
Abstract
Aims:
Synaptic deficits are known to underlie the cognitive dysfunction seen in Alzheimer's disease (AD). Generation of reactive oxygen species (ROS) by β-amyloid has also been implicated in AD pathogenesis. However, it is unclear whether ROS contributes to synaptic dysfunction seen in AD pathogenesis and, therefore, we examined whether altered redox signaling could contribute to synaptic deficits in AD.
Results:
Activity dependent but not basal translation was impaired in synaptoneurosomes from 1-month old presymptomatic APPSwe/PS1ΔE9 (APP/PS1) mice, and this deficit was sustained till middle age (MA, 9–10 months). ROS generation leads to oxidative modification of Akt1 in the synapse and consequent reduction in Akt1-mechanistic target of rapamycin (mTOR) signaling, leading to deficiency in activity-dependent protein translation. Moreover, we found a similar loss of activity-dependent protein translation in synaptoneurosomes from postmortem AD brains.
Innovation:
Loss of activity-dependent protein translation occurs presymptomatically early in the pathogenesis of AD. This is caused by ROS-mediated loss of pAkt1, leading to reduced synaptic Akt1-mTOR signaling and is rescued by overexpression of Akt1. ROS-mediated damage is restricted to the synaptosomes, indicating selectivity.
Conclusions:
We demonstrate that ROS-mediated oxidative modification of Akt1 contributes to synaptic dysfunction in AD, seen as loss of activity-dependent protein translation that is essential for synaptic plasticity and maintenance. Therapeutic strategies promoting Akt1-mTOR signaling at synapses may provide novel target(s) for disease-modifying therapy in AD. Antioxid. Redox Signal. 27, 1269–1280.
Introduction
A
The major finding in this study is that protein translation at the level of postnuclear supernatant is unaltered in Alzheimer's disease (AD), but activity-driven protein translation at synapse is compromised from an early stage of disease progression. We also provide evidence for compromised synaptic Akt1-mechanistic target of rapamycin (mTOR) signaling cascade leading to this effect. Furthermore, we show that this deficit can be reversed by ectopic overexpression of Akt1, indicating a target for potential therapeutic intervention to retard the progression of AD. The major results from the study are outlined as follows: (i) Loss of activity-dependent protein translation, critical for synaptic plasticity and maintenance, was observed in cortical synaptoneurosomes from APP/PS1 mice and postmortem AD brains. (ii) This is caused by increased reactive oxygen species (ROS) generation in synaptosomes leading to redox modification of Akt1, resulting in compromised Akt1-mTOR signaling. (iii) Although there is overall generation of ROS, synaptic phosphorylated Akt1 is specifically compromised because of redox modification. (iv) Activity-dependent protein translation can be modulated by Aβ-generated ROS through Akt1 thiol modification. (v) ROS-mediated synaptic pathology in AD starts very early in life, whereas overt behavioral and pathological symptoms are evident much later.
Oxidative stress and synaptic dysfunction are considered to be among the primary events in AD pathogenesis (7, 31). Structural changes seen as loss of synapses correlate with cognitive dysfunction in human subjects and mice that carry human mutations of familial AD (35). However, the molecular underpinnings of synaptic deficits and the targets of reactive oxygen species (ROS)-mediated neurodegeneration are poorly understood.
Protein translation (basal and neuronal activity-dependent) occurring at the synapse is critical for synapse maintenance and plasticity (10, 36). Akt1-mammalian target of rapamycin (mTOR) signaling mediates cap-dependent protein translation, which accounts for most of the protein translation occurring in the cell (30). In particular, it is critical for activity-dependent local protein synthesis at the synapse in neurons (39). Akt (or PKB) is a cell survival kinase whose activity requires phosphorylation at Thr308 in the activation loop of the kinase domain and at Ser473 in the C-terminal hydrophobic domain (18). Phosphorylated Akt1 (pAkt1) indirectly activates mechanistic target of rapamycin (mTOR) by phosphorylating TSC1/2 complex, the inhibitory regulator of mTOR, thereby activating mTOR pathway that culminates in phosphorylation of 4EBP1 at Thr46/47 and S6K at Thr389, both important regulators of cap-dependent protein translation (34). Although S6K phosphorylates S6 ribosomal protein and induces protein translation, 4EBP1 is a translational repressor that binds to eukaryotic translation initiation factor 4E (eIF4E) and regulates binding of mRNA to small ribosomal subunit and phosphorylated (pmTOR)-mediated phosphorylation dissociates it from eIF4E, hence removing its inhibitory effects on translation initiation complex (13).
Interestingly, although dysregulation of Akt1 signaling has been observed in postmortem brain tissue from AD patients and in animal models of AD (9, 16, 23, 26, 27, 33, 34a, 37, 38, 46), two critical questions remain unanswered. On one hand, we do not have a clear understanding of the molecular mechanisms that lead to defective Akt1 signaling. On the other, the functional consequences of this deficit in the perspective of neurodegeneration are yet to be discerned. We now present evidence that defective Akt1 signaling is mediated by oxidative modification of Akt1, occurring presymptomatically as early as 1 month of age in the APP/PS1 transgenic mouse model of AD. The loss of Akt1 kinase occurs in the synaptic compartment and consequently affects activity-dependent protein translation, leading to potential synaptic dysfunction.
We analyzed local protein translation in synaptoneurosomes isolated from frontal cortical tissue samples of postmortem brains from persons with and without AD. Although basal protein translation was found to be unaffected, KCl-stimulated protein translation was found to be defective in neocortical tissue samples from postmortem brains of AD patients. Furthermore, to determine the molecular mechanism responsible for this dysfunction, we used a mouse model of AD, APPSwe/PS1ΔE9 (APP/PS1) mice, and studied the Akt1-mTOR signaling pathway that is a critical regulator of protein translation and homeostasis at the synapse (15, 19, 28, 36, 44). Our studies were carried out in three ages of mice: adolescent (ADL) animals (1–1.5 months old), young adults (YA, 3–4 months old) before the onset of pathological hallmarks and memory dysfunction, and MA mice (9–12 months of old) when both these features of the disease are evident. In addition, using cultured primary cortical neurons from APP/PS1 mice, we sought to determine whether ectopic overexpression of a constitutively active Akt1 [myristoylated Akt1; (42)] could rescue the stimulated protein translation at the synapse.
Results
Activity-dependent translation is repressed in synaptoneurosomes of APP/PS1 mice from a very young age
We assessed basal and activity-dependent protein translation in synaptoneurosomal (Fig. 1A–C) and postnuclear supernatant (PNS) (Fig. 1E) fractions isolated from APP/PS1 mouse brain cortex at 1, 3, and 9–11 months of age. Upon stimulation of synaptoneurosomes isolated from cortex of wild type (WT) mice with KCl, we observed increased S35-methionine incorporation as has been previously reported [Fig. 1F; (32)]. S35-methionine incorporation was absent in the presence of cycloheximide. Absence of radioactivity from the last methanol wash of the trichloroacetic acid (TCA) precipitate provided evidence for removal of all unbound S35-methionine from the reaction mixture (Fig. 1F), indicating that the measured radioactivity from TCA pellets was contributed solely by S35-methionine incorporated into nascent proteins. Unlike the WT synaptoneurosomes, KCl-stimulated protein translation was absent in synaptoneurosomes isolated from APP/PS1 mice at all ages examined and it was similar to basal translation seen in the absence of KCl (Fig. 1A–C). To validate the aforementioned, we examined activity-dependent changes in expression of the cytoskeletal regulatory protein, Arc, by immunoblotting because it is used as a marker for coupling neuronal activity to synaptic plasticity (5, 17, 29). We observed increased levels of Arc protein after KCl stimulation of synaptoneurosomes isolated from 1-month old WT mice, but not from APP/PS1 mice (Fig. 1D and Supplementary Fig. S6). Surprisingly, protein translation in cortical PNS samples of APP/PS1 mice as assessed by S35-methionine incorporation remained unaltered at all ages examined (Fig. 1E).
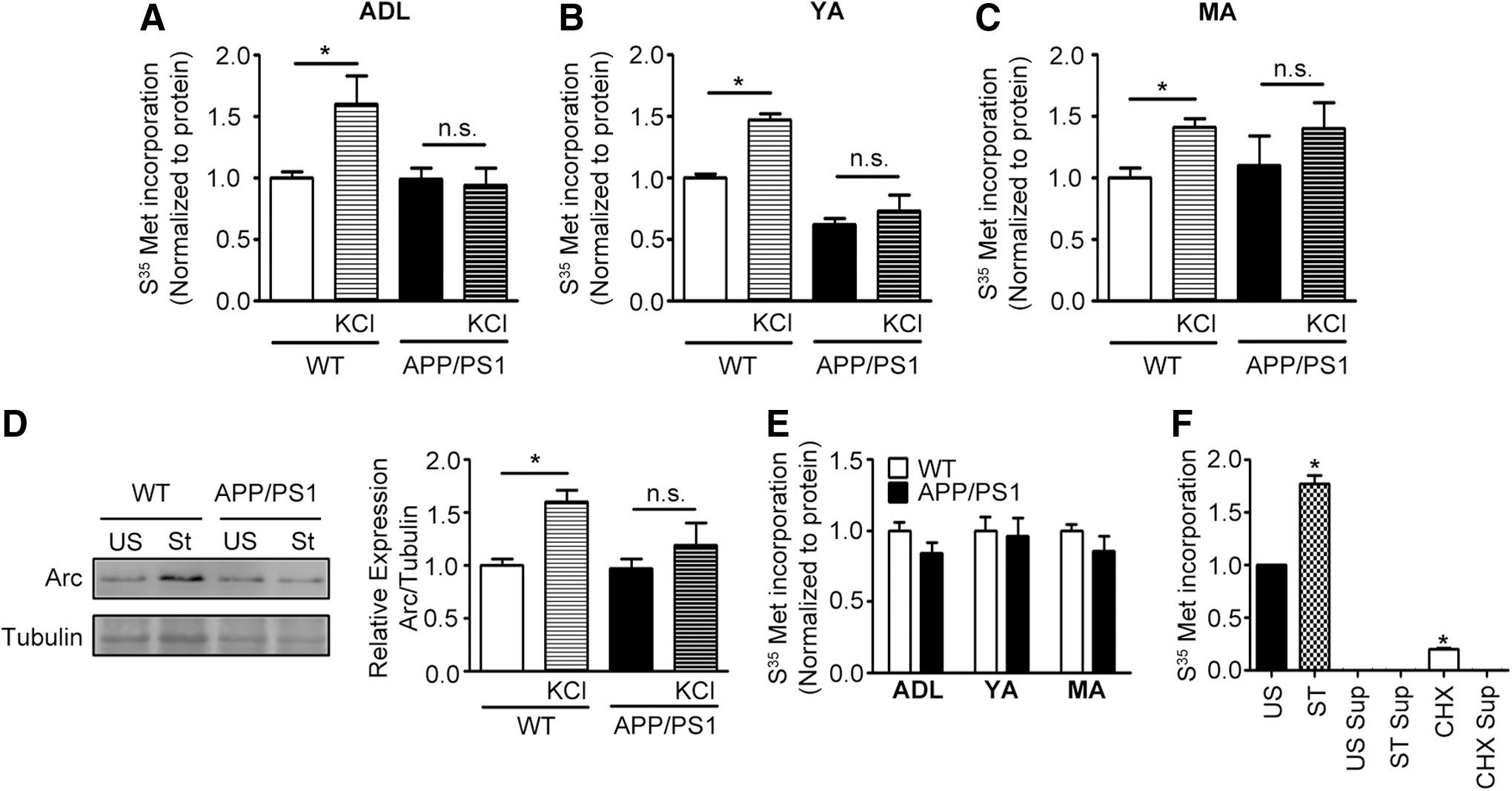
Akt1-mTOR signaling is diminished presymptomatically in synaptosomes of APP/PS1 mouse brain cortex from 1 month of age
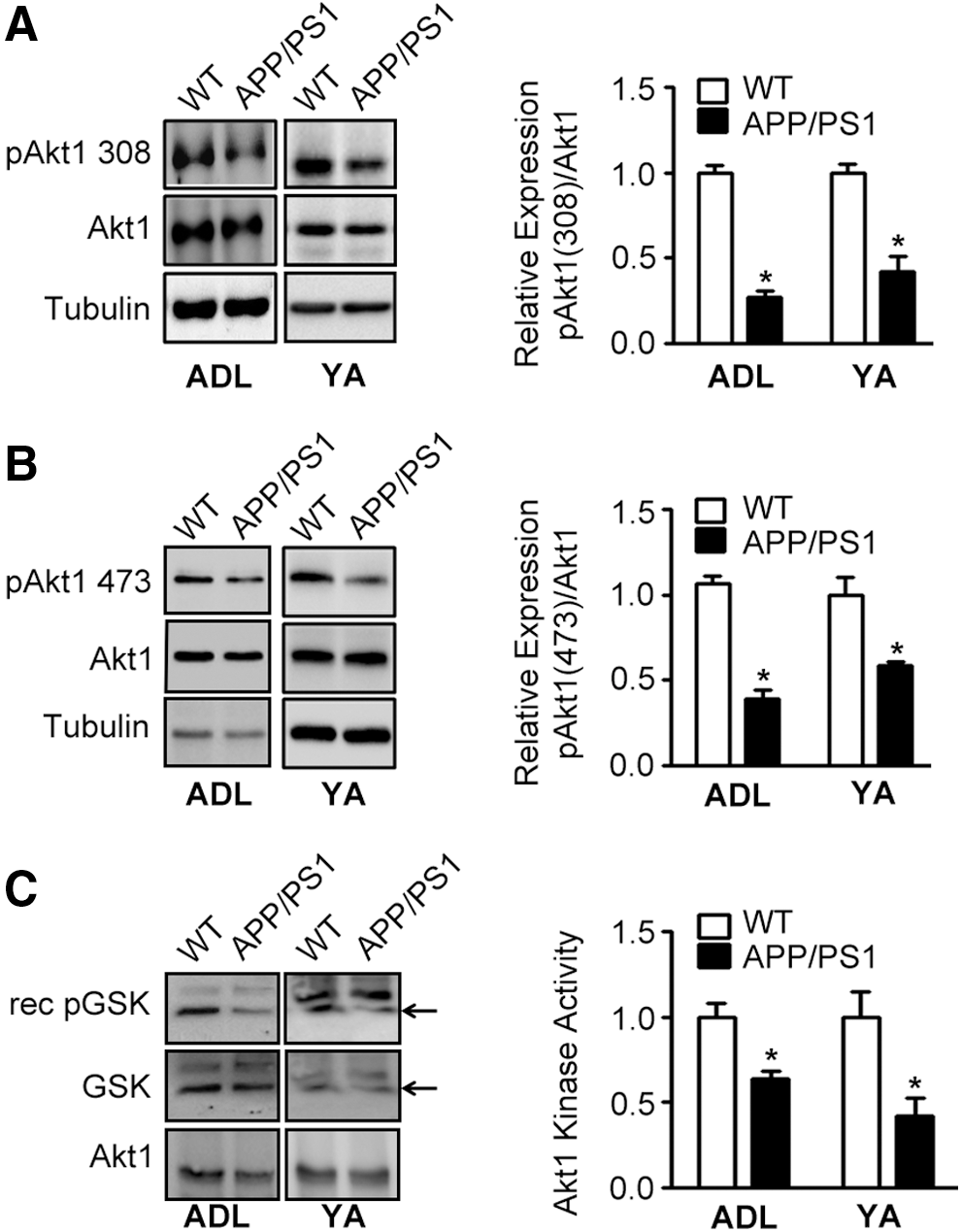
Akt1-mTOR signaling is a key component of activity-dependent local dendritic protein translation (15, 44, 48). The pathway is critical in regulating mRNA translation initiation, thus influencing cell growth in general, and dendritic arborization during neuroplasticity in particular (22). Hence, to understand the molecular mechanisms behind the loss of activity-dependent protein translation at synapses, we assessed the status of several players of Akt1-mTOR signaling cascade in synaptosomes isolated from APP/PS1 mice.
First we assessed the levels of pAkt1 in synaptosomes of APP/PS1 mice at 1, 3, and 9–11 months of age and compared them with the levels in age-matched WT controls. As Akt1 is activated by phosphorylation at Thr308 and Ser473, we analyzed the levels of both pAkt1 forms. We observed considerable loss of pAkt1 at both Thr308 and Ser473 in synaptosomes of APP/PS1 mice compared with WT mice at all ages examined (Fig. 2A, B and Supplementary Figs. S1A, B, S7–S10, S25, and S26). Loss of pAkt1 in synaptosomes of transgenic mice was confirmed by assaying Akt1 kinase activity using recombinant truncated GSK3β as substrate after immunoprecipitation of Akt1 from synaptosomes (Fig. 2C and Supplementary Figs. S11 and S12). Our results demonstrate that Akt1 signaling is reduced very early at 1 month of age and is sustained till 9–11 months, when the behavioral and pathological symptoms of AD are seen.
mTOR is a rapamycin-sensitive serine/threonine protein kinase that is downstream to Akt1 and is indirectly activated by pAkt through phosphorylation of TSC1/2, the inhibitory regulator of mTOR (18). Activated mTOR, in turn, stimulates cap-dependent protein synthesis in neurons by phosphorylating several mRNA translation factors, including eukaryotic initiation factor 4E-binding protein-1 (4E-BP1) and p70 ribosomal S6 kinase [S6K; (34)].
The levels of pmTOR (Ser2448) were decreased in cortical synaptosomes isolated from 1, 3, and 9-month-old APP/PS1 mice compared with those of age-matched WT controls (Fig. 3A and Supplementary Figs. 2A, S13, S14, and S27). Moreover, concomitant decrease in levels of downstream effectors of pmTOR, namely pS6K (Thr389; Fig. 3B and Supplementary Figs. S2B, S15, S16, and S28) and p4EBP1 (Thr37/46; Fig. 3C and Supplementary Figs. S2C, S17, S18, and S29 and), was observed in synaptosomes of APP/PS1 mice at all ages examined. In conclusion, dysregulation of Akt1-mTOR signaling occurring at the synapses of APP/PS1 mice from an early age of 1 month onward could potentially contribute to the reduction in activity-dependent protein translation observed earlier (Fig. 1).

Increased oxidation of Akt1 leads to increased association with the phosphatase, PP2A
To address the mechanism for downregulation of Akt1 phosphorylation and thence its kinase activity, we first measured total Aβ42 levels in cortical homogenates from APP/PS1 mice, and also examined the presence of Aβ42 oligomers in synaptosomes. Total Aβ42 levels were 1.35 and 5.30 pmol/mg protein in the cortical lysates of ADL (1–1.5 months old) and YA (3–4 months old) APP/PS1 mice, respectively, which were 6.43 and 13.95-fold more than the respective age-matched WT mice (Fig. 4A). Using immunoblotting, we detected appreciable amounts of trimers of Aβ in cortical synaptosomes prepared from APP/PS1 mice from age 1 month onward, whereas synaptosomes from WT mice had negligible amounts of Aβ oligomers (Fig. 4B and Supplementary Figs. S19, S20), indicating that Aβ42 is present in appreciable quantities at the synapse even in the presymptomatic stage of the disease.
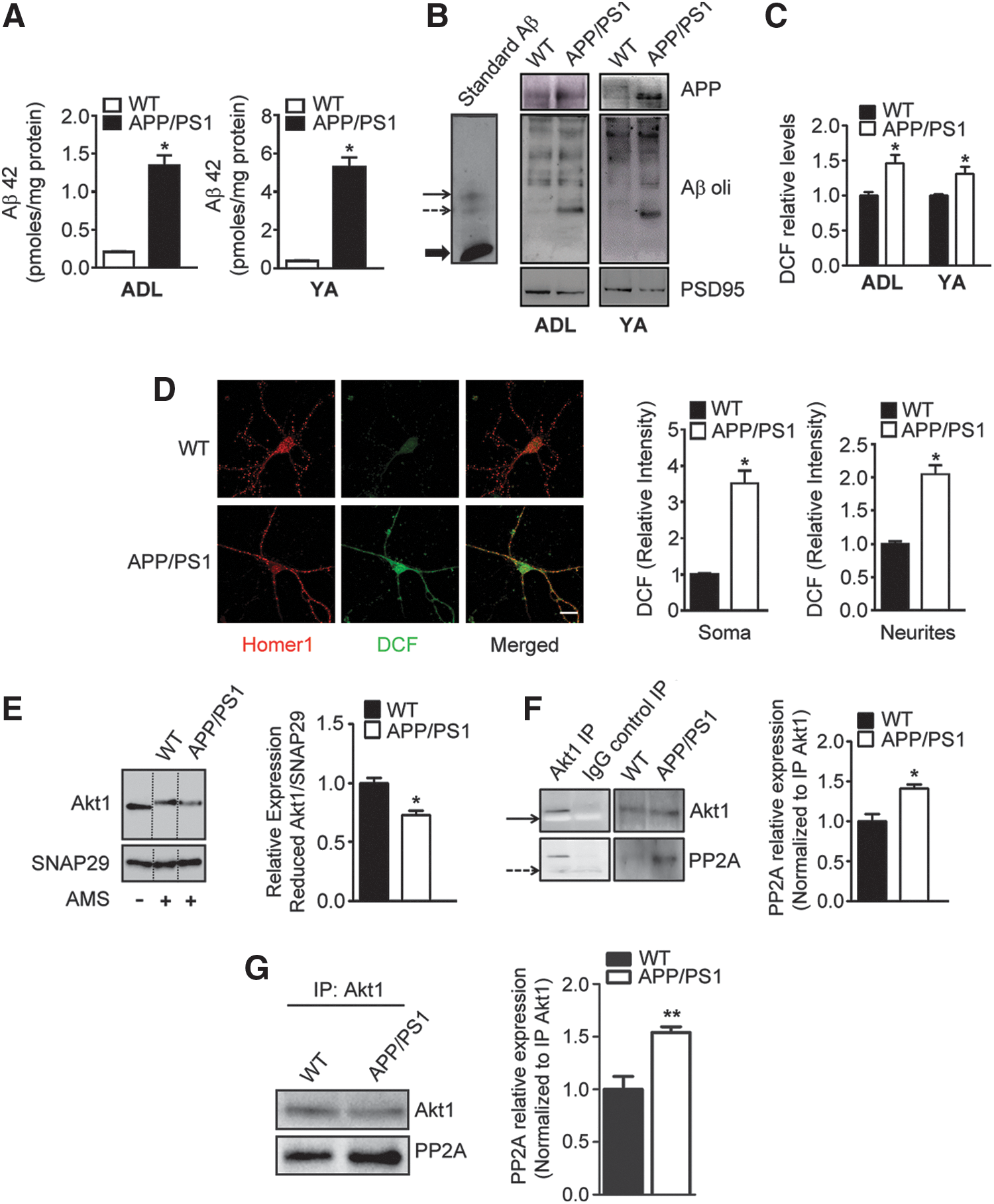
Akt1 is a redox-sensitive protein and its kinase activity is regulated by the redox milieu (1, 11). Oxidation of the cysteine moieties at 296 and 310 positions (flanking the regulatory domain of Akt1) leads to its increased association with the protein phosphatase, PP2A, resulting in dephosphorylation and termination of kinase activity of Akt1 (1, 11). Oxidative stress has been proposed to be a critical mechanism contributing to AD pathogenesis, and Aβ42 peptide is known to generate ROS (7, 31). Indeed, we detected increased ROS levels in synaptosomes isolated from APP/PS1 mice brain cortices at 1 and 3 months (Fig. 4C and Supplementary Fig. S4). Furthermore, using a combination of 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) reaction and Homer1 immunohistochemistry, we were able to detect significantly higher amounts of ROS in both soma and neurites of primary cortical neurons from APP/PS1 mice than those from WT mice (Fig. 4D and Supplementary Fig. S5).
We hypothesized that decrease in synaptic Akt1 activity in APP/PS1 mice could be a consequence of increased Akt1 oxidation. We assessed the levels of reduced Akt1 after its derivatization with 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid, disodium salt (AMS). Reduced Akt1 levels were found to be diminished in synaptosomes (Fig. 4E and Supplementary Fig. S21) isolated from 1-month-old APP/PS1 mice, indicating enhanced oxidation of Akt1 at the synapse. AMS-derivatized blots were normalized to SNAP29 because of its lack of cysteine residues, which could potentially react with AMS. Importantly, we also observed enhanced association of Akt1 with PP2A in synaptosomes from 1-month-old APP/PS1 mice (Fig. 4F and Supplementary Figs. S22 and S23), which persisted up to 9 months of age (Fig. 4G and Supplementary Fig. S24), suggesting that increased oxidation of Akt1 could indeed have resulted in its increased affinity for PP2A, thereby contributing to loss of its phosphorylation and kinase activity (Fig. 2A–C).
Overexpression of myristoylated Akt1 rescues the deficits in brain-derived neurotrophic factor-stimulated protein translation in primary cortical neurons derived from APP/PS1 mice
Primary cortical neurons were isolated from WT and APP/PS1 mice and brain-derived neurotrophic factor (BDNF)-stimulated protein translation was measured using puromycin incorporation as a marker. BDNF and not KCl was employed for stimulation of primary neurons to prevent the consequences of osmotic changes in extracellular milieu that have undesirable effects on cultured neurons.
Treatment with BDNF caused stimulation of protein translation in primary neurons obtained from WT mice but not from APP/PS1 mice, observed as increased puromycin incorporation (Fig. 5). This deficit in stimulated protein translation was observed in both soma and neurites of APP/PS1 neurons.

Next, we overexpressed myristoylated Akt1 [myr-Akt1; a constitutively active Akt1 form (42)] in primary neurons obtained from APP/PS1 mice using lentiviral transduction (Supplementary Fig. S3). Stimulated protein translation after BDNF treatment of APP/PS1 neurons transduced with myr-Akt1 was similar to that of WT neurons (Fig. 5). Thus, myr-Akt1 overexpression was able to rescue the deficit seen in primary neurons from APP/PS1 mice.
Activity-dependent protein translation is diminished in synaptoneurosomes isolated from postmortem brain neocortical samples from human brains
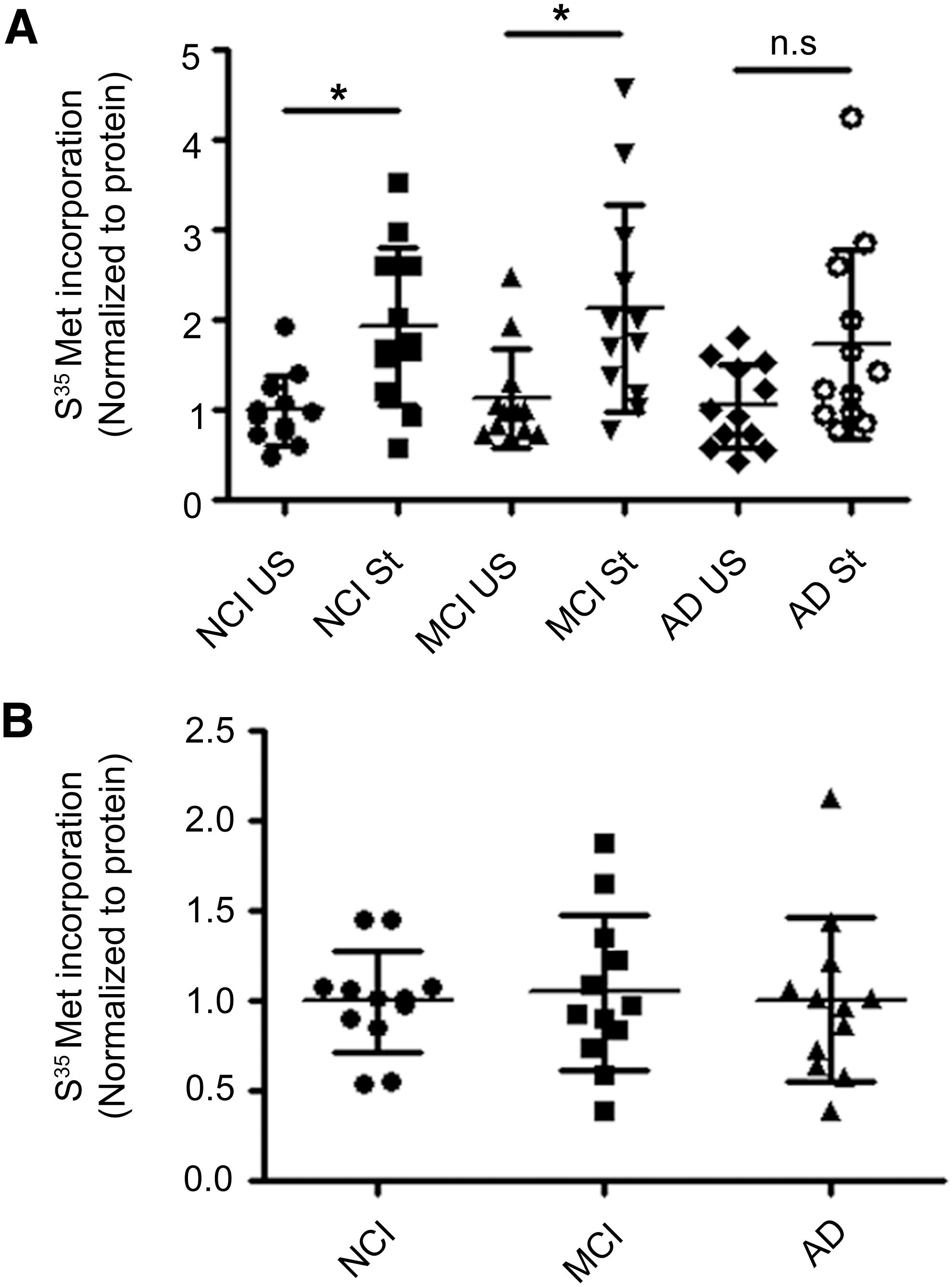
To extend our findings using APP/PS1 mice, we sought to assess protein translation rates in synaptoneurosomes isolated from human postmortem tissue. Synaptoneurosomes prepared from postmortem frozen human brain tissues have previously been shown to have intact mRNAs and protein translation machinery and have also been used successfully for in vitro translation assays (49). Hence, we measured protein translation rates in synaptoneurosomes and PNS isolated from postmortem brain prefrontal neocortical tissue samples of persons with AD using S35-methionine incorporation assay and compared them with samples from subjects with no cognitive impairment (NCI) and mild cognitive impairment (MCI). Basal protein translation rates in synaptoneurosomes were similar among AD, MCI, and NCI groups (Fig. 6A, B). Upon stimulation of synaptoneurosomes, we observed increased S35-methionine incorporation as has been previously reported (49) for samples from both NCI and MCI subjects (Fig. 6A, B). However, activity-dependent protein translation was compromised in synaptoneurosomes prepared from AD subjects (Fig. 6A, B). In contrast, protein translation as measured by S35-methionine incorporation in PNS fraction was unaltered in subjects with AD compared with that in subjects with NCI or MCI (Fig. 6C, D).
Discussion
Synaptic dysfunction is a primary player in the pathogenesis of AD; however, the underlying mechanisms are not clear. Activity-dependent synaptic protein translation is a robust indicator of synaptic function and plasticity (10, 36, 48). Dysfunction of activity-dependent translation very early in the pathogenesis of AD would have far-reaching implications for synapse maintenance and plasticity culminating into deficits in memory and cognitive abilities. We, therefore, examined the status of activity-dependent translation in synaptoneurosomes from APP/PS1 mice to determine whether this was compromised in the disease process. We observed significantly compromised KCl-stimulated activity-dependent protein translation in synaptoneurosomes isolated from APP/PS1 mice from 1 month onward (soon after the mice were weaned from their mothers), which persisted up to MA (Fig. 1A–C). We further demonstrate that ROS-mediated thiol oxidation of Akt1 in the synaptosomes leads to deficient Akt1 kinase activity, resulting in decreased Akt1-mTOR signaling and consequent loss of activity-dependent protein translation.
Akt1-mTOR signaling has been shown to be critical for activity-dependent local dendritic translation (15, 44, 48). Indeed, loss of activity-dependent translation was accompanied by diminished Akt1-mTOR signaling in synaptosomes seen as decreased levels of pmTOR and its downstream substrates, pS6K and p4EBP1 (Fig. 3 and Supplementary Fig. S2). Furthermore, activity of Akt1, the upstream kinase that activates mTOR by phosphorylating TSC1/2 (inhibitory regulator of mTOR), was decreased as evidenced by loss of kinase activity and levels of pAkt1 (Thr308 and Ser473; Fig. 2 and Supplementary Fig. S1). In conclusion, using a mouse model of AD, we demonstrate that activity-dependent translation is inhibited in synaptoneurosomes early in the pathogenesis of AD because of loss of Akt1 kinase activity and resultant inhibition of Akt1 signaling. This is supported by our data showing reversal of this deficit by ectopic overexpression of persistently active Akt1. Thus, loss of Akt1, the major upstream kinase of this pathway, could potentially be the primary cause of dysfunctional protein synthesis seen in synaptoneurosomes. Furthermore, we also observed attenuation of synaptic Akt1 signaling and activity-dependent translation in synaptoneurosomes from neocortical tissue samples of postmortem brains of AD patients (Fig. 6).
Interestingly, basal protein translation seen in the absence of KCl was not significantly different in synaptoneurosomes of both APP/PS1 mice and postmortem AD brains. This indicates that basal translation may potentially occur even with limiting amounts of Akt1-mTOR signaling. Under basal conditions, pAkt1 levels, per se, may not be rate limiting, whereas enhanced translation occurring after neuronal activity, such as KCl stimulation, would require optimal levels of activated Akt1 (including de novo phosphorylation) at the site of translation, which is not available during AD pathogenesis. Indeed, neuronal activity is known to robustly stimulate local Akt1 signaling (12). This is supported by our observation wherein overexpression of constitutively active myristoylated Akt1 rescues the deficits in activity-dependent translation (Fig. 5).
An interesting aspect of our study is that the presence of Aβ42 oligomers in synaptosomes from mice aged 1 month onward (Fig. 4B) indicates that even if Aβ42 accumulation in the brain does not reach pathogenic levels, the presence of toxic Aβ42 oligomers at the synapse can potentially disrupt synaptic functions, thus initiating pathogenic processes before overt symptoms appear. Excessive ROS generation has been considered to be one of the early events that contribute to AD pathogenesis (7, 31). We quantified ROS in synaptosomes isolated from cortices of APP/PS1 mice and observed increase in ROS levels in synaptosomes from APP/PS1 mice from age 1 month onward (Fig. 4C). Cysteine residues at 296 and 310 positions in Akt1 are very sensitive to redox modulation, and such redox modifications have been shown to increase its association with the phosphatase PP2A, leading to its dephosphorylation (1). Hence, we quantified reduced levels of Akt1 and found they were decreased in synaptosomes isolated from 1-month-old APP/PS1 mice compared with those from WT controls (Fig. 4E). As expected, this led to increased association of synaptosomal Akt1 with PP2A in APP/PS1 mice (Fig. 4F, G), leading to compromised Akt1 kinase activity and consequently Akt1-mTOR signaling.
Thus, using a mouse model of AD, namely APP/PS1 mice, we demonstrate for the first time that activity-dependent translation is inhibited selectively in synaptoneurosomes early in the pathogenesis of AD because of ROS-mediated loss of Akt1 kinase activity and resultant inhibition of mTOR signaling. Importantly, we could also rescue the deficits in BDNF-stimulated protein translation by overexpression of myristoylated Akt1 in primary cortical neurons from APP/PS1 mice (Fig. 5). Thus, loss of Akt1, the major upstream kinase of Akt1-mTOR pathway, is a primary cause of dysfunctional protein synthesis seen in synaptoneurosomes in AD.
Another important aspect of this study is that regulation of Akt1 kinase, involved in cell survival signaling, is different and manifests according to age and/or extent of Aβ accumulation in human patients (28). It is important to note that most prior studies on Akt1-mTOR signaling in AD models or human postmortem tissues have been carried out using whole cell lysates and we do not have a clear understanding of the status of Akt1-mTOR signaling at the synapse in AD pathogenesis, especially during the early stages of disease progression. Moreover, previous studies have not delineated the underlying mechanisms involved in disrupted Akt1-mTOR signaling in AD. Hence in this study, we provide evidence of redox-mediated disruption of synaptic Akt1 function early in the disease progression, which persists up to MAs when overt symptoms of AD become evident in the mouse model.
Our results are in agreement with earlier reports that enhancing Akt1 kinase activity offers protection in cellular and animal models of neurodegeneration, often attributed to its prosurvival function. Indeed, increased expression of recombinant Akt1 or active pAkt1 has been shown to underlie the neuroprotection mediated by neurotrophins (33), estrogen (43), and lipoic acid (38) in AD model systems. Moreover, our findings are significant because synaptic plasticity and its consolidation are spatially limited and require both precise and dynamic changes in protein translation, trafficking, and organization within the microdomain of the synapse. Spatiotemporal regulation of synaptic proteins (namely neurotransmitter receptors, signaling proteins, and cytoskeletal and scaffold elements) is critical for precise functioning and plasticity of synapse. This is, in part, dependent on dendritic mRNA transport and its local translation at the synapse (48). Furthermore, dysregulation of the plastic changes in neurotransmission through dysfunction of the machinery used for activity-dependent synaptic translation could potentially culminate into cognitive deficits (6, 14, 21, 48). It is not surprising then that dysregulated activity-driven synaptic translation has been implicated in disorders such as Fragile X syndrome and autism (47). In fact, several cases of autism spectrum disorders have been found to be associated with mutations in genes encoding components of the Akt1 signaling pathway (24).
In conclusion, we demonstrate that increase in ROS levels in synaptosomes occurs very early in the disease process, leading to loss of Akt1 kinase activity and resulting in deficiency of activity-dependent synaptic protein translation, with potentially far-reaching implications for synaptic plasticity, learning, and memory. Furthermore, manifestation of these deficits very early in life indicates that the prodromal stage of AD may start very early and thus provides a window for intervention, particularly in familial AD cases.
Methods
Antibodies and reagents
Primary antibodies against pAkt1 (Ser473), pAkt1 (Thr308), Akt1, pGSK3β (Ser9), GSK3β, pmTOR (Ser2448), mTOR, pS6K (Thr389), S6K, p4EBP1 (Thr46/47), and 4EBP1 were purchased from Cell Signaling Technology. Anti-glycogen synthase kinase (GSK) antibody was from Santa Cruz Biotechnology; anti-Arc, anti-SNAP29, and anti-MAP2 antibodies were from Abcam; anti-Homer1 antibody was from Synaptic Systems; anti-Puromycin antibody was from Millipore; and anti-β-tubulin antibody was from Sigma-Aldrich. Akt1 was immunoprecipitated using Dynabeads A (Life Technologies) and Akt1 kinase assay was performed using the nonradioactive kinase assay kit from Cell Signaling Technology. AMS, a thiol reactive reagent used for derivatization of protein samples for redox immunoblots, was procured from Life Technologies. Antibodies against Aβ42: 12F4 (monoclonal) and H43 (polyclonal), were procured from BioLegend and Santa Cruz, respectively. Antibody against APP (epitope 3–8) 6E10 was procured from BioLegend. 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate kit and secondary antibodies: anti-rabbit and anti-mouse conjugated with horse-radish peroxidase (HRP), were purchased from Vector Laboratories. S35-
Animals
Transgenic mice B6C3-Tg [APPSwe/PS1ΔE9)85Dbo/J;
Human tissues
Frontal neocortical tissue from postmortem brains of age-matched persons with AD dementia and MCI and control subjects with NCI were procured from Rush Alzheimer's Disease Centre, Chicago. All participants signed an informed consent as well as an Anatomical Gift Act for brain donation. The study was approved by the Institutional Review Board of Rush University Medical Center. All experiments involving human postmortem tissues were performed in accordance with institutional guidelines and after approval from the ethics committee. Details of the clinical and neuropathological evaluation have been previously reported (2a).
Synaptoneurosome preparation and S35-methionine incorporation assay
Preparation of synaptoneurosomes from mouse cortical tissue (32) or neocortical tissue samples from postmortem human brain (49) and S35-methionine incorporation assay were performed as described previously (32). In brief, tissue was homogenized in translation buffer containing 118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl2, 1.53 mM KH2PO4, 212.7 mM glucose, and 1 mM 1,4-dithiothreitol (DTT) (pH 7.4), and supplemented with protease and phosphatase inhibitors, 200 μg/ml chloramphenicol, and 30 U/ml RNAse inhibitor. The homogenate was then passed sequentially through two 100 μm and one 10 μm membrane filters (Millipore). The filtrate obtained was then centrifuged at 1500 g at 4°C for 10 min and pellet containing synaptoneurosomes was resuspended in translation buffer. Stimulation of synaptoneurosomes was carried out by incubation with 50 mM KCl at 37°C for 15 min in the presence of 50 μCi S35-
Synaptosomal preparation
Synaptosomes were prepared as described previously (3) with some modification. In brief, brain tissue was homogenized in 10 volumes of homogenization buffer (5 mM HEPES buffer, pH 7.4, containing 0.32 M sucrose, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 2 μg/ml aprotinin, 10 μg/ml leupeptin, 7 μg/ml pepstatin A, 100 μg/ml of phenylmethanesulfonyl fluoride (PMSF), and 10 μl/ml of protease inhibitor cocktail) in a Potter–Elvehjem homogenizer. The homogenate was centrifuged at 1000 g at 4°C for 10 min and the PNS was obtained. PNS prepared from mouse cortex (as already described) was centrifuged again at 12,000 g at 4°C for 15 min, and the pellet was resuspended in 5 mM Tris (pH 8.1) containing 0.32 M sucrose along with protease and phosphatase inhibitors. The resuspended pellet was then layered over discontinuous sucrose gradient (0.85–1.0–1.2 M) and centrifuged at 85,000 g for 2 h at 4°C. Synaptosomal fraction obtained at the interface of 1 and 1.2 M sucrose was collected, washed twice in 5 mM Tris (pH 8.1), and resuspended in homogenization buffer for further experiments.
Enzyme-linked immunosorbent assay
Total Aβ42 levels in brain were quantitated by enzyme-linked immunosorbent assay as previously described (41) using 12F4 as a capture antibody and H43 as a detection antibody. Signal was detected using an HRP-labeled secondary antibody and TMB as a substrate.
Immunoblotting
Synaptosomal and PNS samples were resolved on SDS-PAGE, electroblotted, and immunostained using primary and secondary antibodies. Immunoreactive chemiluminescent signals were detected and the intensities of the bands were quantified using Bio-Rad Imager. In some cases, the samples were denatured with SDS and incubated with 15 mM AMS for 3 h at room temperature under nitrogen atmosphere. The reaction was then quenched with 60 mM glutathione (GSH) and the samples were immunoblotted as already described. SDS-solubilized synaptosomes were also resolved on 10–20% tris-tricine gels for separation of Aβ oligomers (40), transferred to PVDF membranes, and probed with 6E10 antibody that recognizes both APP and Aβ42 species.
Immunoprecipitation of Akt1
Immunoprecipitation of Akt1 was carried out using Protein A-conjugated Dynabeads (Life Technologies) according to the manufacturer's instructions. In brief, Protein A-conjugated Dynabeads were incubated with anti-Akt1 antibody in PBS (pH 7.4) containing 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, and 0.1% (v/v) Igepal CA360 for 3 h at room temperature. The antibody-linked Dynabeads were washed with PBS and incubated overnight at 4°C with synaptosomes in PBS containing 1% (v/v) Triton X-100 and protease and phosphatase inhibitors. The beads were washed twice with PBS containing 0.5% (v/v) Igepal CA360 at 4°C and eluted using Laemmeli's SDS loading buffer and subjected to SDS PAGE.
Akt1 kinase assay
Akt1 kinase assay was performed according to the manufacturer's instructions (Cell Signaling Technology). In brief, synaptosomes were incubated with Sepharose beads cross-linked with antibody against pAkt1 (Ser473) at 4°C overnight for immunoprecipitation. After washing with PBS, the beads were added to kinase assay buffer containing 25 mM Tris (pH 7.5), supplemented with 25 mM MgCl2 and 0.2 mM ATP. Recombinant truncated GSK was added as substrate and incubated at 37°C for 30 min. The reaction was stopped by addition of Laemmeli's sample buffer followed by SDS-PAGE and immunoblotting.
DCFH-DA-based ROS assay
DCFH-DA, an oxidant-sensitive dye that is converted to 2′,7′-dichlorofluorescein (DCF) by oxidation and after de-esterification, was employed to measure ROS levels as described elsewhere (1). In brief, synaptosomes were incubated with 10 μM DCFH-DA in 0.1 M phosphate buffer (pH7.4) for 5 min at room temperature. Fluorescence was measured using excitation at 488 nm and emission at 525 nm every 30 s for 1 h. A standard curve of DCF was always used for quantification of ROS levels in independent experiments (Supplementary Fig. S4).
Primary cortical neuronal cultures
Primary neuronal cultures were established and maintained as previously described (2). In brief, cortical tissue was dissected out from WT and APP/PS1 P1 mice. The tissue was then enzymatically dissociated using trypsin. The cell suspension obtained after trituration was centrifuged and pellets were resuspended and plated in Neurobasal-A Medium (GIBCO, Life Technologies) containing 1% 100 × Glutamax (GIBCO), 1% 100 × penicillin–streptomycin (GIBCO), and 2% 50 × B27 (GIBCO).
ROS measurements in primary neurons
DCFH-DA was used to observe ROS production in primary neurons. Using HEK293T cells, we confirmed the ability of DCF assay to monitor ROS generation in cell culture systems (Supplementary Fig. S5). Primary neurons (DIV 15) grown on coated coverslips were incubated with 10 μM DCFH-DA for 20 min at 37°C, washed with PBS, and fixed using cold acetone for 10 min. The cells were then immunostained for Homer1, mounted on slides, and imaged immediately. Homer1 immunostaining was performed to create a “mask” for image analysis.
Protein translation assay in primary neurons
Protein translation in primary neurons was assayed as described elsewhere (45). In brief, primary cortical neurons (DIV 15) were stimulated with 50 ng/ml of BDNF (Life Technologies) for 1 h at 37°C, followed by treatment with 3 μM puromycin (Sigma) for 7 min at 37°C. After brief washing, treatment was terminated by fixation with 4% (w/v) paraformaldehyde and 4% (w/v) sucrose for 20 min at room temperature. This was followed by permeabilization with 0.3% (v/v) Triton X-100. Cells were then coimmunostained with antibodies against MAP2 and puromycin for 1 h at room temperature.
Overexpression of myristoylated Akt1 in primary neurons
In some experiments, primary cortical neurons from APP/PS1 mice were transduced with myristoylated Akt1 (42) lentiviral particles to observe the effects of Akt1 overexpression on BDNF-stimulated protein translation. The pBSFI-myr-akt1 was a gift from Dr. Peter Vogt (Addgene plasmid# 49186); pMD2.G and psPAX2 were a gift from Dr. Didier Trono (Addgene Plasmid# 12259, 12260). The pRRLsinPPTeGFP plasmid (pLenti-GFP) was kindly provided by Dr. Philip A. Barker (McGill University, Montreal, QC, Canada). Mouse myr-akt1 open reading frame was amplified by PCR to encode BamHI and SalI restriction sites at the 5′ and 3′ ends, respectively. These products were gel purified and ligated into a pRRLsinPPTeGFP vector to generate pRRLsinPPTmyr-Akt1. Lentiviral particles were produced using second-generation lentiviral systems in HEK293T cells; particles were purified by ultra centrifugation and resuspended in Neurobasal-A medium, and the amount of active particles was determined by titration in HEK293T cells. Primary cortical neurons from APP/PS1 mice were transduced on DIV13 and experiments were performed on DIV15-16. Untransduced cultures from the same batch were used as controls. Akt1 overexpression was confirmed by immunostaining for Akt1.
Images were acquired using Zeiss LSM780 and analyzed using Metamorph software (Molecular Devices LLC).
Statistics
Outliers from the data set were removed by employing a widely used and accepted statistical method of outlier removal based on median absolute deviation (25). Statistical comparisons were made by using unpaired two-tailed Student's t-test. Multiple groups were compared using one-way analyses of variance followed by post hoc tests with Newman–Keuls correction.
Footnotes
Acknowledgments
We thank Prof. D.N. Rao for help with the S35-methionine incorporation assay. This work was supported by grants from Tata Trusts and Departments of Science & Technology and Biotechnology, India. The study was supported, in part, by NIH grant R01AG17917 (DAB).
Author Disclosure Statement
The authors declare no conflict of interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.