
Abstract
Aims:
Ceruloplasmin (CP), a ferrous oxidase enzyme, plays an important role in regulating iron metabolism and redox reactions. Previous studies showed that CP deficiency contributes to Parkinson's disease by increasing iron accumulation and oxidative stress in the substantia nigra. However, the role of CP in Alzheimer's disease (AD) is unclear. We hypothesized that the lack of CP gene expression would affect the pathogenesis and damage of AD by promoting abnormal iron levels and oxidative stress.
Results:
AD mouse models were induced in CP knockout mouse either by injection of Aβ25–35 into the lateral ventricle of the brain or transgenic APP expression. CP levels were decreased significantly in the hippocampus of AD patients, as well as Aβ-CP+/+ and APP-CP+/+ mice. Compared to control AD mice, CP gene deletion increased memory impairment and iron accumulation, which could be associated with elevated reactive oxygen species (ROS) levels and lead to cell apoptosis mediated through the Bcl-2/Bax and Erk/p38 signaling pathways in Aβ-CP−/− and APP-CP−/− mice. In contrast, the restoration of CP expression to CP −/− mice through injection of an exogenous expression plasmid into the brain ventricle alleviated Aβ-induced neuronal damage in the hippocampus.
Innovation:
CP alterations in iron contents were mediated through DMT1(−IRE) and changes in ROS levels, which in turn attenuated the progression of AD through the Erk/p38 and Bcl-2/Bax signaling pathways.
Conclusion:
Our results show a protective role of CP in AD and suggest that regulating CP expression in the hippocampus may provide a new neuroprotective strategy for AD. Antioxid. Redox Signal. 28, 1323–1337.
Introduction
A
Ceruloplasmin (CP), a multicopper enzyme, was first described as a ferroxidase in the 1970s (28). In the central nervous system, CP is mainly expressed in astrocytes (25) and Schwann cells (29) and exists in a bound, glycosylphosphatidylinositol (GPI)-anchored form (35). CP is thought to have multiple physiological functions, including those associated with copper transport and biogenic amine oxidation (27). CP can also convert toxic ferrous iron into the nontoxic ferric form and facilitate iron release from cells by binding to apotransferrin in the plasma, thus minimizing cellular amounts of dangerous ferrous iron (19, 37, 43). Adult CP-knockout (CP−/− ) mice were shown to have iron overload in the central nervous system with aging (18, 26). Aceruloplasminemia patients in middle age were also observed to have similar traits, showing iron accumulation and neurodegeneration in the brain (14).
This study showed ceruloplasmin (CP) expression was decreased in Alzheimer's disease (AD) brain. CP deficiency was associated with DMT1(−IRE)-mediated increase in cellular iron content, which elevated reactive oxygen species (ROS) levels. The increased ROS levels accelerate AD progression through Erk/p38 and Bcl-2/Bax signaling pathways. On the other hand, CP deficiency could increase the expression of BACE-1, which enhanced APP cleaved into Aβ. In turn, aggregation of Aβ further aggravated CP deficiency, which formed a vicious cycle so as to exacerbate the cell apoptosis. Meanwhile, CP overexpression could decrease iron levels and cell apoptosis through a Bcl-2/Bax pathway, which in turn attenuates AD pathogenesis.
Many studies showed that decreases in CP activity may be related to neurodegenerative diseases (35). Some studies confirmed that CP concentrations and oxidative activity in serum were significantly lower in Parkinson's disease (PD) patients (20, 36). Furthermore, about 80% of CP activity was lost in the substantia nigra (SN) of PD patients (1). Analysis of CP−/− mice showed high levels of iron in the SN, which facilitated oxidative stress level and resulted in more severe cell apoptosis following treatment with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (41), whereas exogenously expressed CP could alleviate this damage (1).
Meanwhile, studies found that although AD patients have serum CP concentrations that are similar to healthy individuals, the CP activity is significantly lower in AD patients (20). This finding suggested that CP function is related to AD, although the detailed mechanisms involved in this relationship remain unclear. In this study, we first found that CP expression was decreased in AD mouse models and patients. Therefore, we hypothesized that the lack of CP gene expression would affect AD pathogenesis and damage by promoting abnormal iron levels and oxidative stress. We further analyzed the role of CP in AD by knocking out CP gene in two mouse models of AD, which were induced either by injection of Aβ25–35 into the lateral ventricle of the brain or the presence of APP transgenic mouse. Memory impairment, oxidative stress, iron level, cell apoptosis, and signaling pathways were analyzed in these mice to explore the role of CP in the occurrence and progression of AD.
Results
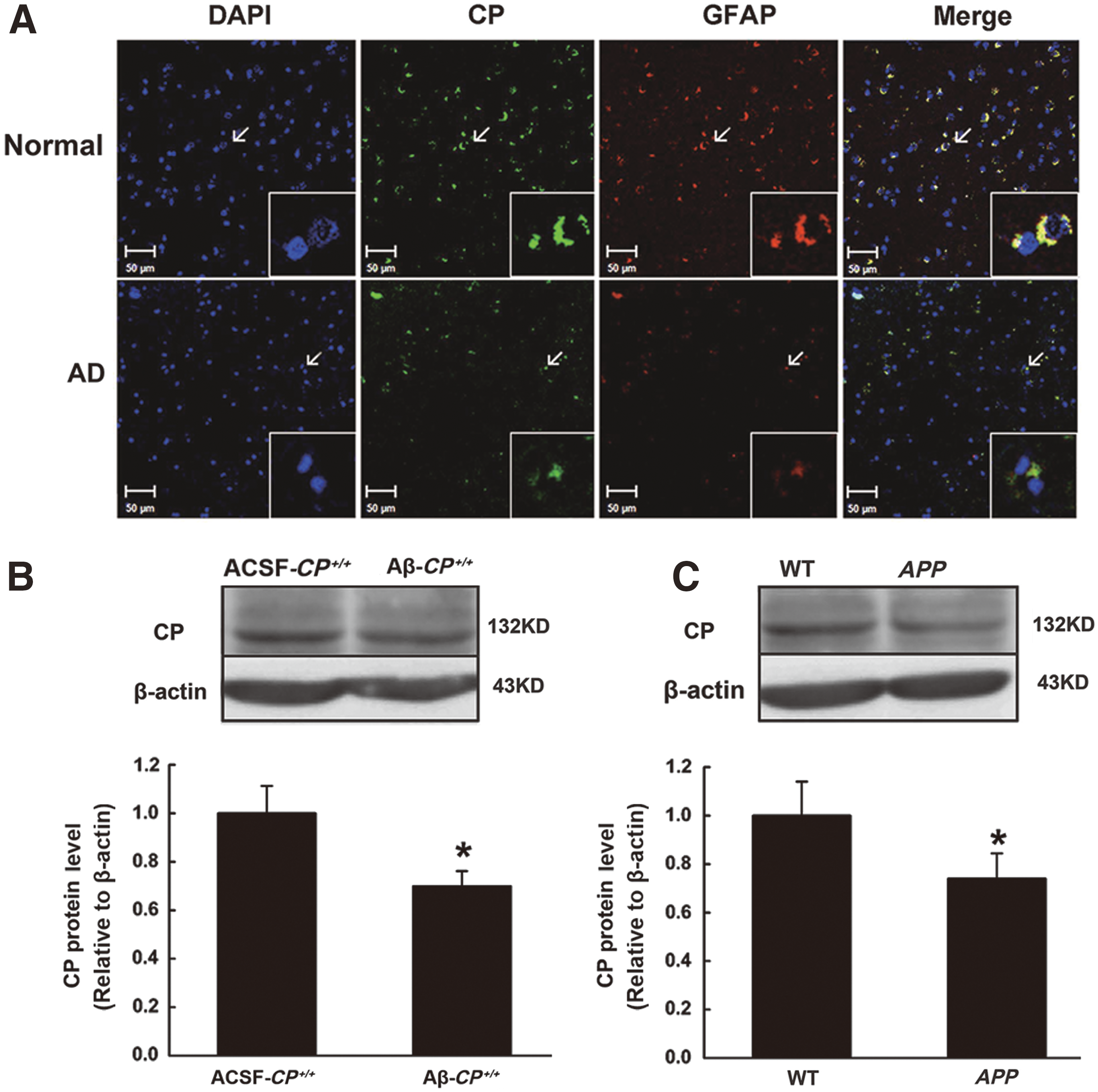
CP protein expression is decreased in the hippocampi of AD patients and AD mice
GPI-CP is the main form of CP expressed in astrocytes (17). Therefore, we first performed immunohistochemistry on human and mouse hippocampus samples with antibodies specific to CP (green staining) and the astrocyte marker glial fibrillary acidic protein (GFAP, red). We saw a decrease in CP expression in astrocytes in the hippocampus of AD patients (Fig. 1A). In addition, Western blotting analysis showed that CP protein levels were also decreased in the hippocampi of the two AD animal models: mice given a lateral ventricle injection of Aβ25–35 (10 μg/mouse) and APP transgenic mice (Fig. 1B, C and Supplementary Fig. S1A; Supplementary Data are available online at
CP deficiency exacerbates learning and memory impairment in an Aβ-induced AD mouse model
To explore the possibility that CP deficiency aggravates AD-related dysfunction, we established the Aβ25–35-induced AD model. We examined spatial learning and memory using the Morris water maze test. After 2 weeks of treatment with Aβ25–35, the escape latency time was increased in the Aβ-CP+/+ and Aβ-CP−/− groups compared to their respective artificial cerebral spinal fluid (ACSF)-injected groups (Fig. 2A). In particular, for the Aβ-CP−/− group relative to Aβ-CP+/+ group, the escape latency time was increased on day 1 and day 7, respectively (Fig. 2A). Meanwhile, the escape latency distance increased on day 1, 2, 3, 5, and 7 for the Aβ-CP+/+ group and on day 1, 2, 5, and 7 for the Aβ-CP−/− group relative to their respective ACSF groups (Fig. 2B). In the last day, Aβ-CP−/− mice showed an obvious decrease in the number of passing times by a hidden platform within 120 s (Fig. 2C). These results indicated that the lack of CP aggravated Aβ25–35-induced learning and memory impairment.

CP deficiency aggravates Aβ25–35-induced neuronal cell apoptosis
To study the mechanism underlying the learning and memory impairment induced by Aβ25–35 in CP−/− mice, apoptosis in the hippocampus was detected by the TUNEL assay. An obvious increase in the number of apoptotic cells was seen for hippocampi from the Aβ-CP+/+ group compared with the ACSF-CP+/+ group. Most importantly, the number of apoptotic cells in the Aβ-CP−/− group was three- and twofold higher than that of the ACSF-CP−/− group and Aβ-CP+/+ group, respectively (Fig. 3A). These results indicated that the lack of CP enhanced Aβ25–35-induced neuron apoptosis in the hippocampus.
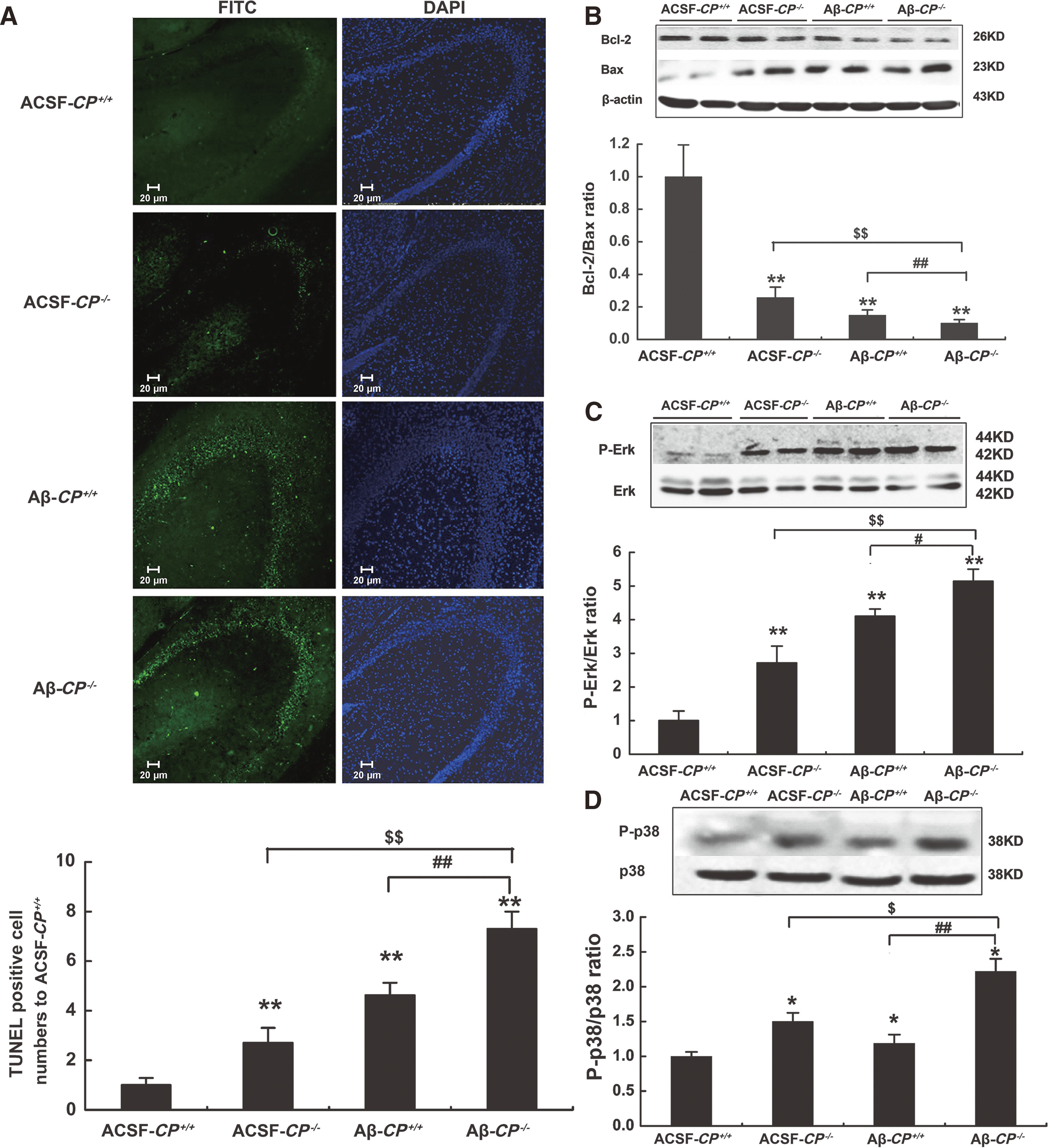
Bcl-2 and Bax are anti- and proapoptotic Bcl-2 family proteins, respectively, which play important roles in regulating cell apoptosis. The ratio of Bcl-2/Bax proteins has been widely used to monitor the degree of cell apoptosis. In this study, we used Western blotting to examine the expression levels of Bcl-2 and Bax in hippocampi from the AD model mice to calculate the Bcl-2/Bax ratios. Compared to the ACSF-CP+/+ group, the Bcl-2/Bax ratios were significantly reduced by 74.2% and 85.0% in the ACSF-CP−/− and Aβ-CP+/+ groups, respectively. Meanwhile, in the Aβ-CP−/− group, the Bcl-2/Bax ratios were decreased by about 70% and 40% relative to that of the ACSF-CP−/− and Aβ-CP+/+ groups, respectively (Fig. 3B). These results suggest that a lack of CP exacerbated the reduction in the Bcl-2/Bax ratio in the hippocampi of AD model mice.
Since activation of the mitogen-activated protein kinase (MAPK) pathway is involved in oxidative stress-induced cell death (39), we next investigated the effect of CP deficiency on MAPK signaling activation. Phosphorylation levels of p38 and Erk, two classical MAPKs, were detected using Western blotting of hippocampus tissue from the different experimental groups. Aβ25–35 injection induced Erk and p38 activation in both wild-type (WT) and CP−/− mice. For Aβ-CP−/− mice, levels of activated Erk and p38 were significantly increased by 47.9% and 89.4%, respectively, relative to that of the Aβ-CP+/+ group (Fig. 3C, D). Thus, the Erk and p38 activation induced by Aβ25–35 injection was further enhanced by the lack of CP, which could lead to changes in downstream signaling.
Increase in ROS in hippocampi from CP−/− mice after Aβ25–35 treatment
Cell apoptosis can be induced by many factors, and ROS are a key apoptosis inducer (39). To understand the molecular mechanism of apoptosis in the context of CP expression, levels of superoxide dismutase (SOD), malondialdehyde (MDA), and lipid peroxide (LPO) were determined to examine whether the increased level of oxidative stress was responsible for cell apoptosis in hippocampi from AD mice lacking CP. The level of SOD declined, whereas that of MDA and LPO increased significantly in the Aβ25–35-treated groups. In particular, the level of SOD was the lowest in hippocampi from the Aβ-CP−/− group, whereas MDA and LPO levels were the highest in these mice (Fig. 4). The data implied that increased ROS in the hippocampus may be an important contributing factor to the increase in cell apoptosis seen in Aβ-CP−/− mice.

CP affects Aβ25–35-induced iron deposition in the hippocampus
Iron is an essential cofactor for many proteins, particularly those involved in oxidative metabolism. However, excess free iron can contribute to enhanced generation of ROS and oxidative stress (42). To clarify the mechanisms by which CP deficiency promoted ROS levels in the hippocampus following Aβ25–35 injection, the total iron content was measured using inductively coupled plasma mass spectroscopy (ICP-MS) (9). Compared to that of the ACSF-CP+/+ group, the total iron in the hippocampus increased significantly in both the Aβ-CP+/+ and Aβ-CP−/− groups. However, the total iron content in the Aβ-CP−/− group was about 1.5-fold higher than that of the Aβ-CP+/+ group (Fig. 5A). Ferritin (FT) consists of two subunits (H and L) and is a ubiquitous and highly conserved iron binding protein that serves as a major means of nonheme iron storage in cells (15). In this study, the level of the ferritin light chain (FTL) increased significantly in the hippocampi in the Aβ-treated groups compared with the corresponding vehicle groups. Notably, the FTL level in the Aβ-CP−/− group was significantly higher than that of the Aβ-CP+/+ group (Fig. 5B).

To investigate whether the increased iron in the hippocampus was associated with iron transport proteins, the expression levels of several proteins were detected by Western blotting in the hippocampi from AD mice. Divalent metal transporter 1 (DMT1) is an important transmembrane ferrous iron uptake protein that is present as DMT1(−IRE) and DMT1(+IRE) isoforms (12). DMT1(−IRE) levels increased significantly in the hippocampi of the Aβ-CP+/+ and Aβ-CP−/− groups compared with those of the ACSF-CP+/+ and ACSF-CP−/− groups (Fig. 5C). The level of DMT1(−IRE) in the Aβ-CP−/− group was about 1.5-fold higher than that for the Aβ-CP+/+ group (Fig. 5C).
Transferrin receptor 1 (TfR1) is a key protein that facilitates ferric iron transport into brain cells, and ferroportin 1 (FPN1) is a unique iron efflux protein (10). In this study, the TfR1 level was decreased significantly in the hippocampi from the Aβ-CP+/+ and Aβ-CP−/− groups relative to their respective ACSF group; moreover, the TfR1 level was decreased significantly in the Aβ-CP−/− group compared to the Aβ-CP+/+ group (Fig. 5E). When compared to the ACSF-CP+/+ group, there were no changes in the levels of FPN1 and DMT1(+IRE) in the hippocampi of the other three groups (Fig. 5D, F). These results demonstrated that the increase in iron content in mouse brains after Aβ25–35 injection may have been caused by an increase in DMT1(−IRE)-mediated iron uptake. Therefore, CP deficiency could increase Aβ25–35-induced iron deposition in the hippocampus, and iron may play a critical role in the elevation of oxidative stress.
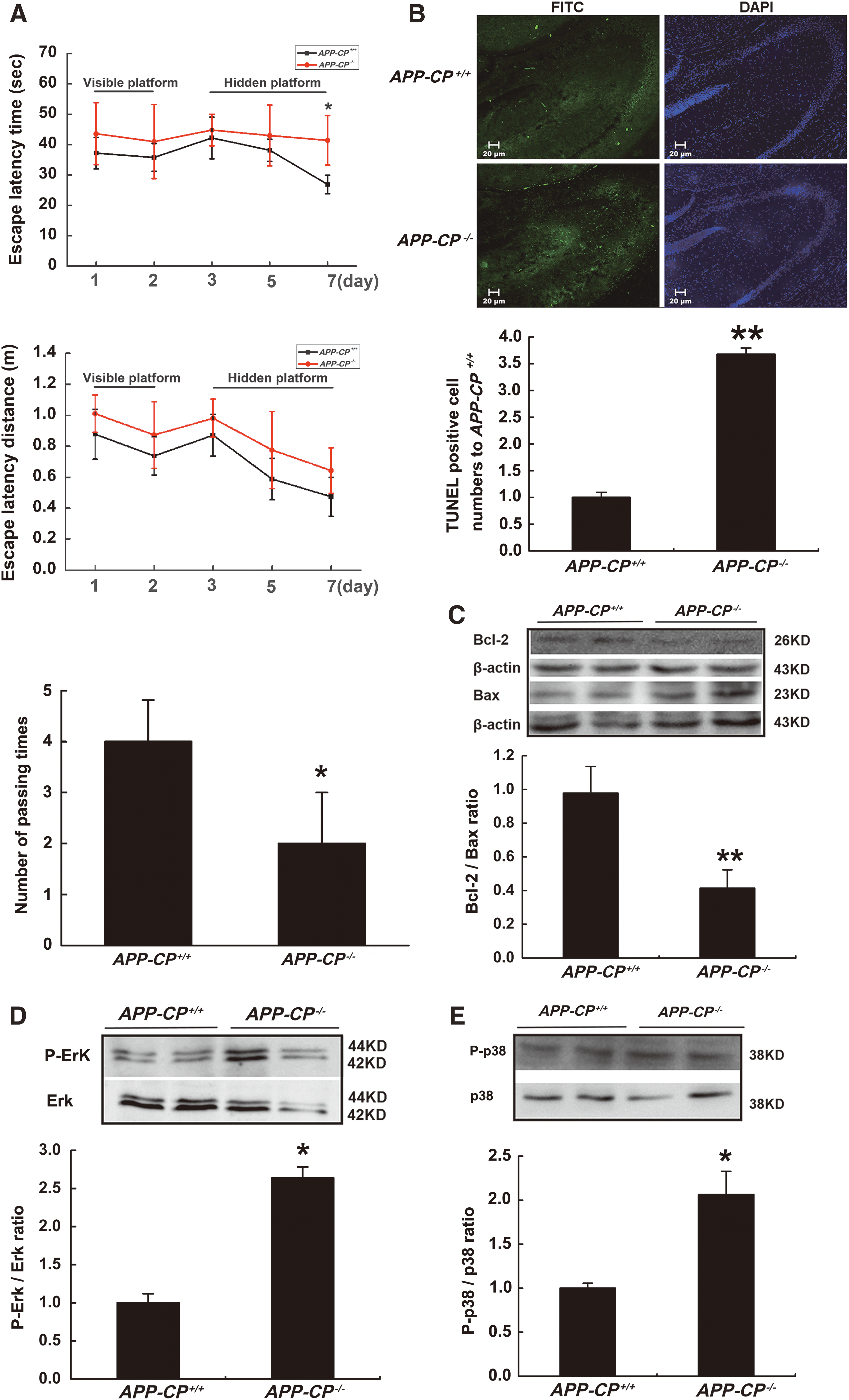
CP deficiency aggravates neuronal apoptosis in the hippocampi of APP transgenic mice
To further confirm that CP deficiency could aggravate AD disease, another AD model was constructed, the APP transgenic mouse model. In the Morris water maze assessment, CP deficiency could increase neurobehavioral dysfunction in 6-month-old APP mice, as indicated by the increased escape latency time and distance, as well as a decrease in the number of passing times by a hidden platform within 120 s (Fig. 6A). The number of apoptotic cells in the hippocampi from APP mice was about 3.7-fold higher for the APP-CP−/− group compared to the APP-CP+/+ group and the Bcl-2/Bax ratio was also significantly decreased in APP mice that lacked CP (Fig. 6B, C). The ratios of P-Erk/Erk and P-p38/p38 were higher in the hippocampi of APP-CP−/− mice versus that of APP-CP+/+ mice (Fig. 6D, E). These results further confirmed that the lack of CP could aggravate cell apoptosis in the hippocampi of AD model animals.
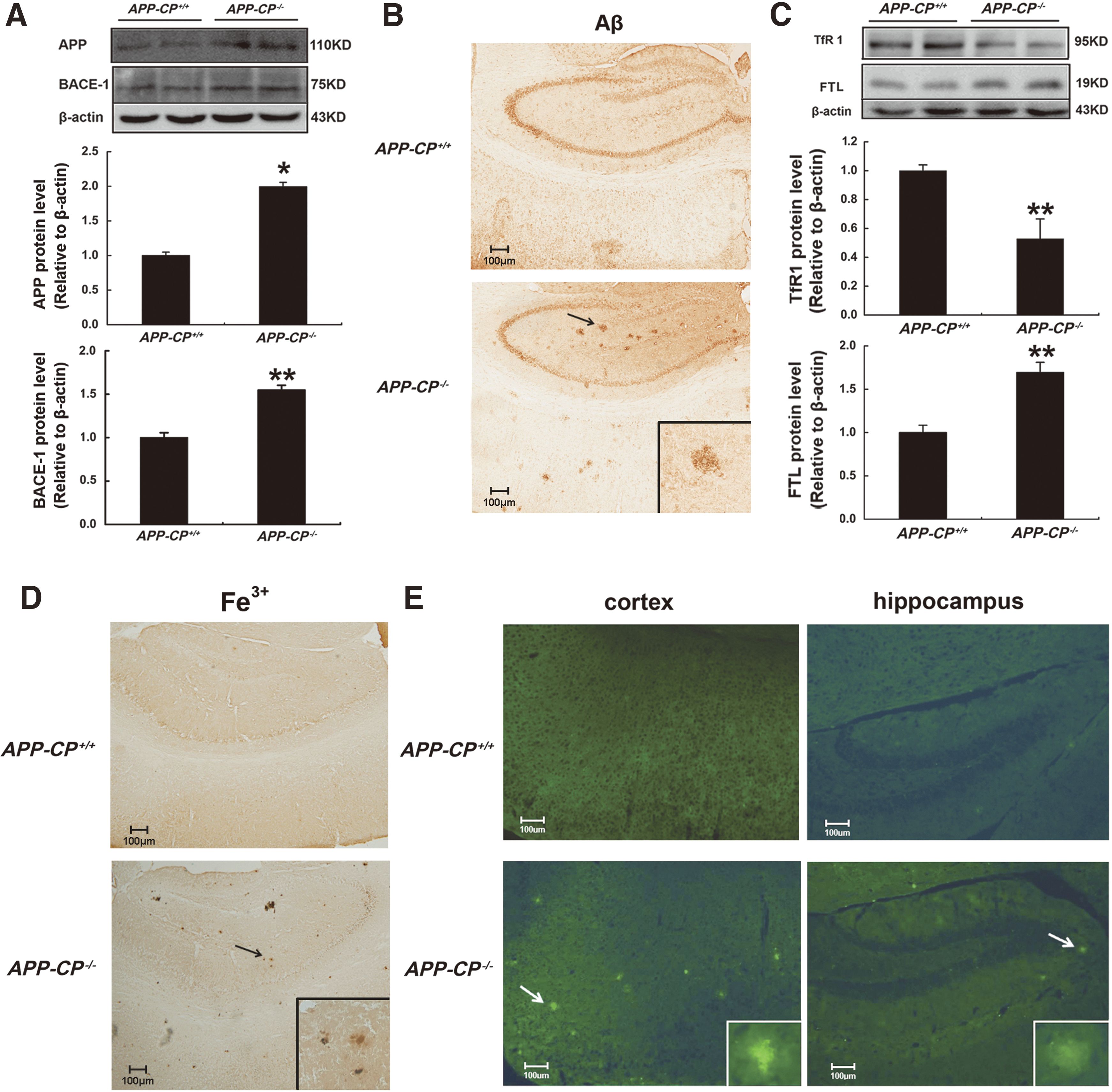
To address whether AD-related neurological damage was associated with deposited neuritic plaques in APP-CP−/− mice, the distributions of APP and beta-secretase 1 (BACE-1) were detected. BACE-1, also known as beta-site APP cleaving enzyme 1, can cleave APP into Aβ fragments and eventually increase neurotoxic injury (44). Western blotting results showed that the level of APP and BACE-1 protein was increased significantly in the hippocampi of APP-CP−/− mice when compared with APP-CP+/+ mice (Fig. 7A). Moreover, Aβ was widely deposited in the cortex and hippocampi of APP-CP−/− mice, whereas no obvious aggregated neuritic plaques were seen in APP-CP+/+ mice at 6-month old (Fig. 7B). Decreased TfR1 and increased FTL protein levels were found in the hippocampi of APP-CP−/− mice (Fig. 7C), indicating a state of iron overload. Images taken after iron staining confirmed that ferric iron aggregates were deposited in the cortex and hippocampi of APP-CP−/− mice (Fig. 7D), similar to the Aβ neuritic plaques. In addition, thioflavin staining of brain tissue revealed that amyloid plaques were present in the cortex and hippocampus of APP-CP−/− but not APP-CP+/+ mice (Fig. 7E). Therefore, CP deficiency could increase iron deposition and promote cleavage of APP into Aβ plaques in the hippocampus.
CP overexpression decreases Aβ-induced cell damage in the hippocampus
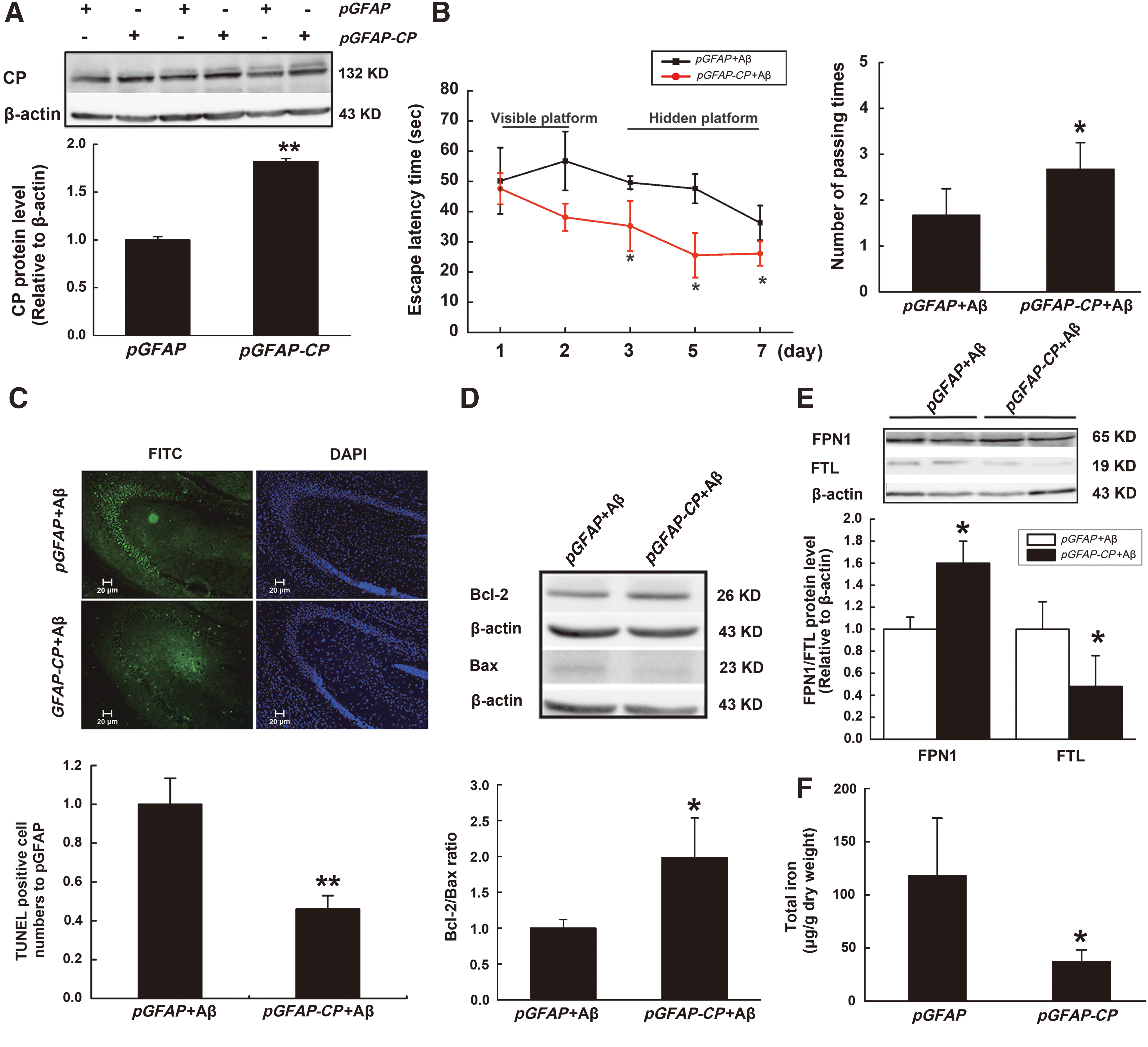
As all the above results showed that CP loss increased cell damage, we reasoned that CP overexpression could prevent AD-related injuries. To test this hypothesis, we constructed a plasmid, pGFAP-CP, which specifically expresses CP in astrocytes. Lateral ventricle injection of this plasmid significantly increased the expression of CP protein in the hippocampus (Fig. 8A). Morris water maze assessments revealed that pretreatment with the CP plasmid could ameliorate the memory impairment induced by Aβ25–35 (Fig. 8B). The increase in the number of apoptotic cells and changes in the Bcl-2/Bax ratio induced by Aβ25–35 were also reduced by administration of the CP plasmid (Fig. 8C, D). We found that the amounts of FTL decreased in pGFAP-CP+Aβ group, indicating that iron level was decreased. FPN1 was increased obviously, implying that FPN1 contributed to the decreased cellular iron level through promoting iron efflux (Fig. 8E). Furthermore, ICP-MS results showed that CP overexpression could significantly decrease the total levels of iron in the hippocampus (Fig. 8F). These results revealed that CP overexpression reduced neuronal damage in the hippocampus induced by Aβ25–35.
Discussion
GPI-anchored CP is the major form of CP in the brain and is expressed abundantly in astrocytes (25). Previous reports revealed that CP is essential for iron homeostasis and antioxidant defenses (35). Indeed, iron accumulation in the central nervous system and increased free radical-mediated damage have been shown in CP−/− mice (26). In aceruloplasminemia patients, iron accumulates in the basal ganglia (22) and SN (40) of the central nervous system, and these patients are more prone to PD. Thus, CP deficiency can aggravate the pathogenesis of several neurodegenerative diseases. In this study, we knocked out CP gene to generate CP−/− mice on two AD backgrounds. One AD model involved injection of Aβ25–35 into the lateral ventricles of WT or CP−/− mice, whereas the other was APP transgenic mice that either expressed or lacked CP. These mice were used to investigate the effect of CP on AD pathogenesis and related mechanisms. CP deficiency exacerbated the neurological damage in the hippocampus of Aβ-induced and APP transgenic mouse models of AD, confirming that CP deficiency is detrimental in AD.
To our knowledge, this study is the first to demonstrate a decrease in CP expression in the hippocampus of AD patients. This finding builds on that of a study by Snaedal et al. showing that CP activity in the serum of AD patients was lower than that of healthy individuals (33). The activity of CP in cerebrospinal fluid (CSF) was also demonstrated to be decreased in AD patients (5). However, the extent to which CP expression levels are altered in brain tissues of AD patients has not yet been reported. Using double immunofluorescence labeling, we found that CP expression in astrocytes was remarkably decreased in the hippocampus of AD patients (Fig. 1A). In line with this observation, lower levels of CP were also detected in the hippocampi of mice with AD induced either by Aβ25–35 injection or expression of an APP transgene (Fig. 1B, C). The results showed that a lack of CP influenced the pathogenesis of AD.
AD patients exhibit memory loss followed by persistent mental decline, as well as aphasia and disorders in judgment and movement (38). The Morris water maze test is widely used in behavioral neuroscience to study spatial learning and memory. CP deficiency increased the escape latency time and decreased the number of passing times in the Aβ-induced and APP transgenic AD mice models (Figs. 2A, C, and 6A). On the contrary, overexpression of CP could weaken the increased escape latency time and decreased number of passing times induced by Aβ (Fig. 8B). These results demonstrated that CP plays important roles in attenuating the deficits of spatial learning and memory in AD.
Aβ-induced neuronal injury is reported to trigger transcriptional and posttranscriptional processes that regulate neuronal fate, including activation of MAPK pathways by neurotrophins and neurotransmitters, as well as the activation of pro- and antiapoptotic proteins (e.g., Bcl-2 family members) (39). Our results showed that activation of Erk/p38 MAPK signaling and decreases in the Bcl-2/Bax ratio in the hippocampus of the Aβ-CP−/− group and APP-CP−/− group (Figs. 3 and 6) caused memory impairment that was likely mediated by apoptotic processes. Together, these findings indicated that CP deficiency aggravated neuronal cell apoptosis in the hippocampi of AD models via an MAPK pathway. However, previous studies have not revealed the mechanisms by which CP−/− activates the MAPK pathway.
Aβ-associated oxidative stress can lead to mitochondrial impairment, loss of synapses and neurons, and ultimately neuronal cell death. Antioxidants are capable of preventing or slowing AD progression, suggesting that oxidative stress plays a key role in this disease. Our results showed that the Aβ-CP−/− group had a significantly reduced SOD level and elevated MDA and LPO activity compared with the Aβ-CP+/+ group (Fig. 4). In recent years, many lines of evidences point to the critical role of iron overload in pathological conditions associated with oxidative stress, such as AD (39) and aceruloplasminemia (14). Clinical evidence shows that certain brain regions in AD patients accumulate large amounts of iron (23), which is a key element in the production of free radicals through biochemical reactions. These free radicals can promote peroxidation of cellular lipids and in turn neuronal injury or death (3). Our data showed that the total iron content and FTL protein levels increased significantly in the hippocampi of Aβ-CP−/− mice and APP-CP−/− mice (Figs. 5B and 7C). DMT1(−IRE) expression was also greatly increased, which would enhance cellular iron uptake and increase the labile iron pool (LIP) level. Excessive LIP may donate electrons during the generation of ROS and lipid peroxidation, thus inducing oxidative damage in brain tissues (39), which might activate the MAPK pathway.
APP, a glycosylated protein, is abundant in the brain and can be cleaved into several polypeptides by intracellular secretases (45). In AD patients, a 42-amino acid β-peptide accounts for most of the cleavage fragments and forms the amyloid core of neuritic plaques (20). APP expression increases following iron exposure, likely due to the presence of an iron response element in the 5′ untranslated region of APP mRNA (32). In addition, in a cell model, iron could accelerate the cleavage of APP into fragments (13). The APP transgenic mouse is a typical AD model, in which older animals exhibit many plaque deposits in the brain (8, 34). To further verify that CP deficiency could aggravate brain damage in AD, we generated APP-CP−/− mice by hybridization. Our results showed that although 6-month-old APP mice did not develop neuritic plaques, APP-CP−/− mice presented with significant plaques in the cortex and hippocampus of the brain accompanied by iron deposition (Fig. 7). Previous studies indicated that at 6 months of age, CP−/− mice presented with a high level of iron in the brain (26). In the current study, we also detected a high level of iron accumulation in the hippocampi of APP-CP−/− mice, characterized by increased FTL protein levels, iron staining, and decreased amounts of TfR1 protein (Fig. 7). Therefore, we speculated that the lack of CP promoted the deposition of iron as well as APP translation and plaque formation to ultimately intensify AD pathogenesis.
Notably, CP overexpression was found to have a protective effect on Aβ-induced AD. We observed that the CP produced by plasmid pGFAP-CP alleviated Aβ-induced memory dysfunction in Morris water maze assessments. Recent studies showed that high activity of CP could stabilize FPN1, an iron transporter on the cell surface that exports ferrous iron from the cytosol across the plasma membrane into the blood (7). We found that overexpression of CP could significantly decrease the total iron level in the hippocampus, and that pretreatment with CP plasmid could increase FPN1 and reduce FTL expression induced by Aβ (Fig. 8), thus indicating the important role of FPN1 in lowering iron levels in neuronal cells. Neurons that have decreases in iron levels may be less likely to undergo cell death due to attenuation of Aβ-induced oxidative stress and activation of Erk/p38 MAPK signaling pathways.
In conclusion, our results demonstrated that CP protein expression was decreased in the hippocampus of AD patients and AD mouse models. Furthermore, CP deficiency exacerbated iron overload and resulted in an increase in ROS in the hippocampi of AD model mice. In the absence of CP, iron was overload, which might be associated with ROS generation; increased ROS activated the MAPK pathway leading to cellular apoptosis in the hippocampus and impairment of learning and memory. Meanwhile, CP deficiency increased the expression of BACE-1 and then exacerbated the aggregation of Aβ, hence aggravated CP deficiency induced iron overload and resulting ROS activated MAPK pathway, which formed a vicious cycle so as to exacerbate the cell apoptosis. Overexpression of CP had a prominent protective role against Aβ- and APP-induced cell damage in the hippocampus. These findings clearly revealed that CP could be a target to develop therapies that prevent and treat AD (Fig. 9).

Materials and Methods
Human specimens
Postmortem paraffin sections of the hippocampus from three patients (male, average age 80 years) with a definitive diagnosis of AD and three control individuals (male, average age 75 years) were obtained from the laboratory of Zhanyou Wang (Northeastern University, Shenyang, China). Each section was cut into 5-μm-thick slices. CP protein expression was detected by double immunofluorescence staining as described below.
Experimental animals and grouping
All procedures were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Animal Care and Use Committee of the Hebei Science and Technical Bureau in China. All animals were housed in pairs in stainless steel cages at 21°C ± 2°C and provided free access to food and water. Rooms were humidity controlled under a 12-h light and 12-h dark cycle.
The CP−/− mice were obtained from Kunihiro Yoshida (Department of Brain Disease Research, Shinshu University Hospital, Japan) and Shin'ichi Takeda (National Center of Neurology and Psychiatry, National Institute of Neuroscience, Japan). These CP−/− mice were backcrossed onto a BALB/cJ x 129SvJ background. The mice were bred and the genotype was identified as previously described (Supplementary Fig. S1B) (6). Ten-month-old male WT mice and CP−/− mice were randomly divided into two groups, and 10 μg aggregated Aβ25–35 (2 μg/μL, 5 μL) or 5 μL ACSF was infused into the right lateral cerebral ventricle (0.5 mm posterior, 1.0 mm lateral, and 2.0 mm ventral to bregma). In addition, the CP plasmid, which was designed to yield tissue-specific expression in astrocytes, was injected into the left lateral ventricle, and the control group was injected with an empty plasmid (pGFAP). After injection of GFAP-CP/pGFAP plasmids for 48 h, Aβ25–35 was injected into the right lateral cerebral ventricle to induce AD.
Behavioral tests were performed 2 weeks after injection using the Morris water maze assessment to evaluate the learning and memory capacities of the animals.
APP transgenic mice obtained from the laboratory of Zhanyou Wang were backcrossed onto a c57BL/6 background and crossed with CP+/− mice to generate APP-CP+/+ and APP-CP−/− mice. The primers for genotype identification are listed in Table 1. Six-month-old male APP-CP+/+ and APP-CP−/− mice were subjected to behavioral analysis using the Morris water maze, and other tests were performed on hippocampal tissues.
CP, ceruloplasmin.
Morris water maze assessment
The Morris water maze assessment is a widely accepted method for testing memory function in rodents. The test was performed using the SMART-CS (Panlab, Barcelona, Spain) program, and two methods were used (30, 38). A circular plastic pool (height 35 cm, diameter 100 cm) painted a dark color was filled with water at 22°C–25°C. An escape platform (height 14.5 cm, diameter 4.5 cm) was submerged 0.5–1 cm below the surface of the water.
Morris water maze assessment was performed as previously described by Wang et al. (38). At first, mice were trained for 2 days to remember the visible platform, which was placed in the center of the northwest quadrant of a tank that was filled with opaque water. From the third to seventh day, the platform was placed just below the water surface (hidden platform) for the place navigation test, and each mouse was subjected to three trials per day with an intertrial interval of 1 min. For each trial, the latency to escape to the hidden platform and the path length were recorded. On the eighth day, the platform was removed from the tank for the probe trial. The number of times the animal crossed the center of the northwest quadrant within 120 s was recorded. Finally, differences in results for the escape latency, path length, and number of passes between groups were analyzed statistically.
pEGFP-GFAP-CP constructs and transfection
A human cytomegalovirus (CMV) immediate promoter-driven plasmid named pEGFP-N1 (U55762.1) was used. The human GFAP promoter was amplified by polymerase chain reaction (PCR) using primers of the sequence 5′-GAGCTCCCACCTCCCTCTC-3′ and 5′-CCTGCTCTGGCTCTGCTC-3′ to transform the CMV promoter to the human GFAP promoter (pEGFP-GFAP). The full-length mouse CP coding sequence was amplified by PCR and cloned into the NotI (XbaI) and multiple cloning sites (MCSs) of the pEGFP-GFAP vector (pEGFP-GFAP-CP). The pEGFP-GFAP-CP plasmid was injected into the left lateral ventricle of mice followed by electroporation of instantaneous current (100 V, 10 ms) with an Electro Square Porator ECM 830 (BTX, San Diego, CA). The control group was injected with an empty plasmid (pGFAP). Forty-eight hours after the injection of GFAP-CP/pGFAP plasmids, Aβ25–35 was injected into the right lateral cerebral ventricle to induce the AD model. CP protein levels were detected 2 weeks after the Aβ25–35 injection.
Brain dissection and slide preparation procedures
Mice were anesthetized by sodium pentobarbital (40 mg/kg, i.p.). For histological and biochemical staining, mice were initially perfused with ice-cold normal saline (0.9% NaCl), until the effluent from the right atrium was clear of blood, and perfusion was then continued with 4% paraformaldehyde in 0.1 M phosphate buffer (PB, pH 7.2–7.4). At the end of the perfusion, the hippocampus was removed from the same coordinates and continuously postfixed in the same fixative solution. After rinsing in 0.1 M PB (pH 7.2–7.4), the hippocampus was submerged in 30% sucrose in 0.1 M PB for 2 days until the organ sank to the bottom of the container. Next, brain sections were cut coronally to a 20 μm thickness using a freezing microtome (Leica CM1900, Germany). For molecular biological analysis and measurement of iron and ROS, the mice were perfused with ice-cold normal saline (0.9% NaCl) in distilled water only until the effluent from the right atrium was clear of blood, and then hippocampi were dissected and used for detection.
Immunohistochemistry and immunofluorescence
Three days after the Morris water maze test, all the mice were perfused and the tissues were fixed as described above. The hippocampus sections were subjected to immunohistochemistry as described previously (38, 41). The sections were treated with 3% hydrogen peroxide (H2O2) for 20 min to reduce endogenous peroxidase activity. After rinsing with phosphate-buffered saline (PBS), the sections were treated with mixed serum (bovine serum:horse serum = 1:1) for 60 min. The sections were incubated overnight at 4°C with a rabbit anti-mouse Aβ antibody (1:150; Abcam, Cambridge, United Kingdom) and then incubated with biotinylated goat anti-rabbit serum (1:200; Zymed Laboratories, South San Francisco, CA) for 60 min at 37°C. Thereafter, sections were treated with avidin-biotinylated horseradish peroxidase complex (1:200; Zymed Laboratories) for 60 min at 37°C and then stained using a 3, 3′-diaminobenzidine tetrahydrochloride (DAB) kit.
For the immunofluorescence assay, rabbit anti-mouse GFAP antibody (1:100; Millipore, Temecula, CA) and goat anti-human CP antibody (1:200; Sigma-Aldrich, St. Louis, MO) were used as primary antibodies. FITC-conjugated and rhodamine-conjugated secondary antibodies were used. Negative controls were processed using the same procedures, but normal serum instead of specific antibodies was used.
Finally, brain sections immunostained with Aβ antibody were photographed with a Leica Aperio CS2 microscope (Germany), and microscopic images were processed with the software Aperial Scanner Console (Ver 102. 0. 4. 6). Brain sections with TUNEL assay were photographed with a Zeiss Imager A2 microscope (Germany). Parallel areas were selected for imaging in the hippocampus. The photographs were analyzed using Image-Pro Plus software, and the Automated Optical Inspection was set as the whole image.
Thioflavin staining
Thioflavin staining of brain tissue reveals amyloid plaques and neurofibrillary tangles associated with AD. Brain sections were first washed three times for 5 min each in distilled water and then incubated in filtered 1% aqueous Thioflavin-S for 8 min at room temperature in darkness. The sections were then rinsed thoroughly in PBS and dehydrated through a series of graded alcohols (5 min each in 70% and 80%, followed by 10 min each in 90%, 95%, and 100% ethanol) before being immersed in xylene (twice for 10 min each). The treated sections were then coverslipped with Canada balata. Every step after thioflavin staining was performed in darkness.
Detection of apoptosis
The presence of apoptosis in the hippocampus was assessed by the terminal deoxynucleotidyl transferase-mediated FITC-dUDP nick-end labeling (TUNEL) method as previously described (41). Nuclei were stained with DAPI. The number of TUNEL-positive/DAPI-stained neurons was counted. The counting area was based on the hippocampus tissues from the control groups, which was located at the same position as in the other groups.
Western blotting
Protein expression was assessed by Western blotting as previously described (41). The following primary antibodies were used: FTL from Epitomics (Cambridge, MA); DMT1(+IRE), DMT1(−IRE) and FPN1 from Alpha Diagnostic International (San Antonio, TX); TfR1 from Zymed Laboratories; P-Erk/Erk, P-p38/p38, and APP from Cell Signaling Technology; Bcl-2 and Bax from Santa Cruz Biotechnology (Santa Cruz, CA); BACE-1 from Proteintech (Chicago, IL). The primary antibody was incubated with the samples at 4°C overnight, and then, the corresponding horseradish peroxide-conjugated IgG (Amersham, United Kingdom) was added for incubation at room temperature for 90 min. All experiments were conducted at least three times. Relative light densities of the positive bands were calculated and expressed as a ratio to β-actin (Sigma-Aldrich).
Measurement of iron content in hippocampus tissues
The total iron content in the hippocampus was determined using ICP-MS (Thermo Fisher, X Series, FL) as described previously (9). Before the experiments, all of the containers were soaked with 15% nitric acid for 24 h, washed with deionized water, and then rinsed with ultrapure water before drying. Approximately 20 mg of each hippocampus sample was added to 1 mL ultrapure nitric acid (69.9%–70.0%; J.T. Baker, Phillipsburg) in Teflon digestion tubes and digested using a microwave digestion system for 2 h at 100°C and at 180°C until dry. The digested samples were diluted to 10 mL with ultrapure water. Standard curves ranging from 0 to 100 ppb were prepared by diluting the iron standard (1 mg iron/mL) with blanks prepared from homogenization reagents in 0.2% nitric acid. Standards and digested samples were analyzed in triplicate by ICP-MS.
Ferric iron in the hippocampus was assessed using Perl's iron staining with modifications (24). The sections were first treated with 3% H2O2 for 30 min. After rinsing, the sections were immersed in the mixed reaction liquid (4% potassium ferrocyanide:4% hydrochloric acid = 1:1) at 37°C for 12 h and then stained using a DAB kit.
Measurement of LPO, MDA, and SOD levels
The levels of MDA, LPO, and T-SOD were measured using commercial kits according to the manufacturer's instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). MDA as a marker of lipid peroxidation (11) was measured with thiobarbituric acid as previously described (39). LPO is a proven index to measure oxidative stress in cells and tissues, and it is closely related to the pathological process of several diseases (27). SOD can catalyze the dismutation of superoxide into oxygen and hydrogen peroxide, which provides an important antioxidant defense in cells exposed to oxygen (21). The level of SOD was measured with the hydroxylamine method. The hippocampus tissue was homogenized in 1:9 (w/v) ice-cold saline. The homogenate was centrifuged at 3000 g for 15 min (4°C), and the supernatant was collected to measure the level of MDA, LPO, and T-SOD. Finally, the absorbance was detected with a microplate reader at the wavelengths of 532, 586, and 550 nm, respectively. Blank and control samples were treated in parallel and used for calculation.
Statistical analysis
All experiments were performed at least in triplicate. Data were calculated as mean ± standard error of the mean. Differences between means were determined using one-way ANOVA (data in Figs. 2, 4, and 6A) and Student's t-test (the data in the other figures). A probability level of 5% (p < 0.05) was considered significant.
Footnotes
Acknowledgments
We thank Kunihiro Yoshida (Shinshu University Hospital, Japan) and Shin'ichi Takeda (National Institute of Neuroscience, Japan) for providing the CP−/− mice. This work was supported by the National Natural Science Foundation of China (31520103908, 31471035, 31528013, and 31271473).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.