
Abstract
Aim:
Chronic airway diseases such as asthma are associated with increased production of reactive oxygen species (ROS) and oxidative stress. Endogenous NADPH oxidases are a major source of superoxide in lung, but their underlying role in asthma pathology is poorly understood. We sought to characterize the involvement of NADPH oxidase in allergic asthma by studying the role of CYBA (p22phox) in human asthma and murine house dust mite (HDM)-induced allergic airway inflammation.
Results:
Increased expression and localization of p22-PHOX were observed in biopsies of asthmatic patients. HDM-treated wild-type mice possessed elevated p22phox expression, corresponding with elevated superoxide production. p22phox knockout (KO) mice did not induce superoxide and were protected against HDM-induced goblet cell hyperplasia and mucus production and HDM-induced airway hyperresponsiveness (AHR). IL-13-induced tracheal hyperreactivity and signal transducer and activator of transcription (STAT)6 phosphorylation were attenuated in the absence of p22phox or catalase pretreatment.
Innovation:
Our study identifies increased expression of p22phox in lungs of asthmatic patients and in experimental model. The induced AHR and mucus hypersecretion are a result of increased ROS from the p22phox-dependent NADPH oxidase, which in turn activates STAT6 for the pathological feature of asthma.
Conclusions:
Together with the increased p22phox expression in lungs of asthmatic patients, these findings demonstrate a crucial role of p22phox-dependent NADPH oxidase for the development of mucus hypersecretion and AHR in HDM-induced model of asthma. This suggests that inhibition of functional NADPH oxidase by selective interference of p22phox might hold a promising therapeutic strategy for the management of asthma. Antioxid. Redox Signal. 27, 1460–1472.
Introduction
A
Increased reactive oxygen species (ROS) signaling and oxidative stress are strongly linked to the asthma pathogenesis, but to date, little is known about the NADPH oxidase role in development of asthmatic phenotype. We show for the first time increased p22phox expression in bronchial biopsies of asthmatic patients. We demonstrate that oxidative stress/ROS from the p22phox-dependent NADPH oxidase is involved in signaling for AHR via activation of STAT6 in experimentally induced asthma, suggesting that selective interference of p22phox might hold a promising therapeutic strategy for the management of asthma in clinical settings.
Inflammatory diseases such as asthma are often associated with an increase in endogenous reactive oxygen species (ROS) (14, 31). Exposure to allergens and other stimulants has been shown to stimulate ROS production, which can lead to oxidative stress-induced cell damage and mitigate the physiological function of structural cells (48). NADPH oxidase and mitochondria are the major contributors of basal endogenous ROS generation in the lung. The NADPH oxidase family consists of one of the membrane-bound cytochrome isoforms (NOX1–4), the adaptor protein cytochrome b-245, alpha polypeptide (Cyba, also known as p22phox), and additional cytosolic regulatory proteins. p22phox functions as an adaptor molecule permitting the assembly and function of active NADPH oxidase and is therefore crucial for NADPH oxidase-derived ROS production (2, 34).
NADPH oxidases are enzyme complexes that generate ROS in the form of hydrogen peroxide or superoxide by transfer of electrons from NADPH to molecular oxygen. Assembly and activation of NADPH oxidase require translocation of the cytoplasmic p40phox, p47phox, and p67phox subunits to the membrane-bound gp91phox and p22phox subunits. The expression and cellular localization of NADPH oxidase determine its function in various cell types. In phagocytic cells, NADPH oxidase-derived ROS is critical for the elimination of invading bacteria via induction of the oxidative burst. In structural cells, NADPH oxidase is involved in several cellular processes such as signal transduction, differentiation, and proliferation (6, 38). In the pulmonary system, production of ROS from NADPH oxidase has been implicated in several diseases such as asthma, chronic obstructive pulmonary disease, fibrosis, and pulmonary hypertension (16).
Currently, there is limited evidence available on the functional contribution of NADPH oxidase in asthma pathogenesis. Haplotype analysis has revealed the association of single-nucleotide polymorphisms (SNPs) in p22phox with bronchial asthma (24). Recently, an increased NOX4 expression has been observed in airway smooth muscle bundles and in isolated smooth muscle cells from asthmatic patients (44). Studies using animal models have, however, given conflicting results. In one study, mice deficient in NOX2 displayed reduced allergic airway inflammation following ovalbumin challenge (42), while other studies reported that NOX2-deficient mice exhibited the reverse phenotype with increased inflammation and airway hyperreactivity (3, 4, 27).
To delineate the role and characterize the involvement of ROS generating NADPH oxidase in allergic airway inflammation and remodeling, we have utilized the house dust mite (HDM) model in mice defective in the key NADPH oxidase adaptor molecule p22phox. We hypothesized that the lack of p22phox will inhibit functional NADPH oxidase complex, thereby decreasing ROS generation and result in decreased allergic asthma. These data provide an understanding into p22phox-dependent NADPH oxidase function(s) for the development of asthma.
Results
Asthmatic patients exhibit increased NADPH oxidase subunit p22phox
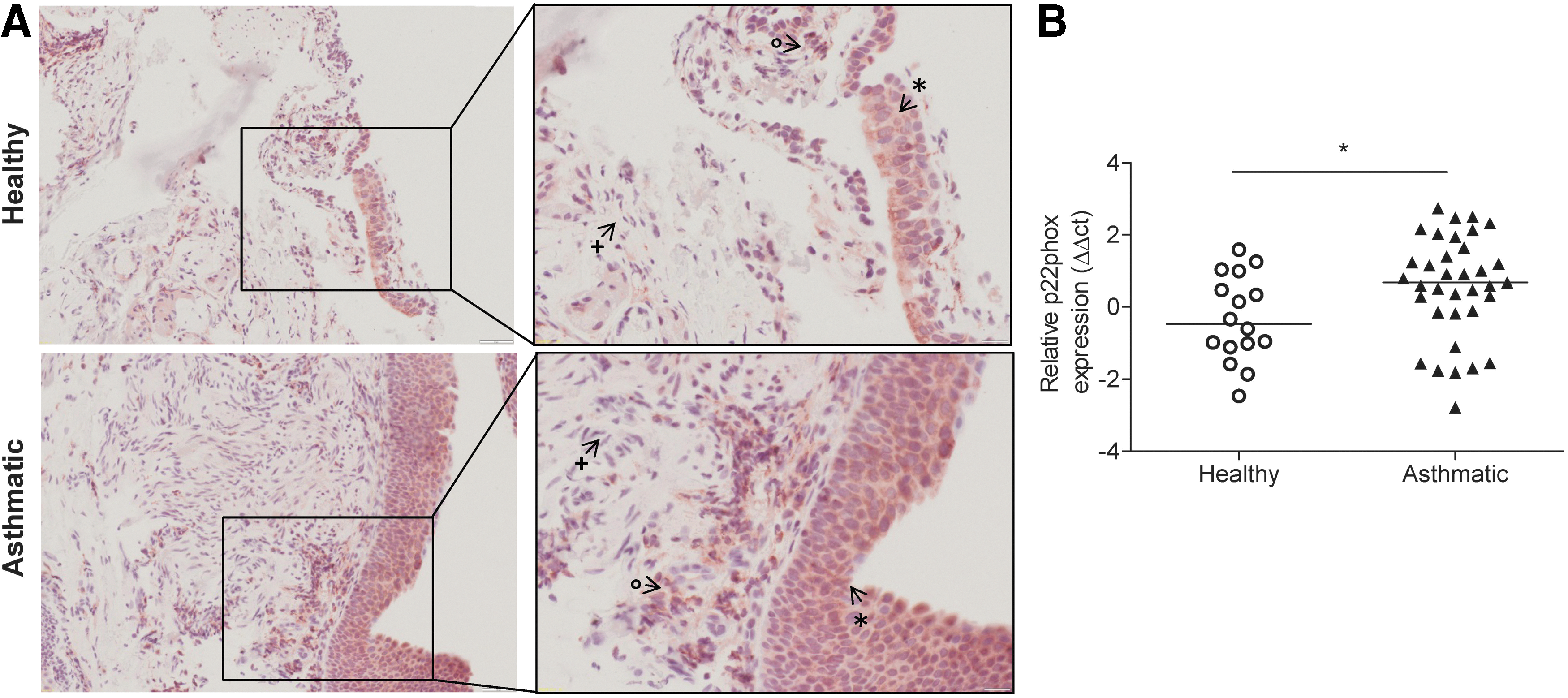
As p22phox is a vital subunit for the functional NADPH oxidase system to generate ROS, we investigated the expression of p22phox in human asthma samples by immunohistochemistry and real-time polymerase chain reaction (PCR). In healthy control biopsies, positive immunoreactivity for p22phox was observed in the bronchial epithelium and lymphocytes. In biopsies from asthmatics, more intense p22phox staining was observed in the bronchial epithelium and underlying smooth muscle layers (Fig. 1A). The negative control for the immunohistochemistry is shown in Supplementary Fig. S1; Supplementary Data are available online at
HDM treatment induces p22phox expression in mice
We next investigated the expression of p22phox in a murine model of experimental asthma (Supplementary Fig. S2). In naïve mice, expression of p22phox was predominately localized to bronchial epithelium and lung parenchymal cells (Fig. 2A). Intranasal HDM treatment induced stronger p22phox immunoreactivity that was localized to areas of peribronchial inflammation, bronchial epithelium, and airway smooth muscle cells (Fig. 2A). Dual immunofluorescence revealed p22phox expression in both epithelial cells and in smooth muscle cells (Fig. 2B, C). To determine whether p22phox was also regulated in HDM-treated mice, we performed Western blotting. Increased p22phox levels were observed in HDM-treated mice in comparison with controls (Fig. 2D). All negative controls for immunostaining and Western blotting are shown in Supplementary Figure S3. We also analyzed the expression of NOX enzymes (NOX2, NOX4) and subunits (p40, p60, p67, NOXO1, and NOXA1) by real-time PCR (Supplementary Fig. S4). In this study, we also observed a significant upregulation of the noxa1 activator subunit after HDM treatment.

HDM-induced ROS production is dependent on p22phox
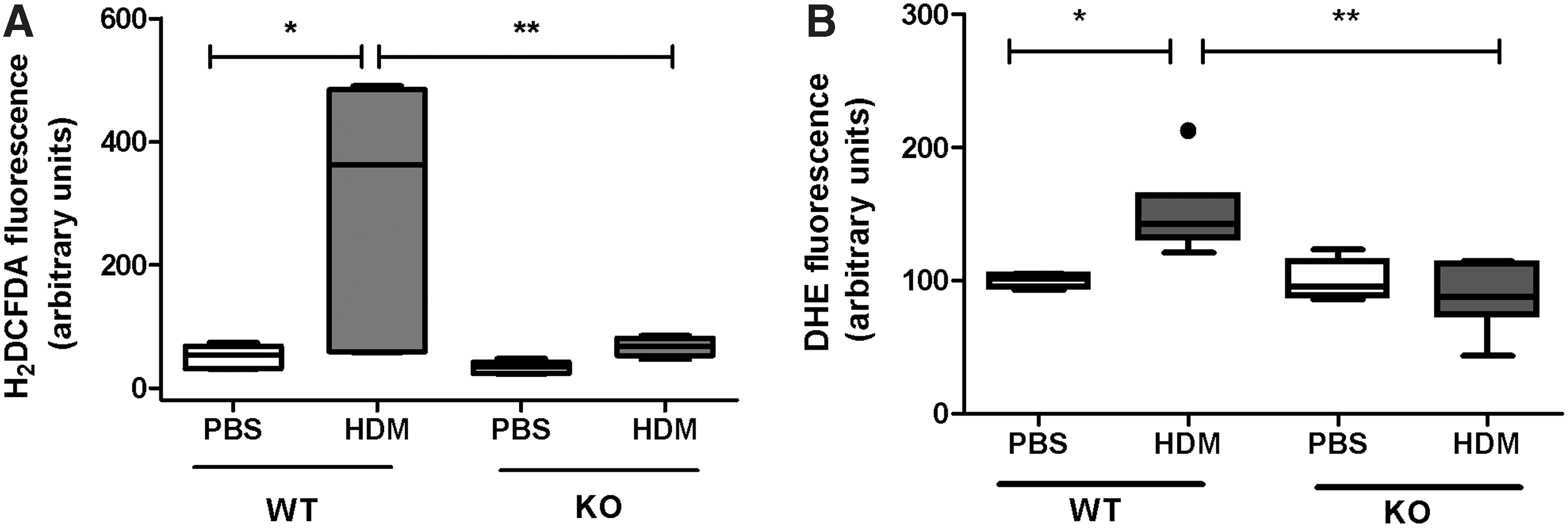
As p22phox is integral for ROS produced from NADPH oxidase, we investigated intracellular ROS production by measuring the superoxide levels in wild-type (WT) mice and mice defective in p22phox expression following treatment with phosphate-buffered saline (PBS) or HDM. Mice defective in p22phox (Cybanmf333) contain a point mutation within the p22phox gene, which results in the absence of p22phox protein (34), (Supplementary Figure S3) and are here onward called p22phox knockout (KO) mice. In WT and KO PBS-treated mice, minimal levels of ROS were observed, while increased ROS levels were observed in WT mice treated with HDM (Fig. 3). In contrast, p22phox KO mice treated with HDM did not show any significant increase in ROS production, demonstrating the importance of p22phox in generating HDM-induced ROS production in the lung. The superoxide dismutase (SOD) quenchable signal is shown in Supplementary Figure S5. We next examined the expression of the antioxidative system in WT and p22phox mice treated with HDM (Supplementary Fig. S6). We observed a general trend for the decreased expression of catalase, SOD, thioredoxin (THX), glutathione peroxidase (GPX), and peroxiredoxin (PRDX) isoforms, whereas PRDX6 was significantly lower expressed in HDM-treated KO mice (Supplementary Fig. S6).
p22phox KO mice exhibit an altered inflammatory profile in response to HDM
We first confirmed HDM sensitization in WT and KO mice by measuring circulating HDM-specific immunoglobulins. HDM treatment showed that elevated IgG1a and IgG2c levels in the serum were comparable between WT and KO mice. Furthermore, HDM treatment significantly increased IgE levels only in WT mice; however, in KO mice, this increase did not reach significance (Supplementary Fig. S7). To characterize the inflammatory profile, the cellular composition of bronchoalveolar lavage fluid (BALF) was analyzed by flow cytometry. Treatment with HDM resulted in an increase in total cell counts in the BALF in both WT and KO mice (Fig. 4A). Analysis of the cellular composition revealed higher numbers of eosinophils following HDM exposure in WT mice, this increase did not reach significance in KO mice. Elevated numbers of neutrophils, B cells, T cells, and CD4+ and CD8+ T cell subsets were observed in p22phox KO mice treated with HDM compared with their WT counterparts.

Analysis of the inflammatory cell recruitment by flow cytometry in lung homogenate samples revealed similar changes, with significantly increased eosinophils in WT mice and increased neutrophils in KO mice treated with HDM (Supplementary Fig. S8). Analysis of cytokine levels in lung homogenate samples revealed elevated IL4 and IL13 in WT mice treated with HDM, in HDM-treated KO mice, these changes were not significant (Fig. 4B). In contrast, IFNγ was only elevated in p22phox KO mice treated with HDM, this increase was absent in the WT mice treated for HDM (Fig. 4B). We additionally profiled a number of cytokines by real-time PCR (Supplementary Fig. S9). Both WT and KO mice possessed increased mRNA expression of the Th2 cytokines IL-5 and IL-13. HDM-treated KO mice possessed elevated levels of IL-10 and IL-17 mRNA. The increased IL-17 was also accompanied by elevated Cxcl1 (Kc) and Cxcl2 (Mip-2) mRNA in KO mice treated with HDM in comparison with WT and control KO mice (Supplementary Fig. S9).
p22phox KO mice are protected from structural alterations in the airway epithelium
Due to the strong increase in p22phox levels in HDM-treated mice, we next investigated whether deficiency in p22phox affects the development of goblet cell hyperplasia and mucus production following HDM treatment. Analysis and quantification of periodic acid-Schiff (PAS)-stained lung sections revealed predominant goblet cell hyperplasia and mucus production in HDM-treated WT mice. However, in p22phox KO mice, HDM treatment resulted in significantly reduced levels in comparison with HDM-treated WT mice (Fig. 5). The PBS-treated p22phox WT and KO mice did not show any changes in goblet cells and mucus production at basal levels.

Airway hyperreactivity is attenuated in p22phox KO mice
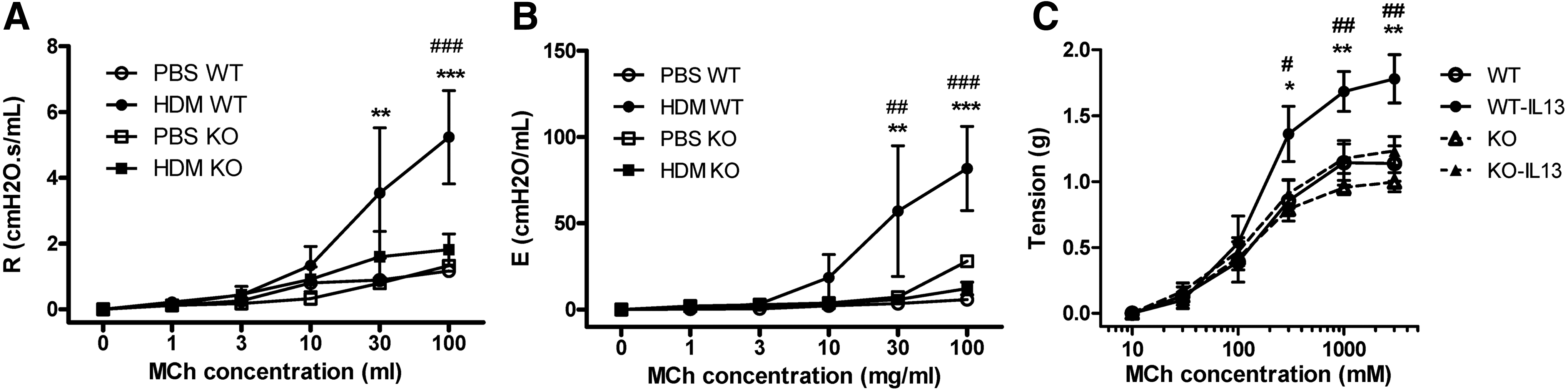
We next assessed an additional feature of asthma, AHR, as assessed by lung function measurements in response to increasing concentrations of inhaled methacholine (MCh). HDM treatment of WT mice resulted in increased AHR at both 30 and 100 mg/ml MCh as demonstrated by increased airway resistance and elastance compared with PBS-treated mice (Fig. 6A, B). Moreover, p22phox KO mice treated with HDM exhibited significantly decreased AHR compared with HDM-treated WT mice (Fig. 6A, B). PBS-treated p22phox WT and KO mice exhibited similar response to the increasing MCh concentrations.
p22phox KOs are less responsive to IL-13
As IL-13 is one of the key molecules controlling AHR (19), we investigated whether p22phox KO mice were protected from IL-13-induced AHR. To this end, trachea ring segments were incubated overnight with IL-13, then an MCh dose–response curve was performed using a wire myograph. In the absence of IL-13 preincubation, both WT and KO tracheas produced a similar concentration-dependent increase in tracheal tension with increasing MCh concentrations. Pretreatment of isolated WT tracheas with IL-13 strongly potentiated the response to increasing MCh doses. Similar to the in vivo AHR data, IL-13 incubation did not enhance AHR in KO mice (Fig. 6C). To further understand the decreased IL-13 response, we analyzed signal transducer and activator of transcription (STAT)6 activation in isolated tracheas with and without overnight IL-13 stimulation. In WT tracheas, IL-13 induced a robust phosphorylation of STAT6; however, in KO tracheas, activation of STAT6 in response to IL-13 was not significantly increased (Fig. 7A, B). We also examined whether these results could be replicated by the use of ROS inhibitors. Pretreatment with catalase, but not N-acetyl-

Discussion
In this study, we assessed the contribution of NADPH oxidase in the development of allergic asthma. The NADPH adaptor subunit p22phox (CYBA) was significantly upregulated in asthmatic patients and mice treated with HDM. Due to the essential role of p22phox in assembly and function of all major NOX isoforms (NOX1-4), loss of p22phox function inactivates NADPH oxidase and consequently NADPH oxidase-derived ROS generation. Employing mice that lack the p22phox protein, we provide strong evidence that NADPH oxidase plays a crucial role in the HDM-induced development of AHR and goblet cell hyperplasia. We also show that IL-13-induced hypercontractility and signaling were significantly attenuated in bronchial rings from p22phox KO mice ex vivo. These protective effects could be attributed, in part, to the inability of p22phox KO mice to generate ROS (34). These findings support the notion that increased oxidative stress is integral in asthma pathogenesis and requires an active NADPH oxidase system.
ROS derived from NADPH oxidase helps maintain vascular tone and can regulate important processes such as cytoskeletal organization, cell migration, growth, proliferation, and apoptosis (18). In allergic animal models, increased levels of oxidative stress and expression of antioxidant enzymes have been described (5, 9). Accordingly, we here show that mice treated with HDM strongly induce the production of ROS. Elevated ROS species such as H2O2 have previously been measured in the exhaled breath condensate of asthma patients and correlate with disease severity (15, 33). Due to this increased oxidative stress, antioxidants such as peroxiredoxins, catalase, and SOD maintain ROS levels in the lung and maintain the redox environment (20). In our study, deficiency of p22phox was accompanied by loss of ROS production and associated with decreased goblet cell hyperplasia and AHR. Our results support the data of Sevin et al., who analyzed the ovalbumin response in gp91phox (NOX2) KO mice and observed decreased goblet cell hyperplasia, AHR, and eosinophilic inflammation (40). We expand on these observations by demonstrating elevated expression of p22-PHOX in asthmatic bronchial biopsies and that p22phox essentially contributes to mucus hypersecretion and AHR in vivo and ex vivo. Recently, Patel et al. demonstrated that NADPH oxidases are required for mucin secretion in intestinal goblet cells (37). Increased NOX4 expression has been reported in airway smooth muscle bundles and isolated smooth muscle cells of asthma patients. Furthermore, the hypercontractility phenotype of asthmatic SMCs could be abrogated by anti-NOX4 siRNA or NOX inhibition (44). The presence of different SNPs in p22phox has been shown to associate with asthma; a homozygous 640A allele confers an increased risk, while 640G a decreased risk (24). SNPs in the p22phox gene that result in lower expression or activity may reduce the prevalence of asthma, while mutations that increase expression could promote asthma. It should also be considered that most of the patients presented in this study were being treated with inhaled corticosteroids, about half of them on oral corticosteroids.
One of the key Th2 cytokines involved in asthma pathogenesis is IL-13; neutralization of IL-13 signaling strongly attenuates allergen-induced AHR, mucus production, and eosinophilia (19, 49). In this study, we show that NADPH oxidase-derived ROS is essential for the IL-13-driven AHR response. IL-13 can augment airway smooth muscle contractility via upregulation of RhoA and RhoA kinase (11, 17), and activated Rho kinase phosphorylates myosin light chain and thereby enhances SMC contraction. Accordingly, application of the Rho kinase inhibitor, Y-27632, leads to a significant reduction in AHR (39, 45). Our in vivo observations are further supported by the wire myograph experiments where direct incubation of tracheal rings with IL-13 failed to produce a hyper-response in p22phox KO mice. As we observed only strong induction of IL-13 in HDM-treated WT mice, the protective effect observed in p22phox mice in AHR is most likely due to reduced IL-13 and IL-13 signaling within structural cells of the airways. This premise is supported by the decreased IL-13-induced activation of STAT6 in tracheal rings when p22phox is absent. Moreover, pretreatment with ROS inhibitors was able to diminish the IL-13-induced activation of STAT6. Along these lines, it has been demonstrated that ROS inactivates the protein tyrosine phosphatase, PTP1B, which negatively regulates IL-4 receptor signaling via dephosphorylation of STAT6 (29, 41). Therefore, loss of p22phox and consequently NADPH-dependent ROS production may promote dephosphorylation of pSTAT6 and thereby reduce IL-13 signaling.
The HDM-induced increase in ROS levels could result from either recruited inflammatory cells or from the resident structural cells (e.g., epithelial or smooth muscle cells). One limitation of our study is that we could not differentiate between these two options as the presence of ROS was measured in the entire pulmonary lysate. Despite the protective effect that loss of p22phox has on AHR and goblet cell hyperplasia, KO mice possessed an altered inflammatory profile following HDM treatment. WT mice possessed high levels of the Th2 proteins, IL-4 and IL-13, which were accompanied by strong eosinophil recruitment in the BALF and the lungs. On the other hand, in KO mice, these parameters were not significantly increased; instead, elevated numbers of neutrophils and lymphocytes were observed. The elevated levels of IFNγ or IL-17 in the HDM-treated KO mice could provide a mechanistic explanation for these observations. However, further experiments are required to decide between these two options. Earlier work by Snelgrove et al. has shown that a defect in gp91 phox results in a skewed T cell response (43). T cell receptor ligation induces the production of hydrogen peroxide via the activation of NOX2. Hydrogen peroxide in turn activates the Th2 differentiation factors, GATA-3 and STAT6, and inhibits the TH17 differentiation factor, STAT3 (7, 42, 46). Therefore, loss of gp91phox or p22phox would alleviate STAT3 inhibition and promote production of IL-17. Furthermore, IL-17 has been shown to increase the expression and stability of Cxcl1 mRNA, thereby promoting neutrophil recruitment (13, 28). These results are in line with our findings where the increased IL-17 expression observed in HDM-treated KO mice was associated with elevated Cxcl1/2 mRNA expression and neutrophil recruitment.
During the last decade, studies have underpinned the importance of ROS as a crucial secondary signaling messenger in many biological processes required for cellular homeostasis. The use of general ROS blockers such as NAC might give conflicting results as it scavenges ROS regardless of their source (mitochondria/NADPH oxidase). A chronic increase in ROS levels should be tackled; however, there is the caveat to not completely inhibit the basal ROS and affect the redox potential in the cell, which can alter cellular homeostasis (47). The use of p22phox inhibitors might overcome these issues. Unfortunately, there are no commercially available p22phox inhibitors, but highlighting the importance of p22phox in asthma pathogenesis could further encourage their development.
In conclusion, these findings demonstrate the crucial role of p22phox-dependent NADPH oxidase for the development of mucus hypersecretion and AHR in a mouse model of asthma. Elevated p22-PHOX expression in bronchial biopsies confirms the importance of ROS generation from the NADPH oxidase system in asthma pathogenesis.
Materials and Methods
Human samples
Collection and use of bronchial biopsies from asthmatic patients and healthy control subjects were approved by the Southampton and South West Hampshire Joint Local Research Ethics Committee and written informed consent obtained from all study participants. Biopsies were obtained via bronchoscopy from healthy controls (n = 16) and patients with asthma (n = 35); this mixed cohort consisted of 24% of mild cases and remaining 76% were severe asthmatic cases, some patients were included in a previous study (30). Bronchial biopsies obtained were either immediately fixed in cold formalin and embedded in paraffin for further histological staining or immediately homogenized in TRIzol reagent and further processed for RNA isolation. The study subjects' characteristics of healthy controls and asthmatic patients are summarized in Table 1.
In accordance with the British guideline on the management of asthma (British Thoracic Society [BTS] and Scottish Intercollegiate Guidelines Network) Thorax 69 (Suppl 1): i1–i192, 2014. Step 1: Inhaled SABA as required. Step 2: SABA+ICS 200–800 μg beclomethasone dipropionate or equivalent/day. Step 3: SABA + ICS + inhaled LABA. Step 4: Up to 2000 μg/day ICS + LABA + others. Step 5: Up to 2000 μg/day ICS + oral steroids (OCS).
Mann–Whitney U test.
Student's t-test (p < 0.05).
FEV, forced expiratory volume; ICS, inhaled corticosteroids; LABA, long-acting ß2-agonists; OCS, oral corticosteroids; SABA, short-acting ß2-agonists.
Animals and treatment protocol
p22phox KO mice (Cybanmf333) were obtained from Jackson Laboratories and bred in-house. Mice were maintained under pathogen-free conditions in isolated ventilated cages with 12-h light/12-h dark cycles. p22phox KO mice contain a T to C point mutation, which substitutes tyrosine for histidine at amino acid position 121, resulting in the inactivation and loss of the p22phox protein (34). Mice were maintained as a heterozygous colony; to reduce animal numbers, experimental mice were obtained by crossing homozygous parents to produce homozygous offspring. Water and chow were supplied ad libitum. All mouse experiments met EU guidelines 2010/63/EU and were approved by the Federal Ministry of Science, Research and Economics, Vienna, Austria. All measures were taken to keep animal suffering to a minimum. Mice were treated intranasally with a crude extract of HDM (50 μg/25 μl in PBS; Greer, Lenoir, NC; lot #187753) once per week for 6 weeks, while control mice received PBS. Analysis of lung function parameters and organ collection was performed 72 h after the last challenge (Supplementary Fig. S2).
Immunohistochemistry and immunofluorescence
Paraffin-embedded bronchial biopsies and isolated perfused mouse lungs were cut into 3-μm sections for histologic analysis. Sections were deparaffinized in xylene, followed by decreasing concentrations of ethanol. PAS staining was performed according to standard protocols. Immunostaining against p22phox (1:1000; Human—Abcam, Cambridge, United Kingdom; 1:1000; Mouse—Santa Cruz, Heidelberg, Germany) was performed overnight using sodium citrate pH 6-treated sections. The specificity of the immunohistochemistry and immunofluorescence staining was confirmed with p22phox WT and KO lung tissue (Supplementary Fig. S3). p22phox primary antibodies were detected by the immPRESS α-Rabbit Ig (peroxidase) polymer detection kit using NovaRed as the substrate (Vector Laboratories, Burlingame, CA); counterstaining was performed with Hemalaun (from Mayer). An Olympus VS120 slide scanning microscope was used to obtain images.
For double immunofluorescence, the mouse lung sections were deparaffinized at 60°C overnight and incubated in pH 6 antigen retrieval solution at 95°C, followed by blocking with 10% bovine serum albumin (BSA). Then, samples were incubated overnight at 4°C with antibodies anti-p22phox (Abcam) along with anti-αSMA (#EB06450; Everest Biotech, Upper Heyford, United Kingdom) or pan-Cytokeratin (Biolegend, London, United Kingdom) in a concentration of 1:100 in 10% BSA, followed by incubation with Alexa Fluor-488 and −555-labeled secondary antibodies (Life Technologies, Carlsbad, CA; 1:500 in 0.1% BSA) for 45 min at room temperature. Sections were counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma-Aldrich, Dorset, United Kingdom) to visualize nuclei. Negative controls were performed alongside in each experiment where the primary antibody is omitted (Supplementary Fig. S3). Images were taken using a laser scanning confocal microscope (Zeiss LMS 510 META; Zeiss, Jena, Germany) with Plan-Neofluar (40 × /1.3 Oil DIC) objective. Brief details for p22phox staining, PAS, were made according to standard protocols.
Quantitative histology
The percentage of goblet cells and mucus volume was quantified on PAS-stained sections using the NewCast software (Visiopharm, Hoersholm, Denmark) on automatically selected random regions from the 20 × scanned images. The goblet and epithelial cells intersecting the airway basement membrane were counted and presented as percentage goblet cells; the volume of mucus was determined by point counts and compared with the surface area of the airway basement membrane as determined by line probe intersections (1, 23).
Western blotting
Total proteins were isolated from lung homogenate or tracheal samples using RIPA buffer (Sigma) and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane (GE Healthcare, Vienna, Austria). After blocking with 5% nonfat dry milk in Tris-buffered saline-Tween (0.1%) buffer, the membrane was incubated overnight at 4°C with the following antibodies: anti-p22phox (1:100; Santa Cruz), p-STAT6 (1:1000), or anti-α-tubulin (1:4000; all from Cell Signaling, Danvers, MA). Horse radish peroxidase-conjugated goat anti-rabbit secondary antibodies together with the ECL prime (GE Healthcare) developing solution were used to detect primary antibodies. Equal protein loading and transfer were controlled by normalizing to α-tubulin. The p22phox protein size was confirmed by the Western blot with p22phox WT and KO lung tissue (Supplementary Fig. S3). Uncropped Western blots are given in the supplementary data (Supplementary Fig. S11).
Intracellular ROS measurements
The levels of hydrogen peroxide or superoxide were determined by changes in intracellular H2DCF-DA or dihydroethidium (DHE) (Life Technologies, Vienna, Austria) fluorescence levels, respectively. Briefly, single-cell lung tissue homogenates were prepared by digesting the lower right lube with collagenase (200 ng/ml) and DNAse (200 ng/ml) for 40 min at 37°C. The tissue homogenate was passed through a cell strainer (100 μm) for single-cell suspension and, if necessary, erythrocytes were lysed with erythrolysis buffer (2.6 mM NH4Cl, 0.09 M KCO3, 0.6 M Titriplex III). Cells (100,000) were loaded with H2DCF-DA (10 μM) or DHE (10 μM) for 45 min at 37°C. Fluorescence activation of H2DCF-DA was measured at 504 nm excitation and 529 nm emission; for DHE, the measurement was observed at 535 nm excitation and 635 nm emission. Blank (cell fraction without H2DCF-DA or DHE) readings were subtracted from loaded sample readings. SOD quenchable signal was also measured in PBS and HDM-treated lung homogenate; the SOD inhibited the HDM-induced increase in ROS both in H2DCFDA and DHE fluorescence levels (Supplementary Fig. S5).
Cytokine and immunoglobulin detection
Blood was collected from the vena cava and serum levels of HDM-specific immunoglobulin (Ig)E, IgG1a, and IgG2c were measured by enzyme-linked immunosorbent assay (ELISA). In summary, 96-well plates (Maxisorb; Greiner, Kremsmünster, Austria) were coated overnight with HDM at a concentration of 200, 5, or 50 μg/ml for IgE, IgG1, or IgG2, respectively, in coating buffer containing 0.84% NaHCO3 in H2O (pH 8.3). Serum samples were diluted in PBS containing 0.1% Tween and incubated overnight. Specific immunoglobulin subtypes were identified using a biotinylated anti-IgE, IgG1, or IgG2 antibody (BD Biosciences, Heidelberg, Germany). Following incubation with streptavidin conjugated with horse radish peroxidase, the chromogenic substrate BM Blue POD Substrate (Roche, Mannheim, Germany) was used for development. For quantitative measurement of mouse IL-4, IL-13, and IFNγ (eBioscience, Vienna, Austria), in mouse lung homogenate, ELISAs were performed according to the manufacturer's instructions.
Bronchoalveolar lavage fluid
After animals were sacrificed, BALF was obtained using 1 ml PBS containing protease inhibitor cocktail (Roche).
Flow cytometry
Bronchoalveolar lavage and single-cell lung tissue homogenates were analyzed using an LSRII flow cytometer and analyzed with the FACSDiva software (BD Biosciences). Cells were identified as follows: neutrophils (CD11b+, CD11c−, Gr-1+), macrophages (CD11b low, CD11c+, Siglec F+), dendritic cells (CD11b+, CD11c+, MHC-II high), T helper cells (CD3+, CD4+), cytotoxic T cells (CD3+, CD8+), B cells (CD19+), and eosinophil cells (CD11b+, CD11c−, Siglec F+). Antibody details are provided in Table 2.
Cells were identified as follows: neutrophils (CD11b+, CD11c−, Gr-1+), macrophages (CD11b low, CD11c+, siglec-F+), dendritic cells (CD11b+, CD11c+, MHC-II high), T helper cells (CD3+, CD4+), cytotoxic T cells (CD3+, CD8+), B cells (CD19+), and eosinophils cells (CD11b+, CD11c−, Siglec F+).
RNA isolation and real-time PCR analysis
Real-time analysis of p22-PHOX (CYBA) expression in human samples used the TaqMan probe Hs03044361_m1 (Life Technologies, United Kingdom), glyceraldehyde-3-phosphate dehydrogenase (GADPH) was used as the reference gene (PrimerDesign, Southampton, United Kingdom). Data are presented as ΔΔCt. For the analysis of mouse samples, total RNA was isolated from lung homogenate samples using a peqGOLD Total RNA Kit (Peqlab, Erlangen, Germany). cDNA synthesis and real-time PCR were performed as described previously (21). Briefly, total RNA was reverse transcribed using the iScript™ cDNA Synthesis kit (Bio-Rad Laboratories, Hercules, CA), according to manufacturer's instructions. Real-time PCR was performed using a LightCycler® 480 System (Roche Applied Science, Wien, Austria). The PCRs were set up using a QuantiFast® SYBR® Green PCR kit (Qiagen, Hilden, Germany) using the following protocol: 5 min at 95°C (5 s at 95°C, 5 s at 60°C, and 10 s at 72°C) × 45. Due to the nonselective double-strand DNA binding of the SYBR Green I dye, melting curve analysis and gel electrophoresis were performed to confirm the specific amplification of the expected PCR products. Pbgd and B2m were used as the reference genes. The difference in threshold cycle (Ct) values for each target gene was calculated as follows: ΔCt = meanCt reference genes − Ct target gene. Primer sequences are given in Table 3.
Assessment of airway hyperactivity
Seventy-two hours after the final HDM or PBS challenge, mice were anesthetized, intubated, and mechanically ventilated for the measurements of airway resistance and elastance (reciprocal of compliance) using a FlexiVent (SciReq, Inc., Montreal, Canada). Changes in airway resistance and elastance were calculated as response to increasing concentrations of MCh (0, 1, 3, 10, 30, and 100 mg/ml; Sigma Aldrich) as previously described (26). Briefly, mice were deeply anesthetized with 150 mg/kg ketamine and 20 mg/kg xylazine and were ventilated at 150 breaths/min, tidal volume of 10 ml/kg, and a positive end expiratory pressure of 2 cmH2O. Each data point represents the average of 12 snapshot perturbations recorded over a 3-min period after each MCh dose; before each set of perturbations, two deep inflation maneuvers were performed to normalize lung volume.
Tracheal stimulation and Wire myograph
For tracheal stimulation, the samples were cleaned of surrounding adipose and connective tissue is incubated with or without IL-13 (50 ng/ml) in the presence and absence of NAC (1 μM), catalase (10 μ/ml), and H2O2 (200 μM) for an hour, and the tissue samples are snap-frozen in liquid nitrogen and stored at −80°C until protein isolation. For isometric tension measurements, tracheal samples were cleaned of surrounding adipose and connective tissue and cut into segments ∼2 mm in length for use in isometric tension measurements. The tracheal tissues were incubated with or without murine IL-13 (Ebioscience, Vienna, Austria) overnight in Dulbecco's modified Eagle Medium supplemented with 10 mM HEPES and antibiotics. Tracheas were positioned between two adjustable pins in a myograph chamber (Multi Wire Myograph System-620M; Danish MyoTechnology A/S, Aarhus, Denmark) containing physiologic salt solution and continuously aerated with 95% O2, 5% CO2, at 37°C. The myograph chambers were connected to force transducers for isometric tension measurements (PowerLab® 8/35; ADInstruments, Dunedin, New Zealand) (25). Tracheas were incubated at 37°C, and a basal tension of 2 mN was applied and allowed to stabilize for 45 min. Physiologic salt solution containing 120 mM KCl was used to determine viability and adequate contractility of the tissues. Tracheas were stimulated three times using 120 mM KCl to obtain reproducible contractions. Tracheas that did not respond to these repeated stimuli were not included in the study. MCh was introduced in 4-min intervals in accumulative doses.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5/6 software. Data are expressed as mean ± standard error of the mean or box-and-whisker plots. For normally distributed data, intergroup variants were compared by Student's t-tests, otherwise by Mann–Whitney U test. Multigroup comparisons were made with one-way analysis of variance (ANOVA) with Tukey's post hoc test. For comparison of dose–response curves or progressive measurements between groups, two-way ANOVA was used with Bonferroni's post hoc test. p Values <0.05 were considered as statistically significant.
Footnotes
Acknowledgments
Excellent technical assistance from Sabrina Reinisch, Camilla Götz, Thomas Fuchs, Eva Grasmann, and Sabine Halsegger is appreciated. The authors would like to thank Grazyna Kwapiszewska for fruitful discussions. L.M. and H.M.H. are members of COST (Cooperation in Science and Technology) Action BM1201. H.M.H. was supported by a Medical Research Council (MRC) Clinician Scientist Fellowship (G0802804) and human samples were studied from the Wessex severe asthma cohort, which was supported by an MRC grant (G0800649) to P.H.H. A.H. received grant support from the Austrian Science Funds FWF (P22521-B18).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.