
Abstract
Aims:
A recently discovered group of conditionally disordered chaperones share a very unique feature; they need to lose structure to become active as chaperones. This activation mechanism makes these chaperones particularly suited to respond to protein-unfolding stress conditions, such as oxidative unfolding. However, the role of this disorder in stress-related activation, chaperone function, and the crosstalk with other chaperone systems is not yet clear. Here, we focus on one of the members of the conditionally disordered chaperones, a thiol-redox switch of the bacterial proteostasis system, Hsp33.
Results:
By modifying the Hsp33's sequence, we reveal that the metastable region has evolved to abolish redox-dependent chaperone activity, rather than enhance binding affinity for client proteins. The intrinsically disordered region of Hsp33 serves as an anchor for the reduced, inactive state of Hsp33, and it dramatically affects the crosstalk with the synergetic chaperone system, DnaK/J. Using mass spectrometry, we describe the role that the metastable region plays in determining client specificity during normal and oxidative stress conditions in the cell.
Innovation and Conclusion:
We uncover a new role of protein plasticity in Hsp33's inactivation, client specificity, crosstalk with the synergistic chaperone system DnaK/J, and oxidative stress-specific interactions in bacteria. Our results also suggest that Hsp33 might serve as a member of the house-keeping proteostasis machinery, tasked with maintaining a “healthy” proteome during normal conditions, and that this function does not depend on the metastable linker region. Antioxid. Redox Signal. 27, 1252–1267.
Introduction
T
Hsp33 is a first line of defense chaperone that protects organisms ranging from bacteria to green algae from the toxic effects of oxidative stress, which leads to protein unfolding and aggregation. The activation of Hsp33 is triggered by oxidation of its C-terminal redox-sensitive domain, which leads to partial unfolding of Hsp33 via its adjacent metastable linker region. Here, we revealed that the metastable linker region of Hsp33 is a regulatory inactivator, preserving its monomeric architecture and mediating crosstalk with the synergistic chaperone system DnaK/J. We demonstrate a dual cellular function of Hsp33, and its dependence on the metastable linker region.
Historically, molecular chaperones are classified into two groups based on their energy requirements: ATP-dependent chaperones, which use ATP hydrolysis for substrate folding, and ATP-independent chaperones, which provide a binding platform for unfolding proteins, thereby preventing protein aggregation. The ATP-independent chaperones employ structural plasticity for activation, client binding, and release. This class of chaperones is usually stress dependent; it is activated by elevated temperatures (e.g., small Heat Shock Proteins [sHSPs]) (11, 12, 19, 26), a decrease in pH (e.g., HdeA and HdeB) (10, 27, 45), osmotic shock (late embryogenesis abundant proteins) (6, 9, 20, 28, 33), or oxidative stress (e.g., Hsp33, Get3) (24, 34, 36, 40, 46, 52). The modes of activation utilized by these ATP-independent chaperones are as diverse (3, 42). In some cases, activation requires assembly into high-molecular-weight oligomers (19, 41, 46, 50), whereas in others, activation demands the unfolding of metastable domains, which expose potential client binding sites (e.g., in Hsp33 and HdeA) (3, 10, 42, 45). However, the role of structural plasticity in chaperone function and the effect it has on the crosstalk with other chaperone systems are not yet clear.
In this study, our model ATP-independent chaperone is the redox-regulated Hsp33, which uses order-to-disorder transitions of half of its structure for its chaperone activity. Hsp33 plays a crucial role in the protection of bacteria against oxidative stress-induced unfolding and aggregation of cellular proteins (24, 52). Under reducing non-stress conditions, Hsp33 is a compactly folded zinc-binding protein with negligible chaperone activity. However, when exposed to oxidative stress conditions, Hsp33 undergoes massive conformational rearrangements, which activate its chaperone function. These changes occur via the C-terminal redox switch domain, which includes four highly conserved cysteines that form two disulfide bonds on oxidation (Fig. 1A).
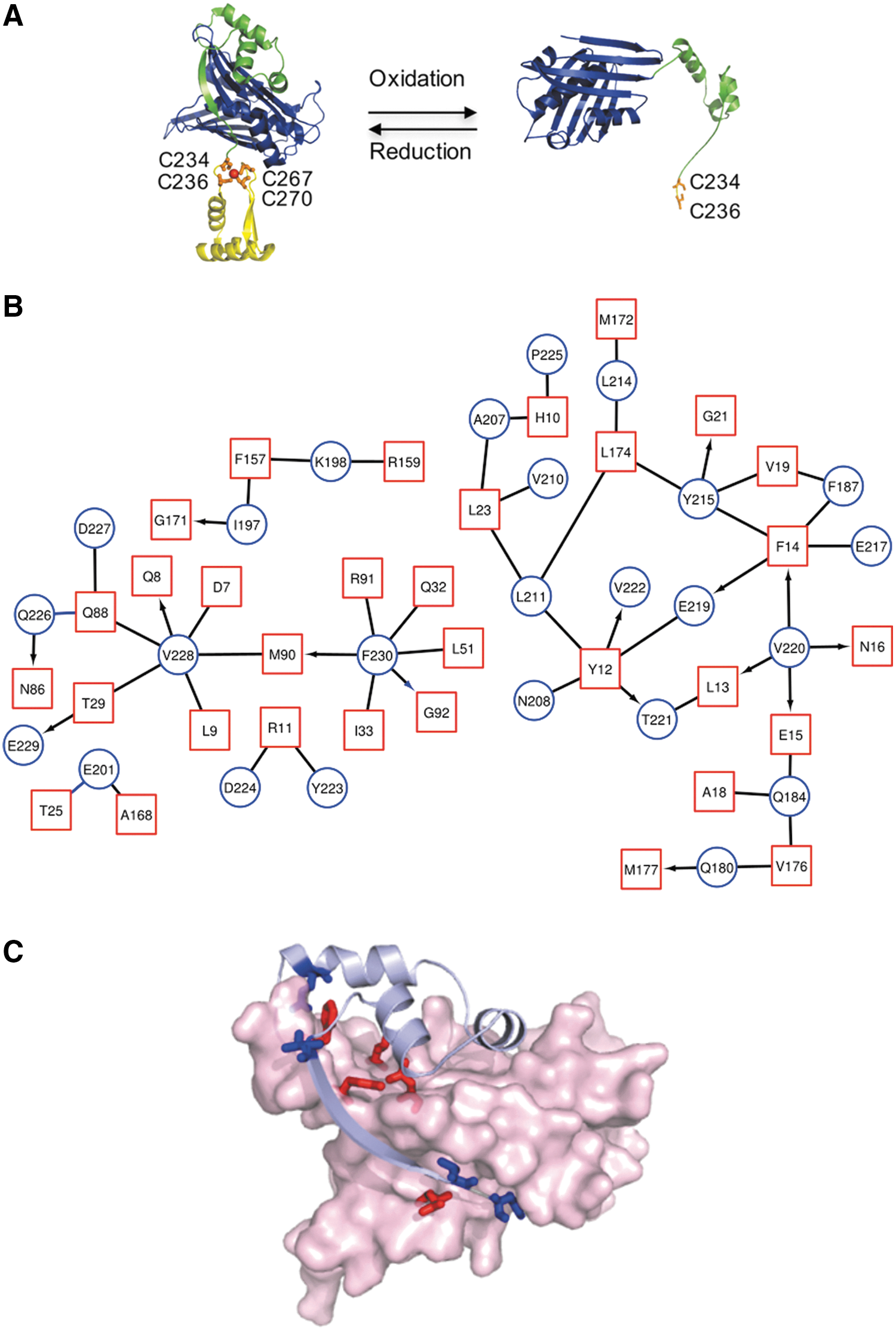
The formation of two disulfide bonds leads to the destabilization of a 50-aa linker region, which is situated adjacent to the redox-sensitive zinc center (8, 23, 39). In turn, unfolding of the linker region, leads to the exposure of hydrophobic surfaces, which likely contribute to the rapid recognition of aggregation-prone substrates (8). Recent studies showed that the linker region mediates interactions with misfolded client proteins (16), suggesting a direct role of the linker region in chaperone function. On its return to reducing non-stress conditions, the redox domain refolds (15), leading to stabilization of the thermostable linker region (8, 23, 39). These conformational changes in the Hsp33 chaperone appear to be accompanied by partial unfolding of the bound client protein, and they mediate its transfer to the synergetic ATP-dependent chaperone system DnaK/J for refolding (21).
Here, we propose a new role for the metastable region of Hsp33 in stabilizing the inactive, folded state of the chaperone, thereby preventing nonspecific binding in the cell under normal conditions. We show that replacing a 50-aa metastable region by non-native sequences does not affect the anti-aggregation activity in vitro but rather its redox-specific activation, oligomeric architecture, and the crosstalk with the synergistic ATP-dependent chaperone system, DnaK/J, to ensure successful client release. Using mass spectrometry, we uncovered a dual function of Hsp33 in vivo and its dependence on the linker region. We suggest that Hsp33 interacts with members of the proteostasis network during normal conditions, preserving a “healthy proteome,” and that these interactions are not linker dependent. However, stress-specific interactions rely heavily on the linker region.
Results
Inter-residue interactions between Hsp33's metastable regions and its N-terminal domain
Hsp33 is a first line of defense chaperone that protects organisms ranging from bacteria (24, 51) to green algae (Chlamydomonas reinhardtii) (40) against the toxic effects of oxidative stress. Hsp33 can be divided into three domains: an N-terminal domain, comprising of a hydrophobic, stable β-sheet platform (Escherichia coli numbering: 1–176; Fig. 1A, blue) and two metastable regions, which undergo destabilization on oxidative unfolding—the redox-sensitive C-terminus, 230–294 aa (Fig. 1A, yellow) and a linker region (Fig. 1A, green).
A map of the inter-residue interactions between the N-terminal domain and the other regions of Hsp33 (linker and C-terminal regions) revealed a dense connectivity network mediated mainly by hydrophobic residues of the linker region (e.g., Val 228, Val 220, and Asn 184), as well as one residue from the C-terminal domain (Phe 230) located before the C234xC236 and C267xxC269 motifs (Fig. 1B). These residues form multiple interactions with the residues of the hydrophobic platform of the N-terminus (Fig. 1C). On oxidation, the linker and the C-terminal domains are destabilized and this connectivity is disturbed (Fig. 1A), allowing the exposure of certain hydrophobic residues and the binding of client proteins. It is tempting to speculate that interactions between the Hsp33 domains, rather than the sequence itself, have been conserved throughout evolution.
The metastable linker defines the redox-regulated activation but not the anti-aggregation activity of Hsp33
Puzzled by the potentially high functional importance of the inter-residue connectivity between the N-terminus and the adjacent domains, we decided to perturb this connectivity by modifying the linker sequence. To do so, we replaced the native 50-aa sequence of E. coli Hsp33 with nonnative, distinct sequences of a similar length and predefined structural features. At first, to preserve sequence properties but to disrupt the secondary structure of the linker domain, we replaced the native linker domain with a fragment comprising the same amino acids as the native linker but in the reverse order relative to the original sequence (Hsp33-L-Rev) (Fig. 2A). Second, we selected fragments with completely unrelated sequences, a fully disordered 52-aa fragment from the human STIL protein (1239–1288 aa) (1) (Hsp33-L-STIL), and a highly stable and well-structured 54-aa domain of the B1 immunoglobulin-binding protein (17) (Hsp33-L-PGBD) (Fig. 2A).

Circular dichroism (CD) measurements of peptides representing either the wild type (WT) Hsp33 linker or the STIL fragment revealed that they are, indeed, unfolded in solution (Fig. 2B), whereas the 54-aa fragment of PGBD is folded and its structure has been determined by nuclear magnetic resonance (NMR) (PDB ID:1GB1) (17).
Aligning the sequences of STIL (1239–1288 aa), PGBD, and the reversed fragments with the WT Hsp33 linker sequence revealed distinct differences in biophysical features (e.g., hydrophobicity and charge) (Fig. 2C, D and Supplementary Fig. S1; Supplementary Data are available online at
Hsp33 possesses two main functions: It prevents the aggregation of misfolding proteins during oxidative stress, and it transfers unfolded substrates to the relevant foldase system, DnaK/J-GrpE, on return to non-stress conditions. These two functions rely on the ability of Hsp33 to convert its structural regions (mainly the linker and the C-terminal domains) into unfolded or partially unfolded fragments (Fig. 1A). The anti-aggregation function depends on the oxidation of C-terminal cysteines and the subsequent unfolding of Hsp33 (8, 23), whereas client transfer requires the reduction of cysteines and the refolding of the redox switch and linker domain (21). Based on these considerations, we reasoned that replacing the 50-aa native segment of the linker region with a completely unrelated sequence would affect substrate binding and/or substrate release.
To test this hypothesis, the chimeric proteins were expressed in a BL21 E. coli strain that lacks the endogenous Hsp33 gene (BL21ΔhslO), and were purified as described in the Materials and Methods section. Other variants lacking either the C-terminus or linker region were also prepared, however, these proteins were insoluble and, therefore, could not be analyzed in this study. The chimeric proteins were first reduced (Hsp33red) by the reducing agent dithiothreitol (DTT) and then oxidized (Hsp33ox) by using H2O2 under unfolding conditions (43°C) (23). The Hsp33 variants were then tested for their ability to prevent aggregation of two commonly used chaperone substrates: thermally unfolded luciferase and chemically unfolded citrate synthase (CS) (Fig. 3, Supplementary Fig. S3). To our surprise, we found that when oxidized, all Hsp33 variants suppressed aggregation of chemically and thermally denatured substrates to about the same degree as oxidized WT Hsp33 (Fig. 3 and Supplementary Fig. S3). Moreover, WT and chimeric chaperones displayed similar concentration dependence for their anti-aggregation activity (Supplementary Fig. S3). Finally, analysis of complex stability over 24 h showed no difference between the WT and the Hsp33 variants (Supplementary Fig. S4). These results suggested that although the linker region was shown to be involved in client binding (16, 39), Hsp33 is able to utilize alternative residues for successful anti-aggregation activity.
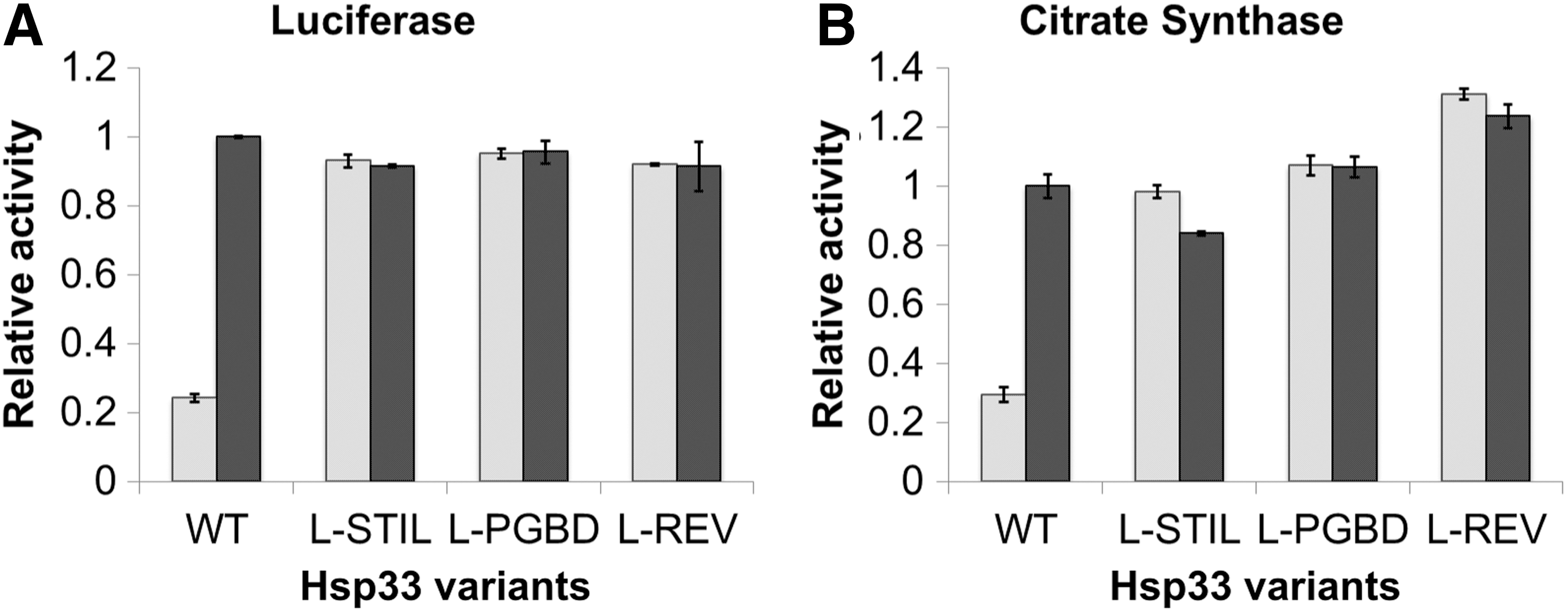
Although replacement of the linker region did not affect client binding, it did have a significant effect on the redox regulation of the chaperone activity. In contrast to strictly redox-dependent chaperone activity of the WT Hsp33, the variants with non-native linker regions (Hsp33-L-STIL, Hsp33-L-PGBD, and Hsp33-L-Rev) prevented protein aggregation to the same extent regardless of whether they were in their reduced or in their oxidized states, showing constitutive anti-aggregation activity (Fig. 3). Thus, the native linker sequence was not evolved to bind client proteins, but to serve as a gatekeeper of the stress-regulated activity. Since the linker region lacks cysteine residues of its own, this gatekeeping activity must be mediated by post-translational changes of the redox-sensitive C-terminus.
The modified Hsp33 chaperones lose their stress-specific unfolding activation mechanism
The chaperone activity of Hsp33 is triggered by disulfide bond-mediated unfolding, which leads to loss of the C-terminal and linker helical and β-strand structures (Fig. 1A) (23, 24). To test the relationship between constitutive chaperone activity and potential structural destabilization, we analyzed the secondary structures of the WT and Hsp33 variants in their reduced and oxidized forms by far-UV CD spectroscopy (Fig. 4). As expected, the WT unfolds in its active, oxidized form, leading to a decrease in chirality and a shift in the CD spectrum, mainly at the helical regions (195 and 210–220 nm) (Fig. 4A, gray traces); these correspond to the C-terminal and linker regions in the inactive, reduced form of Hsp33 (Fig. 1A). These conformational changes lead to the anti-aggregation activity of WT Hsp33 in the oxidized form (23). Modification of the linker region resulted in a loss of structure in both the reduced and the oxidized forms (Fig. 4A, black traces), supporting the constitutive anti-aggregation activity of the chimeric chaperones. Remarkably, the oxidized form of the chimeric chaperone shows even less helical structures (at 195 nm) than the WT chaperone. This might be due to the fact that the linker region is partially folded in the oxidized form as shown in Figure 1A. The CD analysis supports our prediction that the PGBD-containing chaperone retains a slightly higher degree of structure, mainly in the reduced form, than other variants, although it is still significantly less structured than the WT.
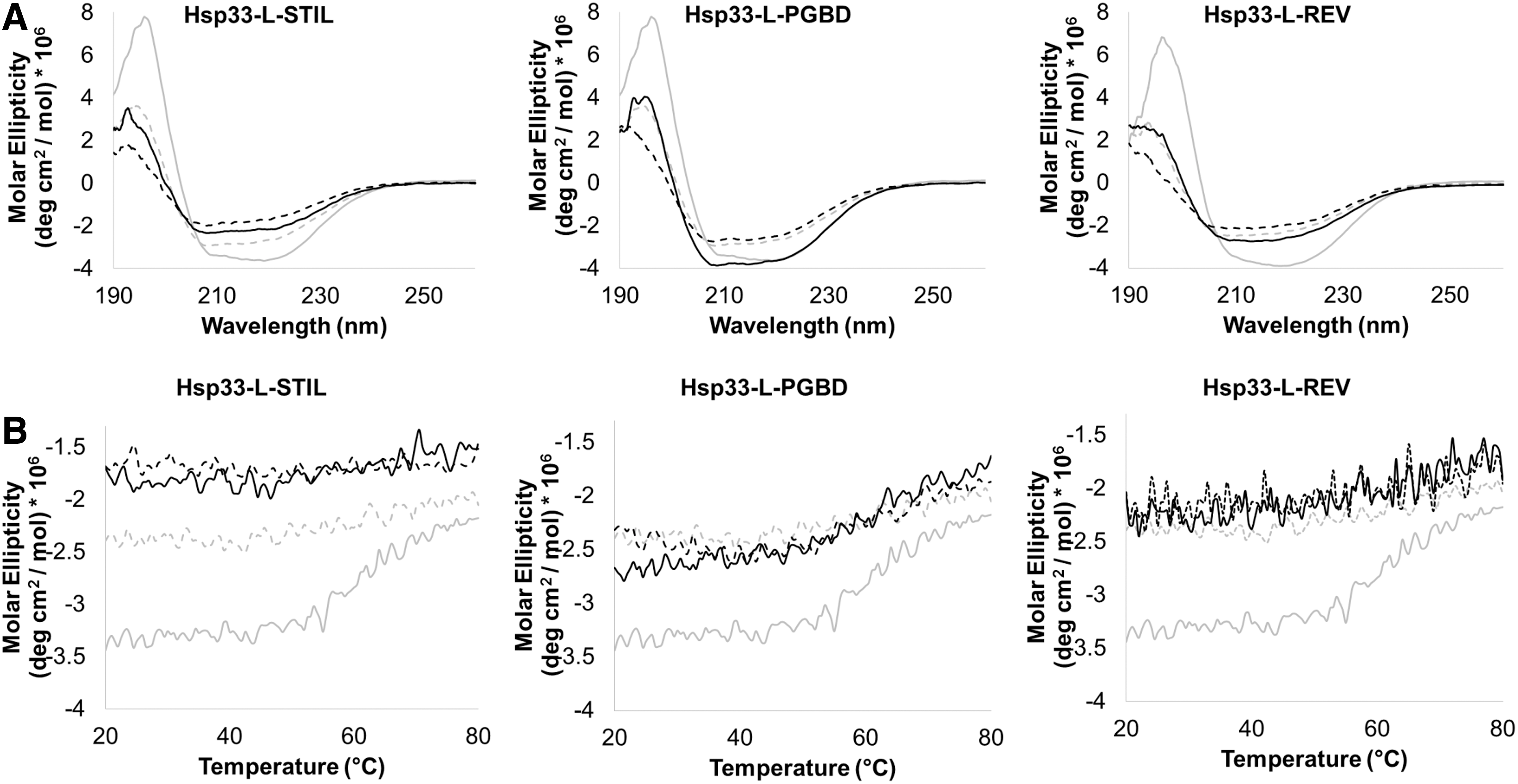
As previously described, inactive, folded Hsp33 is quite thermostable (Tm of ∼60°C), whereas active, oxidized Hsp33 starts to unfold already at ∼40°C (23). Moreover, perturbation of the metastable linker decreases the chaperone's thermostability and leads to constitutive activity (8). To examine the thermostability of the designed chimeras, we monitored the CD signal at 218 nm as a function of temperature (Fig. 4B). As expected, in contrast to WT Hsp33, the thermal stability of the chimeric chaperones was not affected by oxidation. The CD profiles of Hsp33-L-STIL and Hsp33-L-Rev were fully disordered with no clear transition point, at least up to 80°C, whereas Hsp33-L-PGBD showed a moderate loss of structure at 50°C. The WT Hsp33 showed loss of structure at 55°C for the reduced form and no transition for the oxidized form (Fig. 4B).
These results suggest the importance of interactions between the native linker and other Hsp33 domains in the reduced form in determining reversible redox-dependent stability, despite the lack of regulatory cysteine residues in the region.
They also demonstrate that despite low sequence conservation, nature has evolved an efficient self-inactivation mechanism to abolish constitutive binding to partially unfolded proteins under non-stress conditions. This regulatory function is defined by both the sequence and structure of the linker, and not only by amino acid features and the folding state per se.
The metastable linker region prevents self-assembly of Hsp33 and probably affects its crosstalk with the DnaK/J-GrpE system
Hsp33 is unable to refold a substrate by itself, and it instead relies on the successful transfer of the client protein to the ATP-dependent DnaK/J-GrpE system (21). To understand the role that the native linker plays in the transfer process, we monitored the reactivation of thermally unfolded luciferase bound to Hsp33 variants, which were subsequently transferred to DnaK, DnaJ, and GrpE on reduction.
To generate luciferase-Hsp33 complexes, we denatured luciferase at 43°C in the presence of Hsp33 variants (at a fourfold molar excess of chaperones over luciferase). Luciferase lost most of its activity during the 15 min incubation at heat shock temperatures. As previously shown, reactivation of luciferase was strictly dependent on (i) the presence of oxidized wild-type Hsp33 during the unfolding process, (ii) a shift to non-stress temperatures, (iii) addition of the DnaK/J-GrpE/ATP system, and (iv) establishment of reducing conditions (21) (Fig. 5). In contrast to WT Hsp33, none of the chimeric chaperones (Hsp33-L-STIL, Hsp33-L-PGBD, or Hsp33-L-Rev) effectively transferred the substrate on reduction and they achieved only 40–60% of the maximal WT activity (Fig. 5A and Supplementary Fig. S5). In addition, redox regulation of substrate transfer was lost in the Hsp33 variants, showing relatively similar transfer efficiency in the reduced and oxidized states (Hsp33-L-STIL and Hsp33-L-PGBD) or slightly elevated efficiency in the reduced state (Hsp33-L-Rev) (Fig. 5B). This is due to the fact that the Hsp33 chimeric proteins, harboring non-native sequences, are active regardless of their redox status, and they form stable complexes with the misfolded substrate protein.

These results suggest that the evolutionary driving force leading to the native sequence of the linker region was to enable redox regulation of the Hsp33 activity and fruitful crosstalk with the related DnaK/J foldase system, rather than enhancing Hsp33 anti-aggregation activity and client recognition.
One of the possible explanations for a decrease in the client transfer efficiency is a perturbation of the disorder-to-order transition of the C-terminus and the linker region, which are crucial for the unfolding and transfer of the substrate (39). Another possible explanation may lie in the potential influence of the reversed or artificial sequences, STIL and PGBD, on the quaternary structure of Hsp33, and the subsequent effect on substrate release. To test this hypothesis, we characterized the oligomeric structures of the Hsp33 variants.
We used size-exclusion chromatography coupled with multi-angle light scattering (SEC-MALS) to investigate the reduced (inactive in WT) and oxidized (active) forms, which were analyzed on an analytical gel filtration column. To maintain the reduced status of the Hsp33red proteins, the gel filtration buffer was supplemented with DTT. The protein size was evaluated by the refractive index (RI) and 280 nm absorption of the three main fractions as shown in Figure 6 and Table 1. The inactive reduced WT resolved as a single peak (number 3 in Fig. 6) corresponding to a mass close to the monomer size (35.5 ± 3.7). In contrast, reduced Hsp33 variants eluted as two peaks each, one corresponding to a dimer (58 ± 4, 53 ± 6 and 70 ± 9, peak number 3) and the other composed of high oligomeric structures, with a mass higher than 100 kDa (peak number 1 in Fig. 6). RI analysis indicated that the oligomeric structures are highly heterogenic, with an undefined size (Fig. 6, red dots). Interestingly, in contrast to WT Hsp33 whose oxidation did not change the oligomeric state dramatically, with a small amount of dimer and oligomer formation (comprising less than 15% of the oxidized Hsp33 protein), oxidation of the chimeric proteins Hsp33-L-STIL, Hsp33-L-PGBD, and Hsp33-L-Rev resulted in a complete shift of the dimer structures into high oligomers. These results suggested that the native Hsp33 linker region controls the quaternary architecture of Hsp33 most likely by stabilizing the monomeric form via interactions with residues from the N-terminus.
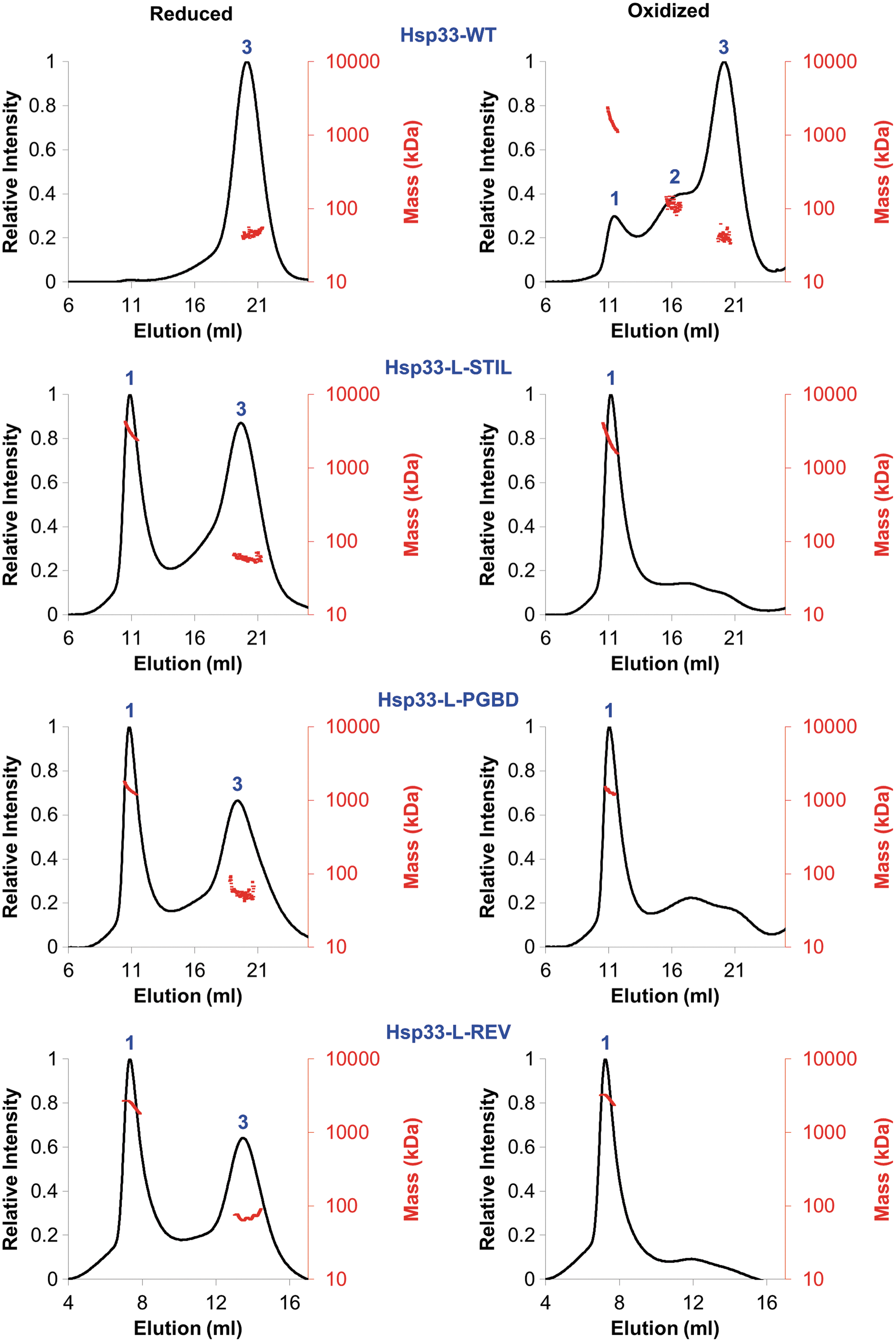
Combining the biophysical and the chaperone activity results, we demonstrate that the native metastable region of the conditionally unfolded chaperone Hsp33 is a regulatory unit that inhibits the tendency of the chaperone to form undesirable oligomeric structures on unfolding. Presumably, due to the hydrophobic regions of Hsp33 that mediate client binding, unfolded Hsp33 tends to form high oligomeric structures that decrease the efficiency of client release. Thus, despite low sequence conservation, nature has evolved an efficient self-inactivation mechanism to ensure successful crosstalk between Hsp33 and the DnaK/J chaperones.
The linker region determines the specificity of substrate recognition in vivo
One of the main objectives of this study was to determine the role of the native linker region in the protection provided by Hsp33 during oxidative stress. Our constitutively active chimeric chaperones represent an excellent model system to investigate the influence of structure on the redox regulation and the Hsp33 interactome during normal and stress conditions in vivo. For this purpose, we transformed BL21ΔhslO E. coli strains with a pET15 vector containing either no Hsp33 (control) or one of three Hsp33 variants: WT Hsp33, Hsp33-L-STIL, and Hsp33-L-PGBD. The cells were cultivated in M9 minimal medium in the presence or absence of 0.75 mM hydrogen peroxide at 37°C. After 60 min of oxidative stress treatment, we harvested the cells and subjected the soluble fraction to immunoprecipitation (IP) by using an anti-Hsp33 antibody. Hsp33-interacting proteins were digested with trypsin and analyzed by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). We applied a label-free quantification (LFQ) strategy to increase the specificity of the isolation by comparing co-precipitated proteins from cells expressing or lacking the Hsp33 gene. All proteins identified in the BL21ΔhslO strain expressing an empty pet15b vector were excluded. Hsp33-interacting proteins (identified in at least two out of three, or three out of four experiments) are summarized in Supplementary Table S1. Note that the identified interacting proteins include both direct and indirect binding partners, and not necessarily misfolded client proteins.
In total, 75 proteins were identified as interacting with WT Hsp33 under oxidative stress conditions. In contrast, 66 proteins were associated with Hsp33-L-STIL and only 41 proteins were associated with Hsp33-L-PGBD (Fig. 7A). As expected, peroxide treatment increased the number of Hsp33-WT interactions (Fig. 7B, dark bars). Almost half of the Hsp33-WT interactome consists of peroxide-specific protein interactions (32 of the 75 interacting proteins identified are peroxide specific). Conversely, only 16–17% of the proteins interacting with the Hsp33 variants are peroxide specific (11 out of 66 for Hsp33-L-STIL, and 7 out of 41 for Hsp33-L-PGBD). This might be due to the observation that the activity of the chimeric Hsp33 proteins is constitutive rather than oxidation dependent.

A consideration of proteins interacting with all three Hsp33 variants identified a subset of 34 proteins common to all three interactomes (Fig. 7C). This indicates that Hsp33 does not recognize this subset of proteins via the linker region. The majority (65%) of this group comprises of proteins that bind Hsp33 during both normal and stress conditions, suggesting that they are house-keeping, non-stress induced interactions. Functional annotation and intersection with a database of known interactions, STRING, revealed that the majority of these linker-independent interacting proteins are part of the protein biogenesis pathway (e.g., Clp and Hfl proteases, tRNA ligases, transcriptional factor G, etc.), and two of them possess redox homeostasis function (Catalase, Glutaredoxin 4, etc.) (Fig. 7D). This finding suggests that Hsp33 has a role as a member of the proteostasis network in bacteria during normal conditions, and not only during oxidative unfolding as previously suggested.
Using the chimeric proteins, we demonstrate that there is another type of Hsp33 interactome, linker-dependent interacting proteins. This subset of proteins is composed of 21 cellular proteins whose Hsp33 interactions showed exclusive dependence on the WT Hsp33 linker sequence. These proteins co-precipitated only with Hsp33-WT, and not with Hsp33-L-STIL or with Hsp33-L-PGBD (Supplementary Table S1). There was no detectable functional enrichment in the linker-specific proteins, suggesting that the majority of them are client proteins of Hsp33 during unfolding conditions. Interestingly, the STIL sequence has a relatively large STIL-specific interactome (12 proteins), which might be due to the unfolded and charged nature of the STIL sequence. It is likely that these interactions are not physiologically relevant for bacterial survival during oxidative conditions. Overexpression of the Hsp33-L-STIL chimeric chaperone led to significant growth defects under oxidative stress conditions (1 mM H2O2, 37°C, overnight growth) in comparison to the WT and the Hsp33-L-PGBD variants (Supplementary Fig. S6). In contrast, normal growth was observed in the absence of oxidative stress. This decrease in stress-related growth might be due to unfavorable interactions with the Hsp33-L-STIL chaperone; however, other explanations are possible.
We conclude that Hsp33 has dual cellular house-keeping or stress-specific functions, which are defined by the metastable region. Despite low sequence conservation, the linker region of Hsp33 is designed to specifically activate the chaperone in the presence of oxidants, and to mediate specific interactions in cells that might be beneficial for survival during unfolding conditions.
Discussion
The metastable linker region acts as a fine-tuning hinge affecting chaperone inactivation and crosstalk with the DnaK/J system
Our results suggest that protein plasticity can play a major role in protein inactivation and can contribute to fine-tuning hinge of chaperone activity. This is demonstrated by the case of Hsp33, in which the metastable 50 aa linker region dramatically affects the redox regulation of Hsp33 chaperone activity, and alters the oligomeric structure of the active chaperone, which, in turn, may influence the crosstalk with the foldase system DnaK/J-GrpE (Fig. 8).
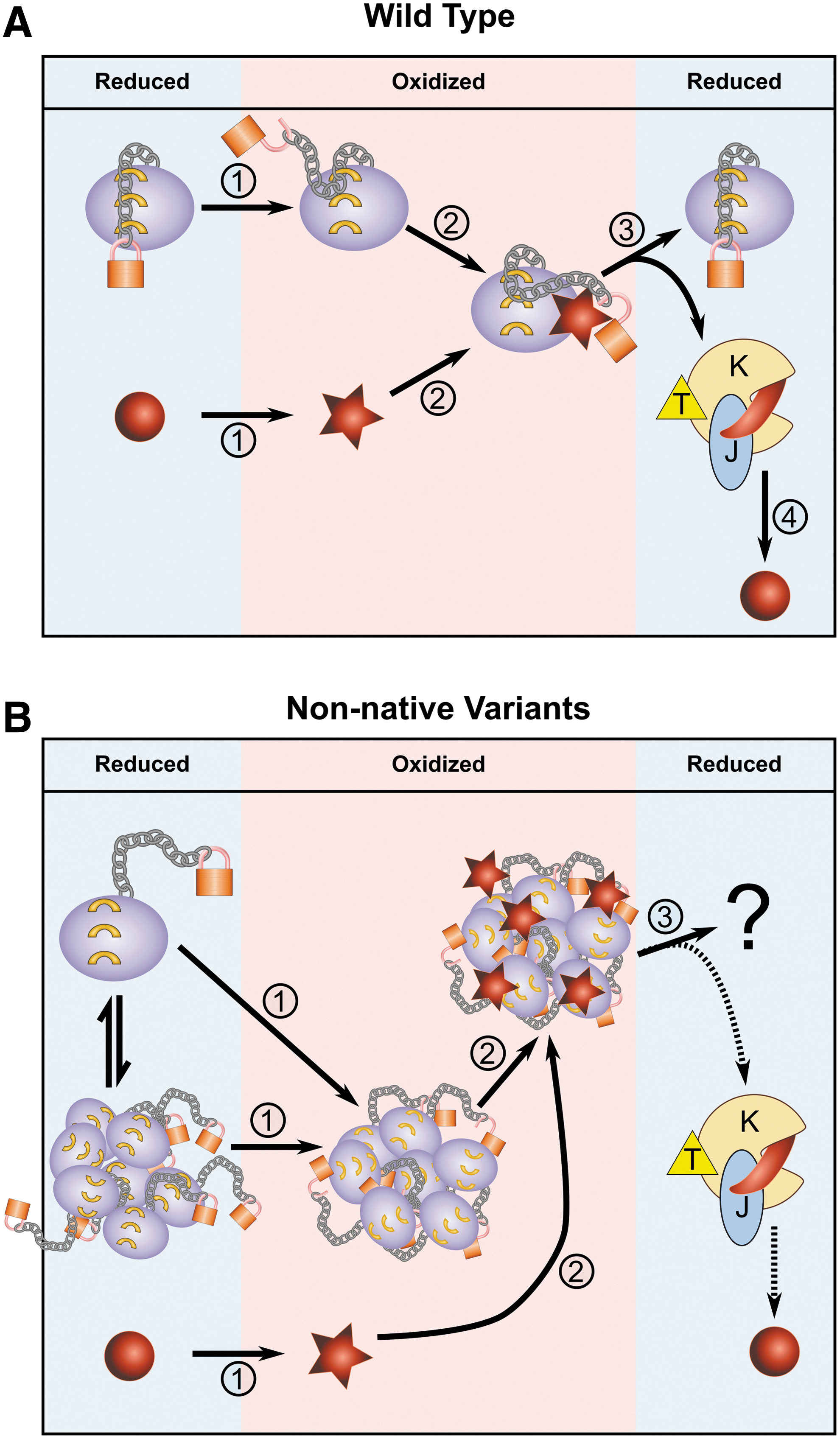
The results of this study suggest that the linker region functions as a gatekeeper for the inactive form of the Hsp33 chaperone by forming an extensive inter-residue network and masking a partially hydrophobic N-terminal β-sheet structure when folded. The helical and β-sheet structures of the folded linker region might also serve as an anchor for the adjacent, intrinsically disordered C-terminal domain, stabilizing it in the reduced form. When the C-terminal redox domain is oxidized, disulfide bonds are formed between the highly conserved cysteines; this leads to destabilization of the linker region and perturbation of its connectivity map with the N-terminus. Replacement of the linker region by a sequence comprising the same amino acids as the native domain but in reverse order led to the unfolding of the chaperone and abolished this WT anchor function, leading to constitutive chaperone activity. The same phenomenon was observed when the linker domain was replaced by other artificial sequences. It is tempting to speculate that interactions between residues from the folded linker and the N-terminal domain have been conserved during evolution, and they may be important in the deactivation of other Hsp33 chaperone homologues. This hypothesis will remain to be tested once there is a sufficient number of solved Hsp33 structures.
The model in Figure 8 posits that perturbation of the linker region destabilizes, and probably detaches, the redox sensitive domain. To test this hypothesis, we monitored the oxidation kinetics of the WT and the chimeric proteins by trapping reduced thiols over the course of 3 h of oxidation. We used a methoxypolyethylene glycol maleimide reagent (∼2 kDa) that modifies free thiols, resulting in a migration shift of the modified proteins in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Supplementary Fig. S7). Consistent with our model, all the chimeric proteins underwent faster oxidation than the WT protein. These results may indicate that the redox domain is more exposed or destabilized upon perturbation of the linker domain.
By modifying the chaperone sequence, we succeeded in uncoupling two functions of Hsp33: anti-aggregation activity and substrate release. Remarkably, replacement of the 50-aa linker region by either a reversed sequence or completely unrelated sequences had no effect on Hsp33's anti-aggregation activity, even though this was experimentally shown to be a potential binding site for substrates (16, 39). On the other hand, replacement of the native linker region did dramatically affect the substrate release properties of Hsp33. We hypothesize that the maintenance of binding illustrates the high binding promiscuity of the intrinsically disordered chaperone, whereas the substrate release and subsequent transfer to the DnaK/J system is unfavorably affected by the formation of higher oligomeric structures. This stands in contrast to the importance of the heat-induced oligomeric structures formed by sHSPs, for their anti-aggregation activity and substrate release (14, 19).
In recent years, different methodologies have been used to map structural changes of Hsp33 on activation, substrate binding, and release, as well as to identify the binding site in Hsp33 with either full-length proteins (16, 23, 39) or peptides (16). Hydrogen/deuterium exchange and limited proteolysis coupled with mass spectrometry are powerful methods that are employed to monitor structural changes and solvent accessibility. However, they cannot accurately map direct interactions between the chaperone and its client protein. These methods suggest that the linker domain undergoes conformational changes on activation (13) and substrate binding, suggesting that this region is involved in substrate recognition. To map the binding site of Hsp33, the Jacob lab used unbiased methodologies, including in vivo and in vitro crosslinking and 19F NMR (16). That study showed that many polar residues of the linker domain are involved in substrate binding both in vivo and in vitro, along with residues of the N-terminal domain. Together with our study, this information suggests that the substrate recognition of Hsp33 is highly promiscuous and involves distant and stretched regions of the chaperone (Fig. 8A), thus enabling extensive perturbation of the amino acids comprising the substrate binding domain. This is consistent with the fact that no distinct sequence motifs corresponding to the binding site with client proteins are conserved among the thousands of Hsp33 homologues.
The Hsp33 interactome consists of linker-dependent and linker-independent interactions
Recent studies showed that Hsp33 recognizes its substrates via several hydrophobic regions, ranging from the N-terminal region, through the linker region to the C-terminus (16). This may explain why simple replacement of the native Hsp33 linker region by artificial sequences did not abolish the ability to prevent protein aggregation in vitro. However, in vivo, perturbation of the native sequence had a dramatic effect on the Hsp33 interactome. Mass spectrometry analysis uncovered two types of Hsp33-interacting proteins: linker dependent, interacting only with the WT Hsp33 chaperone, and linker independent, interacting with all three Hsp33 variants tested (WT, Hsp33-L-STIL, and Hsp33-PGBD). The linker-independent proteins are probably involved in interactions of Hsp33 with members of the proteostasis network in bacteria, including ribosomal proteins, Clp and Hfl proteases, tRNA ligases, as well as other proteins, such as elongation factor G, redox homeostasis proteins, catalase-peroxidase 1, and Glutaredoxin 4 (Supplementary Table S1 and Fig. 7). These interactions expose a previously unexplored and potentially important role of Hsp33 in the house-keeping proteostasis under normal conditions, as was suggested by the report that Hsp33 regulates the stability of the transcriptional factor Ef-Tu and targets it to the Lon protease (5). Given the fact that some misfolded proteins cannot be refolded by the DnaK/J system, and that Hsp33 cannot transfer its substrate to the alternative chaperone system GroEL/S (21), there is a possibility that Hsp33 is involved in substrate degradation. This hypothesis should be investigated in the future.
In contrast to the linker-independent proteins, a subset of 24 proteins was shown to interact in a linker-dependent fashion, binding exclusively to WT Hsp33 (Supplementary Table S1 and Fig. 7). Members of this group cannot be clustered by functional annotation, which suggests the possibility that these proteins are client proteins of Hsp33, mediators of the Hsp33 activity via the linker region, or both.
There is a possibility that changes in the interactome between WT and chimeric proteins are rooted in potential structural changes of the adjacent regions, or the oligomeric structures of the chimeric proteins. Moreover, due to the nature of co-immunoprecipitation analysis, there is no simple way to distinguish between direct and indirect interactions. To precisely identify the role of the linker in the Hsp33 interactome, additional studies such as in vivo crosslinking should be conducted.
From the results of this study, we propose a mechanism of stress-specific inactivation of chaperone activity depending on a metastable segment that adopts multiple structural conformations (Fig. 8). Despite weak sequence conservation, the native sequence of the region must have evolved to be a closely controlled region, which under normal circumstances maintains the chaperone in an inactivated conformation but that can be activated by unfolding of the adjacent redox domain to enable a dual function of Hsp33 in the cell during normal and stress conditions.
Materials and Methods
Strains and Plasmids
The BL21ΔhslO containing an empty pet15 expression vector, and BL21ΔhslO expressing WT hslO (Hsp33) were generously provided by the lab of Ursula Jakob (UM). BL21ΔhslO expressing Hsp33-L-STIL, Hsp33-L-PGBD and BL21 strain expressing GrpE were designed for this study as described in the Results and Supplementary Data sessions. The GrpE plasmid was obtained from the Jakob lab (UM). Strains expressing DnaK, DnaJ were generously provided by Drs. Bukau and Mogk.
Sequence and structural analysis of the Hsp33
To define hydrophobicity and charge (Fig. 2C, D), we calculated the net hydrophobicity and charge scores by using a sliding window of five amino acids. The hydrophobicity of each amino acid sequence was calculated by the Kyte and Doolittle approximation (29). The hydrophobicity of individual residues was normalized to a scale of 0–1 in these calculations, and an average score was calculated for the five residues. The net charge was derived using natural pH (7.0), and an average score was calculated for the five residues.
The inter-residue connectivity map between residues from the N-terminus and the linker region (Fig. 1B) was derived by using modified AQUAPROT software (38) using the default parameters. The structural model of reduced, inactive, E. coli Hsp33 was based on the structure of reduced Bacillus subtilis Hsp33 (PDB: 1VZY) (25) as described in (8). Cytoscape v 3.2.1 was used to draw the connectivity map.
Gene constructs
The Hsp33-L-STIL gene was synthesized by IDT and cloned into the pet15b vector by using a restriction-free cloning approach (48). The STIL fragment amplification with pet15 complementary sequences was performed by using KAPA HiFi HotStart PCR kit (KAPABiosystems). Replacement of the native linker region by the STIL fragment was done by Phusion High-Fidelity DNA Polymerase (New England Biolabs, NEB). Polymerase chain reaction (PCR) was carried out by using the Labcycler PCR machine (SensoQuest).
The Hsp33-PGBD gene was constructed by using restriction-free cloning based on the pET15b-Hsp33-L-STIL vector as a template aligned with extended dsDNA oligomers (geneBlock) synthesized by IDT. The sequence of the PGBD block is TATGTTGTTGCAGGTAATGCCGGCGACCTATAAATTAATCCTGAACGGTAAGACCTTGAAAGGCGAAACCACGACCGAAGCGGTGGATGCGGCCACAGCTGAAAAGGTGTTCAAACAGTATGCCAATGATAATGGTGTTGATGGCGAATGGACCTACGACGACGCTACCAAAACGTTTACGGTCACGGAGGGATCCTTCAAATGCACCTGCTCGC.
The GeneBlock amplification and its insertion into the pet15b-Hsp33-L-STIL plasmid were done with the KAPA HiFi HotStart PCR kit and the Phusion High-Fidelity DNA Polymerase (NEB).
Sequences of Hsp33 and its variants are as follows:
Hsp33-WT-linker: QNAQQDDFDHLATLTETIKTEELLTLPANEVLWRLYHEEEVTVYDPQDVE
STIL C-terminal fragment 1239–1288 (to replace the WT linker):
WQHPEKENEGDITIFPESLQPSETLKQMNSMNSVGTFLDVKRLRQLPKLFGS
PGBD (to replace the WT linker):
TYKLILNGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKTFTVTE
Protein expression and purification
Hsp33 and its variants were expressed and purified from E. coli BL21ΔhslO by using a His-tag as described later. The purified proteins were first reduced by 5 mM DTT and ZnCl2 and then oxidized as described (23). Luciferase and CS were purchased from Promega (E7101) and Sigma (C3260), respectively.
Purification of Hsp33 and variants
Hsp33 constructs were inserted into the E. coli BL21 ΔhslO bacteria strain, selected by ampicillin resistance, and grown to logarithmic phase (optical density [OD] of 0.5–0.6 at 600 nm) in 6 liters of LB media at 37°C. Expression was induced by adding isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM and culture overnight at 18°C. Cells were then harvested from the media; resuspended in 80 ml of 40 mM HEPES, 500 mM NaCl, and 5 mM imidazole, pH = 7.5; and supplemented with 0.2 mg/ml lysozyme, 1 mM AEBSF, and one tablet of Roche's EDTA-free protease inhibitor cocktail. After 30 min of incubation on ice, the cells were disrupted at ∼25,000 psi and the lysate was centrifuged for 45 min at 14,500 rpm in 4°C.
Proteins were purified on a 5 ml HiTrap chelating column (GE Healthcare) that was preloaded with NiSO4 and eluted with a 100 ml gradient of 5–500 mM imidazole in the resuspension buffer (40 mM HEPES, 500 mM NaCl,). Proteins were dialyzed against 40 mM HEPES, 140 mM NaCl pH = 7.5 and concentrated to 10 ml through an Amicon Ultra-15 30K molecular weight cutoff (MWCO) filter (Millipore). Further purification was done on a HiLoad 16/60 Superdex 75 pg gel filtration column (GE Healthcare). Hsp33-L-PGBD required a further purification step, separation by using a hydroxyapatite column (self-packed, 58 ml hydroxyapatite resin from BioRad). Proteins were separated by using a 200 ml gradient of 5–70 mM KH2PO4 pH = 7.5.
Purified proteins were then dialyzed against 40 mM KH2PO4 pH = 7.5 buffer and concentrated.
Protein purity was determined and confirmed by SDS-PAGE, and protein concentration was determined by light absorbance at 280 nm (ɛHsp33-WT = 19,285 cm−1 M−1, ɛHsp33-L-STIL = 17,795 cm−1 M−1, ɛHsp33-L-PGBD = 20,600 cm−1 M−1).
Purification of GrpE
GrpE was purified from BL21 cells expressing the pWKG20 -GrpE vector, which had been generously provided by the Jakob Lab (UM, Ann Arbor). Cells were grown in 2 L of LB media at 37°C to reach an OD 600 nm of ∼0.4. Expression was induced by the addition of 1% L-arabinose and culture for 5 h at 37°C. Cells were harvested from the media; resuspended in 7 ml of 50 mM EDTA and 5 mM DTT pH = 8.0 and 38 ml of buffer A (50 mM Tris-HCl and 10% sucrose pH = 8.0); and supplemented with 0.8 mg/ml lysozyme, 1 mM AEBSF, and one tablet of EDTA-free protease inhibitor cocktail (Roche). After incubation on ice for 20 min, the cells were disrupted at ∼25,000 psi and the lysate was centrifuged at 4°C for 45 min at 14,500 rpm. While on ice, 60% (NH4)2SO4 was added to the supernatant, and after 1 h the pellet was collected by centrifugation at 20,000 rpm at 4°C for 30 min. The pellet was resuspended in 10 ml of buffer B (50 mM Tris-HCl, 10% sucrose, 1 mM EDTA, and 10 mM β-ME pH = 7.2) and dialyzed against the same buffer overnight.
Anion exchange was performed by using three 5 ml HiTrap Q HP columns (GE Healthcare) equilibrated with 5 column volumes (CV) of buffer B. The samples were eluted with a 300 ml gradient of 0–0.4 M NaCl in the same buffer. Fractions containing GrpE were collected and dialyzed against buffer B overnight.
To remove nucleotides, further purification was done by using a 5 ml HiTrap HP Blue column (GE Healthcare), which binds nucleotides but not GrpE. The column was equilibrated with 5 CV of buffer B. Protein samples were concentrated down to a volume of 15 ml by using an Amicon Ultra-15 30K MWCO filter (Millipore), and they were loaded onto the column. Fractions were collected and dialyzed overnight against buffer C (5 mM KH2PO4 pH 6.8).
The sample was further purified on a 58 ml hydroxyapatite column equilibrated with buffer C. The column was eluted with a 400 ml gradient from 5 to 100 mM KH2PO4, and the fractions were concentrated and dialyzed against buffer D (30 mM HEPES, 50 mM KCl, and 5% glycerol pH = 7.0). Protein purity was determined and confirmed by SDS-PAGE, and protein concentration was determined by light absorbance at 280 nm (ɛGrpE = 1490 cm−1 M−1).
Purification of DnaJ
BL21 cells expressing the DnaJ gene from the pUHE21Δ12 plasmid were generously provided by the B. Bukau lab at the Center for Molecular Biology in Heidelberg University.
Cells were grown in 3 L of 2xYT media at 30°C to reach an OD 600 nm of ∼0.5. Expression was induced by the addition of 1 mM IPTG and culture for 5 h at 30°C. Cells were harvested from the media; resuspended in 130 ml lysis buffer (50 mM Tris-HCl, 10 mM DTT, 0.6% Brij58 pH = 8.0); and supplemented with 0.8 mg/ml lysozyme, 1 mM AEBSF, and one tablet of the EDTA-free protease inhibitor cocktail (Roche). After incubation on ice for 30 min, the cells were disrupted at ∼25,000 psi and the lysate was centrifuged at 4°C for 45 min at 14,500 rpm.
The supernatant was then collected and mixed with one volume of buffer A (50 m M NaH2PO4, 5 mM DTT, 1 mM EDTA, and 0.1% Brij58 pH = 7.0), and (NH4)2SO4 (60%) was added. After a 1 h incubation on ice, the sample was centrifuged at 145,000 rpm at 4°C for 50 min, and the pellet was resuspended in 115 ml of buffer B (50 mM NaH2PO4, 5 mM DTT, 1 mM EDTA, and 0.1% Brij58, 2 M Urea pH = 7.0) and dialyzed against the same buffer overnight.
Cation exchange was performed by loading the sample onto four continuously connected 5 ml HiTrap SP HP columns (GE Healthcare) that were equilibrated with 5 CV of buffer B. Elution was performed with a 15 CV gradient from 0 to 0.3 M KCl. Fractions containing DnaJ were dialyzed against buffer C (50 mM Tris-HCl, 5 mM DTT, 0.1% Brij58, 2 M Urea, and 50 mM KCl pH = 7.5).
The sample was further purified on a 58 ml hydroxyapatite column that was equilibrated with buffer C. After loading the sample, the column was washed with 1 CV of buffer C that was supplemented with 1 M KCl, and then with 2 CV of buffer C. Samples were eluted by a 1 CV gradient from 0 to 0.3 M KH2PO4, followed by a 2 CV wash in 0.3 M KH2PO4. Fractions containing DnaJ were collected and dialyzed against buffer D (50 mM HEPES, 100 mM KCl pH = 7.7).
Protein purity was determined and confirmed by SDS-PAGE, and protein concentration was determined by light absorbance at 280 nm (ɛDnaJ = 14,900 cm1M1).
Purification of DnaK
Expression of a DnaK protein with His-tag on its C-terminus was in the BL21 ΔdnaK52 cells that were generously provided by B. Bukau's lab at the Center for Molecular Biology at Heidelberg University.
Cells were grown in 3 L of 2xYT media at 30°C to reach an OD 600 nm of ∼0.5. Expression was induced by the addition of 1 mM IPTG and culture for 4 h at 30°C. Cells were then harvested from the media and resuspended in 40 ml lysis buffer (50 mM NaH2PO4, 300 mM NaCl, and 5 mM TCEP pH = 8.0) that was supplemented with 0.5 mg/ml lysozyme, 1 mM AEBSF, 10 μg/ml DNaseI, 5 mM MgCl2, and one tablet of Roche's EDTA-free protease inhibitor cocktail. After incubation on ice for 30 min, the cells were disrupted at ∼25,000 psi and the lysate was centrifuged at 4°C for 45 min at 14,500 rpm.
The samples were purified on two 5 ml HiTrap chelating columns (GE Healthcare) that were preloaded with NiSO4 and equilibrated with 5 CV of lysis buffer containing 5 mM imidazole and 1 mM ATP. The columns were eluted with a 30 CV gradient from 5 to 250 mM imidazole in the same buffer at pH = 7.5. The samples were then dialyzed overnight at 4°C against 20 mM HEPES, 50 mM KCl, and 5 mM β-ME at pH 7.5 and loaded onto four 5 ml HiTrap Q HP anion exchange columns (GE Healthcare). The columns were eluted with a 12 CV gradient from 50 mM to 0.5 M KCl. The collected fractions were dialyzed against 40 mM HEPES, 50 mM KCl, 5 mM MgCl2, 5 mM β-ME, and 5% glycerol at pH = 7.5. Protein purity was determined and confirmed by SDS-PAGE, and protein concentration was determined by light absorbance at 280 nm (ɛDnaK-C-term-His = 15,930 cm−1M−1).
Analysis of Hsp33-complex stability
Complexes between chimeric or native Hsp33 and thermally unfolded porcine heart, mitochondrial CS (Sigma) were formed as described (53). Briefly, 220 μL of oxidized Hsp33 variants at 5 μM in 40 mM KH2PO4 were shaken at 43°C/600 rpm whereas CS was added in small portions every 15 min (four times total) to a final concentration of 7.5 μM. After the CS reached 7.5 μM, the sample was incubated at 25°C with 400 rpm shaking speed. As a control, we used reduced, inactive Hsp33 to follow the aggregation of CS at 43°C. At time 0 and subsequent designated time points, aliquots of 32 μL were removed and centrifuged at 13,300 rpm/5 min. The pellet was harvested and resuspended in 32 μL 40 mM KH2PO4. After collection, all samples were analyzed together by SDS-PAGE.
Chaperone activity assay of Hsp33
The influence of Hsp33 and variants on aggregation of either thermally unfolded luciferase (150 nM) or chemically unfolded CS (75 nM) was determined as described (15) by using Hsp33 chaperones in various concentrations as described in the figure legends. Light scattering was monitored in 40 mM HEPES, pH = 7.5 at 43°C (for thermal unfolding) or 30°C (chemical unfolding) at 360 nm (excitation and emission wavelengths).
Hsp33-mediated refolding of thermally unfolded luciferase
The protocol for Hsp33-mediated refolding of luciferase (Promega) was based on Ref. (21). Briefly, luciferase (150 nM) was incubated in the presence or absence of 600 nM Hsp33 chaperone in 0.5 ml incubation buffer (40 mM MOPS, 50 mM KCl, pH = 7.5) for 15 min at 43°C. In parallel, aggregation of the luciferase protein was measured by light scattering, to ensure unfolding and aggregation of luciferase and to engage the protective function of Hsp33. The reaction was cooled down to 25°C and diluted twice into the refolding buffer (40 mM MOPS, 50 mM KCl, pH = 7.5 supplemented with 0.1 mg/ml BSA, 2 mM MgATP, 2 mM DTT, 1.5 μM DnaK, 0.3 μM DnaJ, and 1.5 μM GrpE). To evaluate spontaneous refolding, luciferase refolding was measured with or without 2 mM DTT or DnaK/J, GrpE chaperones either together or separately. Aliquots of 5 μl were taken and assayed for luciferase activity by using Luminescence (Modulus II Microplate Multimode Reader, Turner Biosystems) for eight consecutive reads every 20 s. Relative activity was calculated with reference to folded luciferase (100%) and to fully unfolded Luciferase (0%).
Far-UV CD spectroscopy
To determine changes in the secondary structure of WT Hsp33 and its variants, far-UV CD spectra were recorded for 190–260 nm, by using the Jasco J810 spectropolarimeter. For the CD spectra and thermostability measurements, we used 5 μM Hsp33 proteins in 40 mM KH2PO4, pH = 7.5 in a quartz cell with a 1 mm path length at 25°C. All spectra were buffer corrected, and the average of 3–5 spectra was calculated and presented. To determine the thermostability of the mutants, the CD signal at 218 nm was measured while raising the temperature from 20°C to 80°C at a rate of 1°C/min.
Size-exclusion chromatography coupled with multi-angle static light scattering
Protein samples (1.5–2.5 mg/ml) were analyzed by the multi-angle light scattering detector MiniDAWN TREOS (Wyatt Technology) during separation on a SEC column (Superose 12 10/300, GE Healthcare) in 40 mM KH2PO4, pH = 7.5. A 5 mM solution of DTT was added to analyze the reduced Hsp33 samples. Protein masses were calculated with respect to the concentration, absorption at 280 nm, and RI measured by an Optilab T-rEX refractometer (Wyatt Technology).
Immunoprecipitation of the Hsp33-interacting proteins and the sample preparation for MS analysis
BL21ΔhslO cells expressing either empty pet15 bp vector or pet15 b vector with WT Hsp33 or Hsp33-L-STIL or Hsp33-L-PGBD were cultivated (12 ml) at 37°C in M9 minimal medium until an OD600 of 0.5 was reached. Hydrogen peroxide (1 mM) was added, and the cells were incubated at 37°C for 1 h. The cells were then lysed in 1 ml of Lysis buffer (50 mM Tris-HCl pH = 7.5, 0.5 M NaCl, 10% Glycerol) that was supplemented with 0.8 mg/ml lysozyme, 1:1000 benzonase, 0.1% n-Dodecyl-β-D-maltoside (DDM), protease inhibitor AEBSF (1 mM), and one tablet of EDTA-free protease inhibitor mix (Roche). After two freeze-thaw cycles, the lysate was centrifuged at 13,000 g for 30 min at 4°C, and supernatants were collected.
For immunoprecipitation, the lysates were incubated with 2 μl of Rabbit anti-Hsp33 antibody (provided by the Ursula Jakob lab) and rocked for 30 min at 4°C. Thereafter, re-equilibrated Protein A beads were added; the samples were rocked for 3 h at 4°C and then washed five times. The immunoprecipitated samples were analyzed by SDS-PAGE. To prepare samples for the mass spectrometry analysis, on-beads digestion was conducted as described (22, 47). Briefly, the beads were suspended in freshly prepared denaturation buffer (25 mM Tris-HCl, 10 mM DTT, 6.4 M Urea, pH 8.0) and alkylated in the dark for 1 h by using 55 mM iodoacetamide (IAM). After a sixfold dilution, the proteins were digested with 0.3 μg Trypsin (sequencing grade modified trypsin V5111, Promega) in digestion buffer (10% acetonitrile [can], 1.3 M Urea, 25 mM Tris pH 8) overnight at 37°C. The beads were then removed by centrifugation at 13,000 rpm for 5 min. The peptide mixture was desalted by using in-house packed C18 StageTips (37). The StageTips were preconditioned with 100% Methanol and then with 80% ACN. The equilibration was performed with 2 × 0.1 ml 0.1% formic acid (FA) before application of the samples at a total amount of 15 μg total protein per stage tip. The bound peptides were washed with 2 × 0.1 ml 0.1% FA and eluted with 120 μl 80% ACN plus 0.1% FA. The eluted fractions were dried under vacuum and resuspended in 0.1% FA to a final concentration of around 0.4 μg/μl.
Nano-LC-MS/MS analysis
The peptides were injected and then separated on a C18 reverse-phase column coupled to the Nano electrospray, EASY-spray (PepMap, 75 mm × 50 cm, Thermo Scientific) at a flow of 300 nl/min by using a Dionex Nano-HPLC system (Thermo Scientific) coupled online to an Orbitrap Mass spectrometer, Q Exactive (Thermo Scientific). To separate the peptides, the column was applied with a linear gradient at a flow rate of 300 nl/min at 35°C: from 3 to 45% in 60 min, from 45 to 90% in 15 min, and then held at 90% for an additional 30 min, before being equilibrated at 3% for 20 min (solvent A is 0.1% FA, and solvent B is 80% acetonitrile, 0.1% FA). The Q Exactive was operated in a data-dependent mode. The survey scan range was set to 300–2000 m/z, with a resolution of 70,000 at m/z. A maximum of the 12 most abundant isotope patterns with a charge of ≥2 and <7 were subjected to higher-energy collisional dissociation with a normalized collision energy of 28, an isolation window of 1.5 m/z, and a resolution of 17,500 at m/z. To limit repeated sequencing, dynamic exclusion of sequenced peptides was set to 30 s. Thresholds for ion injection time and ion target value were set to 120 ms and 3 × 106 for the survey scans and to 70 ms and 105 for the MS/MS scans. Only ions with a “peptide preferable” profile were analyzed for MS/MS. The data was acquired by using Xcalibur software (Thermo Scientific).
Data analysis and statistics
For protein identification and quantification, we used MaxQuant software (7), version 1.5.3.30. We used Andromeda search incorporated into MaxQuant to search MS/MS spectra against the UniProtKB database of the E. coli proteome (K12) (Uniprot release, Aug 2015), including sequences of the Hsp33 variants, Hsp33-L-STIL and Hsp33-L-PGBD, and the database of potential contaminators (embedded in Andromeda). Enzyme specificity was set to trypsin, allowing cleavage N-terminal to proline and a maximum of two miscleavages. Peptides had to have a minimum length of seven amino acids to be considered for identification. Carbamidomethylation was set as a fixed modification, and methionine oxidation was set as a variable modification. A false discovery rate of 0.05 was applied at the peptide and protein levels. Initial precursor mass deviation till 4.5 ppm and fragment mass deviation till 20 ppm were allowed.
Only proteins identified by more than two peptides were considered. To quantify changes in protein expression, we utilized the LFQ using the MaxQuant default parameters (7). Protein contaminators and proteins that were identified in the control (BL21ΔhslO with empty pet15 b vector) pulldown were removed. A list of Hsp33-interacting proteins that appeared in at least two from three experiments (or at least three from four) is summarized in Supplementary Table S1. The string server (
Footnotes
Acknowledgments
The authors are extremely grateful to Prof. Ursula Jakob for unlimited assistance during this project, and specifically, for exceptional help with establishing the luciferase refolding assays, and for providing them with the BL21ΔhslO strains, GrpE expression vector, anti-Hsp33 antibodies, and the related protocols, as well as for helpful discussions and critically reading the article. They are tremendously thankful to Drs. Bernd Bukau and Axel Mogk for providing them with the DnaK/DnaJ expression system and the purification protocols, and for helpful discussions. They are indebted to the Protein Purification unit of the Life Science Institute, and especially to Dr. M. Lebendiker.
The authors are grateful to the German-Israel Foundation (grant number: I-2332-1149.9/2012), Marie-Curie integration grant (project number: 618806), Abish-Frenkel Foundation, Israel Science Foundation (Grant Number: 1765/13), and Human Frontier Science Program (CDA00064/2014) for financial support, and to the Dean Fellowship for funding O.S. A.F. was supported by grants from the Israel Science Foundation (ISF) and the Israel Cancer Research Foundation (ICRF); H.A. was supported by the Dalya and Dan Maydan Fellowship.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.