
Abstract
Aims:
Dysfunction of neurovascular pericytes underlies breakdown of the blood-brain barrier, but the molecular mechanisms are largely unknown. In this study, we evaluated the role of the transient receptor potential melastatin-related 2 (TRPM2) channel and autophagy during brain pericyte injury both in vitro and in vivo.
Results:
A rapid induction in autophagy in human brain vascular pericytes, in the zinc oxide nanoparticles (ZnO-NP)-induced cell stress model, was paralleled with an increase in the expression of the TRPM2-S truncated isoform, which was abolished by treatment with a nitric oxide synthase inhibitor and a peroxynitrite scavenger. Furthermore, Y1485 in the C-terminus of the TRPM2 protein was identified as the tyrosine nitration substrate by mass spectrometry. Overexpression of the Y1485S TRPM2 mutant reduced LC3-II accumulation and pericyte injury induced by ZnO-NP. Consistently, LC3-II accumulation was reduced and pericytes were better preserved in intact brain microvessels of the TRPM2 knockout mice after ZnO-NP-induced vascular injury.
Innovation and Conclusions:
Our present study has revealed a novel mechanism of autophagy disturbance secondary to nitrosative stress-induced tyrosine nitration of TRPM2 during pericyte injury. Antioxid. Redox Signal. 27, 1297–1316.
Introduction
T
Our study is the first demonstration that transient receptor potential melastatin-related 2 (TRPM2)-dependent autophagy is critically involved in the pathological process of pericyte injury. Our findings have further identified nitrosative stress as an important element in the induction of TRPM2 tyrosine nitration at Y1485 and the autophagy response. Mutational prevention of this significantly reduced the elevation of endoplasmic reticulum apoptotic proteins and LC3-II accumulation during pericyte injury. LC3 accumulation was reduced, and brain pericytes were better preserved in the TRPM2 knockout mice on vascular injury. A full understanding of the molecular mechanisms of pericyte injury is crucial to the development of therapeutic strategies to treat neurovascular dysfunction-related pathologies.
Recent findings have revealed that pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells (35). As a sensor of oxidative stress and a mediator of Ca2+ entry and apoptosis, the transient receptor potential melastatin-related 2 (TRPM2) channels play a crucial role in a variety of physiological and pathological processes, such as regulating endothelial barrier function, increasing lung microvessel permeability and neutrophil sequestration (16, 25, 43, 51, 58). However, it is unclear how pericytes are damaged in response to stress stimulation and whether TRPM2 is involved in stress-induced pericyte injury.
The present study aimed at elucidating the molecular mechanism that is responsible for stress-induced pericyte injury. Using the ZnO-NP-induced stress model, in combination with genetic and pharmacological approaches, we investigated the role of TRPM2 in the crosstalk that couples with autophagy and microvascular pericyte injury. Our results from in vitro and in vivo studies provide compelling evidence to support a role for nitrosative stress in linking the TRPM2 turnover with the disturbance of autophagy in brain pericyte injury.
Results
Spatiotemporal changes of autophagy in stress-induced brain pericyte injury
The autophagic process induced by ZnO-NP is an early event for the evaluation of cell injury (20, 39). The size distribution and zeta potential distribution of ZnO-NP were determined (Supplementary Fig. S1; Supplementary Data are available online at
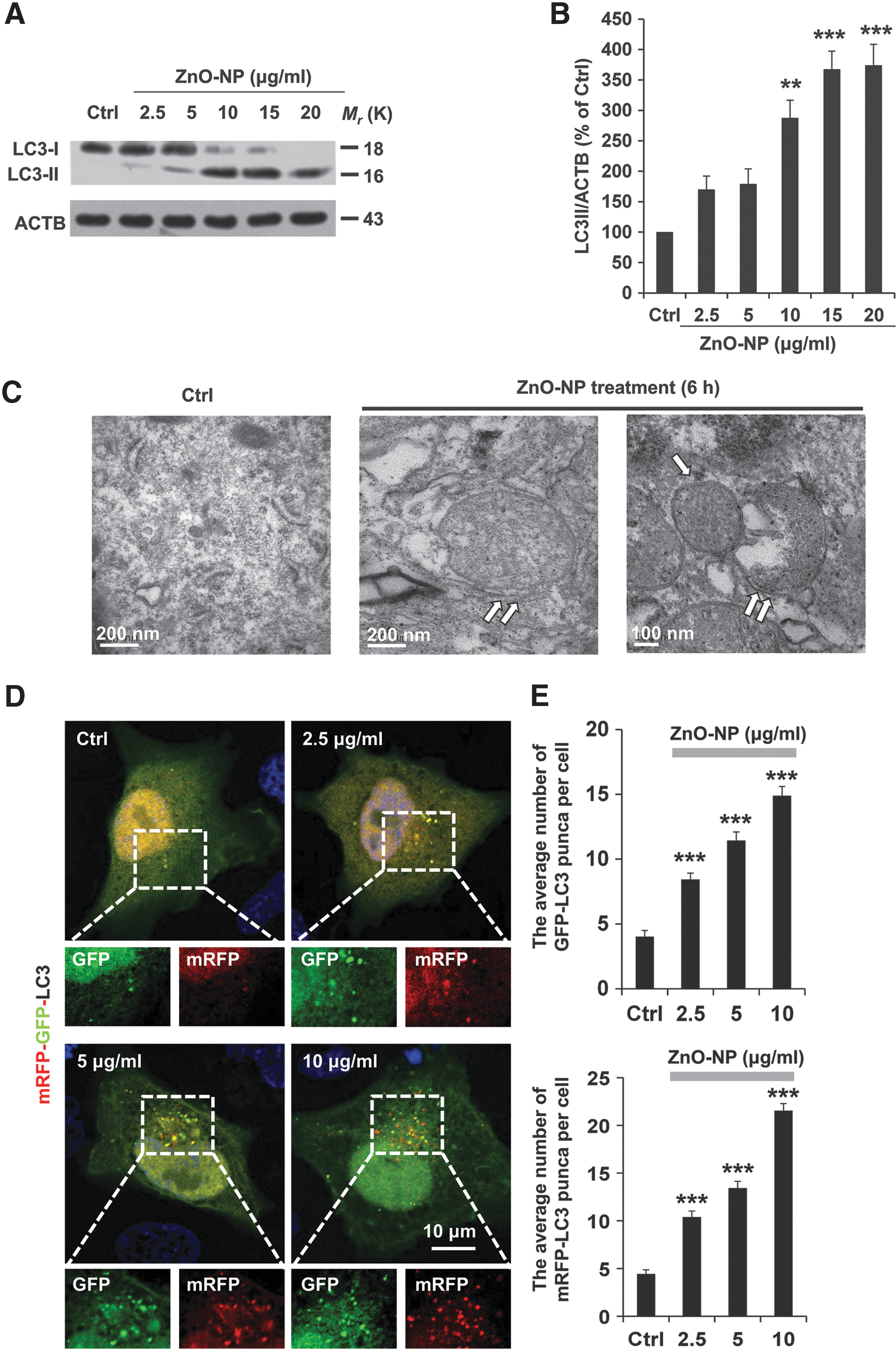

Formation of the double-membrane cistern structures or autophagosomes containing cytoplasmic materials or aberrant organelles is an ultrastructural hallmark of autophagy (19, 36). Consistent with the biochemical data, transmission electron microscopy (TEM) revealed the formation of double-membrane structures with engulfed cytoplasm fractions in pericytes after treatment with ZnO-NP for 6 h (Fig. 1C).
The accumulation of autophagosomes could indicate the dynamic process of autophagy (22). Therefore, we next examined ZnO-NP-induced autophagy flux by using fluorescence confocal microscopy in pericytes transfected with an mRFP-GFP-LC3 tandem construct, which is based on the concept of lysosomal quenching of GFP in GFP-labeled LC3(23, 33). As shown in Figure 1D, exposure to ZnO-NP for 6 h induced the formation of GFP-LC3 and mRFP-LC3 puncta, and an increase in the accumulation of yellow fluorescent puncta, which is indicative of autophagosomes. Such effects were ZnO-NP concentration dependent (Fig. 1D, E). These results were confirmed by immunofluorescence staining with the anti-LC3 antibody (Supplementary Fig. S2) to show an increase in autophagic vesicle accumulation in ZnO-NP-treated pericytes. Taken together, these results provide consistent evidence to indicate that autophagy initiation is increased after ZnO-NP-induced pericyte injury. Furthermore, time-lapse imaging showed that ZnO-NP induced dynamic changes in autophagy flux over a period of 60 min, as indicated by the fluorescence switch between GFP-LC3 puncta and mRFP-LC3 puncta (Fig. 1F and Supplementary Video S1). Autophagic flux can also be reflected by an elevation in the LC3-II level while interrupting the autophagosome-lysosome fusion step. As shown in Figure 1G, after inhibition of the lysosome function by using bafilomycin A1 (50 nM), the LC3-II level in pericytes was further upregulated in response to ZnO-NP stimulation, suggesting increases in autophagy induction and autophagic flux (Fig. 1G, H and Supplementary Fig. S3).
TRPM2 knockdown blocks autophagy during brain pericyte injury
A recent study shows that the TRPM2 channel is important in mediating oxidative stress-induced disruption of lung endothelial barrier function (16). In the present study, we set out to investigate the role of TRPM2 during brain pericyte injury. The temporal changes in the TRPM2 expression in brain pericyte cells after injury were examined by using Western blotting. Treatment with ZnO-NP induced a time-dependent increase in a protein band with a molecular weight of 95 kDa, which was expected for the short TRPM2 protein isoform (TRPM2-S) (Fig. 2A). The expression level of TRPM2-S relative to the full-length TRPM2 (TRPM2-L) was increased as the exposure was extended from 1 to 9 h (Fig. 2A, B). Therefore, our data indicate that ZnO-NP stimulation results in significant effects on the TRPM2 turnover in pericytes.
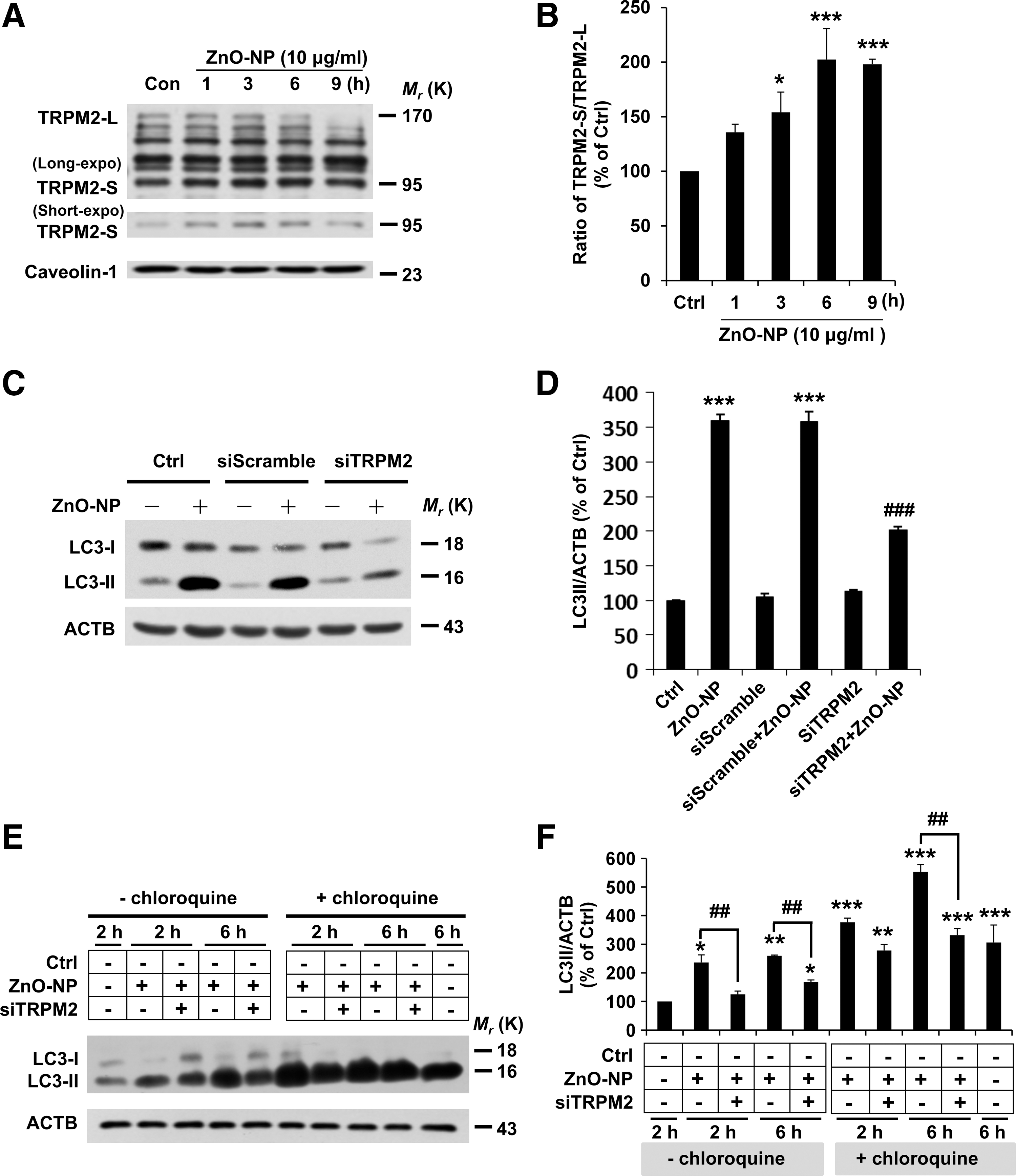
A recent study suggests TRPM2-S to be critically involved in reducing the HIF-1/2α level and mitophagy in SH-SY5Y neuroblastoma cells (4). We next investigated whether alteration in the TRPM2 protein expression affected the autophagy events in pericyte injury by examining the effect of reducing TRPM2 expression on LC3-II accumulation. Our results show that small interference RNA (siRNA)-mediated reduction in the TRPM2 expression significantly decreased LC3-II accumulation (Fig. 2C, D), indicating that ZnO-NP-induced LC3-II formation is TRPM2 dependent. Furthermore, we investigated whether ZnO-NP stimulation also disturbed autophagic flux in a TRPM2-dependent manner. After exposure to ZnO-NP for 2 h or 6 h, LC3-II accumulation was elevated further in pericytes treated with chloroquine (CQ, 25 μM) as compared with that in cells treated with CQ alone, indicating enhanced autophagic flux (Fig. 2E, F). Moreover, siRNA-mediated knockdown of the TRPM2 expression decreased LC3-II accumulation in pericytes in the absence and presence of CQ, indicating that TRPM2 participates in both autophagy induction and enhanced autophagic flux during pericyte injury (Fig. 2E, F).
TRPM2 knockdown reduces ER stress during brain pericyte injury
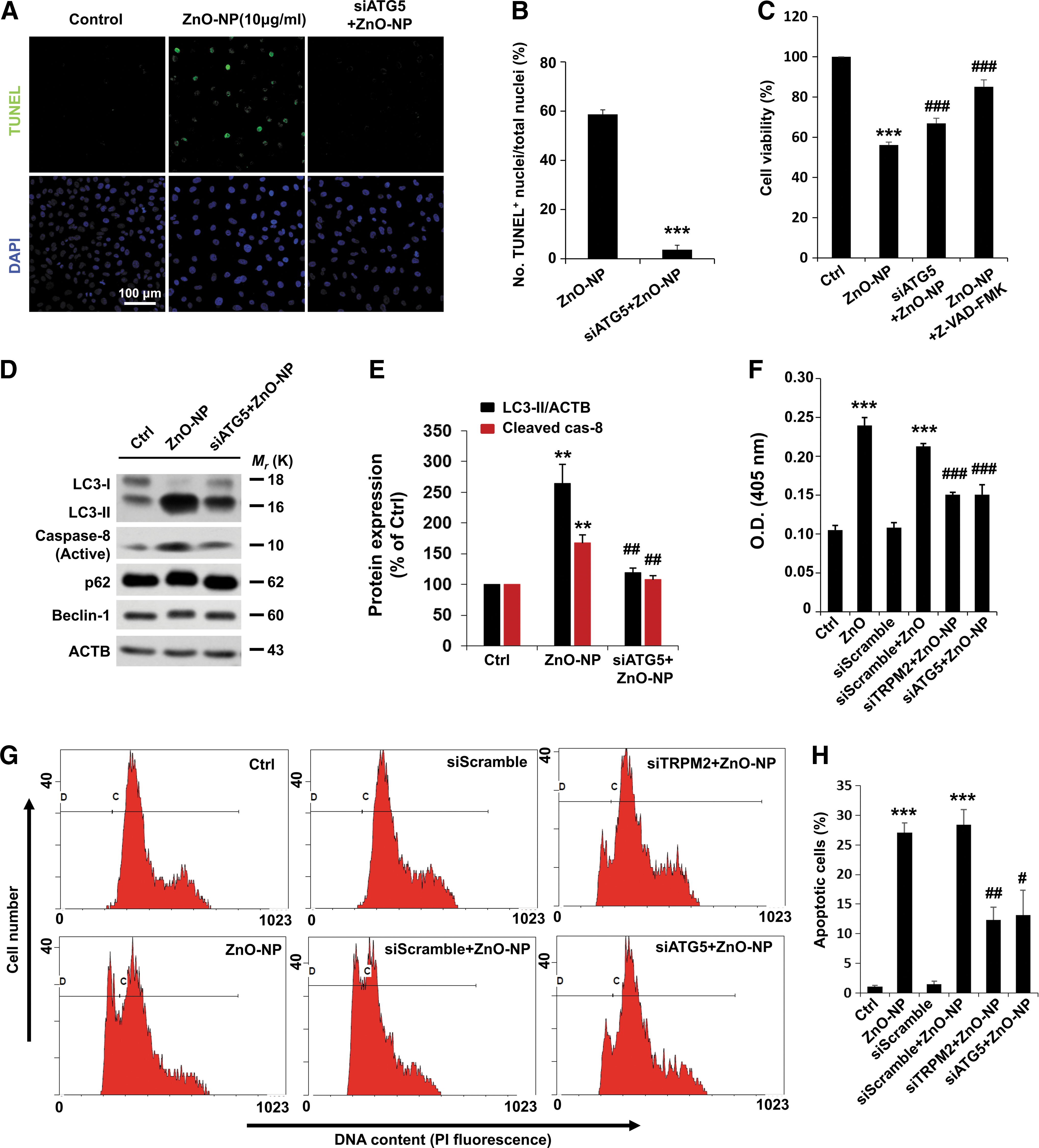
We next addressed the role of TRPM2 in ZnO-NP-induced brain pericyte injury by using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining. ZnO-NP treatment resulted in an increase in the percentage of TUNEL positive pericytes, which was almost completely abolished by treatment with TRPM2-siRNA (Fig. 3A, B), indicating TRPM2-dependent apoptotic cell death.

ER stress is a hallmark of cell injury, reflecting the extent of protein aggregation and promoting cell injury (48). The downstream ER stress markers, such as CHOP, JNK, and PERK, are known to be involved in the ER stress-mediated autophagy and apoptosis pathway (42, 48). To further validate the observation that TRPM2 is involved in pericyte injury, we examined such ER stress signaling proteins. Indeed, exposure to ZnO-NP led to time-dependent upregulation of CHOP, phospho-JNK (Thr183/Tyr185), and phospho-PERK (Thr981) in pericytes (Supplementary Fig. S4). Moreover, ZnO-NP-induced upregulation of ER stress-associated signaling activity in pericytes was significantly inhibited by treatment with TRPM2-siRNA (Fig. 3C–F).
To confirm that the observed TRPM2 turnover and ER stress-autophagy axis were causatively related to the severity of pericyte injury, ER stress-related signaling protein expression was examined. In contrast with the control cells, pericytes treated with ZnO-NP showed a significant increase in CHOP, phospho-JNK (Thr183/Tyr185), and phospho-PERK (Thr981) and again, TRPM2 silencing decreased ZnO-NP-induced elevation in such ER stress signaling proteins in the presence of CQ (Fig. 3G and Supplementary Fig. S5). We further examined whether TRPM2 silencing had a protective effect on the severe damage induced by prolonged exposure to ZnO-NP. Treatment with TRPM2-siRNA resulted in a significant protective effect in pericytes even after treatment with ZnO-NP for 24 h, as revealed by the downregulation of p-JNK and CHOP, along with the reduction in the degradation of IRE-1α and PERK (Fig. 3H). These results show that siRNA-mediated knockdown of the TRPM2 expression effectively reduces ZnO-NP-induced severe cell injury in pericytes.
We performed further experiments to determine whether autophagy events correlate with pericyte injury. As shown earlier, ZnO-NP treatment resulted in an increase in the percentage of TUNEL-positive pericytes. In contrast, siRNA knockdown of ATG5 expression reduced the number of apoptotic cells (Fig. 4A, B), confirming that disturbance of autophagy promoted pericyte cell injury. Moreover, siRNA knockdown of ATG5 or TRPM2 significantly increased the cell viability and suppressed the activation of caspase-8 during brain pericyte injury (Fig. 4C–H).
Nitrosative stress associates with TRPM2-dependent autophagy
To better understand the molecular mechanisms underpinning TRPM2-dependent autophagy during pericyte injury, we examined the possible involvement of the nitrosative stress. It is known that peroxynitrite modifies tyrosine residues in proteins, which accounts for the effects of endogenously produced NO by oxidation and nitration reactions (14, 47). As shown in Figure 5A, Western blotting analysis of nitrotyrosine indicates that significant peroxynitrite formation was observed at 3 h after exposure to ZnO-NP and maintained throughout the treatment for a maximum of 12 h. To gain mechanistic insights, confocal imaging was used to detect the change in the fluorescence intensity of NP3, an ONOO− probe in pericytes during treatment with ZnO-NP (Fig. 5B). There was a significant increase in the NP3 fluorescence intensity in response to treatment with ZnO-NP, which was blunted by treatment with 200 μM uric acid (UA), an ONOO− scavenger, before and during treatment with ZnO-NP (Fig. 5B). The autophagic flux can be morphologically traced with the mRFP-GFP-LC3 tandem construct. As shown earlier, the autophagic flux was increased after 6 h treatment with ZnO-NP. In contrast, treatment with UA led to barely discernible accumulations of yellow and red puncta on pericyte injury (Fig. 5C).
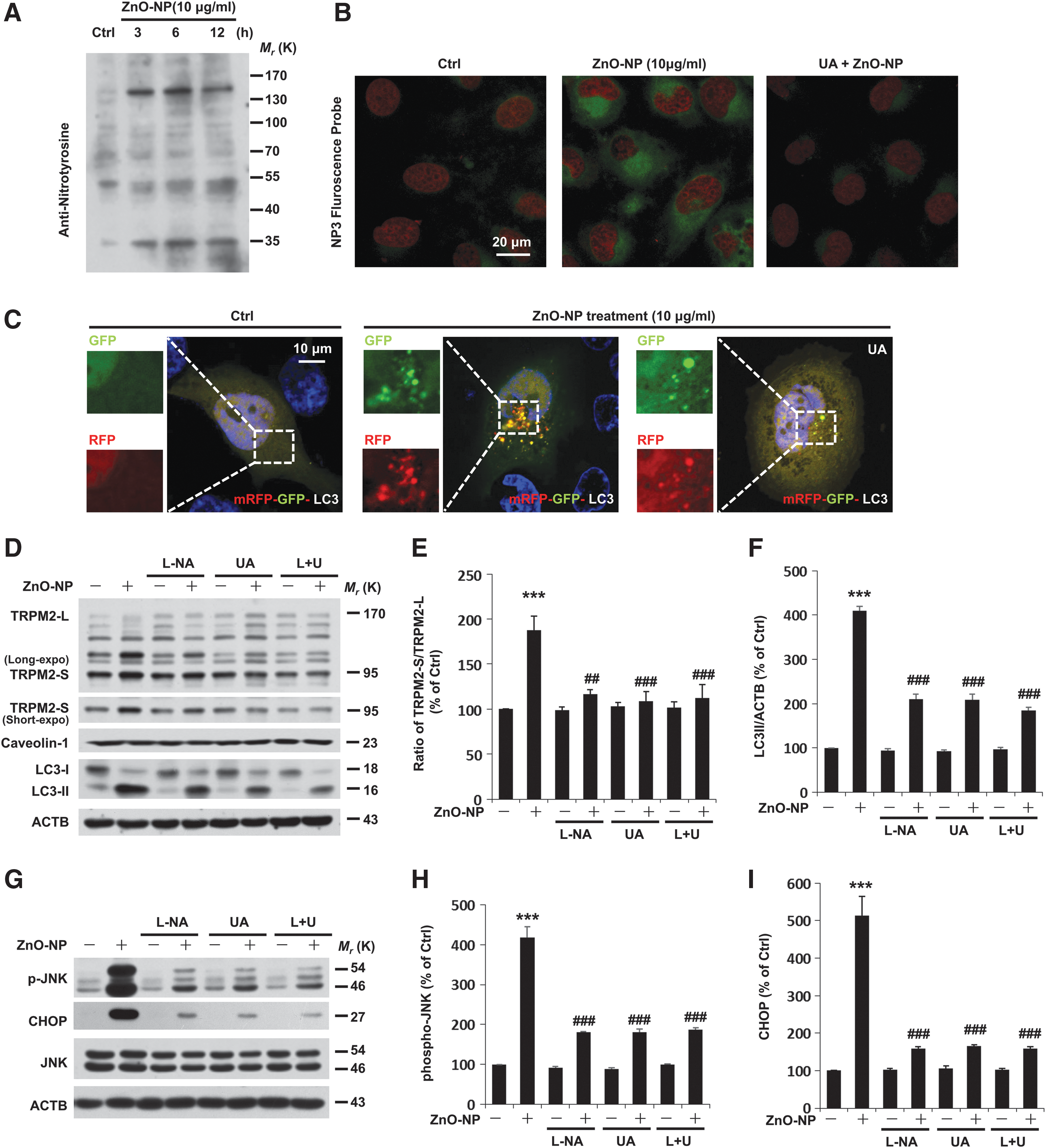
Of note, inhibition of nitric oxide synthase by using N(G)-nitro-L-arginine methyl ester (L-NAME, 100 μM) was also capable of inhibiting ZnO-NP-induced elevation in the TRPM2-S expression in pericytes (Fig. 5D–F). Pretreatment of pericytes with UA alone or together with L-NAME led to similar results (Fig. 5D–F). Next, we assessed the functional relevance of ONOO− formation with autophagy signaling during pericyte injury. Exposure to ZnO-NP for 6 h led to four- to fivefold increases in ER signaling proteins (Fig. 5G–I). To elaborate our finding, the ONOO− was used to further clarify the role of nitrosative stress during pericyte injury. Treatment with ONOO− induced a pronounced increase in ER signaling proteins and excessive autophagy in pericytes (Supplementary Fig. S6). Although a pronounced decrease in ER signaling proteins (namely CHOP and phospho-JNK), and LC3-II was detected in ZnO-NP-exposed cells treated with L-NAME, UA, or both (Fig. 5D–I), in these cells, fewer apoptotic cells were observed by TUNEL staining (Supplementary Fig. S7). On the basis of these data, we conclude that there is a requirement for nitrosative stress signaling in engagement of the autophagy during pericyte injury.
In vitro nitration of TRPM2 protein
The TRPM2-L protein contains several tyrosine residues that are located in the C-terminal region. The assessment of TRPM2 nitration and identification of the nitration sites should provide critical information in unraveling the mechanisms of TRPM2-mediated autophagy and pericyte injury. To directly confirm that nitrosative stress was, indeed, responsible for protein tyrosine nitration of TRPM2, we treated a human TRPM2 (hTRPM2) peptide composed of a majority of the C-terminus (residues 1206–1504) with ONOO−. An examination using MS/MS demonstrated that nitration caused a mass shift of +45 Da in 3-nitrotyrosine-containing peptides, and it further identified that tyrosine nitration occurred at Y1485 residue (Fig. 6).

Mutation of the nitration site in the TRPM2 attenuates pericyte injury
To reinforce the crucial role of TRPM2 protein nitration in mediating pericyte injury, we mutated Y1485 to serine (Y1485S) and transfected pericytes with the plasmid encoding the TRPM2-Y1485S mutant protein (Fig. 7A). Western blotting revealed that CHOP and phospho-JNK were substantially higher in cells transfected with the empty plasmid after 6 h exposure to ZnO-NP but not in cells transfected with TRPM2-Y1485S (Fig. 7B–F). In line with this, overexpression of the TRPM2-Y1485S mutant construct significantly attenuated excessive LC3-II formation during pericyte injury (Fig. 7B–F), as well as ER stress and autophagy in ONOO− treated pericytes (Fig. 7G–J). These results provide clear evidence to support that ZnO-NP-induced insult promotes activation of the ER stress-autophagy axis and pericyte injury and that these events are dependent of tyrosine nitration of TRPM2 at Y1485.
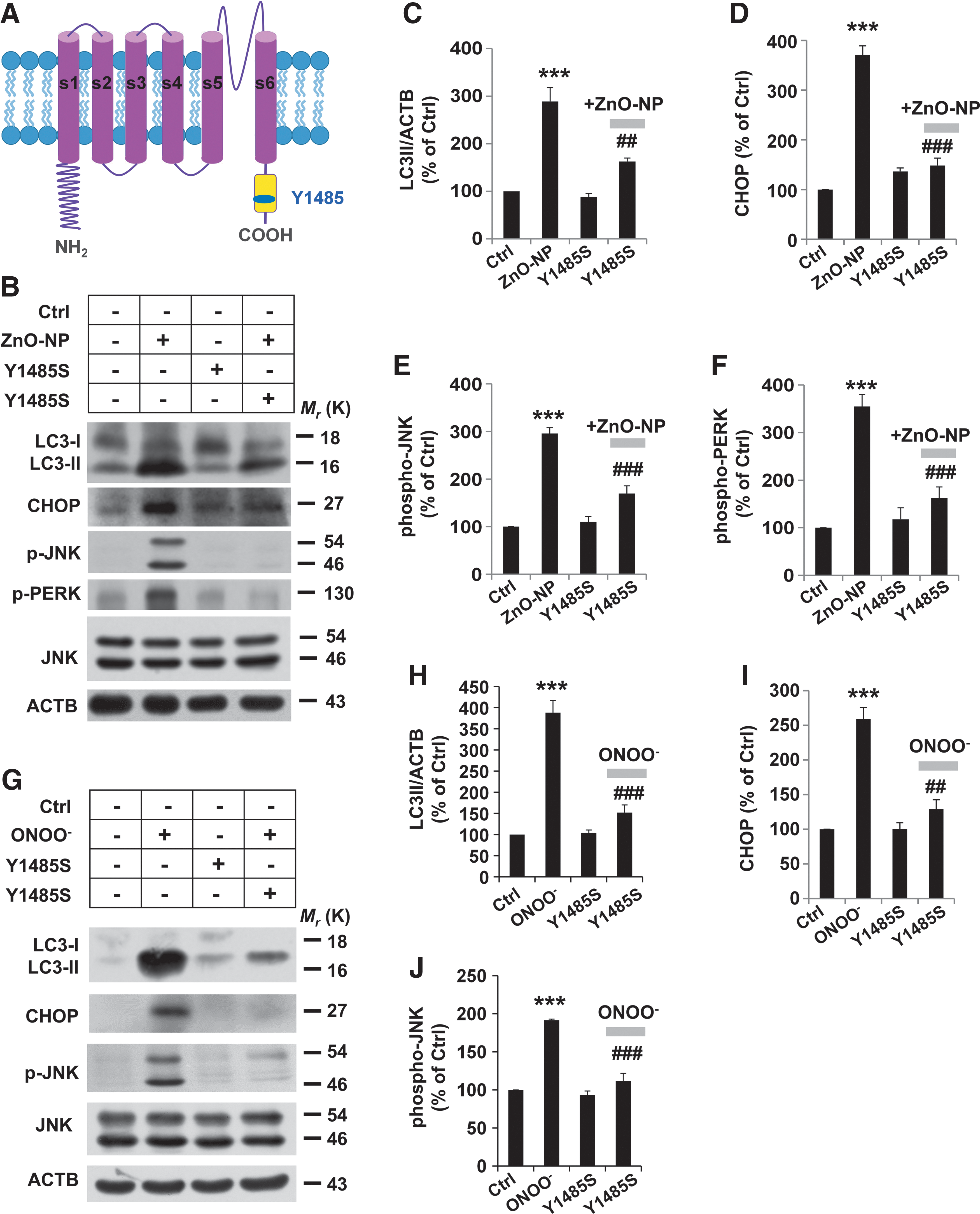
Nitrosative stress affects the TRPM2 channel function
We considered the possibility that tyrosine nitration of the TRPM2 protein regulated the TRPM2 channel function. To test this, ADP-ribose (ADPR)-induced TRPM2 channel currents were recorded in tetracycline-inducible TRPM2 expressing HEK293 cells (Fig. 8A). To examine whether the TRPM2 channel modulation could occur under nitrosative stress, we used SIN-1, a donor of ONOO− and a potent inducer of cell apoptosis, as well as ZnO-NP. Treatment with SIN-1 and ZnO-NP resulted in a significant decrease in ADPR-induced current amplitude (Fig. 8). The decrease induced by ZnO-NP or SIN-1 in the ADPR-induced current amplitude was prohibited by treatment with L-NAME or UA (Fig. 8A, B). Similar downregulation by ZnO-NP or SIN-1 was observed for the wild-type TRPM2 channel but not the TRPM2-Y1485S mutant channel transiently expressed in HEK293 cells observed (Supplementary Fig. S8). Taken together, these results suggest functional downregulation of the TRPM2 channels via nitration of tyrosine 1485.

Deletion of the TRPM2 reduces pericyte injury in mice
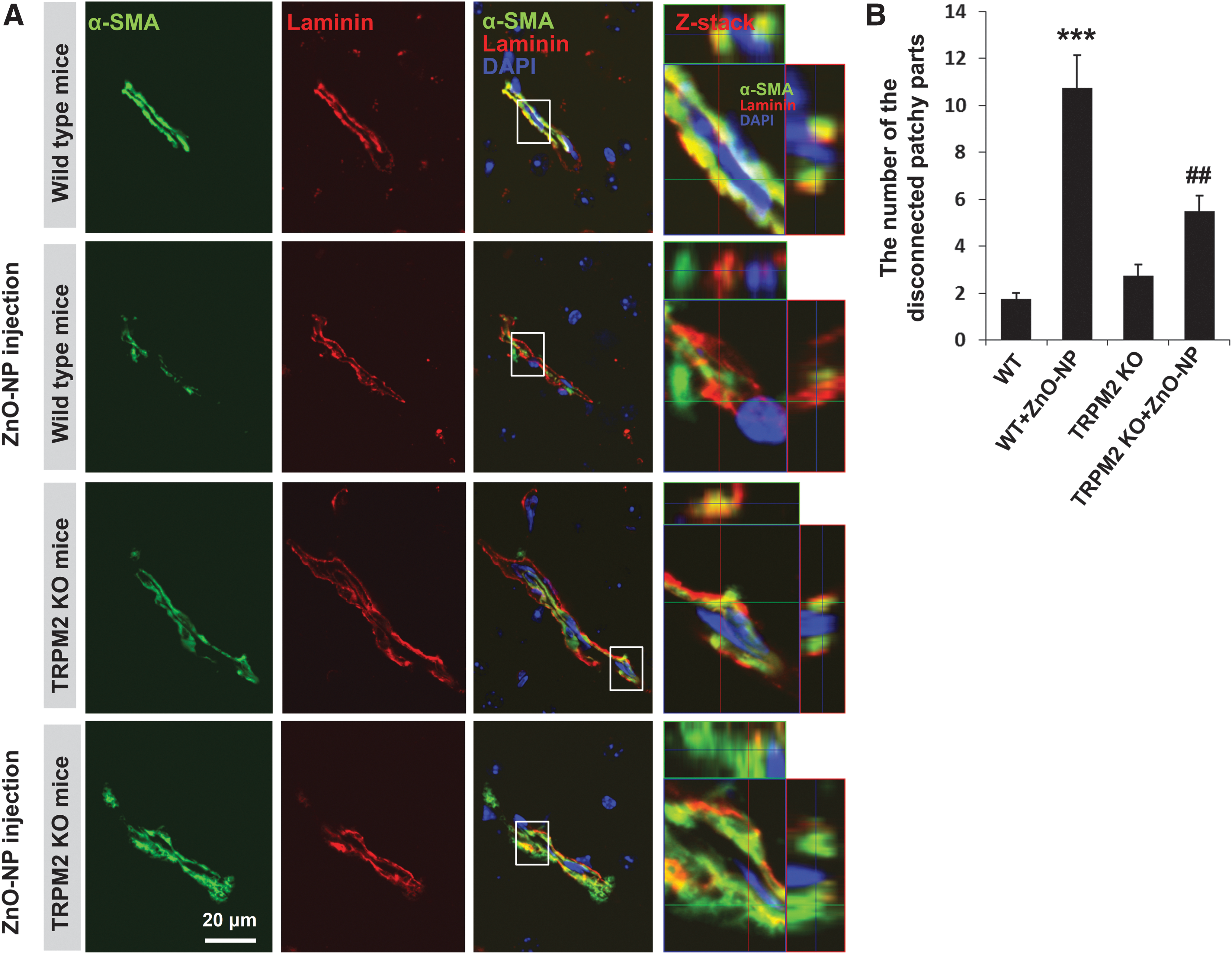
Pericytes are embedded within the basement membrane of microvessels (1, 12) and exhibit a number of characteristics for vessel maintenance and formation (52). To explore the pathophysiological significance of TRPM2-autophagy crosstalk identified from the in vitro studies described earlier, we extended our investigation to the brain pericytes in vivo. We examined pericyte injury in the cerebral vessels in the wild-type and age-matched TRPM2 knockout (KO) mice after ZnO-NP injection, using immunofluorescent confocal imaging of alpha-smooth muscle actin (α-SMA), one of the widely accepted pericyte markers in brain microvessels (8) and laminin in the vascular-specific basal membrane (1). Pericytes were embedded within basement membranes with a continuous and patchy pattern in the capillaries. Double labeling with α-SMA and laminin revealed more α-SMA-positive cells in the TRPM2 KO mice than in the wild-type mice after neurovascular insult (Fig. 9).
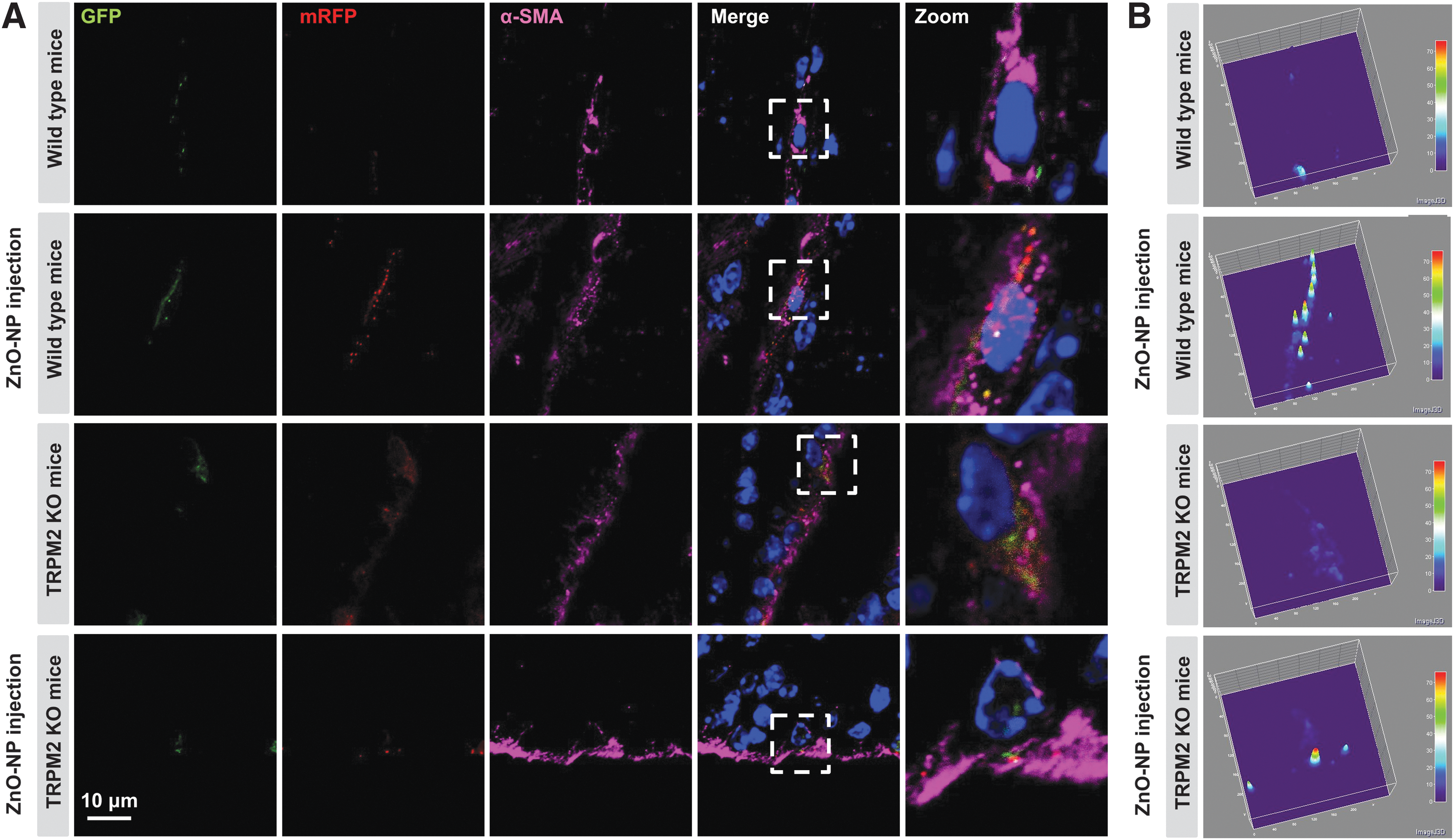
To provide further evidence to support the notion that excessive autophagy is associated with pericyte injury, adenovirus carrying the mRFP-GFP-LC3 construct was injected into the mice brain to visualize the role of autophagy during ZnO-NP-induced pericyte injury. In addition to the weak fluorescence of LC3-GFP in the control mice, more mRFP-LC3 puncta were observed in the brain pericytes in the mice after neurovascular insult (Fig. 10), indicating the increased autophagic process and flux activities. Moreover, the accumulation of autophagic vesicles was partially reduced in the vessels in the TRPM2 KO mice after neurovascular insult (Fig. 10). Therefore, the TRPM2 in pericytes has an important role in mediating excessive autophagy on neurovascular insult.
Discussion
Pericytes dynamically respond to stress that is induced by injury in brain diseases. Recent studies provide compelling evidence that pericytes are essential for the preservation of the BBB function during ischemic stroke and Alzheimer's disease (40, 54). In this study, we have revealed a previously unrecognized role for the TRPM2 channel in promoting autophagy in pericytes and defined a novel mechanism of autophagy disturbance secondary to nitrosative stress-induced tyrosine nitration of TRPM2 protein during stress-induced pericyte injury.
In this study, we demonstrate that TRPM2-dependent autophagy is critically involved in the pathological process of pericyte injury. TRPM2 is a potentially important pharmacological target for inhibiting the pathological increase in endothelial barrier function (17). The importance of TRPM2 in mediating autophagy pericytes is supported by our finding that knockdown of TRPM2 partially inhibits ZnO-NP-induced LC3-II accumulation. Control experiments showed that TRPM2 knockdown did not significantly affect the LC3-II as well as ER stress protein levels during starvation (Supplementary Fig. S9). A recent study has reported that interference with TRPM2-L function modulates HIF-1/2α, mitochondrial function, and mitophagy (4). Other studies have shown that overexpression of TRPM2-L confers protection against oxidative stress-induced cell death (5). A dramatic increase in the TRPM2-S was observed after pericyte injury. Zhang et al. reported that TRPM2-S plays an important role in many tissues because it can modulate Ca2+ influx and cellular responses to oxidative stress (56). Recently, Hecquet et al. have shown that TRPM2-S expression during oxidative stress may mitigate endothelial cell apoptosis and vascular injury and inflammation (17, 18).
To search for a link between TRPM2-dependent autophagy and pericyte injury, we investigated whether TRPM2 or autophagy inhibition can affect cell survival after being subjected to ZnO-NP-induced insult. Here, we showed that either downregulation of endogenous TRPM2 expression in pericytes by siRNA in vitro or TRPM2 KO in mice consistently reduced LC3-II accumulation and pericyte injury. Of particular note, we found that siRNA knockdown of ATG5 markedly attenuated pericyte apoptosis, indicating that autophagy serves as an important cell death mechanism. In this study, we also demonstrated that autophagy during pericyte injury strongly depends on ER stress, which was confirmed by the finding that treatment with salubrinal, a selective inhibitor of ER stress, significantly blocked ZnO-NP-induced LC3-II formation (Supplementary Fig. S10). Moreover, siRNA-mediated knockdown of TRPM2 significantly blunted tunicamycin-induced ER stress and LC3-II formation in pericytes (Supplementary Fig. S11). In addition, our data showed that autophagy and ER-stress signaling were Ca2+ dependent and inhibited by a TRPM2 blocker (Supplementary Fig. S12). Thus, our data suggest that autophagy changes as a regulator of pericyte injury, TRPM2 lies upstream of the ER stress-autophagy axis, and autophagy is functioning primarily as a cytotoxic response to excess ER stress.
How does stress stimulation induce TRPM2-dependent autophagy and subsequently pericyte injury? We have further explored the upstream regulation of TRPM2 channel function underlying pericyte injury. The present study demonstrated that nitrosative stress is an important element in the induction of TRPM2 turnover and the autophagy response. We previously reported that Ca2+/calmodulin-dependent nitrosative stress initiated early cerebrovascular injury and subsequent neuronal damage, indicating that brain microvessels are the most vulnerable and sensitive cellular components of ONOO− (13, 46, 48). In the present study, the Ca2+ sensor protein calmodulin was identified as one of the TRPM2-interacting proteins by using a proteomic approach. Immunoprecipitation confirmed that TRPM2 and calmodulin are associated with each other, whereas this association was decreased in the presence of ZnO-NP (Supplementary Fig. S13). We hypothesize that ongoing nitrosative stress induces the TRPM2 turnover, and, subsequently, induces ER stress to initiate the autophagy pathway. The process described earlier is, at least in part, dependent on nitration of TRPM2 at Y1485. TRPM2 nitration does not directly trigger autophagy and, instead, activates ER stress that provides the critical link to the increased autophagy. In the present study, overexpression of the TRPM2-Y1485S construct significantly reduced the LC3-II formation in tunicamycin-treated pericytes (Supplementary Fig. S14). The autophagy induced by the TRPM2 turnover is likely through ER signaling, and the consequent cascade of reactions is related to the unfolded protein response (15, 24, 38). Our data suggested that nitration of TRPM2 at Y1485 might contribute to TRPM2-S formation because both the NOS inhibitor (L-NAME) and peroxynitrite scavenger (UA) significantly reduced TRPM2-S formation and restored the TRPM2-L channel function. These results further support the notion that a nitrosative stress-dependent mechanism might regulate the TRPM2 turnover during pericyte injury. These steps were in parallel with an inhibition of CHOP and phospho-JNK (Thr183/Tyr185), and an increase in the level of LC3-II in the same context.
The neurovascular unit is the primary target of nitrosative stress in cardiovascular diseases, stroke, and neurodegenerative disorders (2, 46, 47). The toxicity of superoxide is greatly increased by reacting with nitric oxide to form peroxynitrite (44, 47). Here, combined treatments with L-NAME and UA did not further reduce anti-nitrosative stress efficacy after ZnO-NP insult, indicating that the nitrosative stress pathway is specifically involved in the pathological process of pericyte injury. Further support for this conclusion is provided by the observation that overexpression of the TRPM2-Y1485S mutant was sufficient to inhibit the ER stress, coinciding with the inhibitory effect on the elevation of LC3-II accumulation during pericyte injury. Our results together with these observations suggest that inhibition of TRPM2 protein tyrosine nitration protects against pericyte injury via inhibition of autophagy.
A number of studies demonstrate that pericyte-endothelial cell communication is essential to regulate the capillary blood flow and to maintain the function of the BBB (9, 11, 37). Indeed, pericytes are a key component of the neurovascular unit, which wrap around the endothelial cells in the microvessels, eliciting a protective effect on endothelial barrier function (9, 16). To assess the translational relevance in vivo, we addressed the role of pericyte TRPM2 channel during ZnO-NP-induced neurovascular injury. Interestingly, the decreased abundance of pericyte LC3 puncta at 24 h after neurovascular injury in the TRPM2 KO mice coincided with reduced pericyte injury in brain microvessels. Taken together, these genetic and biochemical results support the idea that TRPM2-dependent autophagy plays a crucial role in mediating nitrosative stress-induced pericyte injury in mice. Moreover, our study also raises interesting questions regarding the interrelationships between pericyte-specific TRPM2 channels and other neurovascular components in the pathological process of neurovascular injury. However, this hypothesis remains to be tested in the future.
In summary, our study has established a previously unrecognized mechanism for nitrosative stress-induced TRPM2 protein tyrosine nitration and subsequent disturbance of autophagy, leading to pericyte injury. More importantly, our results provide clues for developing more effective neurovascular therapeutic strategies by targeting nitrosative stress. Ultimately, a full understanding of the molecular mechanisms of pericyte injury is crucial to the development of therapeutic strategies to treat neurovascular dysfunction-related pathologies.
Materials and Methods
Reagents
All chemicals were purchased from Sigma-Aldrich, unless otherwise specified.
Characterization of the size, morphology, and zeta potential for ZnO-NP
ZnO-NP (Sigma; 721077) suspension stock was prepared in phosphate-buffered saline (PBS) at 10 mg/ml and treated with ultrasound to make uniform distribution. The particles size and zeta potential distribution were determined with a Zetasizer (3000HS; Malvern Instruments) after the prepared dispersion performed 100-fold dilutions with distilled water. The particle size and surface morphologies were further characterized by using a transmission electronic microscope (JEM-1230; JEOL). The samples were placed on copper grids for viewing.
Culture of human brain pericytes
Human brain vascular pericytes (#1200) were purchased from ScienCell Research Laboratories and cultured in pericyte growth medium (contains growth factors, hormones, and proteins, ScienCell Research Laboratories; Catalog #1252) in a humidified atmosphere containing 5% CO2 at 37°C. Cells between passages 3 and 7 were used in this study.
Treatment of human pericytes with ZnO-NP
Human pericytes were plated in six-well plates at a density of 2 × 106 cells per well in a final volume of 2 ml. An appropriate aliquot of ZnO-NP was added to achieve a desired concentration. L-NAME (100 μM) or CQ (25 μM) was added into the well with ZnO-NP (10 μg/ml) at the same time, and the cells were incubated for 6 h. UA (200 μM) was added 3 h before addition of ZnO-NP (10 μg/ml). Cells that were treated with indicated concentrations of ZnO-NP for 6 h were used for Western blotting or immunocytochemistry, and cells that were not exposed to ZnO-NP were used as controls.
RNA interference
Human pericytes were cultured in six-well plates in growth medium. Transfection with double-stranded siRNA targeting TRPM2, ATG5, or a control scramble siRNA using Lipofectamine RNAiMAX (Invitrogen, 13778075) was conducted according to the manufacturer's instructions. siRNA of TRPM2 (sense: 5′-UGAUCCAGCAGAAACUGAGCGUGUU-3′ and anti-sense: 5′-AACACGCUCAGUUUCUGCUGGAUCA-3′) and control scramble siRNA were purchased from Thermo Fisher Scientific. siRNA of ATG5 (sc-41445) were purchased from Santa Cruz Biotechnology. The gene-silencing efficacy of siRNA is depicted in Supplementary Figure S15.
Mice
The TRPM2 KO mice were generated in the C57BL/6 background as detailed in our previous studies (53, 58). Both wild-type (C57BL/6 strain) and TRPM2 KO mice were housed under standard conditions with a 12/12 h light/dark cycle and free access to food and water. Eight- to 12-week-old male mice weighing 22–25 g were used in the study and were randomly assigned to each group. All animal use procedures were approved by the Committees at Zhejiang University and Leeds University for the Care and Use of Laboratory Animals. All the experiments were performed at room temperature or as specifically indicated.
CCK-8 assays
A Cell Counting Kit-8 (CCK-8) assay (Dojindo; CK04) was used to measure cell viability. Human pericytes were seeded in 96-well plates at a density of 2.5 × 103 cells per well and incubated overnight to allow for cells to settle down, before being transfected with siRNA-ATG5 or negative control for 48 h. Furthermore, the culture medium was changed to 100 μl fresh medium containing ZnO-NP (10 μg/ml) with or without Z-VAD-FMK (a caspase inhibitor, 10 μM) and incubated for a further 6 h. Cells in each well were incubated for 1 h with 110 μl fresh medium containing 10 μl CCK-8 reagent. Finally, the cell viability was determined by measuring the optical absorbance at 450 nm by using a multimode reader (Beckman Coulter; DTX880).
Injection of ZnO-NP in mice
The procedures were approved by the Committee on Animal Experiments at Zhejiang University, and they conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The male mice were injected with 0.1 ml of 0.5 mg/ml ZnO-NP via the tail vein. The control mice were injected with the same volume of vehicle. For immunohistochemical analysis, mice were anesthetized through diethyl ether inhalation in a chamber 24 h post-injection, and their brains were removed for analysis.
Injection of adenovirus vector carrying mRFP-GFP-LC3 in mice
Adenovirus vector carrying mRFP-GFP-LC3 construct (Hanbio Biotechnology) was injected into the bilateral ventricle over a 10-min duration by using a Hamilton microsyringe with the coordinates of 0.5 mm caudal to the bregma, 1 mm lateral to the midline, and 3 mm depth from the skull surface under the guidance of a stereotaxic instrument. Two weeks after the injection, the male mice were injected with 0.1 ml of 0.5 mg/ml ZnO-NP via the tail vein. The adenovirus batches used for experiments had comparable titers ranging from 1 × 1010 to 1 × 1011 integration units/ml. Virus suspensions were stored at −80°C until use, briefly centrifuged, and kept on ice immediately before the injection.
Western blotting
Western blotting was carried out by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as previously described (48). Briefly, cells were homogenized in the homogenizing buffer containing 50 mM Tris-HCl (pH 7.4), 0.5% Triton X-100, 4 mM EGTA, 10 mM EDTA, 1 mM Na3VO4, 30 mM sodium pyrophosphate, 50 mM NaF, 100 nM calyculin A, 50 μg/ml leupeptin, 25 μg/ml pepstatin A, 50 μg/ml trypsin inhibitor, and 1 mM dithiothreitol. For TRPM2 detection, membrane fractionation from cell lysates was performed as previously described (29). The following primary antibodies were used: nitrotyrosine (1:1000; Merck Millipore, 05-233), TRPM2 (1:1000; Abcam, ab11168), LC3 (1:5000; Sigma Aldrich, L7543), Phospho-SAPK/JNK(Thr183/Tyr185) (1:2000; Cell Signaling Technology, 4668), JNK (1:2000; Santa Cruz Biotechnology, sc-571), IRE1α (1:1000; Cell Signaling Technology, 3294), Phospho-PERK (Thr981) (1:1000; Santa Cruz Biotechnology, sc-32577), PERK (1:1000; Cell Signaling Technology, 5683), CHOP (1:500; Cell Signaling Technology, 5554), caspase-8 (1:1000, active form; Biovision, 3258), caveolin-1 (1:5000; Cell Signaling Technology, 32667), and β-actin (1:5000; Sigma Aldrich, A5441). Protein intensities were analyzed by Image J software (NIH), and they were normalized against the β-actin or caveolin-1 band in the matched experiments. For immunoprecipitation, cells were lysed in the lysis buffer (10 mM Tris-HCl, pH7.5, 0.5% NP-40, 150 mM NaCl, 1 mM EDTA) that was supplemented with a complete protease inhibitor cocktail (Roche). Cell lysates were incubated with 2 μg of indicated antibodies overnight at 4°C, followed by incubation at 4°C with protein A/G-agarose beads for 4 h. Immunoprecipitated samples were washed six times with lysis buffer, electrophoresed on SDS-PAGE, and subjected to Western blot analysis as described earlier. All full unedited blots are displayed in Supplementary Figure S16.
Transmission electron microscopy
The ultrastructure of human pericytes was determined by using TEM. Cells were washed twice with PBS and fixed in 2.5% glutaraldehyde in PBS (pH 7.4). The fixed cells were detached by gentle scraping, pelleted, fixed in 1% osmium tetroxide, and imaged by using a Philips Tecnai 10 transmission electron microscope (Philips).
Immunofluorescence confocal microscopy
Immunolocalization and changes in LC3 in pericytes were examined by confocal microscopy. Briefly, after indicated treatment, cells seeded on coverslips were washed three times in PBS and fixed in 4% formaldehyde. To quantify LC3-positive vesicles, cells were transfected with mRFP-GFP-LC3 plasmid or were fixed and stained with anti-LC3 antibody (Cell Signaling Technology; 2775). Images were acquired by using a Nikon A1R confocal microscope with a × 60 oil immersion lens at 1024 × 1024 pixel resolution. The average number of GFP- or mRFP-LC3 puncta per cell was determined by using an Imaris Imaging Software (Bitplane) (50). For time-lapse confocal imaging of live cells, after being transfected with mRFP-GFP-LC3 plasmid, human pericytes were cultured on glass-bottomed dishes overnight, and they were incubated with or without ZnO-NP (15 μg/ml). The changes in GFP and mRFP fluorescence intensity were captured by an Olympus IX-81 confocal microscope for 120 min that was equipped with a × 60 oil-immersion lens, a polychrome IV light source (Till Photonics), a 505 DCXR beam splitter, and a CCD camera (ANDOR iXon3).
To immunolabel the mouse brain sections, slices were incubated with antibodies against LC3 (1:200; Cell Signaling Technology, 2775), α-SMA (1:250; Abcam, ab7817), and laminin (1:200; Abcam, ab11575) overnight at 4°C. After washing, the slices were incubated with Alexa fluor 488-conjugated anti-rabbit IgG (Invitrogen; A-21206) and/or Alexa fluor 594-conjugated anti-mouse IgG (Invitrogen; A-21203) in Tris-NaCl-blocking buffer (1:400). NP3, a fluorescent switch-on probe, was used to examine the ONOO− formation, as previously reported (26). To minimize nonspecific staining, the experiments include a negative control using the IgG with no primary antibody (Supplementary Fig. S17). Immunofluorescence was captured by using a Zeiss LSM 510 confocal microscope. The number of the disconnected patchy parts was analyzed by Imaris Imaging Software (Bitplane). The 3D filled plots were processed by using ImageJ v1.45 with the accompanied “Interactive 3D Surface Plot” plug-in.
TUNEL assay
Apoptotic cell death was assessed by using TUNEL staining (30, 48). Images were recorded after counterstaining with DAPI, and pericytes were identified by phase images. Five random fields were examined on each coverslip, and the experiments were repeated four times. Apoptotic cell death was presented as the percentage of TUNEL+ cells in the total number of cells identified by DPAI staining. In addition, the caspase-8 activity assay kit (Abcam; ab39700) was also used to study the effect of siRNA-mediated knockdown of Atg5 or TRPM2 expression on cell death. The cell samples were collected under indicated conditions, and cells were suspended in 50 μl of ice-cold cell lysis buffer provided by the assay kit. They were then incubated with reaction buffer and IETD-pNA substrate after transfer of the supernatant. Finally, they were detected on a microplate reader (Beckman Coulter; DTX880) at an optical density of 405 nm as described in the instructions.
Propidium iodide flow cytometry analysis
Flow cytometric assays to evaluate cell death by propidium iodide (PI) (Sigma-Aldrich; P4170) staining were performed essentially as previously described, following the manufacturer's instructions. Briefly, human pericyte cells, after being treated with indicated conditions, were incubated in solutions containing 50 μg/ml PI, 100 μg/ml RNase A, and 0.2% Triton X-100 at room temperature for 30 min in the dark. At least 1 × 104 cells were analyzed for each sample by using an FACS-Calibur flow cytometer (BD Biosciences).
In vitro protein nitration of TRPM2 and mass spectrometry analysis
A 10 μg portion of recombinant TRPM2 was reacted with 100 μM ONOO− for 1 h in 50 mM phosphate buffer (pH 7.4) containing 0.1% Lauryl-β-D-maltoside (28, 46). The mixture was treated with 9% of ethanol at 40°C for 10 min and centrifuged at 75,600 g for 30 min. The ONOO−-treated TRPM2 (10 μg/lane) was subjected to SDS-PAGE and silver staining. The protein band with a size corresponding to that of the recombinant TRPM2 peptide was excised and further prepared for mass spectrometry analysis. Sequence information from the MS/MS data was processed by using the Mascot 2.0 active perl script with standard data processing parameters (28, 46). The database was searched with MASCOT 2.0 (Matrix Science). As compared with the protein untreated with ONOO−, a nitrated peptide was determined by a mass shift of +45 Da to the corresponding y and b ions.
Site-directed mutagenesis, plasmid constructs, and transfection
Polymerase chain reaction-based site-directed mutagenesis was performed as previously detailed (58). In brief, the full-length hTRPM2 sequence with C-terminal Glu-Glu tag in pcDNA3.1 vector was used as a DNA template (32). The forward and reverse primers used were 5′-AGG CGC ATC CCA CTC TCT GCG AAC CAC AAG ACC-3′ and 5′-GGT CTT GT GGT TCG CAG AGA GTG GGA TGC GCC3′, respectively. The mutation was confirmed by sequencing.
Human pericytes cultured in six-well plates in growth medium were transfected with plasmid encoding the wild-type and Y1485S mutant TRPM2 protein, or an empty plasmid as control using Lipofectamine 3000 (Invitrogen; L3000-015) (Supplementary Fig. S18A, B). The transfection medium was replaced with fresh growth medium 6 h later, and the cells were collected for experiments 2 days after the transfection. For the electrophysiological study shown in Supplementary Figure S8, the wild-type and Y1485S mutant TRPM2 channels were transiently expressed in HEK293 cells as described in our previous study (55).
Whole-cell patch clamp recording
Whole-cell patch-clamp recordings were performed by using Axonpatch 200B amplifier as previously described, from tetracycline-inducible HEK293 cells stably expressing the hTRPM2 channel, induced with 1 μg/ml tetracycline for 12–24 h, and also from HEK293 cells transiently transfected with the wild-type or Y1485S mutant hTRPM2 channel (Supplementary Fig. S18C, D). Before recording, cells were exposed to ZnO-NP (10 μg/ml) with or without 3-morpholinosydnonimine (SIN-1, 0.5 mM), L-NAME (100 μM), and UA (200 μM). Change of the extracellular solutions was performed by using an RSC-160 system (Biologic Science Instruments). The membrane potential was held at 0 mV. Voltage ramps with a 500-ms duration from −100 to 100 mV were applied every 5 s. Data were acquired at 10 kHz and filtered offline at 50 Hz. For analysis, the mean of the first three ramps before channel activation was used for leak subtraction of all subsequent current records.
Statistical analysis
Results are shown as mean ± SEM, where appropriate. For all Western blots, immunohistochemistry, cell viability assay, and patch clamp recording experiments unless otherwise specified later, one-way ANOVA followed by Tukey's post hoc test were used for comparisons among three or more groups. For TUNEL assay and other experiments, an unpaired two-tailed Student's t-test was used for comparisons between two groups. Statistical analyses were carried out by using GraphPad Prism 6 (GraphPad Software), and p < 0.05 was considered statistically significant.
Footnotes
Acknowledgments
This work was supported in part by National Natural Science Foundations of China (81120108023, 81573411, 81473202, 31471118); National Basic Research Program of China (2013CB910204, 2014CB910300); The Zhejiang Province Program for Cultivation of High-level Health Talents and New Century 151 Talent Project of Zhejiang Province; and Department of Education, Henan Province and University of Leeds-Zhejiang University Strategic Collaboration Partnership Programmer.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.