
Abstract
Significance:
Cellular metabolic activity impacts the production of reactive oxygen species (ROS), both positively through mitochondrial oxidative processes and negatively by promoting the production of reducing agents (including NADPH and reduced glutathione). A defined metabolic state in cancer cells is critical for cell growth and long-term self-renewal, and such state is intrinsically associated with redox balance. Promyelocytic leukemia protein (PML) regulates several biological processes, at least in part, through its ability to control the assembly of PML nuclear bodies (PML NBs).
Recent Advances:
PML is oxidation-prone, and oxidative stress promotes NB biogenesis. These nuclear subdomains recruit many nuclear proteins and regulate their SUMOylation and other post-translational modifications. Some of these cargos—such as p53, SIRT1, AKT, and mammalian target of rapamycin (mTOR)—are key regulators of cell fate. PML was also recently shown to regulate oxidation.
Critical Issues:
While it was long considered primarily as a tumor suppressor protein, PML-regulated metabolic switch uncovered that this protein could promote survival and/or stemness of some normal or cancer cells. In this study, we review the recent findings on this multifunctional protein.
Future Directions:
Studying PML scaffolding functions as well as its fine role in the activation of p53 or fatty acid oxidation will bring new insights in how PML could bridge oxidative stress, senescence, cell death, and metabolism. Antioxid. Redox Signal. 26, 432–444.
Promyelocytic Leukemia Protein, a Multifunctional Protein Nucleating Nuclear Bodies
P
PML is ubiquitously expressed with more than seven alternative splicing PML variants, PML I and II being the two more abundant isoforms (18). PML isoforms differ in the C-terminal fraction of the protein, but retain RING-finger B-box coiled coil (RBCC) domains characteristic of the tripartite motif protein family (TRIMs; Fig. 1) (53). The RBCC is required for the interaction of PML with the majority of its partners as well as for its ability to multimerize (95). C-terminal parts do not contain domains with specific functions, except for the exonuclease III-like domain in PML I that additionally contains a nuclear export signal (NES) (17, 85).

PML isoforms are slightly differentially distributed in the nucleus when expressed separately in Pml −/− mouse embryonic fibroblasts (MEFs) (18). These discrete localizations are likely enforced by isoform-specific interactions with distinct molecules. PML IV was extensively shown to regulate p53-induced senescence, most likely due to a specific interaction with the ADP ribosylation factor (ARF) tumor suppressor (50). However, all PML isoforms interact with each other through the RBCC and are assembled in nuclear bodies at the endogenous level (18).
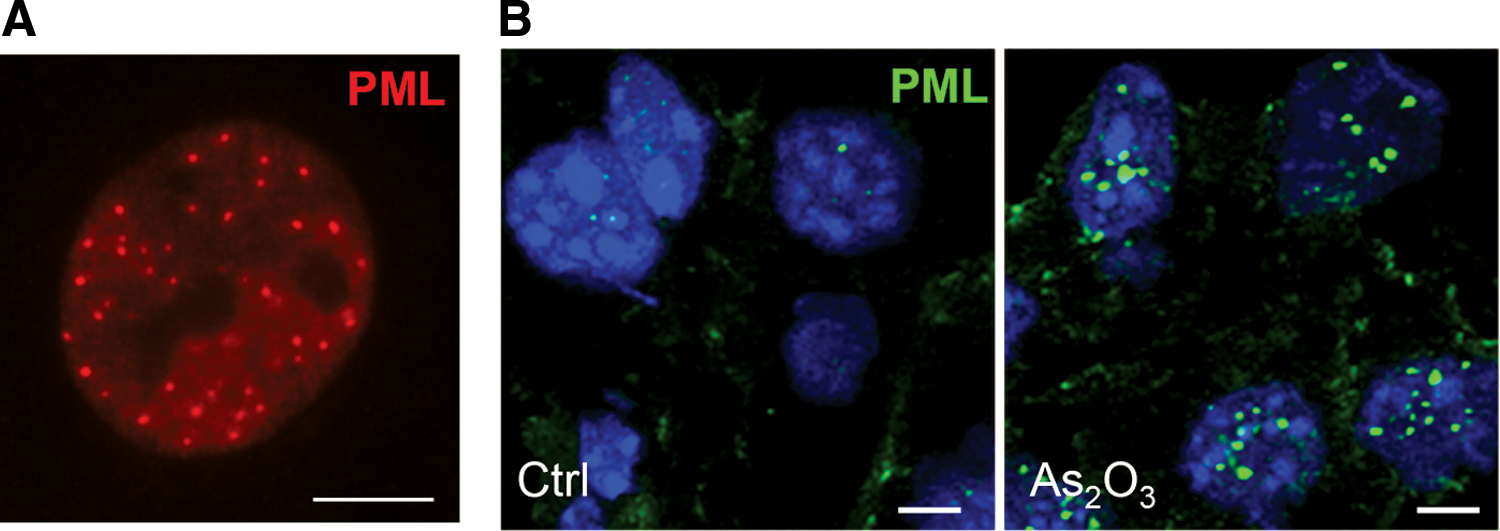
PML nuclear bodies (NBs) are membrane-free subnuclear compartments whose assembly and function are molecularly complex. Their formation is initiated and controlled by PML (4, 67). NBs are spherical subnuclear domains from 0.3 to 1 μm with an abundance ranging from 10 to 30 bodies per nucleus, depending on intrinsic (cell cycle) or extrinsic (cellular stressors) stimuli ex vivo (Fig. 2). At the molecular level, PML proteins that are distributed in the nucleoplasm can multimerize to form the NB outer shell, thus creating a partition within the nucleus. The exact biochemical activity of PML remains obscure, but its structural scaffolding role is central to its biological functions. Indeed, NBs recruit multiple PML partner proteins that concentrate inside the bodies. The number of these unrelated cargos stably or transiently concentrated inside PML NBs, provides a feasible explanation for the involvement of PML in a variety of biological processes. PML NBs have been proposed to fine-tune and optimize protein–protein interactions and post-translational modifications (PTMs), with a remarkable contribution to SUMOylation processes (4, 23).
PML NBs are dynamic structures, and NB-associated proteins rapidly shuttle (within seconds) in and out of the bodies (119). In contrast, the PML shell is stable (few minutes) and belongs to the nucleus heterogeneous scaffold, the nuclear matrix (63, 107). None of the known NB-associated proteins are required for the full assembly of PML bodies, while these proteins are redistributed in Pml knockout cells. These observations formally demonstrate the essential requirement of PML for NB nucleation and cargo proteins recruitment. Interestingly, differences among NB-cargos exist. Some partners, such as p53, BLM, breast cancer 1 (BRCA1), and HP1, are only recruited under particular conditions (stress or specific stage of the cell cycle), while others, such as SP100 nuclear antigen (SP100), homeodomain-interacting protein kinase 2 (HIPK2), death domain-associated protein (DAXX), or CREB-binding protein (CBP), are NB-resident proteins, being constitutively localized to NBs (67). Of note, SP100 and PML stabilize each other, and some specific resident proteins (SP100, promyelocytic leukemia zinc finger protein [PLZF], or eIF4E) can self-aggregate in the absence of PML, forming distinct domains (84, 110).
Discrete PML NBs may contain nascent messenger RNAs (mRNAs) (6, 69). In addition, telomeres being subjected to alternative lengthening also locate to these subnuclear structures in some cancer cells (16). In cell lines, PML NBs localize to the interchromatin space, and the periphery of some of these bodies associates with specific genomic loci (15, 103, 113). Moreover, association between PML NBs and specific histone variants or satellite DNA regions suggests that PML bodies could be involved in pericentromeric heterochromatin organization (72). This idea is also strengthened by the fact that DAXX—a notorious PML-associated protein—is a histone chaperone (100). The diverse observations regarding PML and DNA/RNA proximity might reflect differences in NB partners according to cell types, cell cycle, and/or stress stimuli.
Oxidant-Prone Nuclear Domains for Reactive Oxygen Species-Sensitive Post-Translational Modifications
While PML is ubiquitously expressed in vivo, PML NBs are weakly detected in normal tissues such as endothelial cells, with remarkable abundance in inflammatory conditions (Fig. 2) (62, 109). Cell culture represents a stress condition that likely facilitates PML assembly into NBs. Indeed, NBs are sensitive to cellular stress, and numerous exogenous agents perturb their organization (67). Heat shock, metal, or DNA damage induces PML redistribution into numerous small nuclear dots (4). Interestingly, heat shock or cadmium induces PML NBs to reversibly bud into microspeckles. These insults result in the relocalization of some partners, including DAXX, which is transferred onto centromeric and pericentromeric heterochromatin (28, 78, 83). Thus, disassembly of PML NBs might release factors required for stress response, hence contributing to stress signaling. Heat shock promotes protein SUMOylation (21, 43), an effect that could be involved in partner release from the NBs. Cadmium effects are suggested to rely on mitogen-activated protein (MAP) kinase signaling (83). However, the exact action of this pathway on PML is not fully understood. A potential explanation is related to the fact that cadmium exposure increases reactive oxygen species (ROS) levels, which regulate PML-NB assembly (20).
Oxidant effects on NB formation were revealed by studies on acute promyelocytic leukemia (APL) treatment (23). In this type of leukemia, the PML-retinoic acid receptor alpha (RARA) oncogenic fusion protein is responsible for PML NB disorganization. Arsenic trioxide (As2O3), an efficient APL therapy, restores these bodies, triggering partial differentiation and loss of LICs (1, 130). Importantly, in addition to its effects on APL cells, As2O3 increases the assembly of the PML nucleoplasmic fraction into NBs in nonleukemic cells (Fig. 2) (130) or in those expressing other X-RARA fusion (61). The increased NB assembly is followed by SUMOylation and SUMO-dependent ubiquitination and degradation of PML and PML/RARA (68, 108).
As2O3 is well known to induce oxidative stress (27, 57, 65) by perturbing different cellular functions that balance ROS levels, such as mitochondrial activity and NADPH oxidase—the main sources for ROS production—or glutathione that is used for arsenic metabolism (36, 123). As2O3 promotes PML oxidation and binds directly to PML through its ability to interact with reactive cysteines in vitro and in vivo (52, 128). Although it is difficult to discriminate which of these two effects is responsible for As2O3-enhanced PML NB assembly, there is evidence supporting a role for ROS in PML NB dynamics. Indeed, other oxidants such as paraquat or paracetamol also promote PML NB formation in vivo (98) (Niwa-Kawakita M, August 10, 2015, unpublished data). Paracetamol metabolites titrate glutathione in vivo, whereas paraquat and alpha-tocopheryl succinate (α-TOS) target complex I of the electron transport chain in the mitochondria, hence unleashing the accumulation of ROS. Analysis of therapeutic strategies in APL revealed an effect of α-TOS similar to As2O3 (25). These results imply that ROS accumulation upon complex I inhibition could be a trigger for PML NB reorganization and tumor suppression in APL. An intermediary metabolite of As2O3, the trivalent monomethylarsonous acid [MMA(III)], still binds PML in vitro, but does not elicit NB reassembly in APL cells, suggesting complex effects of As2O3 molecules regarding direct binding versus indirect generation of ROS (116). In vivo, both PML/RARA degradation and NB reassembly have been shown to be absolutely required for APL cure (1, 82).
Pml is a primary transcriptional target of interferon (IFN) signaling (105). A link between IFNs and oxidative stress was first reported in 1990 (106). Recently, Hubackova et al. demonstrated that IFNs can induce the expression of NADPH oxidase 2 and increase ROS levels (46). Thus, in addition to the massive increase in PML expression, oxidative stress could contribute to PML NB formation activated by IFN signaling. All these findings place ROS in the limelight as a major regulator of PML NB formation in vivo. It remains to be clarified whether ROS operates directly or indirectly on PML. A recent study identified a novel mechanism for PML regulation in endothelial cells based on an internal ribosome entry site (IRES) upstream of the PML initiation codon (44). This IRES is regulated by the inflammation-related cytokine tumor necrosis factor α (TNFα) through the mitogen-activated protein kinase (MAPK) pathway.
SUMOylation represents another important way of regulation for PML NB organization. SUMO1–3 are ubiquitin-like polypeptides that covalently modify PML and many NB-cargos (97). Once nucleated in bodies, PML recruits the ubiquitin-conjugating enzyme 9 (UBC9) SUMO-E2-conjugating enzyme, a key player for SUMOylation, leading to efficient PML and partner modification. In details, the ability of SUMOylated PML shells to recruit proteins through their SUMO-interacting motifs (SIMs) explains the diversity of NB-associated proteins, which all contain both SIM and SUMOylation sites. In response to oxidative stress, NBs behave as catalytic sites, promoting SUMOylation of NB cargos by bringing substrates (SIM-containing NB-associated proteins), enzymes (UBC9), and modifiers (SUMOs) in close proximity (Fig. 3). These ROS-responsive bodies could thus behave as macro-SUMO ligases, in turn providing a plausible explanation for the global enhancement of SUMOylation in yeast upon PML expression (93). By studying SUMOylation, our laboratory provided the first direct evidence of global control of a PTM by PML NBs (98). Interestingly, ADP ribosylation factor (ARF) was associated with these bodies in specific settings, such as senescence (see the section “PML and p53 Regulation”). This protein was shown to enhance SUMOylation through its ability to recruit UBC9 (13, 50). Similarly, ZNF451 is a PML NB-resident protein with SUMO-E3 ligase activity (9).
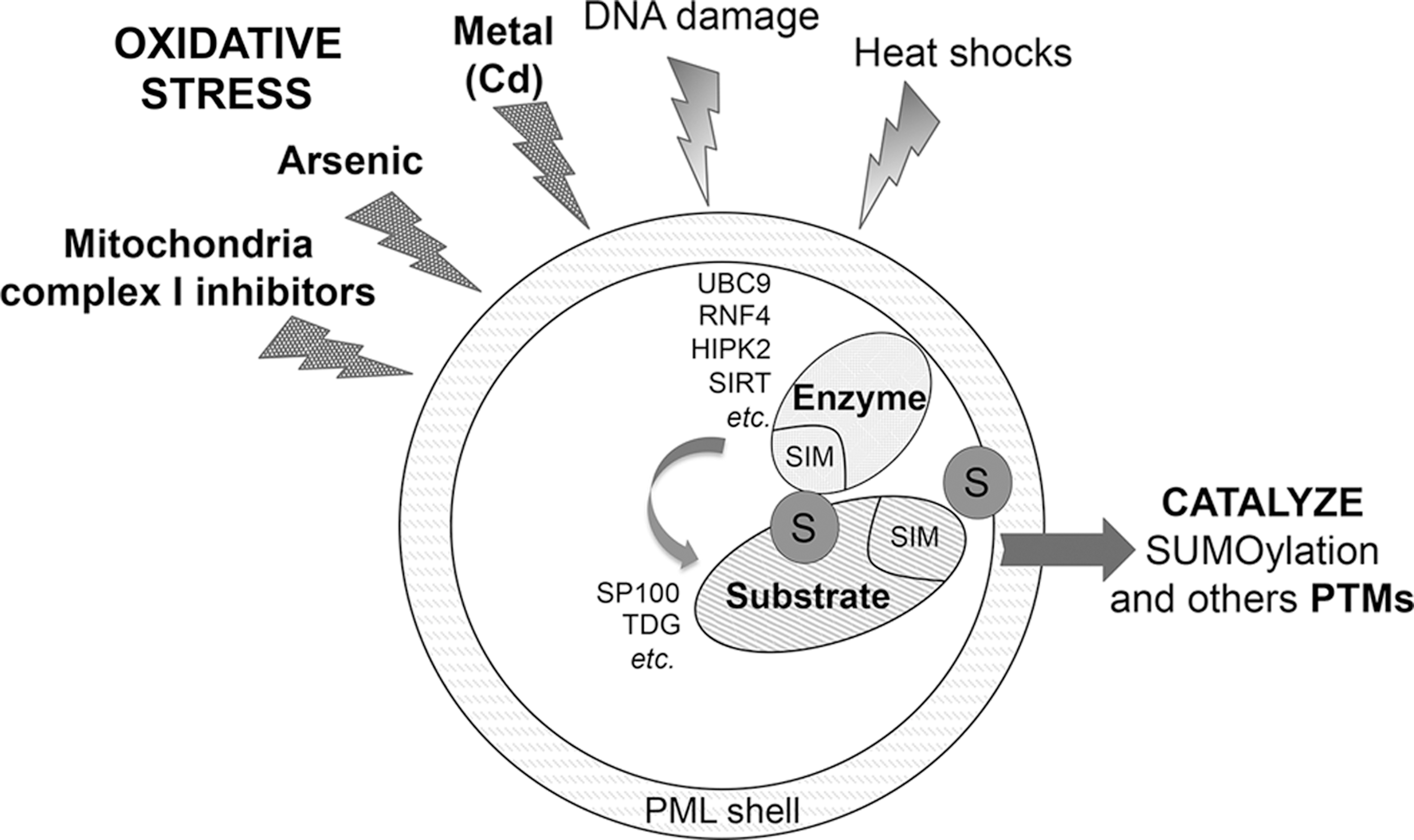
We have proposed that SUMOylation of partners allows their transient retention within PML NBs, in turn promoting additionnal modifications, by other SUMO/SIM NB-associated enzymes (ligases, acetylases, deacetylase, kinases, phosphatases) such as the RNF4 ubiquitin-E3 ligase (97). This SUMO-targeted ubiquitin-mediated degradation is also responsible for PML-RARA catabolism in As2O3-treated APL cells (68).
Many enzymes within the SUMOylation cascade are themselves sensitive to oxidation mainly through their thiol-reactive active sites. UBC9 reacts to strong oxidant exposure such as H2O2 and it is cross-linked to the SUMO-E1-activating enzyme through their respective reactive cysteines (7). Another example is sentrin-specific protease 3 (SENP3), an SUMO protease that cleaves SUMO2/3 specifically and traffics between the nucleoli and PML NBs (35). SENP3 undergoes thiol modification in cell lines exposed to mild oxidative stress and subsequently recruits 90-KDa heat shock protein (HSP90), ultimately leading to its degradation (121). H2O2 also induces SENP3 recruitment to PML NBs (42). These domains may provide a favorable redox environment for thiol enzymes involved in the SUMOylation cascade. Thus, a general function of PML may be to sense the cellular redox status and to modify partner fate accordingly (availability, PTMs, stability, etc.).
PML Outside the Nucleus, Other Links with Oxidation?
Cytoplasmic localization of PML has been described, although its significance is still obscure. The first evidence of a non-nuclear function of PML came from the discovery of an isoform that lacks the exon containing the nuclear localization signal (NLS), in addition to the NES-containing PML I isoform (55, 71). Cytoplasmic PML (PMLc) isoform regulates, in particular, (i) TGFβ signaling (71) and epithelial-to mesenchymal transition in prostate cancer (8), (ii) pyruvate kinase M2 activity (104), and (iii) early step of human immunodeficiency virus 1 replication (114). Some endogenous PML was reported in the cytoplasm, a localization likely driven by the NES of PML I. Indeed, cytoplasmic localization of endogenous PML is found in prostate adenocarcinoma cells, where PML interacts with chromosomal maintenance 1 (CRM1; Exportin-1), a protein involved in nuclear export (8). In addition, subcellular localization of PML in the cytoplasm has been experimentally studied using PML mutants with truncated NLS (56). This study revealed the preferential association of PML with endosomes and lysosomes, where some of the PML target pathways reside (3). Interestingly, endogenous PML NBs were recently shown to associate with early endosomes during mitosis (89).
Mitochondria-associated membranes (MAMs) represent the region where the endoplasmic reticulum (ER) and the mitochondria interact. Importantly, this communication is essential for mitochondrial physiology and cell survival. While studying the localization of PML within the cytoplasm, the group of Pandolfi discovered a discrete accumulation of PML in MAMs (34). The authors could show functional relevance of MAM-PML by using a PML construct tagged with an ER-targeting signal peptide (PML-ER) in Pml knockout cells. This finding provides a possible explanation for the regulation of p53-independent apoptosis by PML. Indeed, PML-ER regulates apoptosis by activating protein phosphatase-2A (PP2A), which leads to opening of the IP3 receptor and increases calcium flux into the mitochondria. Interestingly, it is plausible that ROS production by mitochondria and differences in redox status in the ER could locally modulate PML activity in MAMs and therefore calcium influx and apoptotic response. PML-ER activity is similar to that of nuclear PML, which negatively controls AKT through its recruitment together with PP2A at NBs, triggering AKT dephosphorylation (112). Thus, conserved molecular interactions between the nucleus and the cytoplasm for PML function could be extended to additional non-nuclear activities of this protein.
PML and p53 Regulation
The tumor suppressor p53 is a key player in the oxidative stress response. p53 fine-tunes ROS response by inducing antioxidant program or by controlling ROS-triggered apoptosis. The first demonstration of a role for PML in p53 function came from premature senescence analysis in primary fibroblasts. Different studies converged to show that Ras-induced senescence depends on PML-promoted p53 PTMs (29, 90). At the same time, other laboratories demonstrated the requirement of PML for p53-activated apoptosis (37, 38). Upon the induction of stress, such as activated oncogene expression or irradiation, PML NBs concentrate several p53 regulators such as HIPK2, CBP, or ARF together with p53 itself, enhancing its acetylation or phosphorylation (49, 64). Studies are still ongoing to determine how various PTMs of p53 differentially contribute to the activation of specific target genes regulating apoptosis, senescence, or metabolism (33). Interestingly, among PML isoforms, only PML IV activates p53, leading to senescence when overexpressed (5, 30). PML IV harbors a short specific C-terminus sequence that mediates the interaction with ARF (50). ARF is a nucleolar regulator of p53 that protects the tumor suppressor from MDM2-driven degradation. Moreover, ARF/UBC9 interactions and subsequent SUMOylation play a central role. PML IV concentrates together p53, ARF, and UBC9 in the bodies, promoting SUMO1 conjugation of p53 and its subsequent stabilization (50). This SUMOylation of p53 is required for PML IV-activated senescence. Interestingly, p53 SUMOylation is also required for IFN-induced senescence (22), suggesting an active role for PML NBs upstream p53 in the antiproliferative properties of IFN.
The majority of studies showing that PML regulates p53 PTMs rely on the overexpression of PML or specific p53 regulators. Key experiments demonstrating functional regulation of p53 by PML in vivo were conducted taking advantage of APL mice (1). In this in vivo model, PML NB reorganization following APL treatment leads to p53 reactivation that subsequently triggers elimination of LICs. This effect is associated with a p53-dependent senescence signature in APL cells, and this p53 activation is impaired in Pml-defective mice upon therapies. However, defects in p53 target gene activation have not been unraveled yet, in vivo, in unchallenged Pml−/− mice.
In addition to the negative control of cell cycle progression and survival, p53 participates in the oxidative stress response and the metabolic switch. Thus, PML could regulate the oxidative stress response and act as an ROS sensor with a body-assembled state upon ROS, in turn controlling p53 PTMs and expression of antioxidant proteins, such as TP53INP1 or Sestrins. Upon glucose deprivation, p53 is activated, triggering metabolic changes that include the inhibition of glycolysis and the activation of fatty acid oxidation (FAO) to promote an adaptive response (2, 54). It remains to be established whether PML participates in those p53-controlled processes. Mouse models rather than cell lines could be important to unravel such PML effects in the future.
The Bidirectional Connection Between PML and Nutritional Preferences
Cellular metabolism requires sensing mechanisms to respond to alterations in cellular nutritional demands. In cancer, the seminal idea of core metabolic alterations occurring in transformed cells posed by Warburg et al. (118) has been refined and developed to establish the field of cancer metabolism. Many of the most important cancer genes (oncogenes and tumor suppressors) have been shown to alter the metabolic program. The evidence of cross talk between PML and metabolism has been very limited until recently. Starting from an in vitro simplified system based on MEFs, PML was found to regulate the correct activity of FAO (12). This effect relied on regulation of the transcriptional program elicited by the peroxisome proliferator-activated receptors (PPARs), downstream the PPAR γ coactivator 1α (PGC1α). The PGC1α-PPAR pathway is tightly regulated through the post-translational control of the transcriptional coactivator. PGC1α is regulated negatively by GCN5-mediated acetylation and positively by SIRT1-mediated deacetylation (24). Interestingly, both PGC1α regulators and the coactivator itself localize to PML NBs in a dynamic manner (12, 60). Thus, p53 and PGC1α regulation converges on the NBs, which may link stress responses to metabolic alterations (Fig. 4).
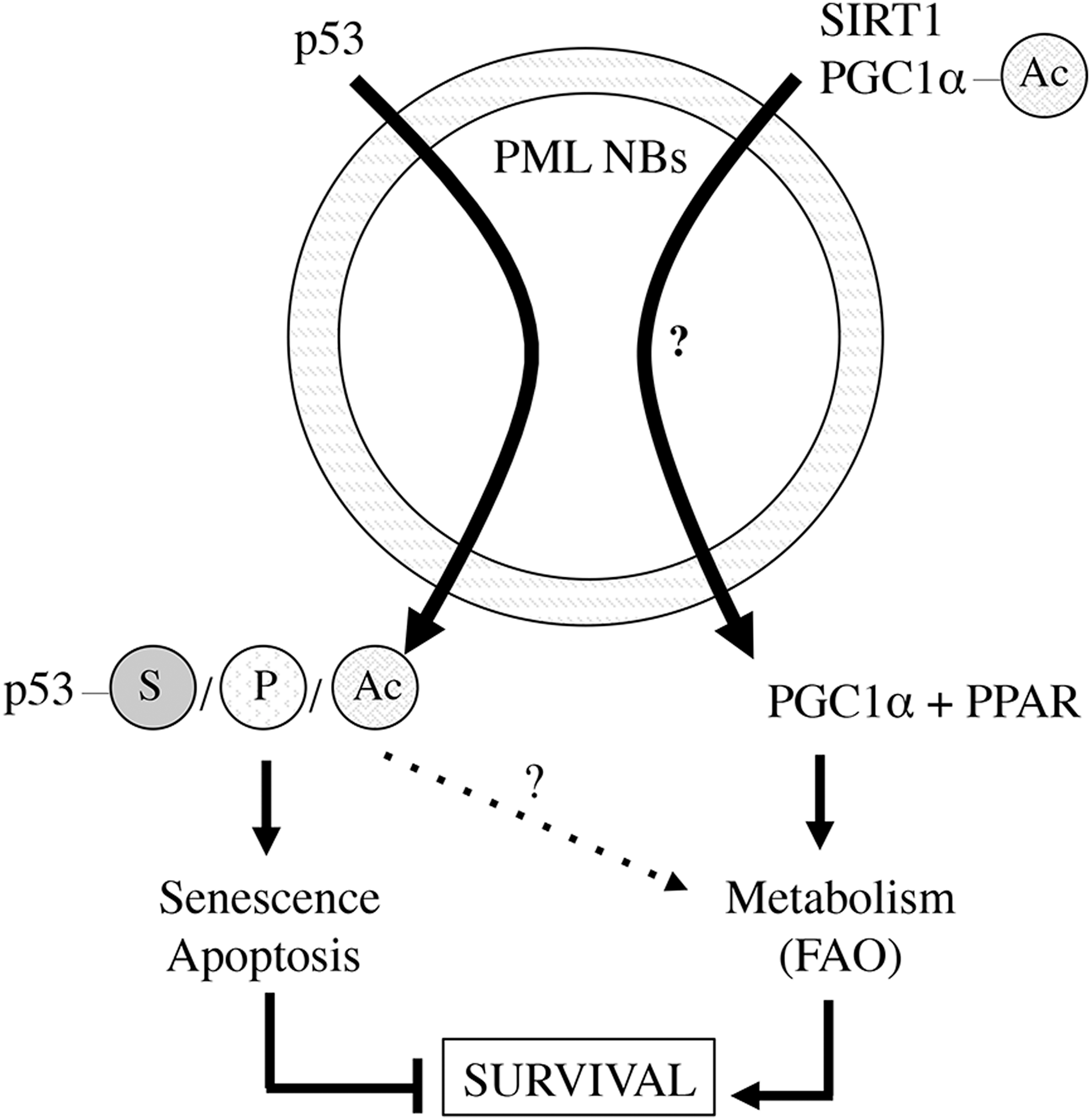
PML metabolic activity was uncovered to provide paradoxical cellular selective advantage. To date, this beneficial activity has been demonstrated in normal hematopoietic cells, LICs, and in breast cancer cells (12, 47). Mechanistically, the metabolic activity of PML is required for both stem cell renewal and breast cancer cell survival (12, 48). In hematopoietic stem cells, PML maintains proper activity of the PPAR delta-FAO pathway to control asymmetric cell division. Indeed, genetic or pharmacological manipulation of this axis resulted in alteration of the asymmetric division versus symmetric commitment equilibrium. In breast cancer, PML upregulation favored the survival of breast cancer cells upon loss of attachment and this was attributed to higher FAO-derived adenosine triphosphate (ATP) production. This observation was coherent with the existence of a breast cancer subset (enriched in triple-negative poor prognosis cases) with elevated PML levels and PPAR signaling. Interestingly, recent findings unraveled the key role of PPAR γ in stem cell maintenance, with therapeutic perspective using PPAR γ agonist to promote LIC exhaustion in chronic myeloid leukemia (CML) patients (92).
In addition to a prosurvival/protumoral role of PML-controlled FAO, Pml loss has been associated with alterations in systemic metabolism. Indeed, Pml-deficient mice exhibit propensity to develop obesity, as shown in two independent studies, under genetic determinants (leptin deficiency) or exposure to hyperlipidic diets (12, 59). The mechanism of this systemic effect was attributed to reduced FAO (12) and/or to increased adipocyte differentiation in the absence of PML (59). It is worth noting that an additional study reported that Pml-deficient mice exhibit resistance to obesity (14), while the environmental or molecular determinants for this discrepancy remain to be elucidated. Interestingly, PML was related to the survival and function of pancreatic β cells upon hyperglycemia-induced oxidative damage (60). PML NBs were shown to serve as a scaffold for the activity of FoxO1 and SIRT1 toward NeuroD and MafA regulation upon H2O2 exposure. PML could thus play a pivotal role from metabolism to oxidative stress response. Most of the work linking PML and metabolism is related to the activation of fatty acid catabolism, an important source of ATP, acetyl CoA, and NADPH. To which extent the effects of PML rely on the energetic balance (ATP), the production of metabolic intermediates (acetyl CoA), or the redox balance (NADPH), remains to be elucidated.
Conversely, nutrient and energy availability may regulate PML expression and NB dynamics. Concerning PML NB dynamics (79, 119), time lapse and FRAP experiments revealed the existence of different NB exchange rates among the overexpressed green fluorescent protein-tagged isoforms. Through the manipulation of mitochondrial metabolism (sodium azide) and glycolysis (a nonglycolytic competitive inhibitor of glucose, 2-deoxyglucose), it has been shown that PML NB dynamics are closely associated with the energetic state of the cell (79, 119). To which extent this response reflects the direct requirement of ATP for PML NB dynamics (active enzymatic processes) versus an energy-sensing mechanism to modulate cell responses, remains to be determined. Both azide and 2-deoxyglucose induce oxidative stress and PML oxidation could also account for changes in PML NB dynamics.
The second aspect of PML regulation by metabolism concerns PML abundance. The level of PML is tightly regulated at the transcriptional, translational, and post-translational level (44, 99). The metabolic contribution to these parameters has been barely explored. Obesity triggers the accumulation of the nuclear protein in hepatocytes, with the appearance of one or few spheres or doughnut-shaped NBs in a fraction of hepatocytes (that were otherwise negative for PML NBs) (12). This observation has been corroborated in two independent genetic mouse models (at the protein level) and in conditions of diet-induced obesity (at the mRNA level) (11, 12). Importantly, the same upregulation was observed in liver extracts from obese individuals, which correlated with the degree of lipid accumulation in the tissue (steatosis) (11). Since obesity triggers ROS and inflammation, TNF induction of PML expression through the p38 MAPK pathway could contribute to the high PML level in steatosis (44). Interestingly, lipid accumulation not only regulates PML abundance but also lipid storage droplets physically associate with PML NBs (86). These data add complexity to bidirectional regulation of PML and lipid metabolism. Taking into account that lipid metabolism regulatory pathways sense lipid abundance and components of these pathways reside or interact with the PML NBs, it is plausible that local lipid droplet signaling in these bodies regulates their dynamics and abundance and determines the metabolic response.
Cross Talk Between PML NBs and Autophagy Regulators
Macroautophagy (from here on referred to as autophagy) is a cellular process consisting in the engulfment and digestion of cellular organelles (31). This process has been involved both in cell survival upon nutrient shortage or stress and in cell death upon prolonged stress through pathways dependent and independent of apoptosis (77). Autophagy is negatively regulated by the mammalian target of rapamycin complex 1 (mTORC1) pathway (58). PML has been shown to inhibit the AKT-mTORC1 pathway in cancers (3, 112). In lymphoid leukemia cells, the cytotoxic response to dexamethasone, which results in PML upregulation through glucocorticoid receptor activation, inhibits the AKT-mTORC1 pathway and unleashes an autophagic response eliciting cell death (Fig. 5) (66). In contrast with the other leukemogenic fusions, such as PLZF-RARA and NPM-RARA, PML-RARA oncoprotein that is dominant negative over PML NBs has also been proposed to upregulate autophagy through Akt/mTOR inhibiton (45), protecting from ARA/C-induced apoptosis.
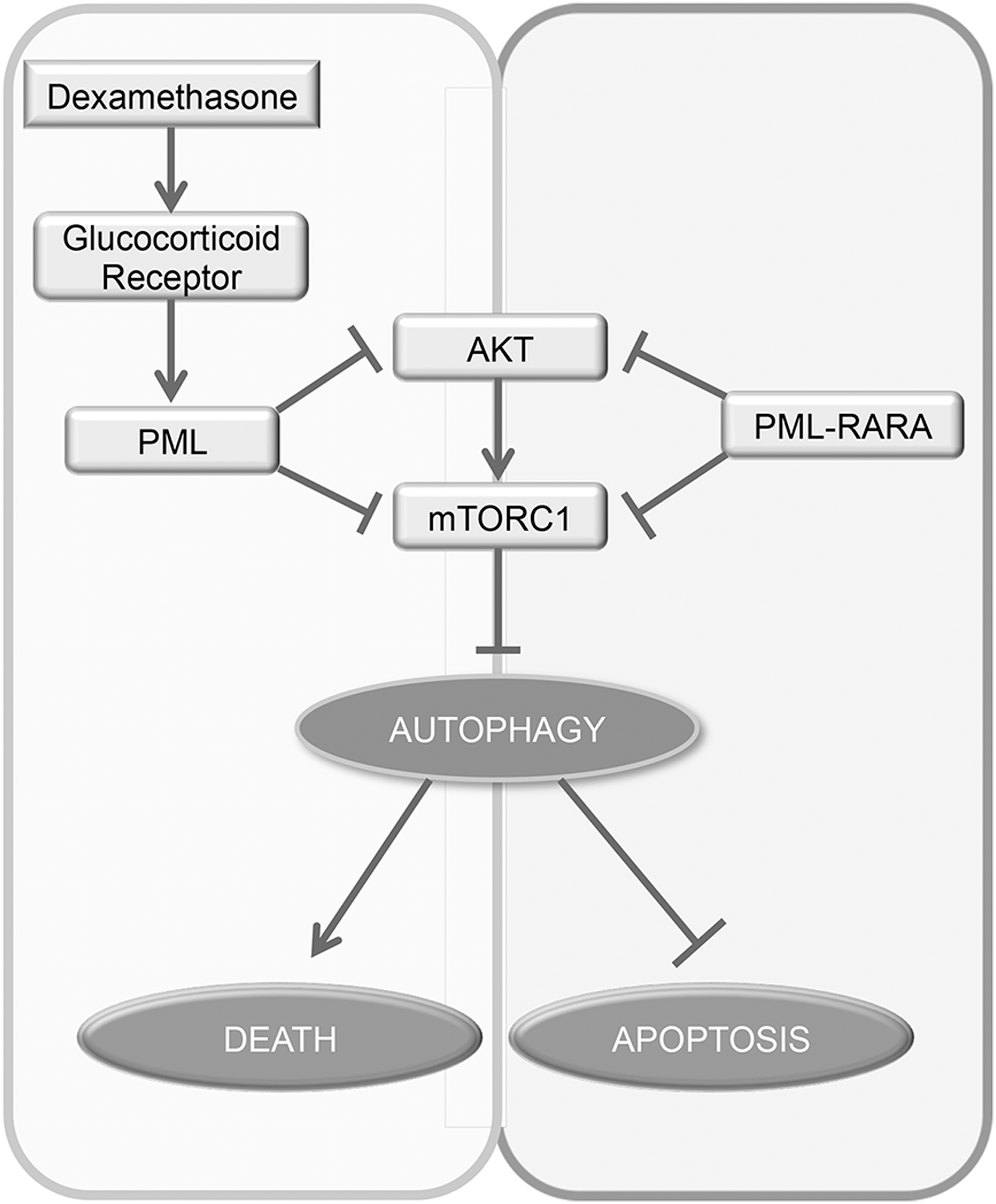
Conversely, autophagy has also been proposed to regulate the abundance of PML-RARA. ROS production leads to activation of proautophagic pathways. First, oxidative stress-induced HMGB1, a proinflammatory factor, could contribute to PML-RARA degradation by promoting autophagy in the APL cell line (122). The upregulation of microRNA (miRNA) miR-125b also correlated with PML-RARA expression in leukemia (126). MiR-125b reduces the expression of DNA damage-regulated autophagy modulator 1 (DRAM1)—a positive regulator of autophagy (19)—leading to the inhibition of autophagy and accumulation of PML-RARA. Conversely, reduction of this miRNA leads to the opposite effects (125). However, to date, no clear evidence was shown for in vivo effects of autophagy on PML-RARA stability and APL treatment outcomes, while a clear demonstration states the requirement of proteasome-mediated PML-RARA degradation (1, 81).
PML and Cancer
PML has been described as a tumor-suppressive protein (76) due to two main observations. First, it is lost in a variety of cancers (40, 62). This reduction in PML is rarely at the genomic level and depends on a variety of pathways, including casein kinase 2 (CK2)-mediated degradative phosphorylation (102) or Notch-dependent, ubiquitin-specific peptidase 11 (USP11) inhibition-mediated destabilization (120). Second, PML regulates a variety of essential pathways in cancer development and progression: ranging from p53-dependent cell death and senescence to the alternative lengthening of telomeres (88). PML also inhibits neoangiogenesis through inhibition of mTORC1 and HIF-1 alpha translation (3). A recent study showed that PML regulates angiogenesis and neuroblastoma cancer relapses through the control of thrombospondin 2 (TSP2) (26). The translational control of PML could also contribute to decrease in angiogenesis and promote TNFα-mediated inhibition of migration of epithelial cells as well as TNFα-induced apoptosis in breast cancer cells (44). At the molecular level, in addition to positive regulation of p53 and negative regulation of AKT/mTOR, the tumor-suppressive activity of PML could be mediated by other activities, including the activation of p73 (87) or regulation of E2F transcription factor 1 (E2F) (74, 115).
Why is PML not lost at the genomic level? The answer to this question might rely on the recently identified advantageous functions of PML in cancer particularly in the regulation of self-renewal. The metabolic effect of PML provides a selective advantage in slow-cycling undifferentiated cells and contributes to survival and tumorigenesis in specific contexts (12, 47, 48). In addition to the hematopoietic system and CML, the essential role of PML in regulating stem and progenitor cell fate has been demonstrated in other tissues and cancers. In the developing brain, PML expression is restricted to neural progenitor cells and controls their proliferation and fate, through the function of essential cell cycle regulators such as pRB (94). This stem cell regulatory and prosurvival activity of PML has been corroborated in glioblastoma, where PML could contribute to mTOR inhibitor resistance and to c-MYC stability (51, 129). In the breast, it has been shown that STAT transcription factors regulate PML levels and PML NBs during the mammary gland developmental cycle (70). In line with this notion, our last report showed that PML targeting hampers breast cancer initiation and metastatic seeding through regulation of Sox9 in breast cancer (75). PML levels impact on the balance of different luminal progenitors. In these neuronal and breast contexts, it remains to be established whether PML role in progenitor cell maintenance is linked with its metabolic activity. Interestingly, there is a resemblance between the paradoxical activity of PML and that of PGC1α, the latter being relevant for cancer stem cell maintenance in pancreatic cancer (101) and tumor suppression in prostate cancer (111). Finally, depending on the cancer type, the pro- versus antitumoral activity of PML may be related to p53 status and PML ability to regulate metabolic-controlled survival/stemness versus p53-dependent senescence/apoptosis.
In perspective, the two faces of PML might not be at all discordant. Tumors are heterogeneous and comprise a majority of highly proliferating cells and a small fraction of slow-cycling self-renewing cells. In this context, the same molecular pathways might exert antagonistic effects on the two compartments. In this respect, tumor-suppressive and growth inhibitory pathways (phosphatase and tensin homolog [PTEN], liver kinase B1 [LKB1]) are required for stem cell function, in contrast to their selected loss in the bulk of cancers (32, 41, 80, 124, 127). Senescent cells express specific cytokines (such as IFNs or interleukin 6 [IL-6]) that maintain senescence state in the microenvironment and promote recruitment of immune cells. However, many factors of this senescence-associated secretory phenotype also have protumorigenic properties (96). IFN promotes hematopoietic stem cell proliferation, leading to a transient expansion of the stem cell pool before it reenters into quiescence (91). With these premises, it is tempting to speculate that equivalent molecular activities exerted by PML in distinct cell compartments might lead to opposing biological outcomes, owing to the intrinsic signaling and metabolic nature of these cells.
Cancer cells cope with chronic stress generated by oncogenes, hypoxia, active oxidative phosphorylation, etc. (36). In these settings, ROS sensors and scavengers are modulated to achieve adaptation. More generally, the contribution of PML to the metabolic switch may be particularly important in tumors where high mitochondrial activity is preferred to promote both survival and adaption to chronic oxidative stress. FAO-induced NADPH production as well as p53 PTMs may counteract this stress. Conversely, Nf-E2-related factor 2 (NRF2)—the main transcription factor controlling antioxidant response—is negatively regulated by PML NBs [inducing NRF2 SUMOylation and subsequent degradation (73)]. Another study recently reported a PML-positive role in mitochondrial complex II activity (39). Loss of PML in some cancers could thus promote ROS and NRF2 adaptive response. Further analysis in this respect will determine the extent of contribution of mitochondrial metabolic dynamics to PML function and the potential exploitation for cancer treatment. To conclude, PML ability to modulate cell fate at the physiological or pathological level is complex. Recent findings linking PML to oxidative stress and metabolism control show that these processes are no exception. This is likely due to the PML scaffolding role for NBs, whose function is to accelerate and control fitness of PTM for several protein complexes whose composition may change according to stress, differentiation, or cell/tissue types.
Footnotes
Acknowledgments
The authors are thankful for all facilities from IUH. Funding: the laboratories are supported by Collège de France, INSERM, CNRS, Université Paris-Diderot, Ligue Contre le Cancer, Institut National du Cancer, ANR (PACRI, SLI, and SUMOPiv projects), Canceropôle Ile de France, and the European Research Council. The work of A.C. is supported by the Ramón y Cajal award, the Basque Department of Industry, Tourism and Trade (Etortek), Health (2012111086), and Education (PI2012-03), Marie Curie (277043), Movember Foundation (GAP1), ISCIII (PI10/01484, PI13/00031), FERO (VIII Fellowship), and ERC (336343). N.M.-M. is supported by the Spanish Association Against Cancer (AECC).