
Abstract
Aims:
Oxidative stress is considered the major cause of tissue injury after cerebral ischemia. The nuclear factor erythroid 2-related factor 2 (Nrf2) pathway is one of the most important defensive mechanisms against oxidative stresses and has been confirmed as a target for stroke treatment. Thus, we desired to find new Nrf2 activators and test their neuronal protective activity both in vivo and in vitro.
Results:
The herb-derived compound, Britanin, is a potent inducer of the Nrf2 system. Britanin can induce the expression of protective enzymes and reverse oxygen–glucose deprivation, followed by reperfusion (OGD-R)-induced neuronal injury in primary cortical neurons in vitro. Furthermore, the administration of Britanin significantly ameliorated middle cerebral artery occlusion–reperfusion (MCAO-R) insult in vivo. We report here the crystal structure of the complex of Britanin and the BTB domain of Keap1. Britanin selectively binds to a conserved cysteine residue, cysteine 151, of Keap1 and inhibits Keap1-mediated ubiquitination of Nrf2, leading to induction of the Nrf2 pathway.
Innovation:
Britanin is a potent inducer of Nrf2. The complex crystal structure of Britanin and the BTB domain of Keap1 help clarify the mechanism of Nrf2 induction. Britanin was proven to protect primary cortical neurons against OGD-R-induced injury in an Nrf2-dependant way. Additionally, Britanin had excellent cerebroprotective effect in an MCAO-R model.
Conclusion:
Our results demonstrate that the natural product Britanin with potent Nrf2-activating and neural protective activities both in vitro and in vivo could be developed into a cerebroprotective therapeutic agent. Antioxid. Redox Signal. 27, 754–768.
Introduction
O
The natural product Britanin is a novel nuclear factor erythroid 2-related factor 2 (Nrf2) activator that potently induces the Nrf2 pathway at low nontoxic doses. In addition, the crystal structure of the complex of Britanin and the BTB domain of Keap1 was characterized, which not only revealed the molecular mechanism of Britanin in Nrf2 induction but also provided structural insight into the regulatory mechanism of Nrf2. Above all, Britanin can protect primary cortical neurons against oxygen–glucose deprivation, followed by reperfusion (OGD-R)-induced injury in vitro, and ameliorate brain ischemia/reperfusion injury in vivo. Hence, Britanin is a promising novel lead for the development of a drug against diseases related to oxidative stress.
The nuclear factor erythroid 2-related factor 2 (Nrf2) pathway is an essential cellular system in place to protect tissues from oxidative stress. Under regular conditions, Nrf2 is negatively regulated to a low level through its interaction with Keap1. However, when cells are exposed to oxidants or electrophiles, the transcription factor Nrf2 is triggered by oxidative stress and it binds to the antioxidant response element (ARE) in enhancer sequence, controlling the transcription of numerous antioxidant enzyme genes (21). Nrf2 protects a variety of organs, including brain tissue (23). Enhancive Nrf2 activity protects against cerebral ischemia in vivo (35). Compared with neurons from wild-type mice, primary cultured neurons derived from Nrf2 knockout mice were more vulnerable to oxidative stress (24). Furthermore, Nrf2−/− mice are notably more likely to have ischemic brain injury than their counterpart mice (37).
Over the past few years, numerous small molecules have been recognized as Nrf2 activators. Some of them are being investigated in clinical trials, such as methyl ester of fumaric acid and RTA408 (10). Meanwhile, some natural compounds have been found to have the capacity to reduce the level of ROS and protect against ischemia/reperfusion injury both in vitro and in vivo (1, 33). Since 2004, our research group has isolated >300 sesquiterpenoids from 10 Inula species distributed across China, and many plants of the genus Inula have long served as folk medicine (40).
In the present study, we found that the natural product Britanin is a potent activator of the Nrf2 pathway. Britanin can protect primary cortical neurons against oxygen–glucose deprivation, followed by reperfusion (OGD-R)-induced neuronal injury in vitro, and ameliorate cerebral ischemia–reperfusion injury significantly in vivo via activation of the Nrf2 pathway.
Furthermore, we report here the covalent complex crystal structure of Britanin and the BTB domain of Keap1, which provides structural insight into the regulatory mechanism of Nrf2 activation. Our results demonstrate that Britanin can covalently bind to the conserved cysteine 151 residue of Keap1, disturbing the capacity of Keap1 to serve as an adaptor for Cul3-Keap1 ubiquitin ligase complex and resulting in activation of the Nrf2 protective pathway. Our findings reveal that Britanin may be used as a lead compound for the discovery of new therapeutic drugs for the treatment of stroke and oxidative stress-related disorders.
Results
Britanin protects primary cortical neurons against OGD-R-induced cytotoxicity
The damage of neurons in ischemic conditions is due to the decrease of glucose and oxygen. OGD-R insult is considered to simulate the pathological process of ischemia in vitro (29). We used the OGD-R model in rat primary cultured cortical neurons to evaluate the neuronal protection mediated by Britanin. To investigate the activity and structure relationship of Britanin, we synthesized an analog of Britain, which was named Br1. Britanin and Br1 have a similar structure, except for the C11–C13 double bond of Britanin, which was reduced to a single bond in Br1 (Fig. 1A). We first evaluated the cytotoxicity of Britanin and Br1 in cortical neurons to determine suitable in vitro treatment doses by using a methyl tetrazolium (MTT) reduction assay. As shown in Figure 1B, no noteworthy cell toxicity was detected after treatment with Britanin below 5 μM. Moreover, the viability of cortical neurons was not affected by treatment with Br1 up to 10 μM. Therefore, 5 μM or less of the compounds was selected for subsequent bioassays.

After culture in neurobasal medium for 7 days, the cortical neurons were exposed to 1.5 h of OGD insult in the following experiments. Cortical neurons were pretreated with Britanin or Br1 for 6 h before OGD insult or post-treated with the compounds at the beginning of reperfusion and maintained for 6 h. The cell viability was measured by MTT assay. As shown in Figure 1C, pretreatment or post-treatment of Britanin (2.5 or 5 μM) protected the neurons against OGD-R-induced damage. In addition, 5 μM Br1 did not improve the survival rate of primary cortical neurons. The cell-released lactate dehydrogenase (LDH) concentration associates linearly with the number of death neurons (19). Thus, neuronal death was evaluated using LDH assay kit. As shown in Figure 1D, Britanin can reduce the amount of LDH released by primary cortical neurons in a dose-dependent manner. However, 5 μM Br1 cannot alter the LDH level in the medium.
OGD-R insult can induce elevation in intracellular ROS levels and mediate caspase-3 pathway activation, which lead to the death of cortical neurons (27). The formation of ROS in primary cortical neurons after OGD-R exposure was investigated using a fluorescent probe, 2′,7′-dichlorofluorescein diacetate (DCF-DA). The DCF fluorescence was subsequently measured by flow cytometry or analyzed by fluorescence microscopy. As shown in Figure 1E and F, the ROS level in cortical neurons was notably elevated after OGD-R insult. Post-treatment with Britanin at the beginning of reperfusion can reduce the level of ROS in a dose-dependent manner. Additionally, 5 μM Br1 did not alter the level of ROS. Our data demonstrated that the OGD-R-induced oxidative stress in neurons was rescued by Britanin. Meanwhile, we also explored the level of proapoptotic protein caspase-3 and Bax by immunoblot assay. The expression of activated caspase3 and Bax was increased by OGD-R treatment, while treatment with Britanin inhibited the upregulation of activated caspase3 and Bax in neurons (Fig. 1G). Taken together, these data indicate that both pretreatment and post-treatment with Britanin protect primary neurons against OGD-R-induced cytotoxicity.
Britanin activates the Nrf2 pathway and induces expression of protective enzymes
To determine whether the ROS reduction effect of Britanin is due to its free radical scavenging activity, we performed a DPPH (2,2-diphenyl-1-picrylhydrazyl) assay. Vitamin E, commonly used as an antioxidant for medical purposes, was used as a positive control. As shown in Figure 2A, Britanin had no free radical scavenging effect, even at a concentration of 1 mM.
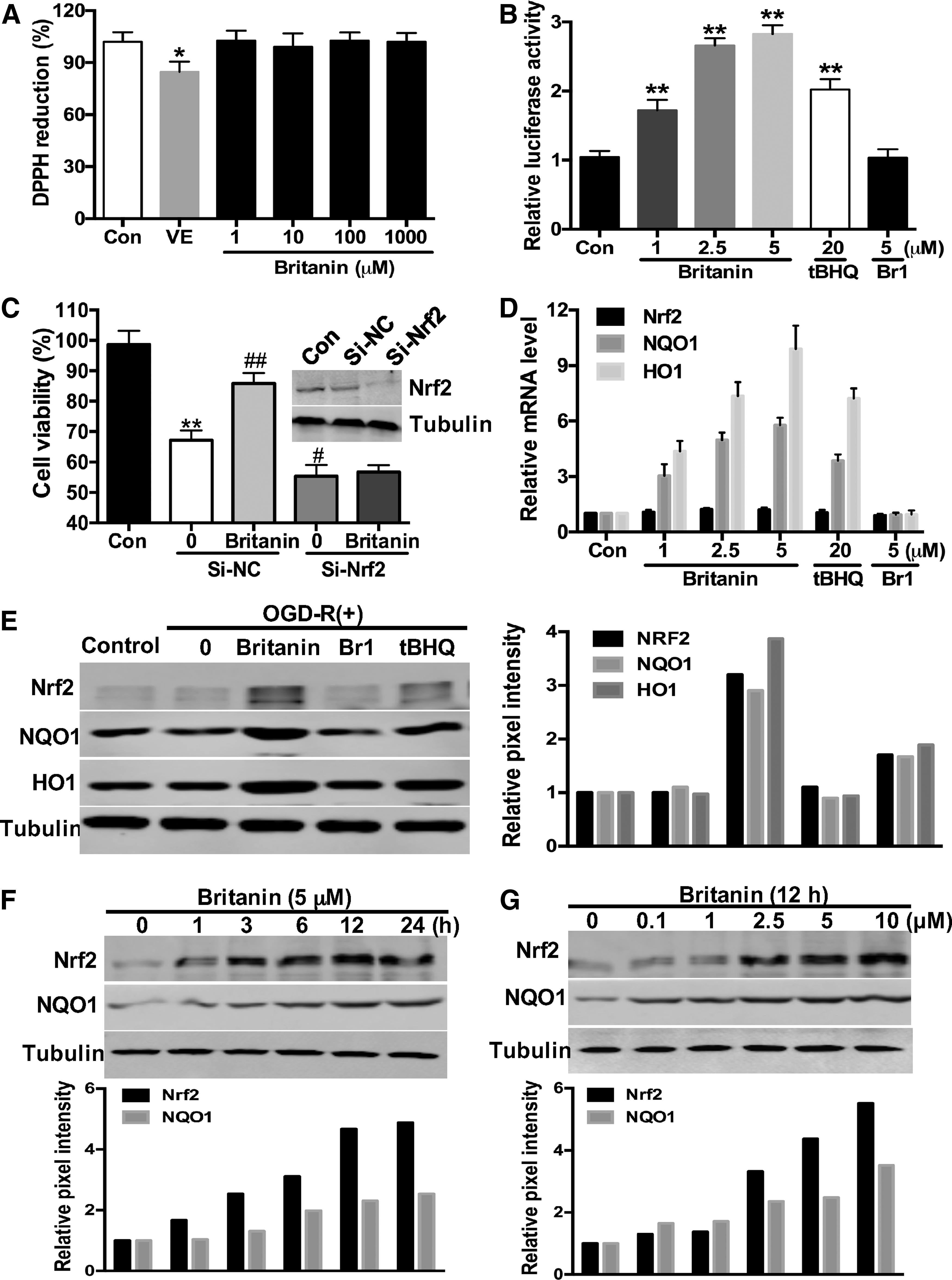
To clarify the ROS reduction mechanism of Britanin, we performed a dual-luciferase reporter gene assay in primary cortical neurons to test the ability of Britanin and Br1 to induce Nrf2 transcriptional activity. Britanin can induce ARE-luciferase reporter gene activity in a dose-dependent manner (Fig. 2B). Compared with tert-butylhydroquinone (tBHQ), a quinone compound that protects human neural stem cells against oxidative stress-induced death via the stabilization of Nrf2 (25), Britanin is a potent Nrf2 inducer with a maximum of nearly threefold induction. Meanwhile, 5 μM of Br1 has no ARE-luciferase inducing activity. These results indicate that Britanin reduces the ROS level by activating the Nrf2 antioxidant pathway.
To investigate whether the neural protective activity of Britanin is Nrf2 dependent, we knocked down the expression of Nrf2 in cortical neurons by specific small interfering RNA. Primary neurons were more vulnerable to OGD-R-induced insult after si-Nrf2 transfection. Meanwhile, the Britanin-mediated protection was lost in neurons transfected with si-Nrf2 (Fig. 2C). These data suggest that the neural protective effect of Britanin is Nrf2 dependent.
As a transcription factor, Nrf2 activates mRNA synthesis and controls the expression of a panel of antioxidant enzymes, such as NAD(P)H: quinone oxidoreductase 1 (NQO1) and heme oxygenase-1 (HO1) (16). Next, we utilized quantitative real-time reverse transcriptase–polymerase chain reaction (qRT-PCR) assay to investigate the mRNA levels of Nrf2, NQO1, and HO1 in neurons treated with the compounds. As shown in Figure 2D, Britanin had no effect on the mRNA level of Nrf2, but it increased the mRNA levels of HO1 and NQO1, indicating that Britanin activated the Nrf2 pathway through upregulation of the protein level of Nrf2.
We performed immunoblot assay to examine the effects of Britanin on the expression of these proteins. Five micromolars of Britanin was more potent than 20 μM tBHQ in elevating the protein expression levels of Nrf2, NQO1, and HO1, while 5 μM of Br1 had no effect (Fig. 2E). A time course study of Britanin (5 μM) showed that the protein levels of Nrf2 and NQO1 were augmented as early as 1 h and reached the maximum level at 24 h (Fig. 2F). Additionally, the protein levels of these proteins increased in a dose-dependent manner after exposure to Britanin for 12 h (Fig. 2G). Taken together, these data indicate that Britanin is a novel Nrf2 inducer and protects primary neurons against OGD-R-induced injury in an Nrf2-dependent way.
Britanin directly targets Keap1 at Cys151
There are two ways to activate Nrf2: phosphorylation of Nrf2 by protein kinases and stabilization of Nrf2 via Keap1 cysteine thiol modification (38). Several upstream protein kinases, such as phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) (34), PRKR-like endoplasmic reticulum kinase (PERK) (9), casein kinase 2 (CK2) (2), protein kinase C (PKC) (15), and mitogen-activated protein kinases (MAPK) (42), can induce Nrf2 phosphorylation and facilitate subsequent transcriptional induction of Nrf2. The activation of Nrf2 by Britanin might come from its direct interaction with upstream kinases. Next, we investigated the interaction between Britanin and different kinases using DiscoveRx's KINOMEscan screening platform at 30 μM in vitro. Britanin showed no direct interaction with any of the protein kinases that may mediate Nrf2 phosphorylation (Supplementary Table S1; Supplementary Data are available online at
The result of kinase screening assay indicated that the Nrf2 activation effect of Britanin may be via Keap1 cysteine thiol modification. Cysteine thiols of Keap1 are covalently modified by thiol-reactive electrophilic centers of many reported small-molecule ARE inducers (38). Keap1 is a cysteine-rich protein and negatively controls the induction of Nrf2. Some biotin-tagged labeling probes make it possible to map reactive cysteines of Keap1, such as iodoacetamide and N-alkylmaleimide (14). The bioactivity data obtained with Britanin and Br1 suggested that the α-methylene-γ-lactone moiety in Britanin was a fundamental thiol-reactive electrophilic center for Nrf2 activation, supporting the assumption that Nrf2 induction by Britanin is quite likely through a Michael addition modification of one of the cysteine residues in Keap1.
We, therefore, synthesized one biotin-labeled probe where a linker was attached to the hydroxyl of Britanin to detect the interaction between Britanin and Keap1 (Fig. 3A). We used the ARE-luciferase reporter gene assay to confirm whether the probe retained the activity to induce the Nrf2 pathway. As shown in Figure 3B, the probe was cell permeable and retained the activity in Nrf2 induction.

To assess whether Britanin directly binds to Keap1, we transfected 293T cells with an Myc-Keap1 plasmid. The cell lysates were incubated with free biotin or probe and then pulled down using streptavidin-coated agarose beads, followed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) separation and Keap1 immunoblotting. As shown in Figure 3C, the band of Keap1 was obviously settled by probe, but not by biotin, with a molecular mass of ∼75 kDa. The band was competed away by high levels of Britanin, but not Br1, demonstrating that the Keap1 protein bound to the probe and Britanin at the same time.
Human His-Keap1 was expressed in BL21 cells for further analyses. We found that the probe interacted with recombinant Keap1 in a dose-dependent manner. The purified His-Keap1 protein was incubated with increasing concentrations of probe, which was followed by SDS-PAGE and biotin immunoblotting. As shown in Figure 3D, a clear band at ∼75 kDa was distinguished using an anti-Biotin antibody in the presence of probe. The signal intensity of this band augmented with growing concentrations of the probe, indicating that the probe can interact strongly with recombinant Keap1.
To further identify the residues that are critical for the binding process, we made an impartial mass spectrometry (MS) analysis to detect cysteine residues modified by Britanin in vitro. The MS analysis of chymotryptic peptide covering Cys151 (amino acids 151–169) showed an increase of 366 in mass, demonstrating that Cys151 was alkylated by Britanin (Fig. 3E).
Apart from Cys151, covalent modifications of Britanin at C77, C226, C288, C368, C489, C583, and C613 were similarly discovered from the MS study, although with less intensity. Covalent modification of these seven cysteines could similarly serve to inactivate Keap1. We separately mutated these cysteine residues into alanine residues to investigate which of these cysteines were indispensable for binding with Britanin. Amid these mutants, C151A was the only mutant that totally lost the capacity to bind with Britanin, while other mutants retained their high binding affinity for Britanin (Fig. 3F), which directed that Cys151 of Keap1 is indispensable for its interaction with Britanin.
The binding mode of Britanin with Keap1 BTB domain revealed by X-ray crystallography
Keap1 comprises five domains, including a Kelch repeat domain, a central linker domain, a BTB domain, an N-terminal domain, and a C-terminal domain (17). The structure of full-length Keap1 (32) and the structure of the Keap1 BTB domain in binding with CDDO (7) have been reported. For the purpose of obtaining greater knowledge of the interactions between Britanin and Keap1, we elucidated the crystal structure of the Keap1 BTB domain in complex with Britanin. A single point mutation was presented at residue 172 (S172A) according to the previous literature (7). The recombinant protein was generated in BL21 cells and was purified as indicated in the Materials and Methods section. The crystals were grown using the hanging drop method. We cocrystallized Keap1 BTB domain with Britanin and determined the crystal structure at 2.19 Å resolution (Fig. 4A).
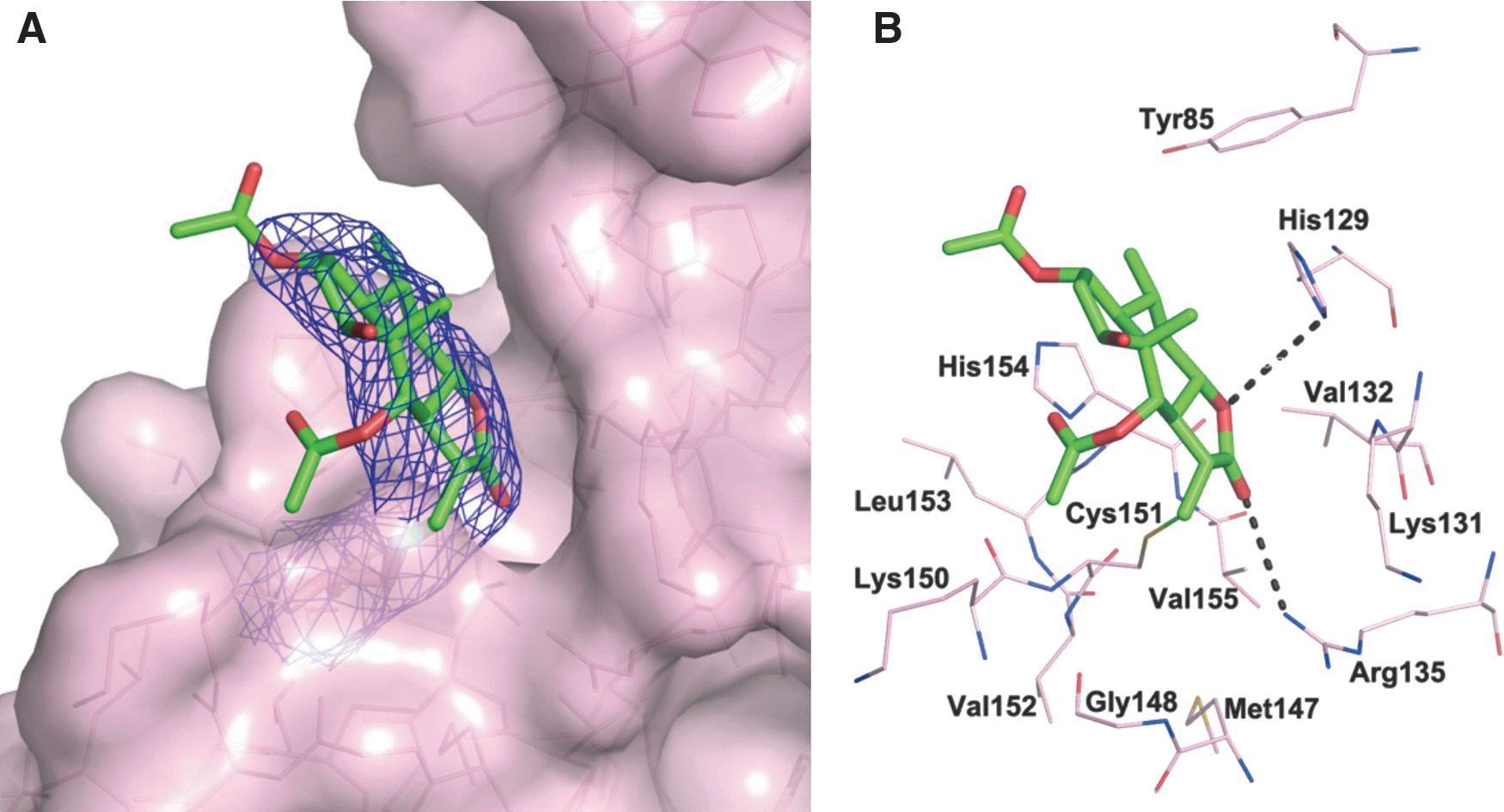
The crystal structure demonstrates that Britanin occupies a shallow groove formed by the residues Tyr85, His129, Lys 131, Val132, Arg135, Met147, Gly148, Cys151, Leu153, and His154 (Fig. 4B). The ring systems of Britanin are accommodated very well in the groove with excellent electron density, but two acetate groups of Britanin are relatively flexible and exhibit very weak electron density. A continuous electron density between Sγ of Cys151 and the Michael acceptor group of Britanin unambiguously indicates that Cys151 is covalently modified by Britanin. In addition to Cys151, there are no covalent modifications for other cysteine residues (Cys77 and Cys171) presented in the crystallographic construction, which are in agreement with the result of cysteine residue mutant assay.
In addition to this covalent bond, hydrogen bonding interactions are involved in the binding of the Keap1 BTB domain and Britanin binding. One hydrogen bond is found between nitrogen of Arg135 and β-oxygen of Britanin (rO…N = 3.5 Å), and another hydrogen bond is formed between nitrogen of His129 and γ-oxygen of Britanin (rO…N = 3.7 Å), which could enhance the binding affinity between Britanin and Keap1 BTB domain. Therefore, the crystal structure of the Keap1 BTB domain in complex with Britanin provides further evidence of Britanin directly targeting Keap1 at Cys151.
Britanin inhibits Keap1-mediated ubiquitination of Nrf2 by disrupting the interaction between Cul3 and Keap1
Evidence has shown that alkylation of the BTB domain in Keap1 by covalent modifiers supports the disruption of the Keap1-Cul3 contact, resulting in Nrf2 induction (13, 21). We then explore how Nrf2 induction was completed by Britanin. We formerly examined the influence of Britanin on the regulation of Nrf2 ubiquitination as Nrf2 stability is controlled through ubiquitin-mediated proteasomal degradation.
293T cells were cotransfected with the expression plasmids for FLAG-Nrf2, ubiquitin (Ub), and Myc-Keap1 and treated with 20 μM of the compounds, along with MG132 (5 μM), for 4 h. Cell lysates were pulled down using anti-FLAG beads, and the immunopurified Nrf2 was examined for the occurrence of ubiquitin by immunoblot analysis. As shown in Figure 5A, both tBHQ and Britanin powerfully repressed Keap1-dependent Nrf2 ubiquitination, while Br1 showed no inhibition effect. When 293T cells were transfected with the C151A-Keap1 mutant vector, neither tBHQ nor Britanin was able to defend Nrf2 from ubiquitination, showing that Britanin prevents Keap1-driven ubiquitination of Nrf2 in a C151-dependent manner (Fig. 5B). In summary, these outcomes reveal that Britanin inducted the Nrf2-mediated protective reaction by hindering its ubiquitination and consequently increasing Nrf2 stability.
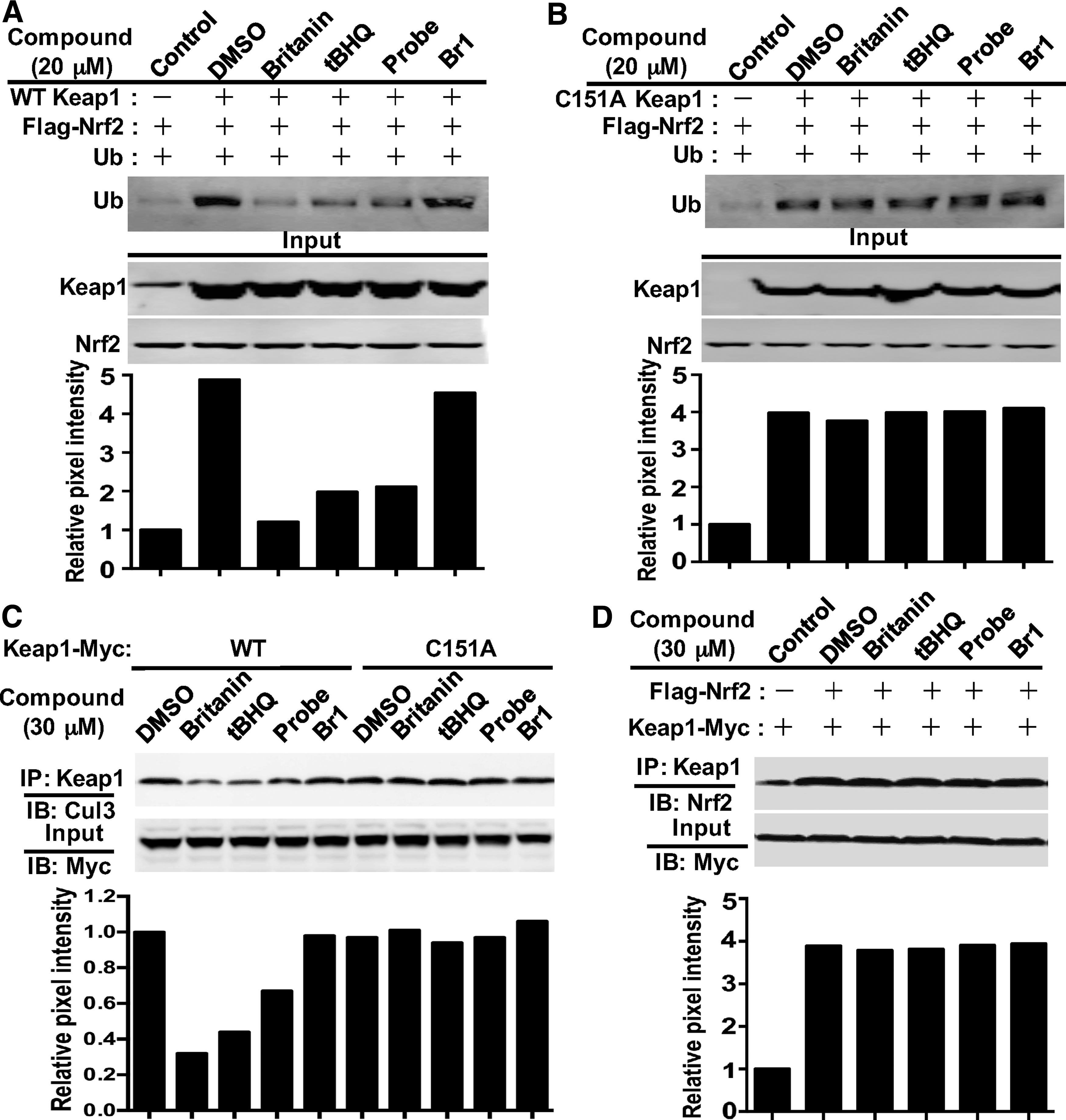
To investigate whether Britanin obstructs Keap1-mediated ubiquitination of Nrf2 by disturbing the contact between Cul3 and Keap1, 293T cells were transferred with vectors encoding Myc-Keap1 and HA-Cul3 and were subsequently treated with 30 μM of the compounds for 4 h. Immunoblotting with anti-Cul3 antibody after being pulled down with anti-Myc beads showed that Britanin, tBHQ, and probe triggered detachment of the Keap1-Cul3 contact, except for Br1. The dissociation of the Keap1-Cul3 contact was not detected when the C151A mutant plasmid was applied (Fig. 5C). This outcome proves that alkylation of Keap1 C151 by Britanin is necessary for separation of the Keap1-Cul3 complex, which is an important requirement for induction of the Nrf2 pathway.
The ability of Britanin to impact the interaction of Keap1 with Nrf2 was studied by directly interrupting the protein interaction of Keap1-Nrf2, and this can also lead to Nrf2 transcription activation (28, 18). We transfected 293T cells with expression plasmids for Flag-Nrf2 and Myc-Keap1 and treated the cells with 30 μM of the compounds for 4 h. The amount of Nrf2 protein was examined by immunoblot study following immunoprecipitation with the anti-Keap1 antibody. As shown in Figure 5D, Britanin, tBHQ, or the probe cannot induce separation of the Keap1-Nrf2 interaction. Thus, these outcomes support that alkylation by Britanin did not trigger separation of the Nrf2-Keap1 complex.
Britanin ameliorates cerebral ischemia–reperfusion injury in vivo
Many animal models of ischemia stroke have been established to simulate human cerebral ischemia. In this experiment, we employed the middle cerebral artery occlusion–reperfusion (MCAO-R) model to mimic ischemia–reperfusion in vivo. Sprague-Dawley (SD) rats were subject to an insult of 2-h occlusion and 24-h reperfusion. After reperfusion for 24 h, the rats were sacrificed and their brains were isolated for the following research. The detailed procedures used with the MCAO-R model and the infarct volume measurement are described in the Materials and Methods section.
First, we conducted a simple dose–response study of Britanin in vivo. SD rats were orally administrated with Britanin at dosages of 25 or 50 mg/kg at the onset of occlusion and dosed twice after reperfusion for 8 h. Infarction was judged by the appearance of white regions after 2,3,5-triphenyltetrazolium chloride (TTC) staining at 24 h after reperfusion. The administration of Britanin at either a low (25 mg/kg) or high (50 mg/kg) dose notably decreased the infarction volume ratios compared with the MCAO group (Fig. 6A, B). Moreover, Britanin treatment significantly ameliorated the neurological deficit score at the two doses investigated (Fig. 6C). Next, we evaluated the capability of Britanin to activate the Nrf2 pathway in vivo. Administrations of Britanin over MCAO-R phase inducted the Nrf2 pathway, as verified by results that the protein levels of Nrf2, NQO1, and HO1 were augmented in the brains (Fig. 6D).

Using malondialdehyde (MDA) offers a convenient way of assessing lipid peroxidation levels in biological materials. As shown in Figure 6E, the content of MDA increased in the brains after MCAO-R treatment, both doses of Britanin significantly reduced the levels of MDA. In addition, cell death was determined using TdT-mediated dUTP-X nick end labeling (TUNEL) staining. As shown in Figure 6F and G, TUNEL staining was undetectable in sham-operated rats, whereas the number of dying cells notably augmented in the infarct region of the MCAO group. Both dosages of Britanin decreased the quantity of dying cells in the infarct region. These results showed that both 25 and 50 mg/kg of Britanin can improve the infarct volume and neurological deficit in rats after MCAO-R treatment. Additionally, there was no significant difference in the effect of the two doses of Britanin treatment.
Next, we investigated the therapeutic time window of Britanin in the MCAO model. Twenty-five milligrams per kilogram of Britanin was given once at different times according to the order shown in Figure 6H. A single-dose treatment of Britanin (25 mg/kg) produced substantial neuroprotection as late as at the onset of reperfusion. Unfortunately, Britanin given at 4 h after reperfusion did not significantly reduce infarct volume (Fig. 6I, J), nor did it significantly decrease the neurological deficit score (Fig. 6K). Taken together, our results demonstrate that Britanin had a notably cerebroprotective role during ischemia-induced injury, but with a relatively narrow therapeutic time window.
Discussion
Enhanced levels of ROS production have been closely related to the pathophysiology of stroke (8). Different pharmacological approaches have been tried to restore such systemic redox imbalances, typically with antioxidant drugs or vitamins. However, intervention trials based on chemical scavenging of pro-oxidant molecules have been mainly fruitless or even harmful (22, 36). Consequently, many investigators have sought to discover new specific pharmacological agents (10). Among them, activating the master redox switch Nrf2 is a possible approach to reinforce redox homeostasis.
In this study, we focus on a natural product called Britanin, which activates the Nrf2 protective pathway by covalent modification of Keap1. Britanin was originally isolated from Inula lineariifolia Turcz, an herb used in the traditional Chinese medicine JinFeiCao (31). This study is the first proof that Britanin can activate the Nrf2 antioxidant pathway via Keap1 cysteine thiol modification. Britanin, a sesquiterpenoid with α-methylene-γ-lactone moiety that serves as Michael acceptor, is a newly discovered Nrf2 activator with a unique structure.
By applying a biotin-labeled probe of Britanin, we discovered that Britanin directly targeted Keap1. Notably, the complex crystal structure of Britanin and the BTB domain of Keap1 provides direct evidence that Britanin formed a covalent bond with cysteine 151 of Keap1. Consistently, our MS and cysteine mutagenesis results demonstrated that Britanin selectively binds to C151. Britanin can disrupt the interaction between Cul3 and Keap1 and inhibit Keap1-mediated ubiquitination of Nrf2.
Nrf2 can protect brain tissue from oxidative injury and play an important protective role in stroke. Many small-molecule activators of Nrf2 have been previously described, but there are only three ester derivatives of fumaric acid and two synthetic triterpenoids that have reached clinical trials. Among them, dimethyl fumarate (DMF) was commercialized for the treatment of relapsing–remitting multiple sclerosis (4, 20), which brings hope for Nrf2 activator research.
Britanin represents a new type of sesquiterpenoid Nrf2 activator with a precise Keap1 modification mechanism. A kinase screening assay confirmed the high selectivity of Britanin for Keap1; thus, it has low off-target toxicity and extreme therapeutic potential. We confirmed that Britanin protected primary cortical neurons against OGD-R-induced neuronal damage in an Nrf2 pathway-reliant way in vitro. Britanin activated Nrf2 and induced expression of protective enzymes in neurons at nontoxic concentrations.
Additionally, our results demonstrated that Britanin can be utilized as a protective phytochemical to protect against ischemic brain injury in a rat MCAO-R model in vivo. Unfortunately, the therapeutic time window of Britanin was relatively narrow. Further research is now underway to expand the therapeutic time window of Britanin. We had synthesized some analogs of Britanin to find compounds with better efficacy and prolonged treatment window after cerebral ischemia. Additionally, it is necessary to clarify the characteristics of absorption, distribution, and metabolism of Britanin in vivo.
In conclusion, further study of Nrf2 activation activities of Britanin is appealing to exploit the uncharacteristic mechanisms of the Nrf2 antioxidant pathway. Additionally, Britanin is a promising pharmacological agent for use in improvement of therapies for prevention or treatment of stroke or other oxidative stress-related diseases.
Materials and Methods
Chemicals
Britanin was isolated from I. lineariifolia Turcz. The syntheses of Br1 and probe was implemented as described in Supplementary Figure S1. The NMR spectra and MS data of the compounds are shown in Supplementary Figures S2–S8. The compounds tBHQ and poly-
Cell culture
Cortical neurons were prepared from embryonic day 18 SD rats. Briefly, cortical tissues were dissected and dissociated with 0.125% trypsin. Then, the neurons were centrifuged and suspended in neurobasal medium (Invitrogen) containing 2% B27 supplement, 0.5% penicillin and streptomycin (50 U/ml penicillin and 50 μg/ml streptomycin), and glutamine (0.5 mM); 1.5 × 105 cells were then plated in poly-
Human embryonic kidney 293T cells were purchased from American Type Culture Collection (Manassas, VA). 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 100 U/ml penicillin and 100 μg/ml streptomycin. 293T cells were incubated at 37°C in a humidified incubator containing 5% CO2.
OGD-R treatment
After culture in neurobasal medium for 7 days, the cortical neurons were washed with glucose-free DMEM for three times. The medium was then replaced with glucose-free DMEM, and neurons were transferred to an incubator humidified with 94% N2, 5% CO2, and 1% O2 at 37°C. After 1.5 h, the medium was changed back to neurobasal medium. The cells were then returned to a normal incubator with 5% CO2 balanced with air at 37°C for 6 h of recovery time. The control cells were kept in neurobasal medium in a normal environment.
Cell viability and cytotoxicity assessment
Cell viability was measured by MTT assay. Primary cortical neurons were cultured with or without OGD-R treatment. A final concentration of 0.5 mg/ml MTT was added to each well and incubated at 37°C for 2 h. The quantity of MTT formazan dissolved in DMSO was measured at 495 nm using a microplate reader.
The LDH concentration in the medium after OGD-R insult was measured according to the protocols of an LDH kit (Dojindo Molecular Technologies). The medium was mixed with a nicotinamide adenine dinucleotide and lactate solution, and colorimetric absorbance was measured at 450 nm with a microplate reader.
Intracellular ROS detection
The fluorescent probe DCF-DA was applied to measure the intracellular ROS concentration. Briefly, the cultured cortical neurons were grown in six-well plates at a density of 1.5 × 105 cells/well. DCF-DA was diluted with neurobasal/B27 media to a final concentration of 10 μM. After OGD-R treatment, the cortical neurons were incubated with 10 μM DCF-DA at 37°C for 30 min in the incubator, and the cells were then washed twice with phosphate-buffered saline (PBS). Single cells were processed by flow cytometry to measure the DCF-DA fluorescence after trypsin digestion. A fluorescence microscope was also used to detect the level of DCF-DA fluorescence. The experiment was repeated three times.
DPPH reduction assay
For the DPPH radical scavenging assay, serial dilutions of the compounds in DMSO were mixed with 40 μg/ml DPPH in methanol in each well of a 96-well plate. The plate was stored in the dark for 15 min at 37°C, after which the absorbance of the solution was recorded at 540 nm in a microplate reader. Vitamin E was used as a positive control.
Cell transfection and dual-luciferase reporter gene assay
For transient transfection, Lipofectamine 2000 (Life Technologies) was used according to the protocol recommended. Si-Nrf2 and si-NC were designed according to the previous literature (6) and manufactured by Invitrogen. The sequences of si-Nrf2 are 5′-UUU GAG UCU AAG GAG UUC AGC UGG C-3′ (forward) and 5′-GCC AGC UGA ACU CCU UAG ACU CAA A-3′ (reverse). After transfection with siRNA for 18 h, cortical neurons were treated with 5 μM Britanin for 6 h before OGD-R insult.
For dual-luciferase reporter gene assay, 1.5 × 105 neurons were plated in six-well plates. The cells were cotransfected with 4 μg of the expression vectors for both ARE-firefly luciferase and TK-Renilla luciferase. Eight microliters of Lipofectamine 2000 (Life Technologies) was used according to the protocol. At 24 h post-transfection, the transfected cells were treated with the compounds at the indicated concentrations for an additional 12 h, and firefly and Renilla luciferase activities were then measured using the dual-luciferase reporter gene assay system (Promega). The firefly luciferase activity was normalized to Renilla luciferase activity.
Antibodies and immunoblot analysis
The antibody for Nrf2 was purchased from Santa Cruz Biotechnology. The antibodies for HO1, activated caspase3, Bax, and α-tubulin were purchased from Cell Signaling Technology. The antibodies for Keap1 and NQO1 were purchased from Abcam. For immunoblot analysis, the total protein was extracted from primary neurons and the ischemic penumbra of the rat cortex using RIPA buffer supplemented with phosphatase and proteinase inhibitors. The cell lysates were fractionated by SDS-PAGE and transferred to polyvinylidene fluoride membranes. The membranes were blocked in 5% nonfat milk TBST buffer for 30 min and incubated with primary antibodies overnight at 4°C. After washing with TBST buffer three times, the membranes were incubated with the appropriate secondary antibody for 1 h at room temperature.
RNA extraction and real-time PCR analysis
Primary cortical neurons were treated with different compounds at the indicated doses for 6 h. The total mRNA was extracted from neurons using TRIzol reagent (Sigma), and 1 μg of RNA was reverse transcribed to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems).
The primers used were synthesized by Sangon Biotech, and the sequence of primers is shown in Supplementary Table S2. Real-time PCR was performed using the SYBR Green Rox Mix (Life Technology), and duplicate reactions were executed for each sample. The figures presented are relative mRNA levels normalized to GAPDH.
Expression and purification of Keap1 BTB
The gene-encoding residues 48–180 of human Keap1 (BTB domain) were inserted into the vector pET28a with an N-terminal tobacco etch virus (TEV)-cleavable hexahistidine tag. A single point mutation was introduced at residue 172 (S172A). The recombinant protein was expressed in BL21 cells. The cells were incubated at 37°C until the value of OD600 reached to 0.8, and the cells were then induced with 0.5 mM IPTG for 18 h at 16°C. The harvested cells were suspended in lysis buffer (50 mM Tris/HCl, pH 8.0, 150 mM NaCl) and disrupted by a high-pressure cracker at 4°C. The cell debris was removed from the crude extract by centrifugation. The crude protein was loaded onto a HiTrap Chelating column (5 ml; GE Healthcare), and fractions containing the BTB domain of Keap1 were eluted with lysis buffer and 150 mM imidazole. The pooled fractions were concentrated to 5 ml and dialyzed against lysis buffer at 4°C in the presence of TEV protease. The protein was successively purified by gel filtration using a Superdex 75 16/60 column (GE Healthcare) equilibrated with 25 mM Tris/HCl, pH 8.0, 150 mM NaCl, and 2 mM TCEP. Fractions containing purified protein were concentrated to 10 mg/ml for crystallization.
Crystallization and data collection
The crystals were grown by the hanging drop method. Drops were formed by mixing precipitant solution (0.2 M lithium acetate and 16% PEG 3350) with an equal volume of Keap1 BTB (10 mg/ml) pre-equilibrated with 3 mM Britanin. Crystals suitable for X-ray diffraction appeared in 3 days after microseeding. The crystals were briefly soaked in mother liquor supplemented with 25% glycerol as a cryoprotectant. The data were collected on BL17U1 at the Shanghai Synchrotron Radiation Facility. The data collection statistics are presented in Table 1.
Structure determination
The raw data were processed using MOSFLM and SCALA program from the CCP4 suite (3, 41). The initial phases were obtained via the molecular replacement method using the PDB entry 4cxi. The complex structure of Keap1 BTB with Britanin was refined using REFMAC (30). After cycles of refinement and model building with Coot (12), the molecular graphic was prepared with PyMOL molecular graphics system to generate Figure 4.
Preparation of the recombinant wild type and the site-mutated Keap1
The His-Keap1 plasmid was purchased from Sino Biological and used as a template for site-directed mutagenesis performed using the QuikChange® site-directed mutagenesis kit (Stratagene). These proteins were expressed and purified using the same method as Keap1 BTB. The mutagenesis primers are described in Supplementary Table S3.
MCAO-R procedure and infarct volume measurement
Rat brain ischemia was produced by transient intracranial occlusion of the right middle cerebral artery, as formerly described (43). Rats were anesthetized with chloral hydrate, and a permanent ligature was employed in the ipsilateral common carotid to prevent blood running into the internal carotid artery. A filament was advanced 16–18 mm from the cervical carotid bifurcation until reaching resistance from the ostium of the MCA. After 2 h of MCAO, the seaming was gently removed to facilitate 24 h reperfusion. Sham rats were exposed to the same operation, but the seaming was not advanced beyond the internal carotid bifurcation. Rats were assessed with neurological scoring using the Longa's method in a blinded manner at 24 h after MCAO (26). After assessment of the neurological deficit, rats were sacrificed. Then, their brains were rapidly dissected on ice and sectioned into 2-mm coronal sections. Six coronal slices were made and subjected to TTC staining. The infarct volumes were measured by morphometric analysis as described (39).
TUNEL assay
Apoptosis of in situ cells was determined by using a fluorescein apoptosis detection kit (Roche) according to the protocol. Positive control slides were prepared by incubation with DNase I (Sigma) for 30 min at room temperature. The negative control slides received 2 μl of distilled water in place of 2 μl of TdT2 in the fluorescein-12-dUTP reaction mixture. Three arenas for each section were selected from the cerebral cortex. TUNEL-positive cells were counted using fluorescence microscopy at × 40 magnification. Total cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Beyotime).
Statistical analysis
The results are expressed as mean ± SD and were analyzed by the GraphPad Prism software. The unpaired Student's t-test was applied to compare the results of two groups. One-way analysis of variance with Bonferroni's correction was used to compare the means of three or more groups. p < 0.05 was considered to be significant.
Footnotes
Acknowledgments
The authors thank Professor Yongjun Dang (Fudan University) for providing Flag-Nrf2 and Ub plasmids and Professor Guoquan Yan (Fudan University) for LC-MS-MS analysis. They thank the staff of the Shanghai Synchrotron Radiation Facility for kind assistance and use of facilities. This work was supported by Professor of Chang Jiang Scholars Program, National Natural Science Foundation of China (81230090, 81520108030, 81573318, 81373301, 1302658, 81302651), Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (10DZ2251300), the Scientific Foundation of Shanghai China (12401900801, 13401900103, 13401900101, 16401901300), National Major Project of China (2011ZX09307-002-03), and the National Key Technology R&D Program of China (2012BAI29B06).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.