
Abstract
Aims:
Methionine sulfoxide reductase A (MsrA), which is abundantly localized in the mitochondria, reduces methionine-S-sulfoxide, scavenging reactive oxygen species (ROS). Cisplatin, an anticancer drug, accumulates at high levels in the mitochondria of renal cells, causing mitochondrial impairment that ultimately leads to nephrotoxicity. Here, we investigated the role of MsrA in cisplatin-induced mitochondrial damage and kidney cell death using MsrA gene-deleted (MsrA–/– ) mice.
Results:
Cisplatin injection resulted in increases of ROS production, methionine oxidation, and oxidative damage in the kidneys. This oxidative stress was greater in MsrA–/– mouse kidneys than in wild-type (MsrA+/+ ) mouse kidneys. MsrA gene deletion exacerbated cisplatin-induced reductions in the expression and activity of MsrA and MsrBs, and the expression of thioredoxin 1, glutathione peroxidase 1 and 4, mitochondrial superoxide dismutase, cystathionine-β-synthase, and cystathionine-γ-lyase. Cisplatin induced swelling, cristae loss, and fragmentation of mitochondria with increased lipid peroxidation, more so in MsrA–/– than in MsrA+/+ kidneys. The ratio of mitochondrial fission regulator (Fis1) to fusion regulator (Opa1) was higher in MsrA–/– than MsrA+/+ mice. MsrA deletion exacerbated cisplatin-induced increases in Bax to Bcl-2 ratio, cleaved caspase-3 level, and apoptosis, whereas MsrA overexpression attenuated cisplatin-induced oxidative stress and apoptosis.
Innovation:
MsrA gene deletion in mice exacerbates cisplatin-induced renal injury through increases of mitochondrial susceptibility, whereas MsrA overexpression protects cells against cisplatin.
Conclusion:
This study demonstrates that MsrA protects kidney cells against cisplatin-induced methionine oxidation, oxidative stress, mitochondrial damage, and apoptosis, suggesting that MsrA could be a useful target protein for the treatment of cisplatin-induced nephrotoxicity. Antioxid. Redox Signal. 27, 727–741.
Introduction
M
The present study shows that cisplatin induces methionine oxidation, oxidative stress, mitochondrial damage, apoptosis, and necrosis, and that MsrA gene deletion accelerates these cisplatin-induced damages. This indicates that MsrA protects kidney against cisplatin-induced acute kidney injury. The understanding of a pivotal role for MsrA in cisplatin-induced kidney injury will be helpful to develop novel therapeutic interventions aimed at minimizing cisplatin-induced nephrotoxicity.
Cisplatin (cis-diamminedichloridoplatinum) is a widely used chemotherapeutic agent for the treatment of solid malignant tumors such as lung, testis, and ovarian cancers (3). However, the utility of cisplatin is often limited because of its side effects, including nephrotoxicity (34). After absorption into the body, cisplatin is excreted via the urine; therefore, renal tubular epithelial cells are exposed to high concentrations of cisplatin (fivefolds higher than in blood) and thus injured, resulting in AKI (42). It has been reported that approximately one-third of patients treated with cisplatin develop AKI (33). Growing evidence has demonstrated that cisplatin accumulates at a high level in the mitochondria; this results in the impairment of mitochondrial redox balance, leading to mitochondrial dysfunction and damage, thereby resulting in renal tubule cell death and dysfunction (9, 32, 42, 43). Several studies have demonstrated that mitochondrial DNA is more susceptible to cisplatin-induced damage than nuclear DNA (15, 34, 49).
Therefore, we investigated the role of MsrA in the pathogenesis of cisplatin-induced AKI. In the present study, we report that MsrA gene deletion increases methionine oxidation and mitochondrial susceptibility to cisplatin, leading to enhanced ROS production, methionine oxidation, oxidative stress, mitochondrial damage, and apoptosis.
Results
MsrA gene deletion exacerbates cisplatin-induced renal morphological and functional impairment
First, we determined the dose-dependent cisplatin-induced nephrotoxicity in MsrA wild-type (MsrA+/+ ) mice and then compared the difference in the nephrotoxicity of cisplatin between MsrA+/+ and MsrA gene-deleted (MsrA–/– ) mice. Cisplatin (10, 15, or 20 mg/kg body weight [BW]) significantly increased plasma creatinine (PCr) and blood urea nitrogen (BUN) levels, and mortality in a dose-dependent manner in the MsrA+/+ mice (Fig. 1A–C). Increases of PCr and BUN concentrations in the MsrA–/– mice were greater than those in the MsrA+/+ mice 3 days after cisplatin (10 and 20 mg/kg BW) injection (Fig. 1D, E). The elevated PCr concentrations began to decrease 5 days after cisplatin (10 mg/kg BW) injection in the MsrA+/+ mice, but continued to increase in the MsrA–/– mice (Fig. 1D). BUN levels continued to increase 5 days after cisplatin (10 mg/kg BW) injection in the MsrA–/– mice, whereas no further increase was observed in the MsrA+/+ mice (Fig. 1E), indicating that MsrA gene deletion worsens cisplatin toxicity. Five days after 10 mg/kg BW of cisplatin injection, all mice had survived with no differences between MsrA+/+ and MsrA–/– mice. However, about 65%, 100%, and 100% of mice were dead 3, 4, and 5 days after 20 mg/kg BW of cisplatin injection, respectively (Fig. 1F). These mortalities did not differ between MsrA+/+ and MsrA–/– mice (Fig. 1F). This indicates that cisplatin nephrotoxicity is dose and time dependent and MsrA protects, in part, the kidneys against cisplatin-induced nephrotoxicity.

Cisplatin injection caused loss of brush borders of tubule cells, and disruption, congestion, dilation, and flattening of tubules to the kidneys of both MsrA+/+ and MsrA–/– mice (Fig. 1G). Tubular cell damage was predominantly found in the proximal tubules relative to the other tubules (Fig. 1G). The tubular cell damage was greater in MsrA–/– than in MsrA+/+ mice (Fig. 1G, H). Taken together, these data indicate that MsrA gene deficiency augments cisplatin-induced kidney injury.
MsrA gene deletion augments ROS production and oxidative stress following cisplatin injection
To determine whether the aggravation of cisplatin-induced kidney injury by MsrA gene deletion is associated with increased oxidative stress, first, we evaluated redox status in the kidneys. Cisplatin significantly increased the formation of superoxide anion and production of hydrogen peroxide (H2O2) in the kidneys of both MsrA+/+ and MsrA–/– mice (Fig. 2A, B). These increases were higher in MsrA–/– mice than in MsrA+/+ mice (Fig. 2A, B). However, the levels did not differ between the vehicle-injected kidneys (Fig. 2A, B). Furthermore, the ratio of oxidized GSH (GSSG) to total GSH increased significantly in the kidneys of both MsrA−/− and MsrA+/+ mice following cisplatin injection with the increase greater in MsrA−/− than MsrA+/+ mice (Fig. 2C). No change in total GSH levels was found in either kidneys following cisplatin injection (Fig. 2D). Total GSH levels were also similar in vehicle-injected MsrA−/− and MsrA+/+ kidneys (Fig. 2D). Levels of 4-hydroxynonenal (HNE, a product of lipid peroxidation) were found to increase in the kidneys of both MsrA+/+ and MsrA–/– mice following the injection of cisplatin (Fig. 2E, F). The cisplatin-induced increase in HNE was significantly higher in MsrA–/– mice than in MsrA+/+ mice (Fig. 2E, F). In addition, the number of cells stained by the anti-8-hydroxy-2′-deoxyguanosine (8-OHdG) antibody, a marker of oxidative damage in DNA (46), was significantly increased in the cisplatin-injected kidneys relative to the vehicle-injected kidneys (Fig. 2G, H). The number of 8-OHdG-positive cells was greater in MsrA–/– mice than in MsrA+/+ mice (Fig. 2G, H). These results indicate that MsrA gene deletion exacerbates cisplatin-induced oxidative stress.

Since MsrA reverses oxidized methionine residues in proteins and therefore prevents the loss of protein functions due to oxidative modification (47), we further determined whether cisplatin-induced nephrotoxicity is associated with oxidation of methionine. Cisplatin increased the level of methionine sulfoxide (MetSO), an oxidized methionine, in the kidney (Fig. 2I, J). The elevation of MetSO level was greater in MsrA–/–
than MsrA+/+
mice (Fig. 2I, J), indicating that MsrA may reduce the loss of protein functions due to oxidative modification. Furthermore, cisplatin led to the reductions in the expressions of antioxidant enzymes; thioredoxin 1 (Trx1), glutathione peroxidase 1 (GPx1) and 4 (GPx4), and manganese superoxide dismutase (MnSOD) (Fig. 3A–E and Supplementary Fig. S1; Supplementary Data are available online at

MsrA gene deletion exacerbates mitochondrial injury in the kidney following cisplatin injection
MsrA is abundantly localized in the mitochondrion (21), which is a major organelle in the production of ROS, as well as highly vulnerable to the effects of these species (16). Therefore, we determined whether MsrA affects cisplatin-induced mitochondrial oxidative stress and mitochondrial damage. Cisplatin increased HNE level in the mitochondrial fractions in both MsrA–/– and MsrA+/+ mice and this increase was greater in MsrA–/– than MsrA+/+ mice (Fig. 4A–C and Supplementary Fig. S1), indicating that cisplatin induces oxidative stress in the mitochondria and MsrA gene deletion exacerbates the cisplatin-induced mitochondrial oxidative damage. As mitochondrial morphology was examined by transmission electron microscopy (TEM), the TEM data showed swelling of mitochondria, with a loss of cristae, and fragmentation of mitochondria in the proximal tubular cells 1 and 3 days after cisplatin injection (Fig. 4D). The aspect ratio (the long axis of mitochondria/short axis of mitochondria, an index of mitochondrial fragmentation) in the kidney cells decreased after cisplatin injection in a time-dependent manner (Fig. 4E). The decrease in mitochondrial aspect ratio was greater in MsrA−/− mice than MsrA+/+ mice (Fig. 4E). Kidney proximal tubule cells in vehicle-treated mice did not show any significant mitochondrial damage (Fig. 4D). These data indicate that mitochondrial damage was more severe in MsrA−/− mice than in MsrA+/+ mice.
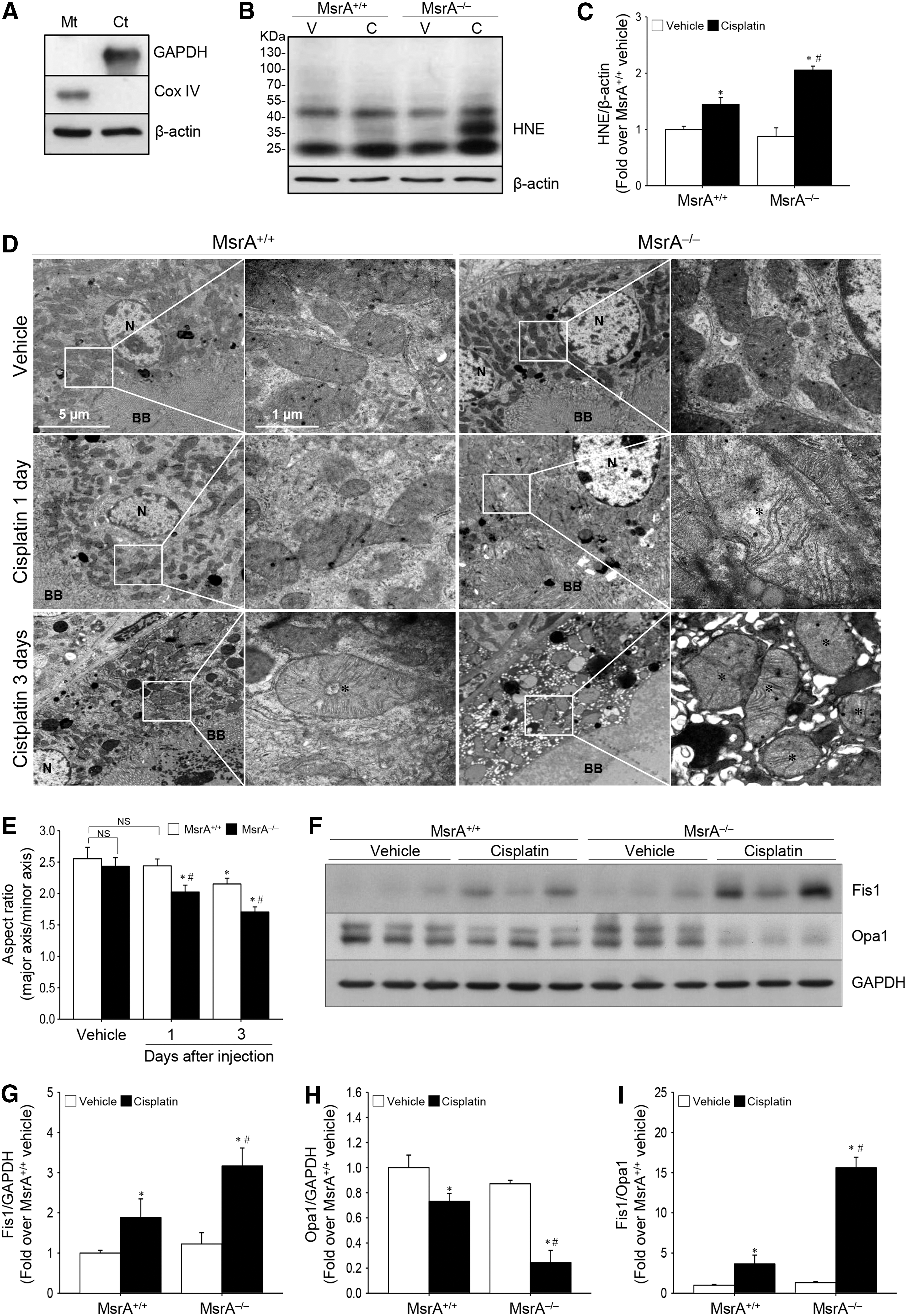
Under normal conditions, the fusion and fission of mitochondria are dynamically balanced (12); an imbalance in these processes, which reflects an impairment in mitochondrial function, leads to cell damage (8, 49). After cisplatin injection, the level of fission 1 protein (Fis1), a mitochondrial fission mediator, increased, whereas those of Opa1, a regulator of mitochondrial fusion, decreased (Fig. 4F–H and Supplementary Fig. S1), leading to the increase in the ratio of Fis1 to Opa1. This ratio increase was greater in MsrA−/− than in MsrA+/+ kidneys (Fig. 4I). There was no significant difference in the ratio of Fis1 to Opa1 between vehicle-injected MsrA−/− and MsrA+/+ kidneys (Fig. 4I), leading to increased fragmentation of mitochondria (Fig. 4D). Taken together, these data indicate that MsrA gene deletion aggravates oxidative stress, impairments of mitochondrial dynamics, and mitochondrial damage after cisplatin injection.
MsrA deficiency accelerates cisplatin-induced apoptosis
Since oxidative stress and mitochondrial injury lead to apoptosis (49), we determined whether MsrA involves apoptosis signaling pathways and apoptosis. Cisplatin caused the increase of the proapoptotic protein Bax expression (Fig. 5A, B, and Supplementary Fig. S1), while simultaneously causing the decrease of the antiapoptotic protein Bcl-2 expression (Fig. 5A, C, and Supplementary Fig. S1). These changes were greater in MsrA–/– than in MsrA+/+ kidneys, resulting in a higher Bax to Bcl-2 ratio in the MsrA–/– mice (Fig. 5D). Increased cleavage of caspase-3, a downstream target of increase of Bax to Bcl-2 ratio, was observed in the cisplatin-injected mouse kidneys and this increase was greater in the MsrA−/− mouse kidneys (Fig. 5A, E, and Supplementary Fig. S1). After cisplatin injection, caspase-3 cleavage was predominant in the proximal tubule cells and the cleavage was greater in the MsrA−/− than MsrA+/+ kidneys (Fig. 5F), indicating that MsrA attenuates the activation of apoptosis signal pathway after cisplatin injection. Indeed, many terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)-positive apoptotic cells were observed in the cisplatin-injected mouse kidneys, more so in MsrA–/– than in MsrA+/+ kidneys (Fig. 5G, H). Almost no TUNEL-positive cells were observed in vehicle-injected MsrA+/+ or MsrA−/− kidneys (Fig. 5G, H). These results indicate that MsrA gene deletion augments the cisplatin-induced apoptosis in kidneys.

Cisplatin reduces MsrA and MsrB activity and expression, and MsrA gene deletion augments cisplatin-induced disturbance of the transsulfuration pathway
To investigate whether cisplatin affects the function of MsrA and MsrBs in the kidney, we determined the activity and expression of MsrA and MsrB. The activity of MsrA was not detected in the kidneys of MsrA–/– mice (Fig. 6A). In the MsrA+/+ mice, cisplatin significantly decreased MsrA and MsrB activities (64% of the MsrA activity of vehicle-injected MsrA+/+ kidneys; 60% and 65% of the activities of vehicle-injected MsrA+/+ and MsrA–/– kidneys, respectively) (Fig. 6A, B). However, there was no difference in MsrB activity between cisplatin-injected MsrA+/+ and MsrA–/– kidneys (Fig. 6B), indicating that cisplatin reduces Msr activities. Consistent with the reductions in Msr activity, MsrA expression in the MsrA+/+ mouse kidneys dramatically decreased after cisplatin injection (Fig. 6C, D, and Supplementary Fig. S2). In addition, MsrB1 and MsrB2 expression also significantly decreased in both MsrA+/+ and MsrA–/– mouse kidneys after cisplatin injection (Fig. 6C, E, F, and Supplementary Fig. S1). However, there were no significant differences between MsrA+/+ and MsrA–/– mice after cisplatin injection (Fig. 6E–F). The levels of MsrB1 and MsrB2 after the vehicle injection were not different between MsrA+/+ and MsrA–/– mice, indicating that MsrA gene deletion did not induce significant changes of MsrB1 and MsrB2 expression (Fig. 6E–F). These results indicate that cisplatin reduces the expression and activity of Msrs in the kidney.
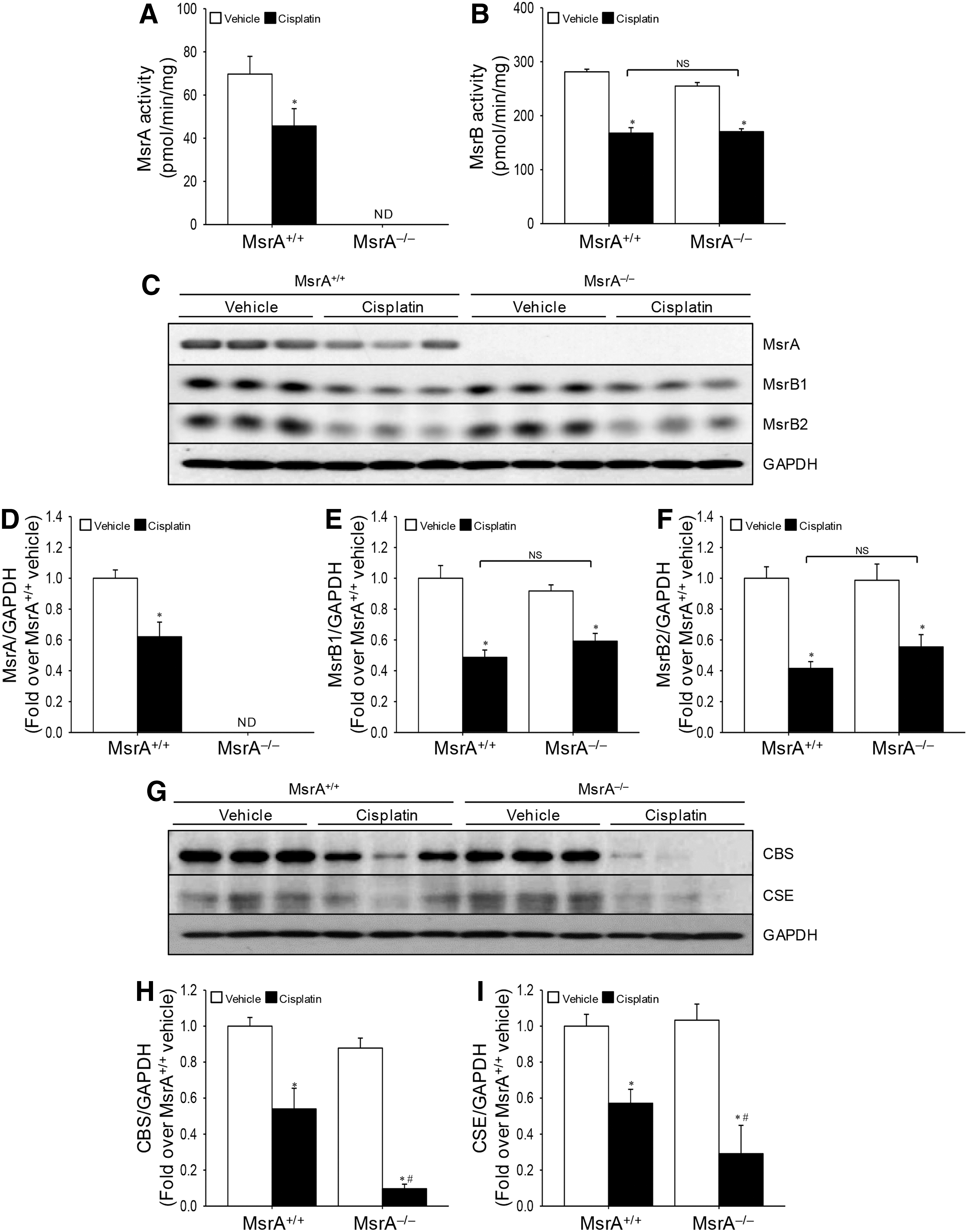
Methionine is metabolized to cysteine, a component of GSH, with the simultaneous production of hydrogen sulfide, an antioxidant gaseous molecule, by cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE) (23). Cisplatin decreased the expressions of CBS and CSE in both MsrA+/+ and MsrA−/− mouse kidneys (Fig. 6G–I and Supplementary Fig. S2). The cisplatin-induced decrease in CBS and CSE expression was greater in MsrA−/− than in MsrA+/+ mice (Fig. 6G–I and Supplementary Fig. S2). These results suggest that MsrA deficiency exacerbates cisplatin-induced disturbance of methionine transsulfuration pathway.
MsrA overexpression protects kidney proximal tubular cells against cisplatin
To investigate whether MsrA protects the kidney tubular epithelial cells against cisplatin, we overexpressed MsrA in the LLC-PK1 cell, a porcine kidney proximal tubule epithelial cell line cell, with either MsrA- (MsrA-Ad) or CMV-adenovirus (Con-Ad) and then evaluated cisplatin toxicity. MsrA-Ad infection increased MsrA expression in the LLC-PK1 cells in a multiplicity of infection (MOI)-dependent manner (Fig. 7A and Supplementary Fig. S3). Cisplatin treatment significantly decreased MsrA expression (Fig. 7B, C, and Supplementary Fig. S3) and increased HNE level (Fig. 7D, E) and the cisplatin-induced elevation of HNE was milder in the MsrA-overexpressed cells than in the control cells (Fig. 7D, E).

MsrA overexpression attenuated increase of Bax expression (Fig. 7F, G, and Supplementary Fig. S3) and decrease of Bcl-2 in the cells after cisplatin treatment (Fig. 7F, H, and Supplementary Fig. S3), leading to decrease of Bax to Bcl-2 ratio (Fig. 7I). Caspase-3 was cleaved after cisplatin treatment in both MsrA-overexpressed cells and control cells and the cleavage was less in the MsrA-overexpressed cells than the control cells (Fig. 7F, J, and Supplementary Fig. S3). These results indicate that MsrA upregulation protects renal proximal tubule cells against apoptosis.
Discussion
The present study shows for the first time that MsrA gene deletion exacerbates cisplatin-induced ROS production, methionine oxidation, and oxidative damage, whereas MsrA overexpression protects proximal tubular epithelial cells against cisplatin. Furthermore, MsrA gene deletion enhances mitochondrial vulnerability to cisplatin, leading to consequently induce kidney cell death. These results suggest that an understanding of the pivotal role of MsrA in cisplatin-induced kidney injury is crucial to the development of novel therapeutic interventions to minimize cisplatin-induced nephrotoxicity.
ROS induces oxidative modification of proteins, which usually cause functional impairments. In this study, we found that cisplatin induced the oxidation of methionine, one of the most readily oxidized residues in proteins by ROS (29), and this oxidation was exacerbated by MsrA gene deletion. It has been reported that MsrA protects cells against oxidative stress by repairing oxidized methionine to reduced methionine, thus protecting protein functions from inactivation (20, 47, 50). This suggests that MsrA may protect protein modification against cisplatin-induced oxidative stress.
Recent studies have demonstrated that Msrs directly scavenge ROS through cyclic oxidation of methionine (22, 29, 45, 47). In this study, cisplatin significantly increased ROS production and oxidative damage to the renal tubule cells. The ROS production and oxidative damage were exacerbated by MsrA gene deletion; in contrast, overexpression of MsrA reduced cisplatin-induced oxidative stress. Furthermore, cisplatin decreased MsrA, MsrB1, and MsrB2, and antioxidant enzyme (Trx1, GPx1, GPx4, and MnSOD) expression in the kidneys, and MsrA gene deletion exacerbated these reductions of proteins. These decreases of antioxidant proteins, including Msrs, after cisplatin injection may be due to tissue damages or functional loss of methionine containing proteins, including transcription factors. In a previous study, we reported that MsrA gene deletion exacerbated I/R-induced decreases of antioxidant enzymes, including catalase (23). Therefore, we speculate that increased oxidative stress in the MsrA gene-deleted mice may be due to both dysfunctions of methionine-containing proteins by oxidation and reduction of ROS scavenging effect. Since GPx4 acts as an antioxidant enzyme to reduce ferroptosis, which is a programmed cell death due to oxidative stress (48), cisplatin nephrotoxicity may be involved in the process of ferroptosis.
The majority of cisplatin is filtered via the glomerulus and excreted in the urine (34). Therefore, kidneys are likely exposed to high concentrations of cisplatin, even when the blood contains nontoxic levels of this compound (15, 27). High amounts of cisplatin accumulate primarily in the kidney proximal tubular epithelial cells and induce kidney tubule cell injury through complex cellular and molecular mechanisms (17). The mitochondrion is recognized as a major site for cisplatin accumulation in kidney tubular epithelial cells (39, 41). Studies have demonstrated that cisplatin induces mitochondrial damage (8, 15, 49); this is most severe in the tubule, particularly in proximal tubule cells, depending on the density of mitochondria in these cells (6). In this study, we found that cisplatin caused lipid peroxidation in the mitochondria and this lipid peroxidation was greater in the MsrA gene-deleted mice than MsrA wild-type mice. This result suggests that MsrA protects mitochondria against cisplatin-induced oxidative stress. Hansel et al. reported that MsrA in the mitochondria contributes to removal of mitochondrial ROS via cyclic oxidation and reduction of methionine residues (18). Superoxide anion is rapidly converted to H2O2 by SOD and then removed by GPx (4). Reduced GSH, a major antioxidant molecule, is required for the scavenging process of H2O2 by GPx in the mitochondria. During this action, reduced GSH is oxidized to GSSG. GSSG is reduced again by glutathione reductase (30). In this study, cisplatin increased the ratio of GSSG to total GSH and this increase, although greater in the MsrA gene-deleted mice, did not change the total GSH amount. It has been reported that cisplatin inhibits cystine/glutamate antiporter (7), leading to reduced uptake of cystine, a source in GSH biosynthesis, and GSH synthesis in cancer cell (31, 40). Therefore, our results suggest that the increased oxidative stress after cisplatin injection may be associated with decrease of GSH-GPx antioxidant system rather than decrease of GSH biosynthesis.
Mitochondria dynamically join and divide by the processes of fusion and fission under normal conditions (5). The disruption of mitochondrial dynamics is implicated in the pathogenesis of various diseases, and aberrant mitochondrial fission is associated with increased ROS production (5). In the present study, we found for the first time that cisplatin disturbs the balance between fission and fusion and is associated with an increased ratio of Fis1 to Opa1, consequently increasing the proportion of fragmented mitochondria of short length. The cisplatin-induced increase in Fis1 to Opa1 ratio was higher in the kidneys of MsrA−/− mice, with a larger proportion of short-length mitochondria relative to MsrA+/+ kidneys. The data indicate that cisplatin induces mitochondrial dysfunction, and that MsrA may be involved in the protection of mitochondrial dynamics against cisplatin. Moskovitz et al. reported that MsrA deficiency induces mitochondrial dysfunction, which may be attributed to a decline in mitochondrial respiration and increased oxidation of methionine residues in mitochondrial proteins (36). The mitochondrial proteome is relatively rich in methionine residues when compared with the cytosolic proteome (2), suggesting that methionine-containing proteins in mitochondria are highly susceptible to oxidative damage. In this study, TEM analysis revealed that cisplatin induces swelling, loss of cristae, and fragmentation of the mitochondria in the proximal tubule cells with greater mitochondrial damage in the MsrA−/− kidneys than in the MsrA+/+ kidneys. These results suggest that the increased cisplatin-induced toxicity in MsrA gene-deleted mice may be attributed to the reduction of the protective effect of MsrA in mitochondria.
Mitochondrial dysfunction and injury and ROS/oxidative stress initiate apoptosis (8, 15, 28). It is known that mitochondrial-mediated intrinsic apoptosis represents one of the major pathways involved in cisplatin-induced nephrotoxicity. The key events in the intrinsic pathway include Bax activation and Bcl-2 inactivation in mitochondria, consequently leading to the release of apoptotic factors, such as cytochrome c, and activation of caspase signals (12). Wei et al. reported that Bax knockout preserves renal function in mice, following cisplatin treatment, by attenuating the cisplatin-mediated release of cytochrome c (35, 46a). Consistent with these findings, in this study, cisplatin increased Bax levels and decreased Bcl-2 levels in the kidneys, leading to the increase in the ratio of Bax to Bcl-2 and the increase in the proportion of TUNEL-positive cells. These changes were more profound in MsrA−/− kidneys than in MsrA+/+ kidneys. However, the levels of Bax and Bcl-2 were not different between vehicle-injected control mice. In this study, MsrA overexpression in the cultured tubular epithelial cells protected cells against apoptosis, which supports the suggestion that MsrA may protect cells against cisplatin-induced apoptosis. In both in vivo and in vitro studies, the overexpression and deletion of MsrA alone did not change the Bcl-2 and Bax expression, although MsrA gene regulation did affect Bcl-2 and Bax expression in the cisplatin treatment. This indicates that changes in the protein expression in MsrA gene-regulated cells may be due to oxidative stress rather than direct regulation of expression.
In a previous study, we found that MsrA gene deletion reduces blood levels of homocysteine, which is converted to cysteine by the consecutive actions of CBS and CSE, along with production of hydrogen sulfide (H2S), an antioxidant gaseous molecule (10, 23, 26). In the present study, cisplatin significantly reduced CBS and CSE expressions; this reduction was more profound in MsrA−/− kidneys than in MsrA+/+ kidneys. These findings suggest that increased cisplatin-induced nephrotoxicity in MsrA-deficient mice may involve reduced production of H2S by impairment of transsulfuration pathways. It has been reported that H2S supplementation reduces cisplatin-induced nephrotoxicity (1, 13).
Taken together, our data demonstrate that the MsrA enzyme plays a critical role in protecting mitochondria against cisplatin-induced oxidative stress and reducing apoptosis, suggesting that MsrA represents a potentially useful target for the treatment of cisplatin-induced nephrotoxicity.
Materials and Methods
Animal preparation
MsrA gene-deficient (MsrA–/–
) and wild-type (MsrA+/+
) C57BL/6 mice were used (24). Eight-week-old male mice, weighing 20 to 25 g, were used for all experiments. The animal study was approved by the Institutional Animal Care and Use Committee of Kyungpook National University and of Yeungnam University. Mice were allowed ad libitum access to water and standard mouse chow. Mice were intraperitoneally injected with cisplatin (10, 15, or 20 mg/kg BW; Sigma, St. Louis, MO) or 0.9% saline (vehicle). Kidneys were harvested at 3 or 5 days after injection, followed by either snap freezing in liquid nitrogen for biochemical analyses or perfusion fixing with PLP solution (4% paraformaldehyde, 75 mM
MsrA and MsrB activity assay
Two hundred micrograms of crude protein was mixed with the reaction solution containing 50 mM sodium phosphate (pH 7.5), 50 mM NaCl, 20 mM dithiothreitol, and 200 μM dabsylated methionine-S-sulfoxide (for MsrA) or methionine-R-sulfoxide (for MsrB). The mixture (100 μl) was incubated at 37°C for 30 min, after which the product of the reaction, dabsyl-Met, was analyzed via HPLC, as previously described (23).
Measurement of renal function
Seventy microliters of blood was obtained from the retro-ocular vein plexus before injection and 3 or 5 days after injection. Concentrations of PCr and BUN were measured using a Vitros250 (Johnson & Johnson, New Brunswick, NJ).
Histology
PLP-fixed kidneys were embedded in paraffin and cut into 3-μm sections. Sections were stained with periodic acid Schiff stain, according to the manufacturer's instructions. Images were captured using a Leica microscope (Leica DM2500, Wetzlar, Germany) and i-Solution software (IMT, Vancouver, Canada). Kidney damage was scored using the following criteria: 0, no damage; 1, mild damage with rounded epithelial cells and dilated tubular lumen; 2, moderate damage with flattened epithelial cells, dilated lumen, and congestion of lumen; and 3, severe damage with flat epithelial cells lacking nuclear staining and congestion of the lumen. Tubular damage was scored in a blind manner. At least 50 tubules were analyzed per kidney.
Western blot analysis
Western blot was performed using antibodies against MsrA (23), MsrB1 (23), MsB2 (24), 4-hydroxynonenal (HNE; Abcam, Cambridge, MA), methionine sulfoxide (MetSO; Cayman, Ann Arbor, MI), thioredoxin 1 (Trx1; Santa Cruz Biotechnology, Santa Cruz, CA), glutathione peroxidase 1 (GPx1; Santa Cruz Biotechnology, Santa Cruz, CA), glutathione peroxidase 4 (GPx4; Abcam, Cambridge, MA), manganese superoxide dismutase (MnSOD; Calbiochem, San Diego, CA), Cox IX (Abcam, Cambridge, MA), cystathionine-β-synthase (CBS; Santa Cruz Biotechnology, Santa Cruz, CA), cystathionine-γ-lyase (CSE; Santa Cruz Biotechnology, Santa Cruz, CA), Fis1 (Sigma, St. Louis, MO), Opa1 (BD Bioscience, Franklin Lakes, NJ), cleaved caspase-3 (Merck Millipore, Darmstadt, Germany), Bcl-2 (Cell Signaling Technology, Danvers, MA), Bax (Cell Signaling Technology, Danvers, MA), β-actin (Sigma, St. Louis, MO), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; NOVUS, Littleton, CO).
Immunofluorescence staining
Immunofluorescence staining was performed using an anti-8-hydroxy-2′-deoxyguanosine (8-OHdG; Abcam, Cambridge, MA) antibody, and 4,6 diamidino-2-phenylindole (DAPI) was used to visualize nuclei. Pictures were taken randomly in the outer medulla. Images were captured using a Leica microscope (Leica DM2500, Wetzlar, Germany).
Immunohistochemical staining
Immunohistochemical staining was performed using anti-cleaved caspase-3 (Merck Millipore, Darmstadt, Germany) antibody. Pictures were taken randomly in the outer medulla. Images were captured using a Leica microscope (Leica DM2500, Wetzlar, Germany).
Measurement of superoxide anion levels in the kidney
Superoxide anion levels in kidney homogenates were determined using dihydroethidium (DHE; Sigma, St. Louis, MO). DHE measurement may contain some artifacts occurred during the conversion of DHE into 2-hydroxyethidium (a red fluorescent product) (19, 51). Briefly, 20 μl of protein lysate was placed in 96-well plates, to which 200 μl of 10 μM DHE was added. Fluorescence intensity was measured using excitation/emission filters of 544/612 nm. Control values were determined using DHE alone. Data are represented as relative fluorescence intensity values.
Measurement of hydrogen peroxide levels in the kidney
A ferric-sensitive dye, xylenol orange (Sigma, St. Louis, MO), was used to determine hydrogen peroxide (H2O2) levels in tissue samples, as previously described (23).
Measurement of GSH levels in the kidney
The ratio of oxidized GSH (GSSG) to total GSH (reduced GSH+GSSG) was measured using an enzymatic recycling method, as previously described (23). The amount of total GSH was determined by analyzing the formation of 5-thio-2-nitrobenzoic acid (TBA) from 5,5-dithiobis (2-nitrobenzoic acid) (DTNB). GSSG was measured by adding 2-vinylpyridine, which inhibits TBA formation when DTNB and glutathione react. Total GSH and GSSG levels were defined as the change in optical density at 412 nm for 1 min at 37°C.
Preparation of cytosolic and mitochondrial fractions
Cytosolic and mitochondrial fractions were prepared as described previously (14). Effective isolation of those fractions was confirmed by Western blot analysis, using anti-Cox IV and GAPDH antibodies for the mitochondrial and cytosolic fractions, respectively (11).
TUNEL assay
A TUNEL assay was performed using an in situ cell death detection kit (Fluorescein; Roche, Basel, Switzerland), according to the manufacturer's instructions. Briefly, 3-μm kidney sections were deparaffinized and rehydrated. Subsequently, sections were incubated with the TUNEL reagent mixture for 30 min and washed three times with phosphate-buffered saline, for 5 min each. Nuclei were stained with DAPI for 1 min and images were observed using a Leica microscope (Leica DM2500; Wetzlar, Germany). DAPI- and TUNEL-positive images were collected separately and then merged using i-Solution software (IMT, Vancouver, Canada). TUNEL-positive cells were counted in 10 fields (0.1 mm2 per field) per kidney (n = 6).
Transmission electron microscopy
One and 3 days after injection of cisplatin or 0.9% saline (vehicle), kidneys were harvested and fixed with 2.5% glutaraldehyde at 4°C for 12 h. The kidney samples were cut into 1-mm3 sections, washed in 0.1 M phosphate buffer, and postfixed in aqueous 2% osmium tetroxide for 90 min. After three washes with the phosphate buffer, the samples were dehydrated through a graded series of 50–100% ethanol and 100% propylene oxide, and infiltrated in 1:1, 1:2, and 1:3 mixtures of propylene oxide:Epon Resin 828 (Polysciences, Inc., Warrington, PA) for 1 h, respectively. The samples were incubated in 100% Epon Resin 828 for 8 h, embedded in molds, and cured at 35°C and 45°C for 12 h, followed by additional hardening at 60°C for 2 days. Ultrathin (60 nm) sections were double stained with 2% uranyl acetate and 1% lead citrate. Sections were visualized using a transmission electron microscope (H-7000; Hitachi, Japan) at 75 kV. Electron micrographs of mitochondria were captured from proximal tubule cells in the cortex. The mitochondrial aspect ratio (major axis/minor axis, 30 mitochondria per cell) was determined using i-Solution software (IMT, Vancouver, Canada).
Cell culture
The LLC-PK1 cells, porcine kidney proximal tubule epithelial cell line cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD). The cells were maintained in high-glucose Dulbecco's modified Eagle's medium (Corning, Tewksbury, MA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY) and 100 U/ml penicillin–streptomycin antibiotics (Welgene, Inc., Gyeongsan, Gyenongsangbuk-do, South Korea) at 37°C with 5% CO2.
Overexpression of MsrA in the LLC-PK1 cells
Recombinant MsrA-adenovirus (MsrA-Ad) and CMV-adenovirus (control-Ad) were prepared as previously described (25). The LLC-PK1 cells (4 × 105 cells) were seeded in the 6-well culture dish. The next day cells were infected with recombinant adenoviruses for 24 h. After infection, the cells were incubated with 50 μM of cisplatin for 16 h.
Statistical analysis
Results are expressed as mean ± SE. Statistical differences among groups were calculated using paired Student's t-tests and one-way ANOVA. Differences were regarded as statistically significant when they had p-values <0.05.
Footnotes
Acknowledgment
This study was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (NRF-2014R1A2A1A11049549, 2015R1A2A1A15052400, and 2016R1D1A1A02936942).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.