
Abstract
Aims:
Cigarette smoke (CS)-mediated acquired cystic fibrosis transmembrane conductance regulator (CFTR)-dysfunction, autophagy-impairment, and resulting inflammatory–oxidative/nitrosative stress leads to chronic obstructive pulmonary disease (COPD)-emphysema pathogenesis. Moreover, nitric oxide (NO) signaling regulates lung function decline, and low serum NO levels that correlates with COPD severity. Hence, we aim to evaluate here the effects and mechanism(s) of S-nitrosoglutathione (GSNO) augmentation in regulating inflammatory-oxidative stress and COPD-emphysema pathogenesis.
Results:
Our data shows that cystic fibrosis transmembrane conductance regulator (CFTR) colocalizes with aggresome bodies in the lungs of COPD subjects with increasing emphysema severity (Global Initiative for Chronic Obstructive Lung Disease [GOLD] I − IV) compared to nonemphysema controls (GOLD 0). We further demonstrate that treatment with GSNO or S-nitrosoglutathione reductase (GSNOR)-inhibitor (N6022) significantly inhibits cigarette smoke extract (CSE; 5%)-induced decrease in membrane CFTR expression by rescuing it from ubiquitin (Ub)-positive aggresome bodies (p < 0.05). Moreover, GSNO restoration significantly (p < 0.05) decreases CSE-induced reactive oxygen species (ROS) activation and autophagy impairment (decreased accumulation of ubiquitinated proteins in the insoluble protein fractions and restoration of autophagy flux). In addition, GSNO augmentation inhibits protein misfolding as CSE-induced colocalization of ubiquitinated proteins and LC3B (in autophagy bodies) is significantly reduced by GSNO/N6022 treatment. We verified using the preclinical COPD-emphysema murine model that chronic CS (Ch-CS)-induced inflammation (interleukin [IL]-6/IL-1β levels), aggresome formation (perinuclear coexpression/colocalization of ubiquitinated proteins [Ub] and p62 [impaired autophagy marker], and CFTR), oxidative/nitrosative stress (p-Nrf2, inducible nitric oxide synthase [iNOS], and 3-nitrotyrosine expression), apoptosis (caspase-3/7 activity), and alveolar airspace enlargement (Lm) are significantly (p < 0.05) alleviated by augmenting airway GSNO levels. As a proof of concept, we demonstrate that GSNO augmentation suppresses Ch-CS-induced perinuclear CFTR protein accumulation (p < 0.05), which restores both acquired CFTR dysfunction and autophagy impairment, seen in COPD-emphysema subjects.
Innovation:
GSNO augmentation alleviates CS-induced acquired CFTR dysfunction and resulting autophagy impairment.
Conclusion:
Overall, we found that augmenting GSNO levels controls COPD-emphysema pathogenesis by reducing CS-induced acquired CFTR dysfunction and resulting autophagy impairment and chronic inflammatory–oxidative stress. Antioxid. Redox Signal. 27, 433–451.
Introduction
C
Cigarette smoke (CS)-induced acquired cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction results in autophagy impairment and inflammatory–oxidative stress that mediates chronic obstructive pulmonary disease (COPD)-emphysema pathogenesis, but underlying mechanisms are still unclear. Our study identifies CS-induced accumulation of CFTR in perinuclear aggresome bodies as a novel causal mechanism of acquired CFTR dysfunction. Moreover, our data demonstrate the potential of S-nitrosoglutathione (GSNO) augmentation strategy for COPD-emphysema as it controls CS-induced acquired CFTR dysfunction, autophagy impairment, and inflammatory–oxidative stress.
We have recently demonstrated the crucial role of inflammatory–oxidative stress and the resulting autophagy impairment in tobacco smoke-induced COPD-emphysema pathogenesis (9 –11, 43, 61). Moreover, CS exposure also leads to a significant decrease in cystic fibrosis transmembrane conductance regulator (CFTR) protein expression (7), and accumulation of CFTR protein in the aggresome bodies (as shown in this study), which correlates with COPD-emphysema severity (10, 64, 65). A recent study shows that CFTR also regulates autophagy responses, as loss of CFTR due to ΔF508-CFTR misfolding leads to ROS-mediated autophagy impairment (38). Interestingly, GSNO is known to regulate the expression, maturation, and function of CFTR protein (17, 71). In fact, CF-patients have reduced GSNO levels and augmenting GSNO levels is a promising therapeutic strategy currently being developed for treating obstructive CF-lung disease (71). The therapeutic benefits of GSNO are evident from the fact that nearly 20 clinical trials have investigated its potency in multiple disease states (13). In recent years, an alternate mechanism of GSNO induction via pharmacological inhibition of the enzyme, GSNOR, has shown therapeutic potential in controlling chronic inflammatory and obstructive lung disease states, such as asthma and CF (51, 63, 71). Out of the several inhibitors tested, the drug of choice is N6022, a potent reversible inhibitor of GSNOR (27, 63). Treatment with N6022 reduces key asthma symptoms in ovalbumin-induced animal models by virtue of its anti-inflammatory, bronchodilatory, and smooth muscle relaxant properties (6). Also, N6022 shows promising therapeutic efficacy and safety profile in animal models of dextran sulfate sodium-induced inflammatory bowel disease (IBD) and a rat model of salt-induced hypertension (19, 27, 63).
The exact role of GSNO in regulating autophagy is still unclear with some reports suggesting its autophagy-inducing potential (44). Thus, the present study was designed to evaluate the impact of GSNO supplementation in regulating CS-induced acquired CFTR dysfunction and the ensuing autophagy responses that regulate chronic inflammatory–oxidative stress in the airways. We hypothesize that GSNO augmentation, either by direct administration of GSNO or treatment with GSNOR inhibitor (N6022), will be therapeutically beneficial for COPD-emphysema subjects due to its ability to increase CFTR maturation/expression by facilitating its rescue from aggresome bodies as a potential mechanism for controlling CS-induced autophagy impairment, chronic inflammatory–oxidative stress, and emphysema progression.
Results
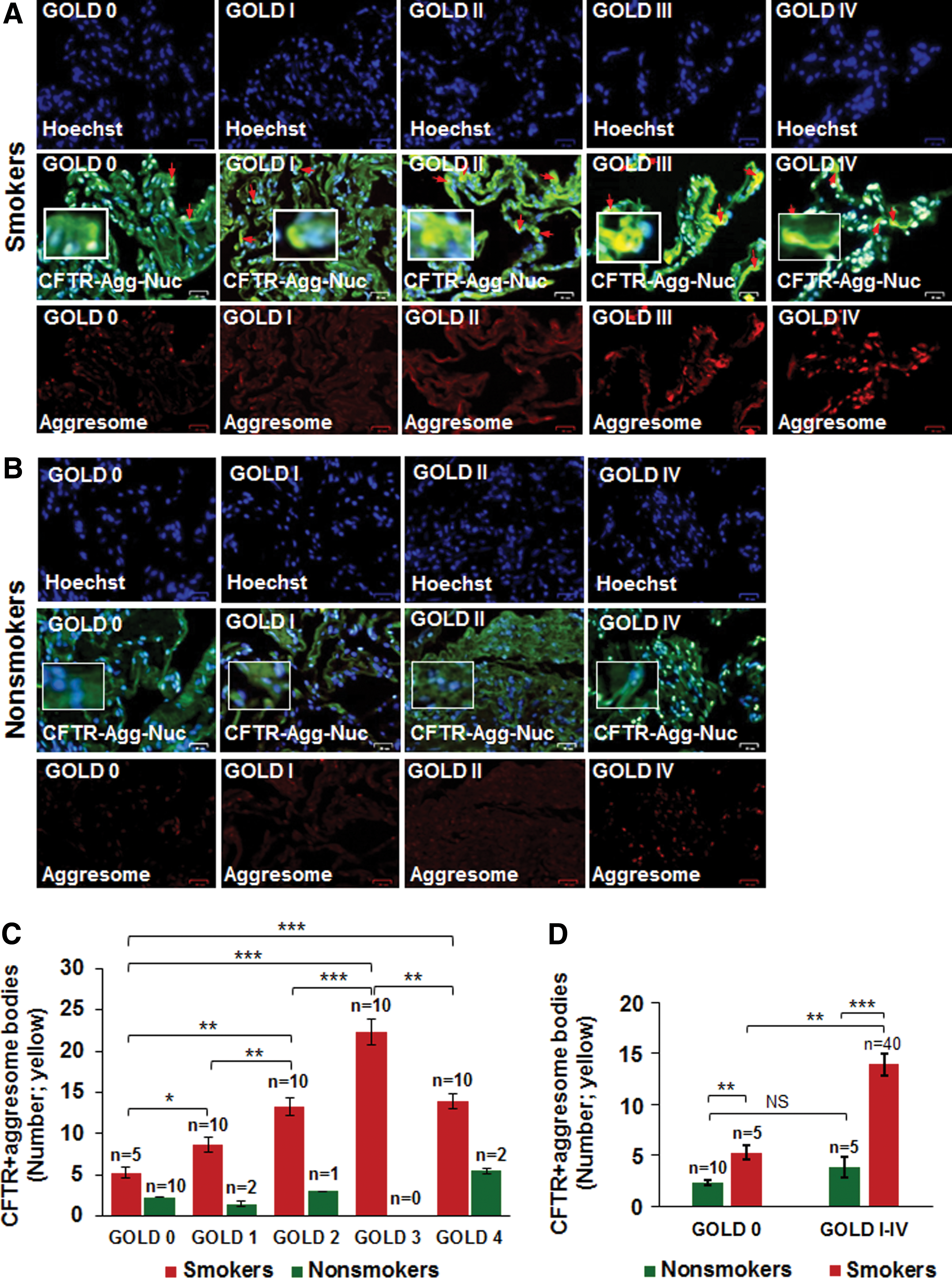
CFTR colocalizes with aggresome bodies in human lung tissues with increasing severity of emphysema
Previous reports from our group have shown that expression of membrane-localized CFTR protein decreases, while the number of aggresome bodies increases in the lungs of COPD subjects with increasing emphysema severity (Global Initiative for Chronic Obstructive Lung Disease [GOLD] I − IV) compared to nonemphysema controls (GOLD 0) (7, 10, 64). We wanted to evaluate whether misfolded-CFTR itself localizes in aggresome bodies as a mechanism of reduced CFTR function (acquired CFTR dysfunction) observed in COPD subjects (24). We found a significant increase in CFTR colocalization within perinuclear aggresome bodies with increasing emphysema severity (Fig. 1A, C, from GOLD 0 to GOLD III, r = −0.88; red arrows: perinuclear CFTR accumulation). Although we observed an increase in CFTR aggresome colocalization in very severe emphysema subjects (GOLD IV) compared to nonemphysema (GOLD 0) and mild emphysema (GOLD I) subjects, the total expression in GOLD IV subjects decreased when compared to GOLD III subjects, as anticipated, due to substantial tissue destruction. Next, we demonstrate that smokers (GOLD 0 or GOLD I − IV) show a significant increase in perinuclear CFTR aggresome colocalization compared to nonsmokers (Fig. 1A−D, *p < 0.05, **p < 0.01, ***p < 0.001). Moreover, the smokers with a greater disease severity (GOLD I − IV) show a comparatively higher increase in CFTR aggresome colocalization compared to GOLD 0 nonemphysema subjects (Fig. 1D, **p < 0.01, ***p < 0.001). Thus, our data suggests that perinuclear accumulation of CFTR into aggresome bodies in smokers and COPD-emphysema subjects serves as a mechanism for acquired CFTR-dysfunction seen in COPD patients (24). Therefore, we next utilized the in vitro and preclinical models of CS-induced COPD-emphysema to verify if rescuing aggresome-localized CFTR could ameliorate CS-induced autophagy impairment, chronic inflammatory–oxidative stress, and resulting lung tissue damage.
Augmenting GSNO levels restores cigarette smoke extract-induced decrease in membrane CFTR by rescuing it from aggresome bodies
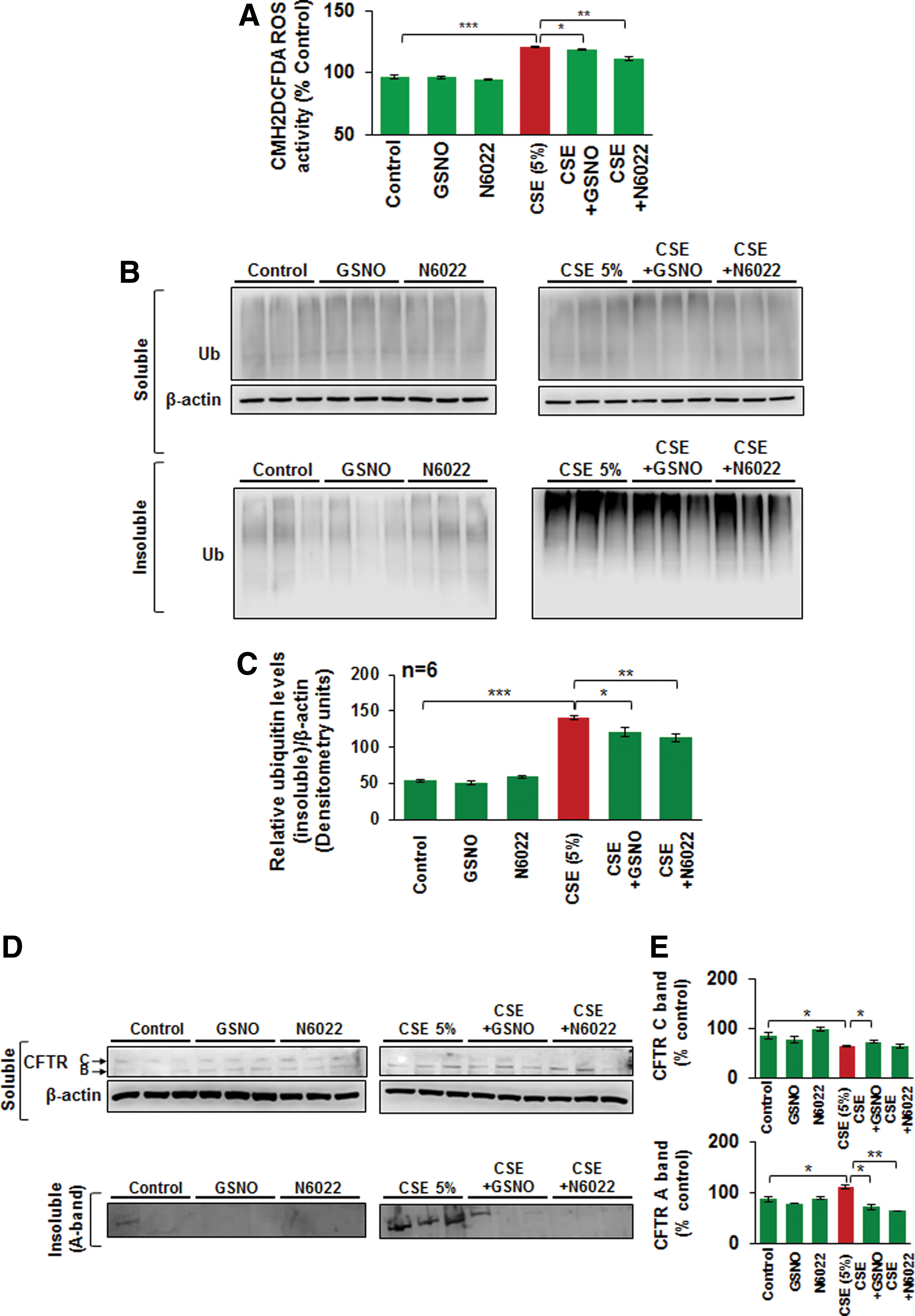
Apart from its known anti-inflammatory properties (21, 63), GSNO has been recently shown to be beneficial in CF lung disease by virtue of its potential to rescue misfolded CFTR to the plasma membrane (69). Since misfolded/defective CFTR promotes aggresome formation (38) and our current human data (Fig. 1) shows that CFTR colocalizes with aggresome bodies with increasing severity of emphysema, we aimed to evaluate if aggresome-localized CFTR is rescued by augmenting intracellular GSNO levels. We demonstrate here that GSNO augmentation using GSNO or GSNOR inhibitor (N6022) rescues the cigarette smoke extract (CSE)-induced decrease in membrane CFTR (Fig. 2A) by significantly inhibiting its aggresome localization (CFTR ubiquitin [Ub]-positive bodies, yellow, red arrows; Fig. 2B, C, *p < 0.05, **p < 0.01, ***p < 0.001). The data imply that restoring GSNO levels corrects CS-induced acquired CFTR dysfunction, suggesting its potential in improving COPD-emphysema-related pathologies.

CSE-induced ROS activation and resulting autophagy impairment are alleviated by restoring intracellular GSNO levels
Exposure to CS leads to ROS-mediated oxidative stress that is known to inhibit autophagy mechanisms (36, 38, 39, 55) that play a crucial role in COPD-emphysema pathogenesis. Thus, we tested the effects of restoring GSNO levels as an antioxidant strategy, in controlling CSE-induced ROS activation. Our data show that CSE (5%)-induced ROS activation in Beas2b cells is significantly reduced by restoring GSNO levels using GSNO or GSNOR inhibitor (N6022) (Fig. 3A, *p < 0.05, **p < 0.01, ***p < 0.001). We have recently shown that tobacco smoke or e-cigarette vapor (eCV) induced ROS activity that initiates autophagy impairment and COPD-emphysema pathogenesis (11, 61). Moreover, treatment with cysteamine (CYS), an antioxidant drug with autophagy-inducing properties, controls tobacco smoke/eCV-mediated inflammatory–oxidative stress and development of COPD-emphysema-like pathologies (10, 11). Here, we demonstrate that restoring GSNO levels by using GSNO or GSNOR inhibitor, N6022, controls CSE-induced autophagy impairment in Beas2b cells, as seen by a significant decrease in CSE-induced accumulation of ubiquitinated proteins in the insoluble protein fractions (Fig. 3B, C, and Supplementary Fig. S1 [Supplementary Data are available online at
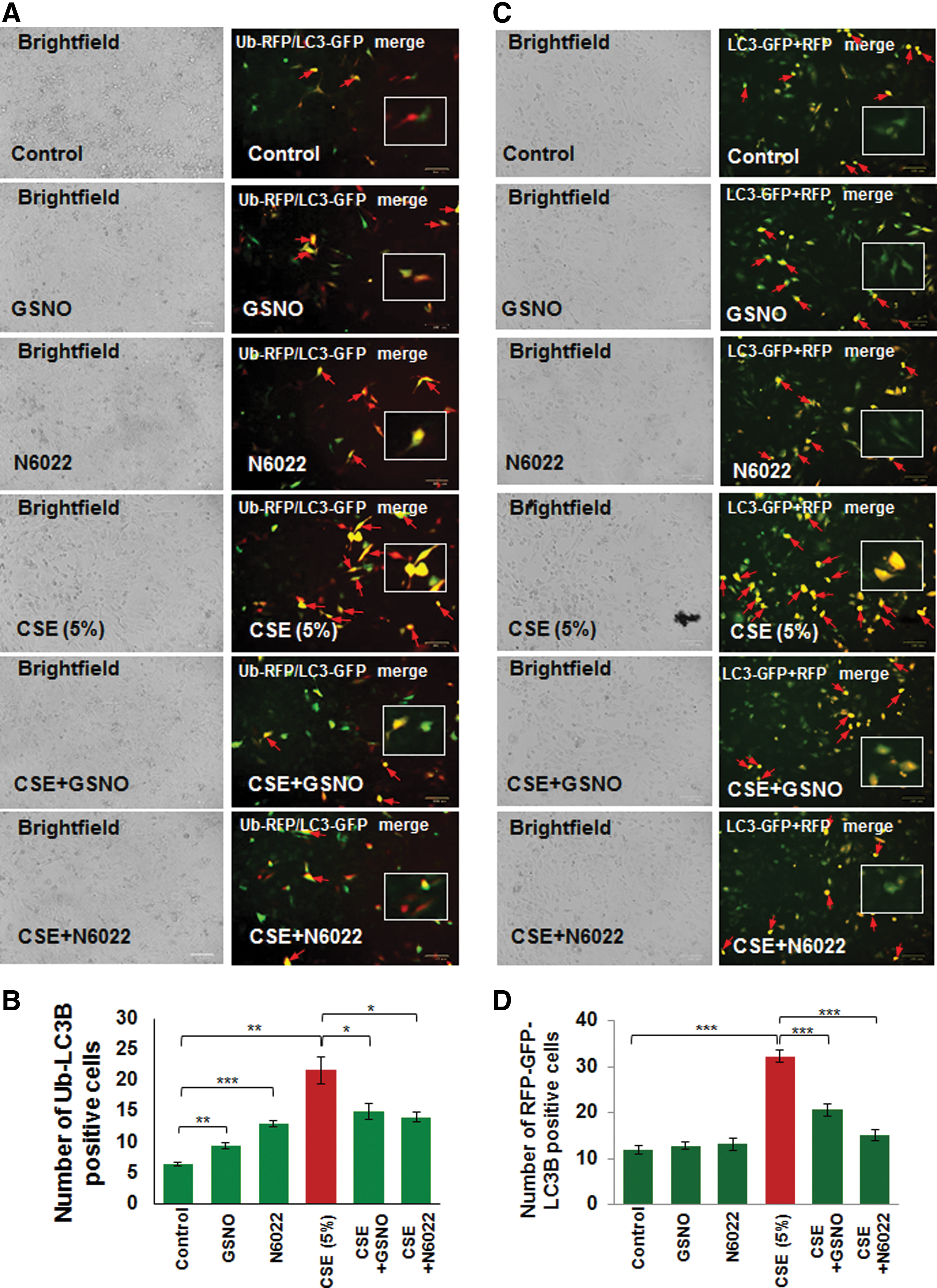
Autophagy inhibition can be quantified by counting the number of cells showing colocalization of ubiquitinated proteins (Ub) and LC3B as previously described (10, 11, 61). Here we report that CSE-induced elevation in Ub-LC3B colocalization (aggresome formation, red arrows) is significantly (p < 0.05) inhibited by restoring intracellular GSNO levels using GSNO or GSNOR inhibitor (N6022) treatment (Fig. 4A, B, *p < 0.05, **p < 0.01, ***p < 0.001). Moreover, we also found that CSE induces green fluorescent protein (GFP)/red fluorescent protein (RFP)-positive autophagosomes (yellow puncta bodies, red arrows), indicative of impaired autophagy flux, which is restored by GSNO augmentation using GSNO or GSNOR inhibitor (N6022) treatment (Fig. 4C, D, ***p < 0.001). Thus, our data help to clarify the contrasting reports regarding the effect of GSNO on autophagy and confirm that GSNO induces autophagy as it protects from CSE-induced autophagy impairment.
Elevated membrane CFTR expression/function corrects CSE-induced autophagy impairment
We and others have shown that diminished levels of membrane-localized mature CFTR protein, due to genetic causes (as in CF) or CS-mediated acquired CFTR-dysfunction, leads to autophagy impairment (8, 38). In this study, we further verified the mechanistic role of CFTR in regulating autophagy responses and demonstrate that CSE induces colocalization of Ub-RFP and LC3B-GFP (yellow, red arrows), indicative of protein misfolding and aggresome formation, which is significantly decreased by treatment with either CYS [an antioxidant drug known to promote membrane trafficking/localization of ΔF508-CFTR protein (23) or VRT-532, a known corrector potentiator of mutant CFTR protein (46) [Fig. 5A, B, ***p < 0.001]). Next, we exposed Beas2b cells to CFTR inhibitor-172 to investigate the specificity of functional CFTR protein in regulating autophagy responses. We observed a significant increase in Ub-RFP and LC3B-GFP colocalization, which was diminished by either CYS or VRT-532 treatment (Fig. 5C, D, **p < 0.01), confirming that CFTR dysfunction leads to autophagy impairment (38). We have recently shown that CS-induced perinuclear accumulation of transcription factor-EB (TFEB), the master regulator of autophagy, into aggresome bodies mediates COPD-emphysema pathogenesis (10). Here we report that treatment of Beas2b cells with CFTR inhibitor-172 leads to increased perinuclear accumulation of TFEB and p62 (impaired autophagy marker) that was significantly rescued by VRT-532 (Supplementary Fig. S3, ***p < 0.001), suggesting the potential mechanism by which CFTR may regulate key autophagy pathways. Thus, we predicted that restoration of CSE-mediated decrease in membrane CFTR expression by GSNO treatment can similarly alleviate autophagy impairment.

Chronic cigarette smoke-induced autophagy impairment is alleviated by augmenting intracellular GSNO levels
Chronic cigarette smoke (Ch-CS) exposure causes autophagy impairment that correlates with emphysema in COPD subjects (10, 64). Although we have shown that restoring the CS-impaired autophagy by autophagy-inducing drugs ameliorates chronic obstructive lung disease and emphysema pathogenesis (10, 64), here we tested a novel hypothesis that restoring intracellular GSNO levels could have a positive effect on CS-induced autophagy impairment and emphysema by controlling the acquired CFTR dysfunction. We exposed C57BL/6 mice to side-stream CS for 18 weeks and performed intratracheal treatments with vehicle (phosphate-buffered saline [PBS]), GSNO, or N6022 (GSNOR inhibitor), as indicated by the scale shown in Figure 6A. We observed that GSNO augmentation significantly decreases Ch-CS-induced accumulation of ubiquitinated proteins (Ub) and CFTR (A-band) in the insoluble protein fractions of murine lungs (Fig 6B, C, and Supplementary Fig. S4, *p < 0.05, **p < 0.01, ***p < 0.001). We confirmed these findings by immunostaining and show that Ch-CS-induced colocalization of ubiquitinated proteins (Ub) and p62 is significantly diminished by GSNO/GSNOR inhibitor (N6022)-mediated increase in intracellular GSNO levels (Fig 6D, F, and Supplementary Fig. S5, **p < 0.01, ***p < 0.001). Moreover, GSNO augmentation significantly reduces CS-induced increase in the number of CFTR-positive perinuclear bodies (Fig. 6E, G, and Supplementary Fig. S5, *p < 0.05, ***p < 0.001) that are aggresomes (Fig. 6E, bottom panel, and 6H and Supplementary Fig. S5, ***p < 0.001). The exact role of GSNO in regulating autophagy responses is elusive and ours is the first report showing that restoring intracellular GSNO levels controls chronic CS-induced CFTR dysfunction and resulting autophagy impairment.

Restoring intracellular GSNO levels mitigates chronic CS-induced aggresome formation and inflammatory response in the airway
We have recently shown that Ch-CS exposure leads to a significant increase in coexpression of ubiquitinated proteins (Ub) and the impaired autophagy marker, p62, in the bronchoalveolar lavage fluid (BALF) cells, which was controlled by treatment with CYS, an antioxidant drug with autophagy-inducing property (10, 66). In the present study, we report that GSNO augmentation by GSNO or GSNOR inhibitor (N6022) could modulate Ch-CS-induced coexpression of ubiquitinated proteins (Ub) and the impaired autophagy marker, p62, in the BALF-cells (Fig. 7A, B, *p < 0.05, **p < 0.01). Specifically, we found that GSNO treatment was more potent in reducing the Ub-p62 coexpression (aggresome bodies), compared to GSNOR inhibitor, at the selected equimolar in vivo doses (4 mg/kg body weight), although both GSNO and GSNOR inhibitor (N6022) were capable of significantly (p < 0.05) reducing the levels of ubiquitinated proteins (Ub). Inhibition of GSNOR showed a trend towards a decrease in p62 protein levels. Since airway inflammation is a crucial mediator of COPD-emphysema pathogeneses (4), we next evaluated the effect of augmenting GSNO levels by GSNO or GSNOR inhibitor (N6022) on changes in the levels of proinflammatory cytokines (interleukin [IL]-6 and IL-1β) in the BALF. An earlier study suggests that treatment with GSNO reduced the expression of inflammatory cytokines and inducible nitric oxide synthase (iNOS) in a rat model of ischemia/reperfusion injury (33). In accord with its known anti-inflammatory property, our results demonstrate that GSNO augmentation significantly decreases the levels of Ch-CS-induced inflammatory cytokines, IL-6 and IL-1β (Fig. 7C, D, *p < 0.05, **p < 0.01), while partially reducing the total number of F/480+ macrophages and CD4+ T cells (data not shown). These data demonstrate the protective role of GSNO augmentation strategy in controlling CS-induced inflammation in COPD-emphysema subjects.

GSNO augmentation controls Ch-CS-induced oxidative-nitrosative stress
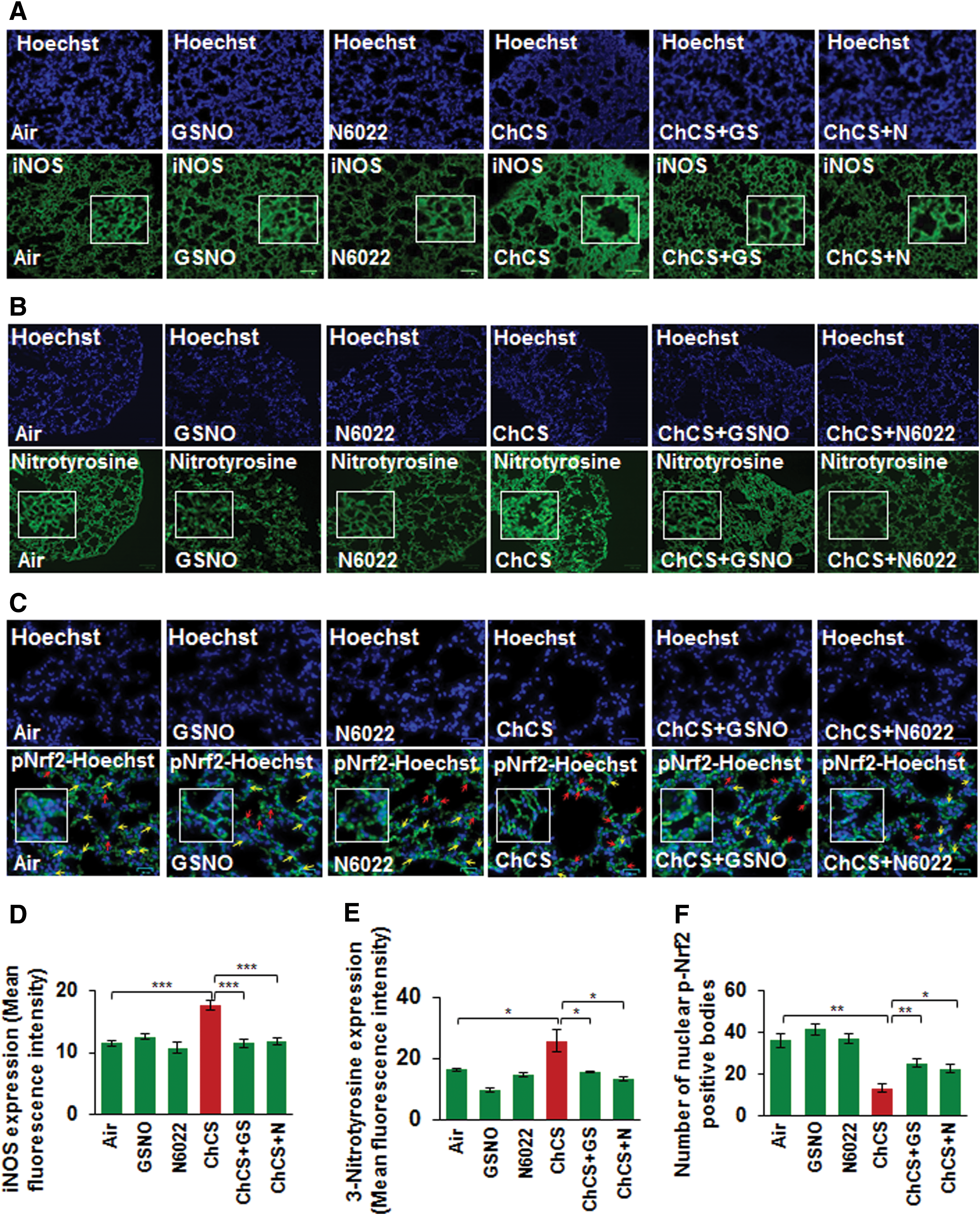
Increased expression of iNOS is implicated in a variety of pathological conditions (15, 40, 48, 68) and elevated oxidative-nitrosative stress triggers high iNOS levels (35). CS exposure-induced oxidative-nitrosative stress elevates iNOS expression that results in generation of toxic NO metabolites, which cause severe lung damage (29). GSNO has been previously reported to diminish iNOS expression levels (33, 59) and in-line with this prior observation, we demonstrate that Ch-CS-induced iNOS expression is significantly controlled by restoring GSNO levels by GSNO or GSNOR inhibitor, N6022 (Fig. 8A, D, ***p < 0.001). We also demonstrate that CS-induced 3-nitrotyrosine expression (a marker of cellular damage due to reactive nitrogen species, [RNS]) was diminished by GSNO or N6022 treatment (Fig. 8B, E, *p < 0.05). Moreover, we show that CS-induced decrease in nuclear localization of p-Nrf2, a critical regulator of antioxidant response pathways (12), is significantly elevated by GSNO augmentation by GSNO or N6022 treatment (Fig. 8C, F, *p < 0.05, **p < 0.01), suggesting its therapeutic potential in controlling oxidative-nitrosative stress.
GSNO ameliorates Ch-CS-induced COPD-emphysema
To further verify the role of GSNO in emphysema pathogenesis, we tested the efficacy of GSNO or GSNOR inhibitor (N6022) in reducing CS-induced alveolar airspace enlargement (Lm) in murine lungs. Our data suggest that augmenting GSNO level results in a significant decrease in CS-induced lung damage (Lm), thus confirming its protective role in COPD-emphysema pathogenesis (Fig. 9A, B, **p < 0.01, ***p < 0.001). In addition, we observed that GSNO augmentation significantly reduces CS-induced cellular senescence and apoptotic cell death in murine lungs (Fig. 9C, D, *p < 0.05, ***p < 0.001), confirming its effectiveness in controlling key pathological mechanisms mediating COPD-emphysema pathogenesis.
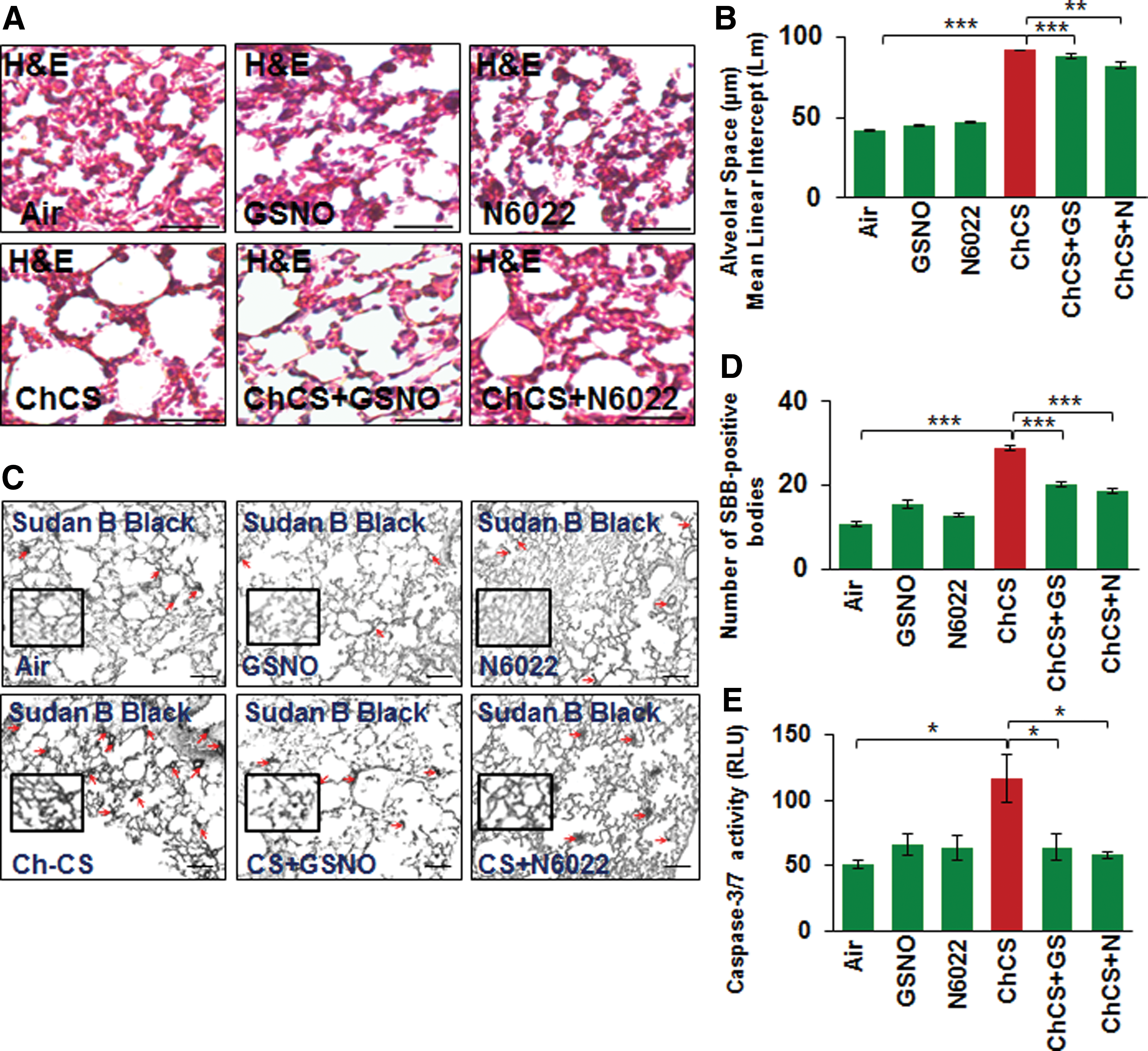
Discussion
Several studies now highlight the protective role of autophagy in tobacco smoke/eCV-induced COPD-emphysema pathogenesis (10, 11, 25, 31, 61). Exposure to CS induces oxidative-nitrosative stress and inflammation in the lungs that plays a crucial role in COPD-emphysema pathologies (34, 36, 47). CS-induced hyperactivation of ROS (oxidative stress) and iNOS (nitrosative stress) results in lung injury, inflammation, and cell death eventually causing lung alveolar cell wall destruction as a mechanism for emphysema pathogenesis (4, 35). We have previously shown that CS-induced autophagy impairment and the resulting aggresome formation are prognostic indicators of emphysema severity in COPD-emphysema subjects (10, 64). Intriguingly, we and others have shown that a dysfunctional CFTR protein leads to autophagy impairment that results in heightened inflammatory–oxidative stress in the airways (8, 38, 65). Also, human lung cells/mice exposed to CSE/CS, and COPD-emphysema subjects demonstrate diminished membrane CFTR expression and function (7, 14, 16, 54), which depicts the acquired CFTR dysfunction in COPD subjects (24, 52). Thus, strategies to reinstate the acquired CFTR dysfunction could have substantial therapeutic benefit in CS-induced COPD-emphysema, by controlling autophagy impairment and resulting inflammatory–oxidative stress.
It is important to note that pulmonary inflammatory–oxidative state and smooth muscle function in the airways is also regulated by NO-signaling (50, 57), which involves endogenous SNOs, such as GSNO (6, 13). The crucial role of GSNO in controlling airway tone and inflammation is evident and its augmentation is currently being utilized as a therapeutic strategy to combat airway inflammation (anti-inflammatory effects) and restore bronchoconstriction (bronchodilatory effects) (19, 27, 53). GSNO is formed by S-nitrosation of glutathione (GSH) and thought to mediate the downstream signaling effects of NO (13). Moreover, GSNO serves as a reservoir of endogenous NO, and its role in several disease states highlights the importance of regulating intracellular GSNO levels (6, 13, 27, 67). The enzyme, GSNOR, catalyzes the degradation of GSNO and helps in maintaining normal cellular GSNO levels (13). Elevated GSNOR activity has been implicated in respiratory diseases such as asthma and CF (6, 26). Moreover, it is perceived that NO generated via oxidative-nitrosative stress mediated pathological manifestations observed in neurodegenerative diseases (49). Thus, exposure to environmental toxins or the normal aging process can trigger higher than normal physiological levels of NO that causes aberrant protein S-nitrosylation, which triggers protein misfolding and aggregation leading to cellular injuries/death in neurodegenerative disorders. In addition, increased NO levels cause elevated generation of RNS, such as peroxynitrite that causes neurodegenerative disease states (45). Thus, generation of peroxynitrite from the interaction between superoxide (O2
We report here that CFTR colocalizes with aggresome bodies with increasing severity of emphysema (GOLD I − IV) in COPD subjects (Fig. 1A–D), while smokers in either GOLD 0 (nonemphysema) or emphysema (GOLD I − IV) group had higher CFTR aggresome colocalization compared to nonsmokers. Our in vitro and murine data demonstrate a novel beneficial role of GSNO augmentation in controlling CS-induced acquired CFTR dysfunction (Figs. 2, 3, and 6), autophagy impairment, and resulting inflammatory–oxidative stress (14, 16) that mediates COPD-emphysema pathogenesis (7, 8, 24). Surprisingly, there are no studies showing the exact role of GSNO in the pathogenesis of COPD-emphysema although many of the pathological manifestations of these obstructive lung conditions are similar. CS exposure induces ROS-mediated deleterious effects on the airway, which includes oxidative stress-mediated protein misfolding (28) and aggresome pathology (10, 38), resulting in cellular senescence (20) and apoptosis (18). Similar to previous reports that have shown the antioxidant effects of GSNO (32, 56), we demonstrate here that CS-induced ROS activation, cellular senescence (Sudan Black B staining), and apoptosis (caspase-3/7 activity) are significantly decreased by GSNO augmentation using GSNO or GSNOR inhibitor, N6022 (Figs. 3A and 9C, D). These findings support our notion that augmenting GSNO levels could protect against CS-induced COPD-emphysema pathologies.
We also evaluated the impact of our GSNO augmentation strategy on the homeostatic autophagy process that is impaired on exposure to CS. Recent reports from our group and others demonstrate that CS exposure induces autophagy impairment marked by formation of perinuclear aggresome bodies (10, 25, 38, 64, 65). Notably, we and others have established the crucial role of ROS activation in protein misfolding, proteostasis and autophagy impairment and resulting aggresome pathology (10, 25, 38, 64). Thus, we postulated that augmentation of intracellular GSNO should at very least protect against CS-induced and ROS-mediated protein misfolding of CFTR and other ubiquitinated proteins. Interestingly, our GSNO augmentation strategy using GSNO or GSNOR inhibitor (N6022) not only significantly elevates CSE/CS-induced CFTR expression and overall protein misfolding (Fig. 2 − 4 and 6) but also autophagy impairment (Figs. 3B, C, 4, 6, and 7A, B), thus demonstrating for the first time that GSNO modulates critical autophagy responses in the airway via restoring CFTR from aggresome bodies. CS exposure also triggers severe pulmonary intrusion of inflammatory cells, which release protein-degrading enzymes (proteases) causing lung tissue destruction and continued inflammation leading to COPD-emphysema symptoms (2 –5). GSNO has been shown to have potent anti-inflammatory properties and thus utilized to suppress inflammation in diverse disease states such as asthma (6, 27, 63), CF (70), ischemia/reperfusion-induced brain injury (32), chronic cerebral hypoperfusion (67), experimental periodontal disease (22), and so on. In the present study, CS-induced increase in levels of BALF-inflammatory cytokines, IL-6 and IL-1β, is significantly reduced by GSNO augmentation that is achieved by treatment with GSNO or GSNOR inhibitor (N6022) (Fig. 7C, D). Thus, we propose that augmenting GSNO levels could have therapeutic benefits in COPD-emphysema by correcting CS-induced acquired CFTR dysfunction and the resulting autophagy impairment and chronic inflammatory–oxidative stress (Fig. 10).
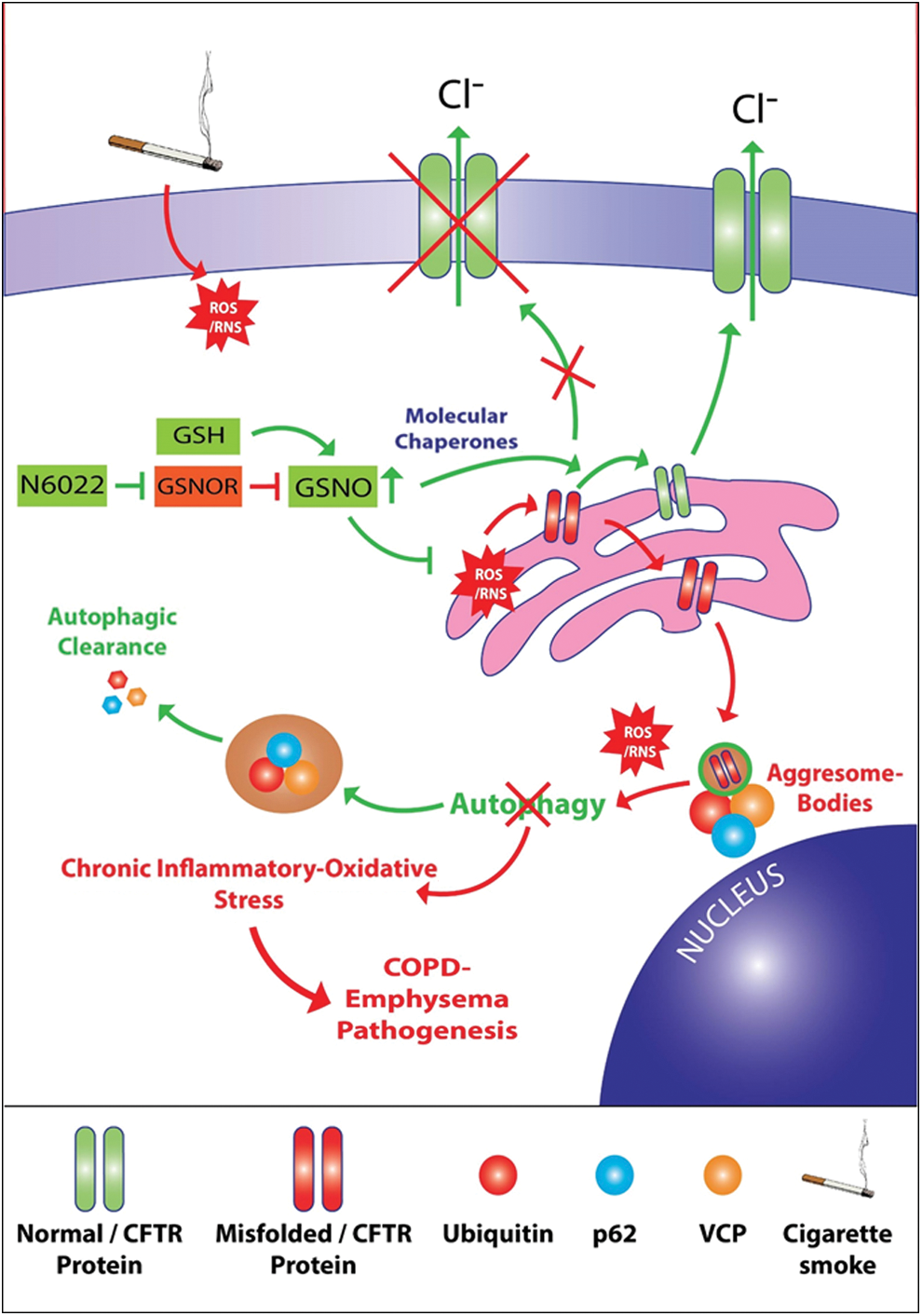
Next, we confirmed potential mechanism(s) by which GSNO might be exerting its protective effects against CS-induced lung pathologies. GSNO has been evaluated by various clinical trials for CF and is reported to augment the maturation and expression of CFTR-protein by modulating CFTR co-chaperones such as Hsp70/Hsp90 (69, 71). We have previously shown that CS exposure decreases membrane/lipid raft CFTR protein expression and a decrease in membrane/raft CFTR expression is not only associated with severity of emphysema in COPD subjects (7) but also critical for emphysema progression in experimental preclinical CF models (8). Our present data shows for the first time that CS induces perinuclear accumulation of CFTR in aggresome bodies that was significantly restored by GSNO or GSNOR (N6022) treatment-mediated augmentation of GSNO levels (Fig. 5E, G, H). The data imply that CS-induced acquired CFTR dysfunction and the resulting autophagy impairment potentiate chronic inflammatory–oxidative stress that could be reverted by GSNO-augmentation, thus suggesting its potential therapeutic utility in COPD-emphysema treatment. Also, smokers with COPD-emphysema have a significantly higher expression of iNOS that is controlled by GSNO (30, 33). We verified that Ch-CS-induced iNOS and 3-nitrotyrosine (marker of cellular damage due to RNS) expression in murine lungs can be suppressed by augmenting GSNO levels (Fig. 8A, B). Thus, our data indicate that GSNO protects against pulmonary emphysema by controlling oxidative-nitrosative stress (p-Nrf2/iNOS/nitrotyrosine expression levels) and acquired CFTR dysfunction.
In conclusion, our study highlights the crucial role of GSNO signaling in ameliorating CS-induced acquired CFTR dysfunction and resulting autophagy impairment and chronic inflammation that triggers COPD-emphysema progression. Thus, we demonstrate the potential of GSNO augmentation as a therapeutic intervention strategy for controlling COPD-emphysema pathophysiology, which warrants further clinical evaluation.
Materials and Methods
Human subject's samples
The paraffin-embedded lung tissue sections were obtained from NHLBI Lung Tissue Research Consortium (LTRC, NIH). The clinical severity, sample size, and classification of COPD subjects were determined on the basis of the stages defined by GOLD (control nonemphysema [GOLD 0] and COPD-emphysema [GOLD I − IV], n = 10–15 for each group with total of 60 samples). The number of smokers and nonsmokers in each COPD GOLD stage is shown in Figure 1C. Our study protocol was approved by the Institutional Review Board (IRB), Central Michigan University and Johns Hopkins University, as “not a human subject research,” under exemption No. 4 and the subjects' lung function data and clinical parameters were obtained from LTRC without disclosing subjects' name and personal information. The classification of the nonemphysema and COPD-emphysema subjects used in this study is previously reported (9). We made sure that nonemphysema or COPD subjects had no other underlying condition other than emphysema for COPD subjects. Although each COPD group (GOLD I − IV) had one patient whose first-degree blood relative suffered from chronic bronchitis.
Murine experiments
We used our CMU IACUC-approved procedures to expose C57BL/6 mice (8 weeks, n = 4) to Ch-CS as previously described (9) and/or treated by intratracheal instillation with GSNO (4 mg/kg body weight), GSNOR inhibitor (N6022, 4 mg/kg body weight), or PBS vehicle control. Briefly, the mice were exposed to room air (control) or Ch-CS (18 weeks) using the TE-2 cigarette smoking machine (Teague Enterprises). The CS was generated by burning 3R4F (0.73 mg nicotine per cigarette) research grade cigarettes (Tobacco Research Institute, University of Kentucky, Lexington, KY) and mice were exposed to direct whole-body smoke for 3 h/day followed by another 2 h in the same chamber without burning more cigarettes. This protocol resulted in an average total particulate matter (TPM) of 150 mg/m3. For treatment, the mice received a total of three doses of the drugs within a 10-day interval (scale, Fig. 5A) before terminal euthanasia following our IACUC-approved protocols. The lung tissues and BALF samples were collected from each group for further analysis by immunoblotting, immunofluorescence microscopy, and flow cytometry.
Western blotting
Western blotting was performed according to our recently described protocol (10, 11) to quantify changes in expression and/or accumulation of ubiquitinated proteins (Ub) and CFTR in the soluble and insoluble protein fractions isolated from murine lungs and Beas2b-cells. The β-actin was used as the loading control. All antibodies were from Santa Cruz Biotechnology with the exception of CFTR (181, rabbit polyclonal) and β-actin, procured from Sigma.
Immunostaining, lung morphometry, and caspase-3/7 assay
The human lung tissue sections were costained with aggresome-specific dye (red, PROTEOSTAT Aggresome Detection Kit; Enzo Life Sciences) and CFTR primary antibody (Santa Cruz Biotechnology) followed by the anti-rabbit CFL-488 secondary antibody (green). The nucleus was stained using the Hoechst stain and images were captured using the Zoe™ Fluorescent cell imager (Bio-Rad). Lung tissues from all the six different murine experimental groups (n = 4; room air, GSNO, N6022, Ch-CS, Ch-CS+GNSO, and Ch-CS+N6022) were collected in 10% neutral buffer formalin. The lung tissue was prepared for histology by preparing paraffin blocks that were cut into 5 μm sections. These sections were then deparaffinized and stained using our previously described immunostaining protocol (9, 10). The primary antibodies used were CFTR (rabbit polyclonal; Santa Cruz Biotechnology), p62 (rabbit polyclonal; Santa Cruz Biotechnology), iNOS (rabbit polyclonal; Santa Cruz Biotechnology), 3-nitrotyrosine (rabbit polyclonal; Santa Cruz Biotechnology), p-Nrf2 (rabbit monoclonal; Abcam), or Ub (mouse polyclonal; Santa Cruz Biotechnology). The secondary antibodies were anti-rabbit CFL-488 (Santa Cruz Biotechnology) and anti-mouse Texas Red (Santa Cruz Biotechnology). In a parallel experiment, these murine lung tissue sections were costained with aggresome-specific dye (red, PROTEOSTAT Aggresome Detection Kit; Enzo Life Sciences) and CFTR primary antibody (Santa Cruz Biotechnology) followed by the anti-rabbit CFL-488 secondary antibody (green). Hoechst stain was used to identify the nucleus. Sections were then mounted on slides using 90% glycerol, and Zoe Fluorescent cell imager (Bio-Rad) was used to visualize tissue areas. These sections were also stained with hematoxylin and eosin (H&E) or Sudan Black B to monitor alveolar tissue destruction by quantifying changes in mean alveolar diameter (Lm), or cellular senescence, respectively, as recently described (10, 64). For the immunofluorescence staining in Beas2b cells, the primary antibodies used were TFEB (Santa Cruz Biotechnology) and p62 (mouse monoclonal; Santa Cruz Biotechnology) followed by anti-rabbit CFL-488 (green) and anti-mouse Texas Red (red) secondary antibodies. The Caspase-Glo™ 3/7 assay kit (Promega) was used to quantify the changes in caspase-3/7 activity in total lung lysates from the same groups of mice using recently described protocol (10).
Cell culture, transfections, autophagy reporter/flux, and ROS assay
Beas2b cells were cultured as recently described (10, 11) and were cotransfected with Ub-RFP and LC3B-GFP plasmids using Lipofectamine® 2000 (24 h; Invitrogen) and treated with control (PBS), GSNO (10 μM), N6022 (10 μM), 5% CSE, CSE+GSNO, or CSE+N6022 for 12 h. In a parallel experiment, functional autophagy was quantified using the Premo™ Autophagy Tandem Sensor RFP-GFP-LC3B assay kit (Molecular Probes) as recently described (10, 61). Briefly, Beas2b cells were incubated with BacMam reagent for 16 h followed by treatment with control (PBS), GSNO (10 μM), N6022 (10 μM), CSE (5%), CSE+GSNO, or CSE+N6022 for 12 h. In a separate experiment, the WT-CFTR protein colocalized with ubiquitinated proteins in Beas2b cells was visualized by transiently transfecting cells with Ub-RFP and/or WT-CFTR GFP plasmids (24 h) followed by treatment with control (PBS), GSNO (10 μM), N6022 (10 μM), 5% CSE, CSE+GSNO, or CSE+N6022 for 12 h. Cells were visualized using the Zoe Fluorescent cell imager (Bio-Rad) as recently described (10, 11). The changes in ROS levels were quantified in the similar experimental groups using the CM-DCFDA ROS indicator dye (Invitrogen) as recently described (11).
Flow cytometry and enzyme-linked immunosorbent assay
Flow cytometry was performed as recently described (10, 11) to monitor changes in the number of Ub-p62+ cells in either Beas2b cells or BALF cells exposed to room air or CS and treated with PBS, GSNO, or N6022. The data were acquired using the BD FACS Aria flow cytometer and analyzed by the FACS Diva Software as recently described (10, 11). To quantify changes in the levels of IL-6 and IL-1β cytokines in BALF samples of each experimental group, standard enzyme-linked immunosorbent assay (ELISA) was performed as previously described (7, 10).
Statistical analysis
Data are shown as mean ± standard error of the mean for n = 3–4 replicates from at least three experiments. A one-way analysis of variance (ANOVA) followed by Tukey's post hoc test or one-tailed Student's t-test was performed to determine significant changes in treatment groups compared to controls. A p-value of ≤0.05 was considered a significant change. In addition, Pearson's correlation analysis was used to determine the correlation between the two data sets and the correlation coefficient is shown as “r” value. Immunoblotting data were quantified using the ImageJ software (NIH).
Footnotes
Acknowledgments
We thank the NHLBI Lung Tissue Research Consortium (LTRC, NIH) for providing human lung tissue sections and Philip Oshel, Director of the Microscopy Core Facility, Central Michigan University, for help with the confocal microscopy experiments. We also thank April Ilacqua, FACS student technician, for assistance during the flow cytometry experiments. The study was supported by the Flight Attendant Medical Research Institute's (FAMRI), Young Clinical Scientist Award (YCSA_082131) to N.V. The funders had no role in study design, data collection and analysis, and decision to publish or preparation of the manuscript.
Authors' Contributions
Conception and design: N.V.; analysis and interpretation: N.V., M.B.; experimental contribution: M.B., N.V., D.S., K.W., K.B.; drafting of manuscript and editing: M.B., N.V.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.