, Mountain View, CA) according to the manufacturer's instructions.
Sirt3 shRNA lentivirus particles were purchased from Santa Cruz (Dallas, TX) and used according to the manufacturer's instructions.
Animal experiments, OGTT, insulin tolerance test, and islet isolation
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals and approved by The Animal Subjects Committee of The Chinese University of Hong Kong. The Sirt3 KO and WT 129 Sv mice were purchased from the Jackson Laboratory (Bar Harbor, ME) (27) and housed in pathogen-free conditions with 12-h light–12-h dark cycle and free access to water and food.
Sirt3 KO and WT mice were fed an STD or HFD (60% kcal% fat; Research Diets) at the age of 4–6 weeks. Body weight and blood glucose levels were measured every 2 weeks between 9:00 and 10:00 am. All mice were fasted overnight before undergoing OGTT. Blood glucose levels were measured by a glucometer (Johnson & Johnson, New Brunswick, NJ) at time points 0, 30, 60, and 120 min after oral gavage of 2 g/kg body weight glucose. During OGTT, blood samples were collected at four time points for measurement of insulin. For insulin tolerance test, mice were fasted for 6 h. Blood glucose levels were measured at time points 0, 30, 60, and 120 min after intraperitoneal injection of 0.5 IU/kg body weight recombinant human insulin (Novo Nordisk, Bagsvard, Denmark). Pancreatic islets were isolated from Sirt3 KO or WT mice as previously described (20). For virus infections, primary mouse islets isolated from Sirt3 KO or WT mice were infected with Ad-Sirt3 or Ad-GFP as control (m.o.i. = 100) and followed by different treatments.
Real-time PCR and immunoblotting
Quantitative reverse transcription-PCR analysis was performed as previously described (20) and the following primers were used: Glucokinase: forward 5′-CCAAGCACCAAGCGGTATCA-3′ reverse 5′- GTCAGTGGGTTGGACTTCTCT-3′; G6pase: forward 5′-CGACTCGCTATCTCCAAGTGA-3′ reverse 5′-GTTGAACCAGTCTCCGACCA-3′; Glut2: forward 5′-TCA GAAGACAAGATCACCGGA-3′ reverse 5′-GCTGGTGTG ACTGTAAGTGGG-3′; Insulin1: forward 5′-CCAGCTATA ATCAGAGACCATCAG-3′ reverse 5′-ACAAAAGCCTGG GTGGGTT-3′; Insulin2: forward 5′-GGAGTCACCTCGGC CTAAAAA-3′ reverse 5′-CAAGTTCAACACTAATGCCA GGA-3′; Pdx-1: forward 5′-CCCCAGTTTACAAGCTC GCT-3′ reverse 5′-CTCGGTTCCATTCGGGAAAGG-3′; Tnfα: forward 5′-TGAAAGGAGAAGGCTTGTGAG-3′ reverse 5′-GGGTAATGGGATGAGTATGGG-3′; and Gapdh: forward 5′-TGGATTTGGACGCATTGGTC-3′ reverse 5′-TTTGCACTGGTACGTGTTGAT-3′.
Total proteins of MIN6 cells or pancreatic islets were extracted and prepared as described (20). The following primary antibodies were used: Sirt3 (1:1000), SOD2 (1:2000), p-JNK (1:1000), JNK (1:1000), cleaved caspase-3 (1:1000), and GAPDH (1:5000) were purchased from Cell Signaling Technology (Danvers, MA), and Ac-SOD2 (1:1000) was purchased from Abcam (Cambridge, United Kingdom). horseradish peroxidase (HRP)-linked anti rabbit IgG (1:2000; Cell Signaling Technology) was used as a secondary antibody. Finally, protein bands were developed by Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA) (Supplementary Fig. S9).
Histological staining
For 3-nitrotyrosine staining, mice were sacrificed and the pancreases were fixed in 4% paraformaldehyde and embedded in paraffin after dehydration. Four-micrometer sections were cut and rehydrated in the following reagents: xylene 5 min, xylene 5 min, 100% ethanol 5 min, 100% ethanol 5 min, 95% ethanol 5 min, 90% ethanol 5 min, 80% ethanol 5 min, 70% ethanol 5 min, and ddH2O 5 min. After rehydration, slides were put in 3% H2O2 for 20 min at room temperature. Then, the sections were treated with 0.01 M sodium citrate buffer (pH 6.0) and heated at ∼100°C for 10 min. After cooling down to room temperature, the sections were blocked in 5% rabbit serum for 1 h. Then, 3-nitrotyrosine primary antibody (1:200, Santa Cruz) was applied to the sections at 4°C overnight. After three times' wash in phosphate buffered saline plus Tween 20, HRP-linked rabbit anti-mouse (1:400; Dako, Glostrup Municipality, Denmark) was applied as secondary antibody for 1 h at room temperature. After that DAB solution (Dako) was dropped on the sections for 3 min. Then, the slides were immersed with H2O and counterstained with hematoxylin (Sigma-Aldrich). Finally, the sections were dehydrated with 95% ethanol 10 s, 95% ethanol 10 s, 100% ethanol 10 s, 100% ethanol 10 s, xylene 10 s, and xylene 10 s, followed by distyrene plasticiser xylene mounting.
For TUNEL staining and Pdx-1 staining in mouse pancreas, the dehydration and rehydration parts were the same as IHC staining, which were described in the former section. After rehydration, sections were treated with 0.01 M sodium citrate buffer (pH 6.0) and heated at ∼100°C for 10 min. After cooling down to room temperature, the sections were washed with phosphate-buffered saline (PBS) and blocked in 1% Bovine serum albumin (BSA) at room temperature for 1 h (for Pdx-1 staining, sections were permeablized in 0.25% Triton X-100 at room temperature for 30 min before blocking). Guinea pig anti-insulin (ready to use; Dako) and rabbit anti-Pdx-1 (1:1000; Abcam) were used as primary antibodies and applied on the sections at 4°C overnight. Then, secondary antibodies, cy3-donkey anti-guinea pig (1:400; Jackson, Bar Harbor, ME) and goat anti-rabbit (Alexa Fluor 488, 1:1000; Invitrogen), were applied to sections at room temperature for 1 h. Then, the slides were washed twice with PBS. For TUNEL staining, 50 μl TUNEL reaction mixture (Roche, Basel, Switzerland) was added to each sample and incubated at 37°C for 60 min. Finally, the slides were mounted with ProLong Gold Antifade Mountant with DAPI (Invitrogen) before microscopic analysis.
For immunocytochemical staining, a round glass coverslip was inserted at the bottom of a 12-well plate before seeding cells. Then, cells were treated with various reagents as described. After treatment, cells growing on the glass slip were fixed with 4% (wt/vol) paraformaldehyde for 1 h. After being permeabilized with 2 M HCl/Triton X-100 at 37°C for 20 min, cells were blocked in 1% BSA for another 30 min. Then, primary antibody rabbit anti-Pdx-1 (1:1000; Abcam) was applied to the cells, followed by the secondary antibody cy3-donkey anti-rabbit (1:2000; Jackson). Finally, the cells were stained with DAPI for 5 min before microscopic analysis. MitoSOX and Mitotracker Deep Red (both purchased from Invitrogen) staining of MIN6 cells was performed according to the manufacturer's instructions.
Insulin ELISA, NADPH, MDA, GPx activity, ROS, ATP, and cAMP measurement
For the GSIS study of MIN6 cells or isolated mouse islets, cells were starved in low-glucose KRBH buffer [129 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, 20 mM HEPES, 24 mM NaHCO3, 0.2% BSA (wt/vol), 0.2% FBS (wt/vol)] containing 5.5 mM (for MIN6 cells) or 2.8 mM (for islets) glucose for 1 h after indicated treatments. Then, the buffer was replaced with low-glucose (5.5 mM for MIN6 cells, 2.8 mM for islets) or high-glucose (25 mM for MIN6 cells, 16.7 mM for islets) stimulation KRBH buffer for 2 h. Finally, the supernatant was collected for insulin enzyme-linked immunosorbent assay (ELISA) (Antibody and Immunoassay Services, Hong Kong University, Hong Kong, China) according to the manufacturer's instructions and cells were harvested for protein amount measurement (Thermo, Waltham, MA).
For measurement of serum insulin, the blood samples were put on ice for 1 h immediately after collection, followed by centrifugation at 1500 g for 10 min. The upper layer of the serum was collected for measurement of insulin using ELISA (Antibody and Immunoassay Services, Hong Kong University).
MTT was purchased from Sigma and dissolved in DMSO to obtain a 5 mg/ml stock solution. After the indicated treatments on MIN6 cells, 10 μL MTT stock solution was added to each well on 96-well plate and cultured for another 3 h. Then, the medium was removed and changed with 50 μl DMSO for absorbance measurement at the wavelength of 570 nm.
The NADPH level was measured by NADP/NADPH Assay Kit, MDA level was measured by Lipid Peroxidation (MDA) Assay Kit, GPx activity was measured by Glutathione Peroxidase Assay Kit, and ROS level was measured by DCFDA-Cellular Reactive Oxygen Species Detection Assay Kit. All assay kits were purchased from Abcam and procedures were performed according to the manufacturer's instructions.
For glucose-stimulated ATP measurement, MIN6 cells or islets were first starved in KRBH buffer containing 5.5 mmol glucose (MIN6 cells) or 2.8 mmol (islets) for 1 h, then stimulated with KRBH buffer containing 25 mmol glucose (MIN6 cells) or 16.7 mmol (islets) for 2 h. The supernatant was collected for ATP measurement by ATP Colorimetric/Fluorometric Assay Kit (BioVision, Milpitas, CA) according to the manufacturer's instructions. Cells were harvested for protein amount measurement (Thermo).
MIN6 cAMP level measurement was the same as previously described (20).
Statistical analysis
Data are presented as mean ± SEM. Differences between groups were analyzed by two-tailed unpaired Student's t-test with Welch's correction or one-way ANOVA, followed by Tukey's post hoc test where appropriate. Statistical comparisons were made using GraphPad (GraphPad Software, San Diego, CA). A p-value <0.05 was considered as statistically significant.
Footnotes
Acknowledgments
This study was partially supported by the Hong Kong Foundation for Research and Development in Diabetes and Liao Wun Yuk Diabetes Memorial Fund under the auspices of The Chinese University of Hong Kong. This study was also supported by grants from the National Natural Science Foundation of China (General Program 21477101); the Research Grant Council of Hong Kong (RGC GRF 463612 and 14104314); Faculty Research Grants from the Hong Kong Baptist University (FRG2/15-16/067); Hong Kong Health and Medical Research Fund (HMRF/03144376); and HKASO research grant 2015–16.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.