
Abstract
Aim:
Myocardial infarction (MI) is one of the leading causes of death in elderly people. Expanding the knowledge of the molecular mechanisms underlying MI is of profound importance to developing a cure for MI. The CUGBP- and ETR-3-like factor (CELF) proteins, a family of RNA-binding proteins, play key roles in RNA metabolism. To determine the functions and molecular mechanisms of CELF proteins in MI, an animal model of acute myocardial infarction (AMI) was used in our study.
Results:
We found that the CUG triplet repeat RNA-binding protein 1 (CUGBP1)/CELF1 expression levels were decreased in AMI-injured hearts, and further studies showed that two highly conserved adenylate-uridylate-rich (AU-rich) elements in the 3′UTR of CUGBP1 were responsible for the decreased CUGBP1 expression. Upon AMI, human antigen R (HuR) was relocated to the cytoplasm from the nucleus and interacted with these AU-rich elements to affect the expression of CUGBP1. Reintroduction of CUGBP1 via gene delivery by recombinant adenovirus improved cardiac function in AMI mice. Our studies also indicated that CUGBP1 protected cardiomyocytes from ischemia-induced injury through the promotion of angiogenesis and inhibition of apoptosis by regulating the vascular endothelial growth factor-A gene.
Innovation and Conclusion:
Our studies indicate a role for CUGBP1 in cardiac disease and reveal a novel MI post-transcriptional gene regulatory mechanism. The reconstitution of CUGBP1 could be developed as a potential therapeutic option for the management of MI. Antioxid. Redox Signal. 27, 1013–1026.
Introduction
M
Similar to transcription, the post-transcriptional regulation of RNA metabolism is an important mechanism of gene regulation. A number of proteins with cardiovascular significance have been shown to be subject to regulation at the RNA level (40). RNA-binding proteins (RBPs) participate in the post-transcriptional processing of RNA by regulating their splicing, polyadenylation, stability, transport, and translation. Hu proteins are some of the most widely studied RNA regulators (17, 49). Human antigen R (HuR) is a ubiquitously expressed member of the Hu proteins. HuR is primarily localized to the nucleus and can shuttle between the nucleus and the cytoplasm in response to various stimuli (12). By regulating target RNAs, HuR participates in numerous cellular events, including proliferation, senescence, differentiation, apoptosis, and survival (15, 17).
Expanding the knowledge of the molecular mechanisms underlying myocardial infarction (MI) is of profound importance to developing a cure for MI. We found that restoration of CUG triplet repeat RNA-binding protein 1 (CUGBP1) expression improved cardiac function in acute myocardial infarction (AMI) mice and protected cardiomyocytes from AMI-induced injury through proangiogenic and antiapoptotic pathways via regulating the vascular endothelial growth factor-A gene. Upon AMI, human antigen R was relocated to the cytoplasm from the nucleus and bound to AU-rich elements in the 3′UTR of CUGBP1, disrupting the expression of CUGBP1. These results provide a new understanding of CUGBP1 in cardiac disease and novel MI post-transcriptional gene regulatory machinery. The manipulation of CUGBP1 could be developed as a potential therapeutic option for MI.
Disruption of RNA metabolism at a variety of steps has emerged as a powerful and dynamic modifier of cardiovascular diseases (6). Several RBPs have been shown to be critical for cardiac functions, including CUGBP- and ETR-3-like factor (CELF) proteins (22). The CELF family of RBPs consists of six members, which includes CUG triplet repeat RNA-binding protein 1 (CUGBP1). They control many aspects of RNA metabolism by binding to GU-rich elements (GREs). In the mouse heart, the CUGBP1 level is developmentally downregulated by post-transcriptional regulation (22). Inducible CUGBP1 re-expression in adult hearts results in an ∼40% reduction in the expression of developmentally upregulated genes (26). CUGBP1 transgenic animals exhibit enlarged hearts (26), and CUGBP1-deficient mice display growth retardation with smaller hearts (27). Cardiac tissue-specific repression of CELF activity causes cardiomyopathy (33). These studies indicate an important role of CUGBP1 in the heart. However, the biological functions of CUGBP1 in human cardiovascular diseases remain poorly understood.
It has been found that GREs in pre-mRNA and mRNA transcripts play diverse roles in the control of gene expression by regulating pre-mRNA processing, mRNA stability, and translation (47). By binding to GREs, CELF proteins direct a variety of post-transcriptional regulatory events (47, 48).
We have performed a computational scanning of GREs using microarray-based expression profiling in a mouse MI model, and the results indicate that ∼14% of all genes with significantly altered expression (see Materials and Methods) contain GREs. This led us to hypothesize that CELF proteins may be important in MI. To determine the function of CELF proteins, an animal model of acute MI (AMI) was used in this study. Decreased CUGBP1 expression was observed in heart samples from AMI mice; further studies showed that the expression of CUGBP1 was mediated by HuR through conserved adenylate-uridylate-rich (AU-rich) elements in the 3′UTR of CUGBP1. Restoration of CUGBP1 expression protected cardiomyocytes from injury induced by AMI, and the impaired cardiac function was significantly improved in CUGBP1 transgenic AMI mice. Detailed analyses demonstrated that CUGBP1 participates in the protection of cardiomyocytes through proangiogenic and antiapoptotic pathways by controlling the expression of vascular endothelial growth factor (VEGF)-A during AMI. Our results reveal a novel MI post-transcriptional gene regulatory mechanism and provide new insights into the role of CUGBP1 in cardiac diseases. The manipulation of CUGBP1 could be developed as a potential therapeutic option for MI.
Results
Expression of CUGBP1 is decreased in AMI hearts
An AMI mouse model was used to study the function of CUGBP1 in MI. Mice were subjected to MI by ligation of the left anterior descending coronary artery (LAD) as described previously (30). To determine the temporal pattern of CUGBP1 expression during heart ischemia in mice, protein extracts from the areas at risk were analyzed in ischemic hearts. As shown in Figure 1, the CUGBP1 was highly expressed in the heart, and its expression decreased gradually during ischemia (Fig. 1A, B). After coronary ligation, CUGBP1 expression levels started to decrease at 4 h postischemia, significantly decreased at 16 h, and reached a plateau at 24 h post-MI. The mRNA expression levels of CUGBP1 were also analyzed by quantitative polymerase chain reaction (qPCR), and the results indicated that the mRNA expression levels of CUGBP1 started to decrease at 8 h and reached the lowest level at 24 h postischemia (Fig. 1C). We further examined the expression of CUGBP1 at the late infarct stage. The expression of CUGBP1 remained low until 14 days post-MI (Supplementary Fig. S1A, B; Supplementary Data are available online at

Given that many patients with AMI receive timely reperfusion therapy, we were interested in whether reperfusion could restore the CUGBP1 level. Samples were therefore collected from the areas at risk in hearts that were subjected to a 2 h coronary ligation followed by a 24 h period of reperfusion. A Western blot (WB) analysis indicated that reperfusion did not restore the CUGBP1 level (Supplementary Fig. S1C).
Next, we examined the subcellular expression of CUGBP1 using immunofluorescence assays. Under normal conditions, CUGBP1 was found in both the nucleus and the cytoplasm (Fig. 1D). Upon ischemia, however, both the nuclear and cytoplasmic expression levels of CUGBP1 were reduced over time. As seen in Figure 1D (the last column from left), at 24 h after infarction, only slight immunoreactivity was detected in the nuclei, and a weak signal was observed in the cytoplasm.
Conserved AU-rich elements are responsible for the decreased CUGBP1 expression in AMI
To investigate the mechanisms by which ischemia regulates CUGBP1 expression, sequence motif analyses were conducted to identify conserved putative regulatory elements between humans and mice. Considering the decreased CUGBP1 RNA expression level, the 5′UTR and 3′UTR regions of CUGBP1 were analyzed. Two highly conserved sequences were found in the 3′UTR of CUGBP1 transcripts from humans, rats, and mice (Fig. 2A). Both of the segments contain AU-rich elements, which are the most commonly found signals in the 3′UTR mediating RNA regulation.
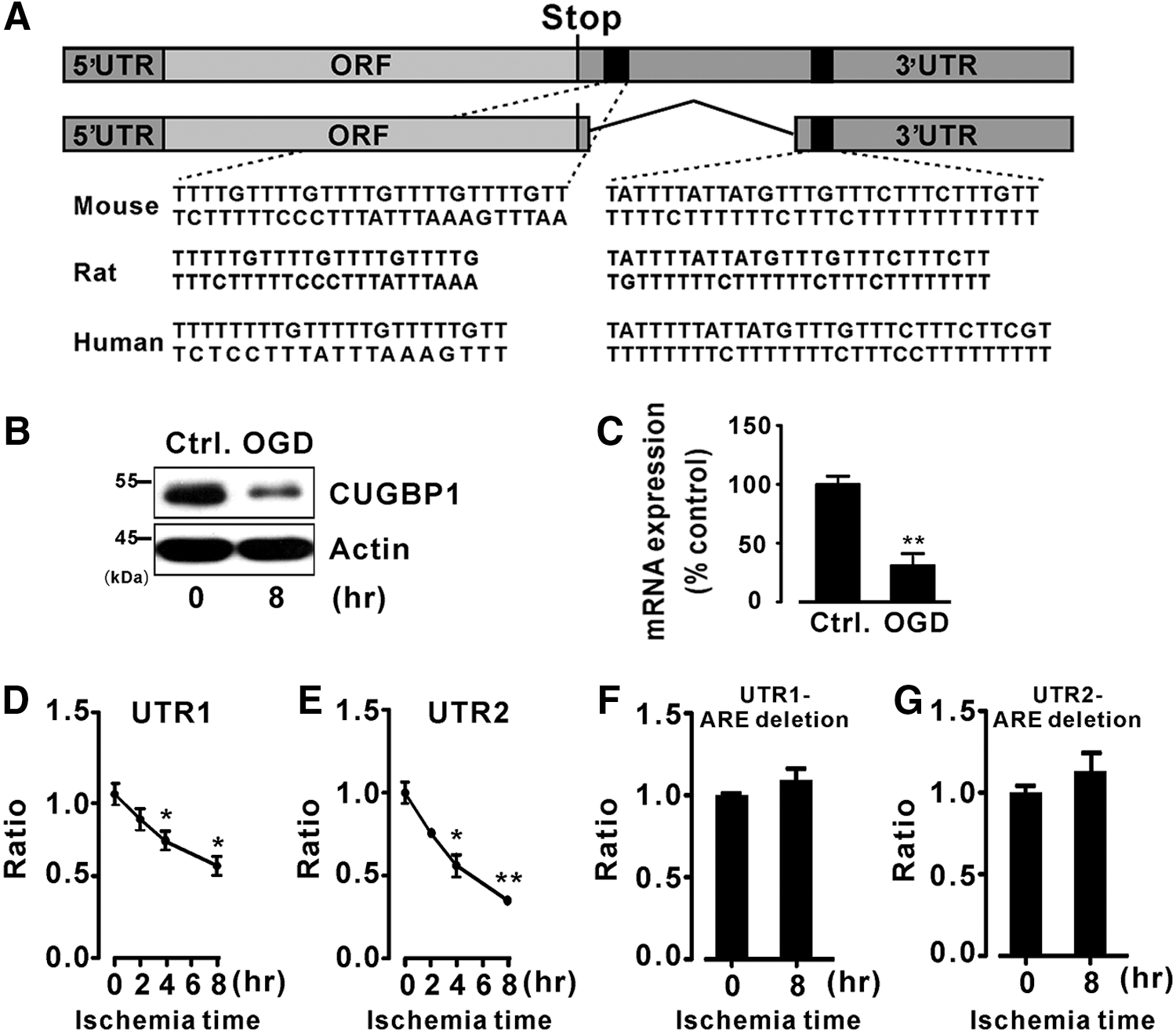
To mimic the ischemic condition, oxygen and glucose deprivation (OGD) was used in the following study. Two luciferase reporter constructs each harboring one of the two segments of the CUGBP1 3′UTR containing AU-rich elements, with lengths of 327 and 312 bp, were generated. Before transfecting these two luciferase reporter constructs into H9c2 cells, a rat cardiomyoblast cell line, we verified that CUGBP1 expression in these cells also decreased during OGD (Fig. 2B, C). We then conducted luciferase activity assays and observed that OGD culture strongly inhibited the luciferase activity from the two reporter constructs (Fig. 2D, E). To determine the importance of these AU-rich elements, we deleted the AU-rich elements from the generated luciferase reporter constructs and conducted additional luciferase activity assays. As expected, OGD treatment no longer inhibited the luciferase activity without the presence of the AU-rich elements (Fig. 2F, G).
The altered CUGBP1 expression in AMI is induced by cytoplasmic HuR relocalization
HuR regulates mRNA metabolism by binding to AU-rich elements (1, 34). Thus, we hypothesized that HuR is responsible for the decreased CUGBP1 expression during ischemia. We first examined the subcellular expression of HuR using immunofluorescence assays in H9c2 cells and ischemic hearts. As shown in Figure 3A (H9c2 cells) and 3B (ischemic hearts), under normal conditions, HuR was found mostly in the nucleus; upon ischemia, HuR was transported to the cytoplasm from the nucleus. WB analyses of nuclear and cytoplasm fractions of neonatal rat cardiomyocytes after 8 h of OGD also confirmed the increase in HuR expression in the cytoplasm (Fig. 3C, D). However, the expression levels of HuR during ischemia were unchanged in both H9c2 cells under OGD (Fig. 3E, F) and ischemic hearts (Fig. 3G, H). Luciferase activity assays demonstrated that HuR overexpression strongly inhibited the luciferase activity of the two CUGBP1 3′UTR reporter constructs (Fig. 4A, B). As expected, when the AU-rich element (ARE) deletion CUGBP1 3′UTR reporter constructs were transfected into cells, followed by HuR overexpression, the inhibitory effects of HuR on the luciferase activity was no longer observed (Fig. 4C, D).

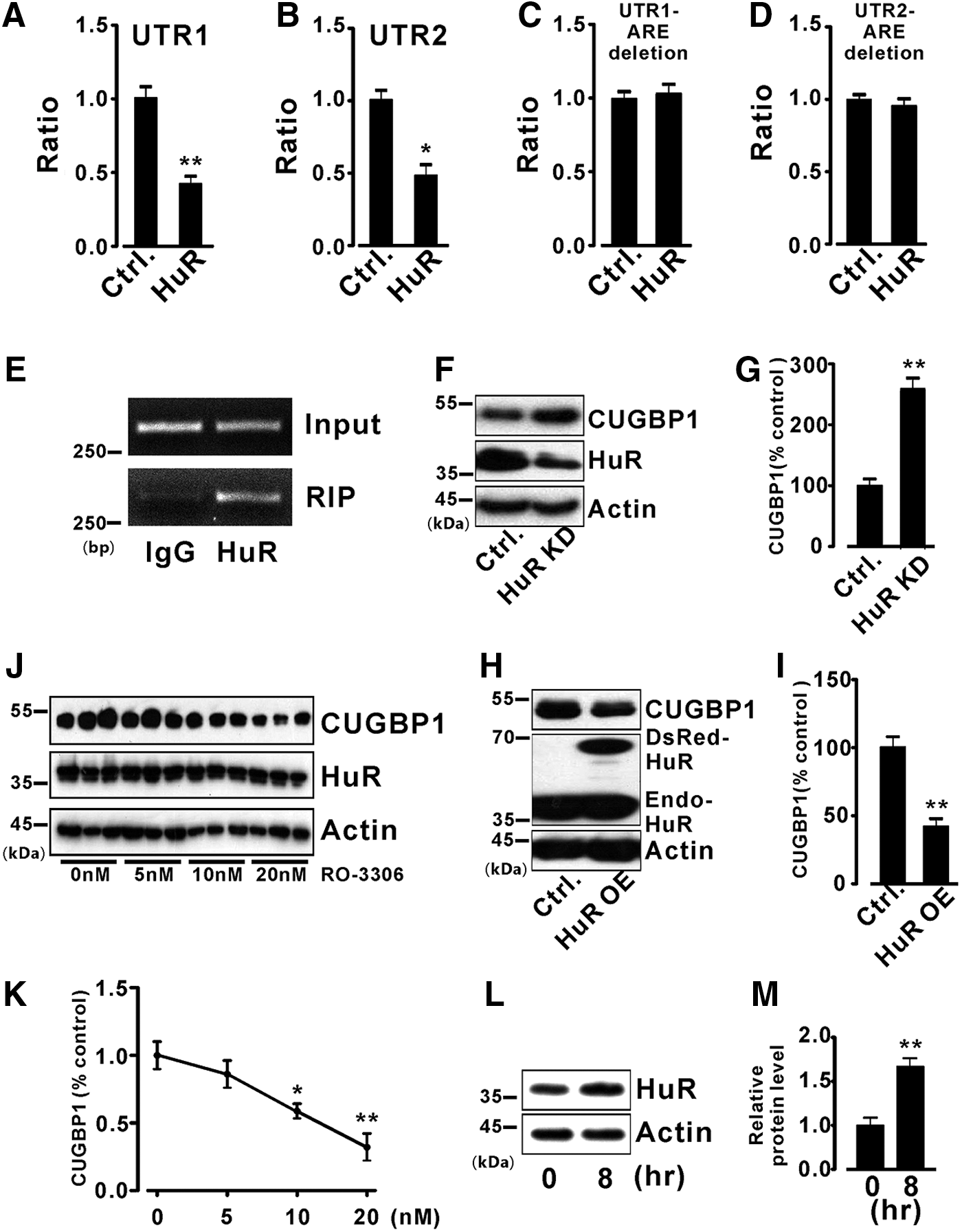
Further studies were performed to provide more evidence of the regulation of CUGBP1 by HuR. First, the physical association between the HuR protein and CUGBP1 RNA was examined by a RNA immunoprecipitation polymerase chain reaction (RIP-PCR) assay using H9c2 cells. The result demonstrated the presence of CUGBP1 RNA after a pull-down assay using an antibody against HuR (Fig. 4E). Second, adenovirus-mediated overexpression or knockdown of HuR was performed in mouse hearts. Knockdown of HuR significantly increased CUGBP1 expression by 2.6-fold (Fig. 4F, G). In contrast, CUGBP1 was downregulated by HuR overexpression (Fig. 4H, I).
Phosphorylation contributes to the nuclear import of HuR (12). Several signaling kinases regulate the HuR subcellular abundance, including Cdk1 (25). Inhibition of Cdk1 elevates cytoplasmic HuR levels (25). To further demonstrate that HuR regulates CUGBP1, a selective Cdk1 inhibitor, RO-3306 (51), was used in our study. As shown in Figure 4J, K, we observed that CUGBP1 expression was decreased in neonatal rat cardiomyocytes treated with RO-3306 (Santa Cruz, Dallas, TX) for 8 h, and the cytoplasmic presence of HuR was strikingly enhanced (Fig. 4L, M). However, the expression levels of total HuR during RO-3306 treatments were not changed. Thus, our studies confirm that CUGBP1 is regulated by the HuR protein.
CUGBP1 re-expression improves cardiac function in MI mice
The reduction of the CUGBP1 expression level in ischemic hearts led us to consider whether CUGBP1 regulates cardiac function after infarction. Therefore, a well-established recombinant adenovirus strategy was used to express CUGBP1 in AMI hearts as described previously (7, 11, 54). Importantly, the expression of CUGBP1 in the heart was not altered by the administration of recombinant adenovirus (Supplementary Fig. S2A) or the LAD operation (Supplementary Fig. S1). We constructed recombinant adenoviruses that expressed DsRed-tagged CUGBP1 or the DsRed protein alone. Hearts infected with adenoviruses were collected, and a fluorescence analysis revealed that CUGBP1 was successfully expressed in the left ventricular (LV) wall (Supplementary Fig. S2C). The Ad-CUBBP1 expression was apparent up to 20 days postinjection. Compared with mice infected with the control virus, the CUGBP1 expression in MI hearts was increased by adenovirus-mediated CUGBP1 expression (Supplementary Fig. S2B).
LV shape variation caused by regional wall motion abnormalities is a universal phenomenon that occurs after MI; therefore, an echocardiography study was performed to analyze the hearts of mice infected with DsRed and DsRed-tagged CUGBP1 4 weeks after infarction. To perform reliable measurements of cardiac function, we calculated the LV ejection fraction (EF%) based on the shape-independent Simpson's rule. In brief, the LV endocardial border was traced in a long-axis view both in systole and diastole, and the volumes were computed from these tracings. Using this method, the LV function can also be measured as the percentage of change in LV area between diastole and systole (fractional area change, FAC%), which correlates well with EF% both in normal and in infarcted hearts (3).
As shown in Figure 5, the decreased postinfarction LV posterior wall thickness at diastole was attenuated (CUGBP1 vs. DsRed: 0.92 ± 0.02 mm vs. 0.72 ± 0.01 mm, Fig. 5A). Accordingly, hearts re-expressing CUGBP1 exhibited a higher FAC (CUGBP1 vs. DsRed: 14.95% ± 1.08% vs. 10.04% ± 1.16%, Fig. 5B) and EF (CUGBP1 vs. DsRed: 27.7% ± 2.10% vs. 18.75 ± 1.90%, Fig. 5C). Echocardiography images and movies showed that re-expression of CUGBP1 attenuated LV dilatation characterized by a smaller endocardial border and thicker ventricular wall in the long-axis view (Supplementary Movies S1 and S2).
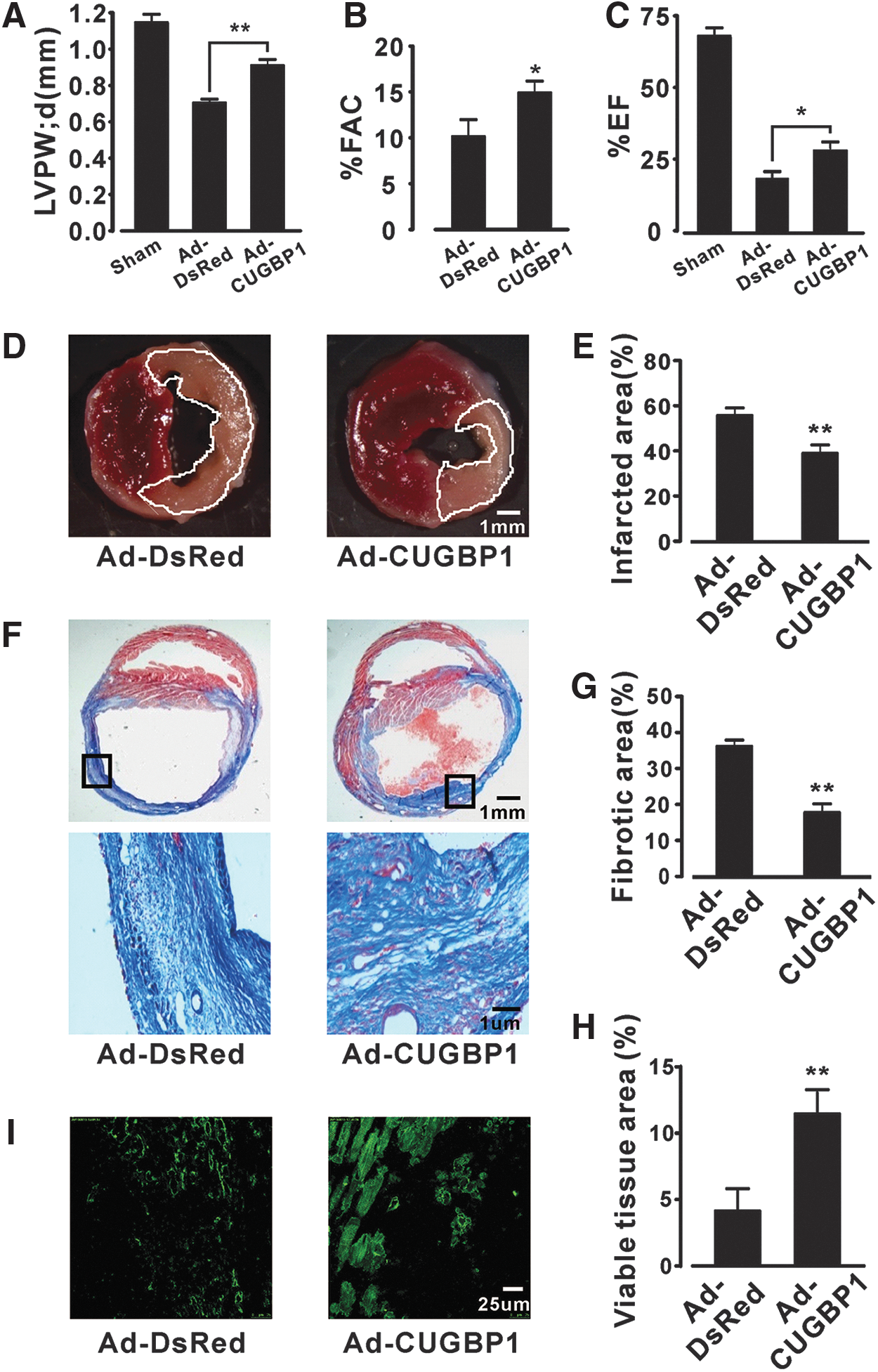
Next, we determined the effects of CUGBP1 re-expression on ischemic injury by histological analysis at 24 h after ischemia. The hearts re-expressing CUGBP1 showed a reduced infarct size (38%) compared with those from control mice (58%, Fig. 5D, E). Tissue fibrosis analysis at 4 weeks post-MI suggested that re-expression of CUGBP1 significantly attenuated the myocardial fibrosis area to 18% (Ad-CUGBP1) from 36% (Ad-DsRed, Fig. 5F, G). Interestingly, an immunohistochemistry analysis of sections from the areas at risk indicated that the amount of viable mass was much higher in the hearts re-expressing CUGBP1 than in control mice (Fig. 5F lower panel and 5H).
It has been demonstrated that the number of viable cardiomyocytes is critical to the recovery of cardiac function after infarction (13). To confirm the identity of the viable mass, we performed immunofluorescence staining of infarction sections with an antibody specific to the cardiomyocyte marker protein actinin, and we observed that hearts re-expressing CUGBP1 exhibited more viable cardiomyocytes than the hearts of control mice (Fig. 5I).
CUGBP1 protects cardiomyocytes from AMI-induced injury by regulating VEGF-A
The formation of new blood vessels around the infarct area is important for the survival of cardiomyocytes (14). Could re-expressed CUGBP1 protect cardiomyocytes from AMI-induced injury by promoting the formation of new blood vessels? In the following experiments, we tested this hypothesis. The sections from the infarcted area at 4 weeks post-MI were immunostained for a marker of endothelial cells (CD31). After CUGBP1 re-expression, the microvessel density in the infarct area was significantly increased from 55/mm2 to 120/mm2 (Fig. 6A, B). Meanwhile, the percentage of the lumen area was significantly increased from 4.5% to 9.4% (Fig. 6A, C). Apoptosis, a highly regulated process, is one of the major forms of cell death that occurs during MI (28). The protective effect of CUGBP1 against apoptosis during MI was investigated in our study. The results indicated that TdT-mediated dUTP nick-end labeling (TUNEL)-positive cells (green nuclei) in the infarcted area were significantly reduced in CUGBP1-re-expressing hearts (7%) compared with that of DsRed-infected controls (21.5%, Fig. 6D, E), suggesting that CUGBP1 expression promotes angiogenesis and decreases apoptosis during MI, increasing the number of viable cardiomyocytes as a consequence.
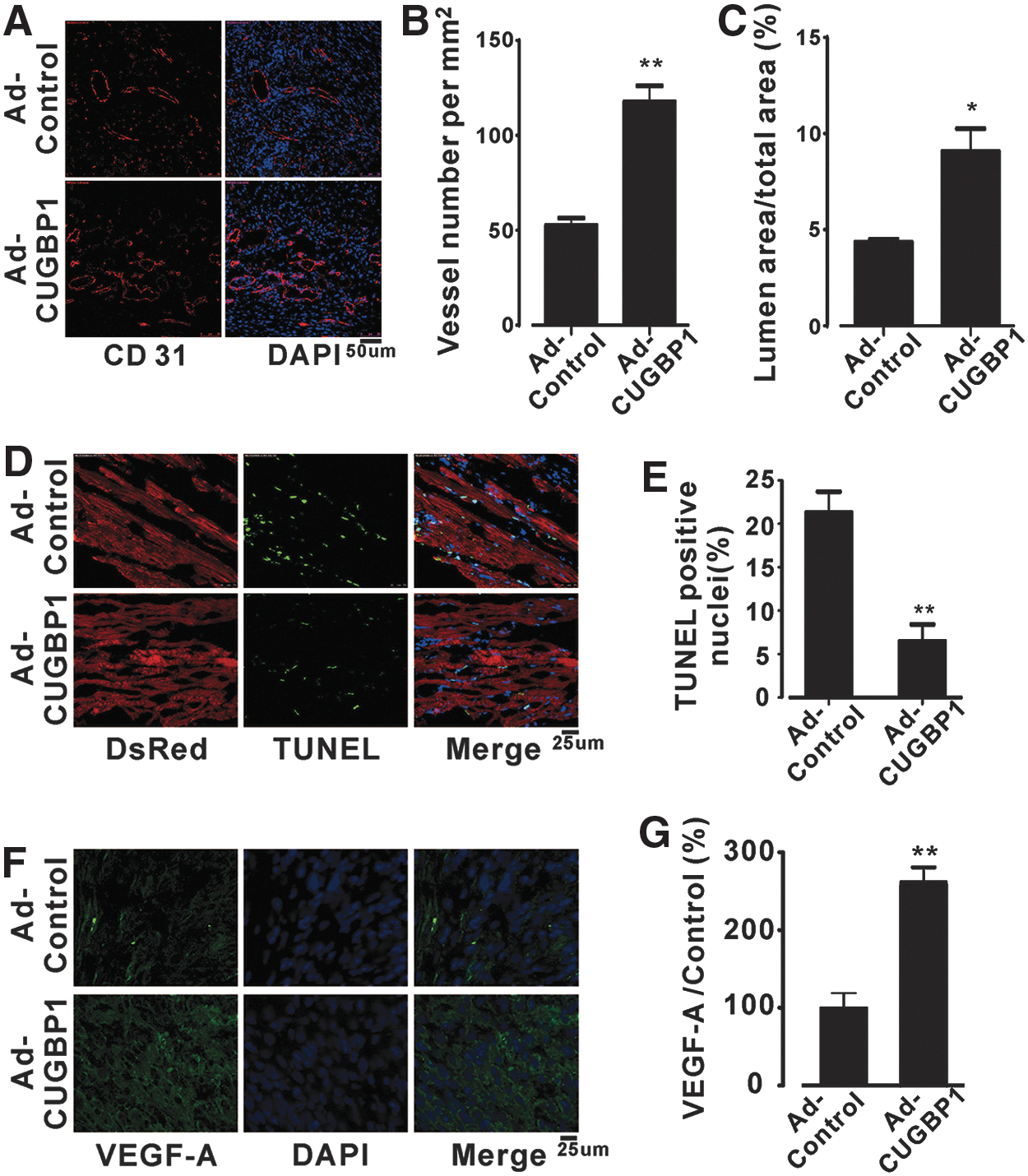
The VEGF gene family is an important angiogenesis and apoptosis regulator (5, 10). Previous studies have shown that the expression levels of VEGF are greatly decreased in the infarcted area (56). Interestingly, our results showed that the expression of VEGF-A in the infarcted area of CUGBP1-re-expressing hearts was significantly upregulated (Fig. 6F, G) at 24 h post-MI. This encouraged us to investigate the possible regulation of VEGF-A by CUGBP1. First, we studied whether CUGBP1 binds to VEGF-A in H9c2 cells. The physical association between CUGBP1 and VEGF-A was examined by an RIP-PCR assay. The result demonstrates that VEGF-A RNA is present in the CUGBP1 pull-down sample (Fig. 7A). Second, we manipulated the levels of CUGBP1 using adenovirus-mediated knockdown or overexpression, followed by WB and qPCR analyses. VEGF-A was strikingly downregulated by the knockdown of CUGBP1 expression in mouse hearts (Fig. 7B, C). In contrast, overexpression of CUGBP1 significantly increased VEGF-A expression by 2.7-fold (Fig. 7E, F). Similarly, a qPCR assay indicated that the mRNA level of VEGF-A was altered as well (Fig. 7D, G).
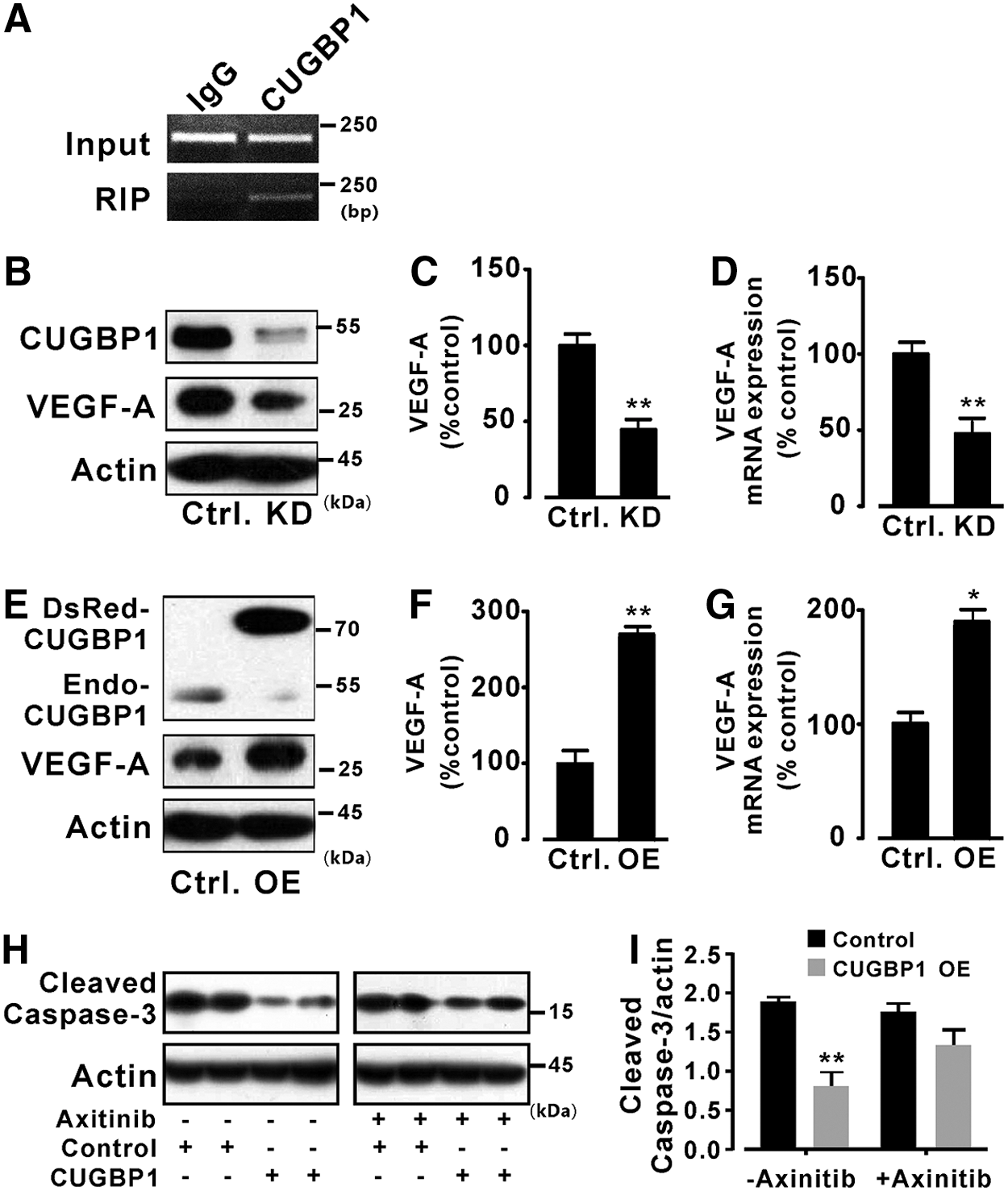
To further demonstrate that CUGBP1 achieves its biological functions through regulating the VEGF-A gene, a potent and selective VEGF signal pathway inhibitor, axitinib, was used in our study (24). As shown in Figure 7, overexpression of CUGBP1 significantly reduced the level of cleaved caspase-3 in H9c2 cells (Fig. 7H, I left panel). In contrast, when H9c2 cells were treated with axitinib, the overexpression of CUGBP1 was only moderately able to decrease the level of cleaved caspase-3 (Fig. 7H, I right panel).
To explain the moderate antiapoptotic effect of CUGBP1 upon VEGF pathway inhibition, we further analyzed the expression of other important apoptosis regulators, such as Bcl-2 family genes (9), p53 (20), and TNF-α (55), which are regulated by CUGBP1. Our results indicated that Bcl-x and Bax were regulated by CUGBP1 re-expression in the heart (Supplementary Fig. S3A, B), whereas Bcl-2, p53, and TNF-α were not affected by CUGBP1 (Supplementary Fig. S3C, D), suggesting that the antiapoptotic function of CUGBP1 is achieved, in part, through regulating Bcl-x and Bax.
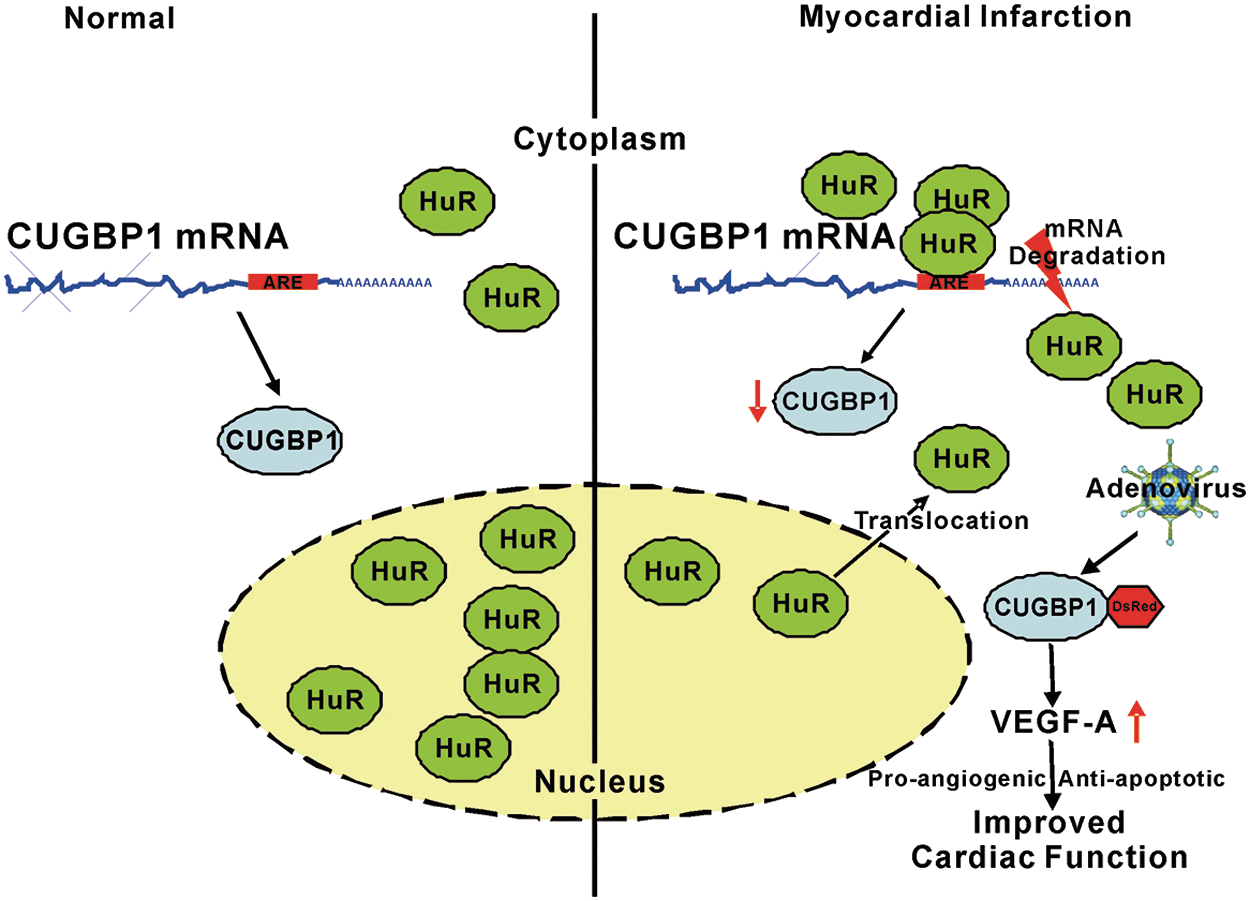
As graphically represented in Figure 8, HuR is mostly located in the nucleus in normal heart tissue. Upon AMI, HuR is transported to the cytoplasm from the nucleus and binds to AU-rich elements located in the 3′UTR of the CUGBP1 mRNA. The expression of CUGBP1 is decreased under the regulation of HuR, resulting in cardiac injury. Restoration of CUGBP1 expression protects cardiomyocytes from ischemia-induced injury via regulating the VEGF-A gene. Thus, cardiac function was improved in the AMI mice expressing the adenovirus-associated CUGBP1 transgene.
Discussion
CUGBP1 in cardiac diseases
As a key regulator, CUGBP1 has been found to play important regulatory roles in the development of muscle pathology, including both myotonic dystrophy type 1 (DM1) and DM2 diseases (35, 43, 45). With increased CUGBP1 expression, >80% of DM1 patients develop cardiac abnormalities (16, 42). Knockout and overexpression studies have shown that CUGBP1 is an important regulator in the heart (26, 27, 33). However, there are no reports regarding the function of CUGBP1 in cardiac infarction. In this study, we provide the first demonstration that CUGBP1 is functionally important in MI. This finding reveals the roles of CUGBP1 in the heart not only during development but also in cardiac disease.
Proper expression of CUGBP1 is important for the heart
During postnatal remodeling, the CUGBP1 is developmentally downregulated (22). This is particularly important as the fetal heart adapts to birth and converts to adult function (22). Overexpression of CUGBP1 or repression of CELF activity in the heart leads to dilated cardiomyopathy (26, 33). Consistently, restoration of altered CUGBP1 expression in the ischemic heart improved heart function in our study. This evidence indicates that maintaining the proper temporal expression level of CUGBP1 is important for cardiac function. Accordingly, earlier restoration of CUGBP1 expression should better mitigate ischemic injury post-MI.
Indeed, multiple levels of CUGBP1 regulation have been described, including transcription (18), protein translation (8, 21), and post-translation modifications (32). Thus, CUGBP1 expression is tightly controlled at every step from its RNA synthesis to protein production.
The opposing functions of CUGBP1 and HuR
Although CELF proteins and Hu proteins are highly similar in overall domain architecture and are grouped within a monophyletic clade (46), they preferentially bind different sequences. HuR proteins bind to AU-rich elements, whereas CELF proteins bind to GREs. However, it has been reported that CUGBP1 also binds to AU-rich sequences (36, 55). Thus, overlapping targets of HuR and CUGBP1 may exist on the same RNA transcript. The results of an RIP-ChIP experiment identified several mRNA targets shared by CUGBP1 and HuR (36).
It had been previously reported that HuR knockdown contributes to improved LV function and cardiac remodeling after MI (29). In our study, re-expression of CUGBP1 in MI hearts resulted in improved LV function and reduced injury. The results that either low HuR expression (29) or high CUGBP1 expression protects infarcted hearts from injury suggest that HuR and CUGBP1 have opposing functions in MI gene regulation. Several studies have indicated that CUGBP1 and HuR negate each other's effects in regulating the occludin and MYC genes (52, 53).
Here, we report that HuR negatively regulates CUGBP1 expression by relocalization to the cytoplasm from the nucleus, which is controlled by different post-transcriptional phosphorylation signaling pathways, including AMPK, p38-MAPK, PKC, and Cdk1 (12). Although Supplementary Data from RIP-ChIP studies of CUGBP1 demonstrate the binding of CUGBP1 to HuR mRNA (2, 36), overexpression of CUGBP1 shows that HuR is not regulated by CUGBP1 (Supplementary Fig. S4). Thus, we suggest that HuR acts upstream of CUGBP1 in regulating the expression of genes related to MI.
Oxidative stress and apoptosis are closely linked physiological phenomena and are implicated in pathophysiology (23). HuR has been demonstrated to have a role in regulating oxidative damage in hippocampal neurons (44). This led us to hypothesize that CUGBP1 protects cells from apoptosis by an antioxidative mechanism. The RIP-ChIP studies of CUGBP1 demonstrate the binding of CUGBP1 to several antioxidative-related genes, such as superoxide dismutases 1 and 2 and heme oxygenase 1, which are also regulated by HuR (31). Further studies are currently being performed in our laboratory to provide more evidence of the potential antioxidant effects of CUGBP1.
Reduced CUGBP1 is responsible for the decreased VEGF-A expression in AMI hearts
VEGF family members have multiple functions in the cardiovascular system (14). The spatial and temporal expression levels of VEGF (mRNA and protein) in the infarcted rat heart have been studied in detail by Zhao et al. (56). VEGF-A expression was found to be significantly increased at the border zone only on the first day post-MI and not in the later stages. However, its expression was consistently suppressed in the infarcted area (56). Importantly, our study shows for the first time that CUGBP1 functions to promote angiogenesis and inhibit apoptosis by upregulating VEGF-A during MI. Considering the correlation between CUGBP1 and VEGF-A expression, decreased VEGF-A expression may be attributed to the suppression of CUGBP1 in the AMI heart.
HuR is an important positive regulator of VEGF-A (37, 41). It directly stabilizes VEGF mRNA in 293T cells (37) and induces its translation in HeLa cells (41). However, our current study suggests that in AMI hearts, HuR negatively regulates the VEGF transcript level through CUGBP1. Therefore, additional studies need to be performed to provide more in-depth mechanistic insights into this seemingly complex regulation.
VEGF-based therapies have already reached clinical experimentation and might be used for new therapeutic applications (14, 38). The transplantation of adipose tissue mesenchymal cells conjugated with VEGF-releasing microcarriers promotes repair in murine MI (38). The activation of VEGF-A expression by CUGBP1 suggests a potential clinical usage of CUGBP1 post-MI.
Potential clinical usage of CUGBP1 post-MI
Importantly, our study identified a novel HuR-mediated CUGBP1 regulation mechanism during MI. As mentioned earlier, CUGBP1 is an important regulator of mouse cardiac development (22), and the overexpression of CUGBP1 induces dilated cardiomyopathy (26). This suggests that abolishing the regulation of CUGBP1 by HuR, thus restoring the endogenous CUGBP1 expression, is a potential therapeutic option. It has been reported that inhibiting HuR binding to an ARE in 3′UTR of TNF-α mRNA abolishes the HuR-mediated regulation of TNF-α (4). So far, several strong and specific inhibitors of HuR binding to TNF-α mRNA have been developed (4). Recently, an FP-based binding assay was established to discover specific, novel, small molecule HuR-mRNA disruptors (50). Thus, the knowledge gained from our study and these new technologies may help pave the way for the development of selective inhibitors for the regulation of CUGBP1 by HuR.
Furthermore, in this study we also found that the blood level of CUGBP1 in mice was decreased during the progression of AMI (Supplementary Fig. S5), which suggests that CUGBP1 may be a promising circulating biomarker for diagnosing and predicting the prognosis of MI in the clinic.
Materials and Methods
Preparation of adenovirus
Adenovirus particles were produced following the manufacturer's protocol (AdEasy™ XL Adenoviral Vector System, Stratagene). The adenovirus was purified by CsCl gradient ultracentrifugation and desalted using a Sepharose CL-4B column (GE Healthcare, Waukesha, WI). The viral concentration was calculated.
Animals
The use of animals and experimental procedures was approved by the Institutional Animal Use and Care Committee of the Institute of Biophysics at the Chinese Academy of Sciences. Eight-week-old C57BL/6 mice were purchased from Weitong-Lihua Co.
For the MI model, mice were subjected to permanent LAD ligation as described previously (30). In brief, the animals were anesthetized using tribromoethanol (1.2%, 0.02 ml/g) and ventilated using a rodent ventilator (DW-3000B; Xinsida, Beijing, China) with 100 breaths per min and a stroke volume of 100 μl. A left thoracotomy was performed, and the LAD was ligated using an 8-0-prolene suture with a mortality rate of 80%. Sham MI mice underwent the same surgical procedure with the exception that the LAD was not ligated.
Adenovirus particles were slowly injected at multiple locations in the LV wall using a syringe attached to a 29-gauge needle. A total of 100 μl of adenovirus (8 × 1010 particles) containing CUGBP1 or control adenoviral vectors were delivered to the heart. No persistent impairments of the heart were observed postinjection. Five days after the administration of the adenovirus, the mice were subjected to LAD or sham operations. The infarction area was defined using Evans blue dye. Samples of ∼20 mg were harvested from the infarction area at various time intervals after the surgery as indicated in figures.
Computational motif analyses
To search for potential binding genes of CELF proteins, we started with the mouse AMI expression database (
Cell culture
H9c2 cells were cultured in Dulbecco's modified Eagle medium (DMEM; GIBCO) with 10% fetal bovine serum (FBS) and antibiotics. The H9c2 cells were stimulated with OGD. In brief, the cells were seeded in six-well plates and incubated in a hypoxia incubator (Thermo, Waltham, MA) containing 1% O2, 5% CO2, and 94% N2 for 2, 4, or 8 h. For these experiments, the cells were cultured in serum-free, glucose-free modified eagle medium (MEM) (GIBCO, Waltham, MA). The cells were then processed for the next study.
Neonatal rat cardiomyocytes were isolated as previously described (39). One-day-old SD rats were killed by cervical dislocation. Cardiomyocytes were isolated by enzymatic disassociation in a Tyrode solution containing 1 mg/ml type II collagenase (Sigma-Aldrich) and without Ca2+ for 30 min at 37°C. A Gilson P200 pipette was used to disrupt the tissue gently until it had dissociated into single cells and small clusters. The isolated cells were cultured in H-DMEM supplemented with 10% FBS and plated on sterile, gelatin-coated plates.
Plasmids
The CUGBP1 (or HuR) and DsRed cDNA sequences were amplified from the pCDNA3.1-hCUGBP1 (or pCDNA3-mHuR) and pDsRed2-N1 vectors, respectively. The CUGBP1 (or HuR) and DsRed-digested PCR fragments were ligated into pShuttle-CMV to generate the pShuttle-CMV-DsRed-CUGBP1 (or HuR) expression plasmid.
The Cas9 gene was obtained from the pLenti-OC-IRES-BSD plasmid. The PCR-amplified Cas9 and DsRed fragments were inserted into the pMD18-T vector. The plasmid was digested, and the Cas9-2A-DsRed fragment was cloned into pShuttle-CMV. The CRISPR design tool was used (
The DNA segments of the CUGBP1 3′UTR were amplified from mouse genomic DNA. The two DNA fragments were digested and individually cloned into the pCS2-luc-3UTR vector (a gift from Jane Ying Wu' laboratory). The two luciferase reporter constructs harboring segments of the CUGBP1 3′UTR were confirmed by sequencing.
Western blotting
Tissues or cells were homogenized and lysed in RIPA buffer (Beyotime Bio Co., Wuhan, China). The proteins were separated by 12% SDS/PAGE, transferred to PVDF membranes (Millipore), blocked, and incubated with antibodies. The primary antibodies used in this study were as follows: anti-CUGBP1 (Eptomics), anti-HuR (Santa Cruz), anti-GAPDH (Invitrogen), anti-β-actin (Beyotime), anti-IgG (Santa Cruz), anti-VEGF-A (Santa Cruz), anti-CD31 (Abcam, Cambridge, MA), anti-actinin (CST, Beverly, MA), anticaspase-3 (CST), anti-Bcl-2 (CW-biotech, Beijing, China), anti-Bcl-x (Beyotime), anti-Bad (CW-biotech), anti-Bax (CW-biotech), anti-p53 (Abcam), anti-TNF-α (Abclonal, Shanghai, China), and anti-Mcl-1 (CW-biotech). The relative expression level of proteins was analyzed by densitometry using the Image J program (Bethesda, MD). See complete unedited blots in the Supplementary Data.
RNA isolation and real-time quantitative polymerase chain reaction analysis
Total RNA was isolated from heart tissue or cells with Trizol reagent (Invitrogen) according to the manufacturer's protocol. RNA was reverse transcribed with the PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara Bio, Inc., Shiga, Japanese). The PCR amplifications were performed using Taqman 7300 (Applied Biosystems, California). The relative mRNA expression of target genes was normalized to the endogenous actin control gene (Applied Biosystems) and represented as fold change versus control.
Echocardiography
Echocardiographs were performed using a 30-MHz probe and the Vevo 770 Ultrasound machine (VisualSonics, Toronto, Canada). The Vevo 770 is equipped with ECG-gated kilohertz visualization software (EKVTM). Mice were anesthetized with tribromoethanol and placed on a thermocontrolled plate (37°C ± 1°C) and were given 10 min to acclimate before echocardiography recording. Blinded measurements and calculations were performed as described in a previously published article (3).
Morphometric studies
The morphometric analyses, including infarct size (%LV area) and percentage fibrosis area (%LV area), were performed by triphenyltetrazolium chloride staining or Masson's trichrome-stained tissue sections using ImageJ 1.30 software (U.S. National Institutes of Health;
TUNEL staining
TUNEL staining was carried out on 4 μm-thick frozen sections according to the manufacturer's instructions (In Situ Cell Death Detection Kit; Roche, Madison, WI). DAPI (Vector Laboratories, Peterborough, United Kingdom) staining was used to count the total number of nuclei. The percentage of apoptotic cells was calculated as the percentage of apoptotic myocyte nuclei/total number of nuclei.
Immunostaining
Slides with heart sections were fixed with 3% paraformaldehyde for 20 min and washed three times with PBST (0.3% Triton X-100 in PBS) for 10 min each. The slides were incubated with a primary antibody overnight at 4°C. Then, the slides were washed three times with PBST and incubated with a secondary antibody for 2 h. The slides were washed three times with PBST, stained with DAPI (Vector Laboratories, Peterborough, United Kingdom) for 10 min, and washed with PBST. The slides were then examined using a fluorescence microscope (Leica, DE).
Nuclear and cytoplasmic extraction
To prepare the protein fractions, neonatal rat cardiomyocytes were lysed using a tissue homogenizer and a nuclear and cytoplasmic protein extraction kit (Beyotime) according to the manufacturer's instructions. The lysates were centrifuged at 12,000 g for 10 min at 4°C, and the supernatants were collected as the cytoplasmic fraction. The pelleted nuclei were resuspended in a buffer containing 1 mM PMSF. After 30 min at 4°C, the lysates were centrifuged, and the supernatants containing the nuclear proteins were stored at −80°C.
RIP assay
The RIP experiment with H9c2 cells was performed as previously described (19). In brief, a specific antibody to CUGBP1 (or HuR) was used for immunoprecipitation (IP). Mouse IgG was used as an IP negative control. Twenty microliters of protein A/G beads (Santa Cruz) was added to the supernatant after the addition of the antibody. The mixture was incubated for 12 h on a roller at 4°C. After washing, the beads were resuspended in 1 ml of Trizol to isolate the coprecipitated RNAs according to the manufacturer's instructions (Invitrogen, Waltham, MA).
Luciferase assay
H9c2 cells were plated in a 24-well plate. The next day, the cells were transfected with plasmid DNA using FuGENE® HD transfection reagent (Roche). The cells were incubated for 24 h before the luciferase activity was measured. The cells were washed and lysed. The supernatant was assayed for luciferase activity with Dual-Luciferase® reporter assay system (Promega, Madison, WI). The produced light was measured at an integration time of 1 s with a GloMax luminometer (Promega).
Statistical analysis
Data are presented as means ± SEM, *p < 0.05 and **p < 0.01 by a two-tailed Student's t-test compared with control or one-way ANOVA, respectively.
Footnotes
Acknowledgments
This work was sponsored by grants from the National Foundation of Sciences and Technology (31371430 to H.W., 31271228 to G.J.) and SRF for ROCS to H.W.
Authors' Contributions
The study presented here was performed in collaboration between all authors. H.W. and G.J. conceived and designed the experiments. L.G., H.W., J.W., and Y.G. carried out the experiments and analyzed the data. Y.T. and L.C. performed the statistical analyses. G.J. and H.L. reviewed and edited the article. All authors have read and approved the final article.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.