
Abstract
Significance:
The family of gasotransmitter molecules, nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S), has emerged as an important mediator of numerous cellular signal transduction and pathophysiological responses. As such, these molecules have been reported to influence a diverse array of biochemical, molecular, and cell biology events often impacting one another.
Recent Advances:
Discrete regulation of gasotransmitter molecule formation, movement, and reaction is critical to their biological function. Due to the chemical nature of these molecules, they can move rapidly throughout cells and tissues acting on targets through reactions with metal groups, reactive chemical species, and protein amino acids.
Critical Issues:
Given the breadth and complexity of gasotransmitter reactions, this field of research is expanding into exciting, yet sometimes confusing, areas of study with significant promise for understanding health and disease. The precise amounts of tissue and cellular gasotransmitter levels and where they are formed, as well as how they react with molecular targets or themselves, all remain poorly understood.
Future Directions:
Elucidation of specific molecular targets, characteristics of gasotransmitter molecule heterotypic interactions, and spatiotemporal formation and metabolism are all important to better understand their true pathophysiological importance in various organ systems. Antioxid. Redox Signal. 26, 936–960.
Gaseous Molecules and Heterocellular Communication
Overview of heterocellular communication
C
Organs comprise multiple cell types that differ widely in morphology, metabolic function, and biochemical and molecular responses. However, tissue cells have evolved to work together to coordinate key functions of the organ system to which they belong. Further complication exists with the combination of different cellular responses to external stimuli within the microenvironment. Importantly, not all types of cells behave similarly in response to certain challenge. Cellular physiological responses are able to adapt to external stressors, resulting in alterations in tissue composition and function. However, when these adaptive responses fail or are dysfunctional, pathological changes ensue. Regulation of adaptive responses and their potential pathophysiology functions has come to light through understanding cellular and molecular signaling mediators between cells.
Different signaling mediators have been identified over the span of biomedical research with varying degrees of activity, longevity, and potency. However, a new class of signaling mediators has emerged, which have existed since the dawn of life on this planet and are still biologically important to this day. The gasotransmitter group of molecules influences multiple cell types and systemic physiological functions, making them essential in the study of heterocellular signaling associated with tissue pathophysiology.
Gaseous signaling molecules, including nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S), fall under the criteria as gasotransmitter family previously suggested by Wang (253). However, it is worth noting that none of these molecules is likely to be present as a gas rather than dissolved solute. Others refer to them as small-molecule signaling agents (69). While the term gasotransmitter may be less than ideal to describe these agents, their gaseous characteristics reveal that they are endogenously produced and freely permeable across cells and tissues with no reliance on classical receptors. In this review, we discuss roles of gasotransmitters in regulating heterocellular signaling responses and their involvement in various pathophysiological functions common between them.
Gasotransmitter family of molecules: NO, H2S, and CO
Chemical properties
To date, three chemicals comprise the gasotransmitter family of molecules, including NO, CO, and H2S. Over the years, thousands of studies have reported important physiological and pathological processes of gasotransmitter molecules with continued research growth (Fig. 1) (96). While these molecules are referred to as gasotransmitters, it is important to understand that the bulk of their biochemical and biological effects do not occur while in a gaseous state (256, 257). Therefore, understanding each molecule's chemical property is useful in knowing how it may react.

NO is a colorless gaseous free radical with an unpaired electron making it highly reactive. NO can readily react with oxygen and water to be converted into nitrogen dioxide and nitrous acid (HNO2), respectively. HNO2 is a weak and monobasic acid with a pKa of 3.39. Nitroxyl (HNO) is the reduced product of NO, and NO dimerization (N2O2) can occur at low temperatures.
CO is a colorless and odorless gas molecule with a triple-bonded carbon and oxygen atom. CO has a dipole moment of 0.122 D since oxygen is more electronegative than carbon (243). CO is highly toxic through its binding to hemoglobin to produce carboxyhemoglobin, rendering hemoglobin ineffective at delivering oxygen throughout the body (87).
H2S is a colorless gas with a pungent odor of rotten eggs and can be oxidized to form sulfur dioxide, sulfates, sulfite, and elemental sulfur. H2S gas is sparingly soluble in water and acts as a weak acid with an acid dissociation constant (pKa1) of 7.04 and pKa2 of 19 at 37°C (215). Consequently, under physiological conditions, the majority of H2S (∼80%) exists as hydrosulfide anion (HS−) in aqueous solution. It is evident that sulfide should be compartmentalized by plasma membrane due to the impermeability of HS−. However, lipid layers have been found not to be a significant barrier for sulfide at the physiological pH (159). The partition of sulfide by lipid membranes decreases even more at an acidic pH, which may be important in ischemic tissues (40). Therefore, the loss of H2S from a compartment may push the equilibrium from HS− toward H2S swiftly. Moreover, HS− can undergo one electron oxidation to form a resulting thiyl radical (HS·) and other reactive sulfur species (RSS) engaging in other radical–radical interactions, which may be a neglected fundamental part of redox biology (47, 186).
All three molecules are permeable across cell membranes with their diffusion rates influenced by the presence of metals (primarily NO and CO), pH (H2S and to some extent NO), and oxygen concentrations. Table 1 lists general chemical properties of gasotransmitter molecules, including classifications and chemical reactions.
CO, carbon monoxide; H2S, hydrogen sulfide; HNO2, nitrous acid; HS−, hydrosulfide anion; NO, nitric oxide.
Formation, metabolism, and catabolism of gasotransmitters
Identification of the physiological roles of NO, followed by CO and recently H2S, led to the novel concept of gasotransmitter signaling. Gasotransmitters are small molecules whose production and metabolism may be enzymatically or nonenzymatically regulated. For several decades, these three gaseous molecules, NO, CO, and H2S, were solely considered as environmental pollutants and extremely toxic for biological systems. These gases were initially thought to be far from playing any major physiological role and have only recently been appreciated for regulating molecular signaling responses. In this section, we review the known source of gasotransmitters in mammalian cells and their fate after generation. To start with, Figure 2 summarizes biosynthetic pathways of NO, CO, and H2S.
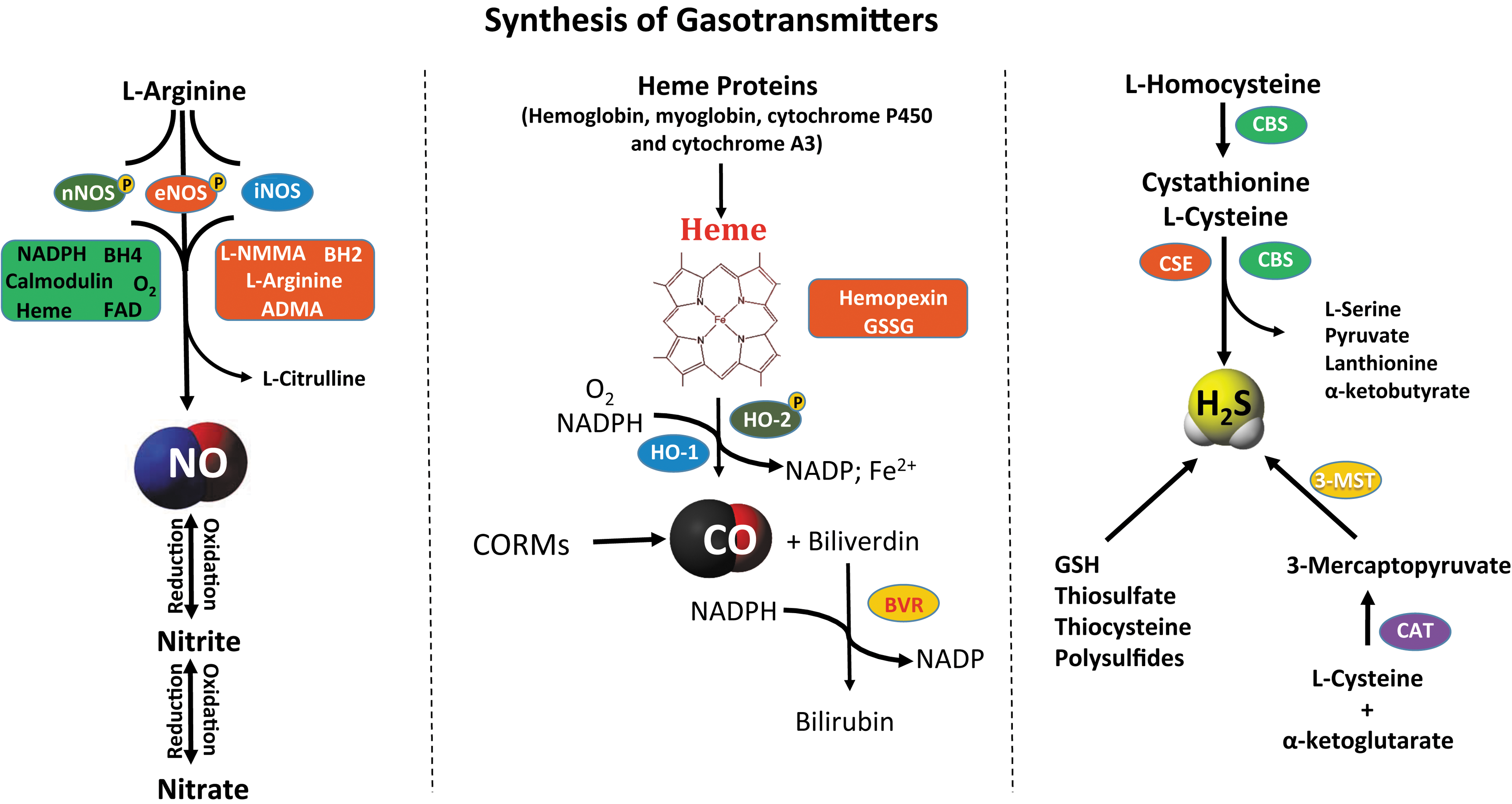
Nitric oxide
The most abundantly studied member of this trio is NO, which was first identified by Joseph Priestly in 1772 as a simple gaseous molecule. However, the elevation of its status from an environment pollutant to an endogenous gaseous substance did not happen until its discovery in the 1980s, further gaining momentum after Nobel-winning work from Robert Furchgott, Louis Ignarro, and Ferid Murad. The novel concept of a gasotransmitter initially came to light from the critical physiologic functions NO plays in regulating a variety of biological processes (154). An understanding of the production and regulating factors that influence it and the rate of metabolism is a prerequisite for elucidating the potential of a signaling molecule in regulating biological functions. However, it is also important to understand localization and cell-based compartmental distribution coupled with reliable analytical assays for its detection (22, 241). The amount of NO produced is dependent on both its rate of formation and its rate of decomposition/metabolism. It is essential to appreciate these components when determining its levels and impact on physiological functions.
NO can be formed through enzymatic as well as nonenzymatic pathways. However, the predominant mechanism of NO production is through nitric oxide synthases (NOSs)—neuronal (NOS 1), inducible (NOS 2), and endothelial (NOS 3) (64). Neuronal NOS (nNOS) and endothelial NOS (eNOS) are considered to generate NO constitutively. However, the emergence of a possible reverse generation of NO through its metabolites such as nitrite and nitrate revealed an alternate mechanism for NO production. Under certain pathophysiological conditions, nonenzymatic generation of NO occurs through nitrite/nitrate reduction mechanisms (117). Even though the medicinal effects of nitrate/nitrite have been known for centuries, these metabolites were considered inert by-products with no definitive biological role (117). However, this concept has changed with nitrite playing a role in maintaining NO bioavailability and its role as a potential biomarker and therapeutic agent in various disease states (77, 117).
NO is produced from all the three isoforms of NOS. The NOS enzyme is a homodimer that has N-terminal oxygenase and C-terminal reductase domains, with calmodulin (CaM) acting as the interdomain linker. Generation of NO through NOS requires several factors, including the substrate
NO formation and decomposition collectively determine its bioavailability and biological effects. NO can be metabolized to various biochemical forms based on its diffusion, location, and reactivity with target molecules in question. A major and commonly observed reaction is with O2 to form nitrite or with oxyhemoglobin to form nitrate (117). However, nitrate may be reduced to nitrite by host commensal bacteria, and nitrite undergoes reduction back to NO under permissive tissue conditions or via proteins and enzymes (e.g., deoxyhemoglobin and xanthine oxidoreductase) (16). NO can react with hemoglobin to form nitrosylhemoglobin or S-nitrosohemoglobin, which is regulated by O2 partial pressure. Reaction of NO/nitrosonium anion and thiols can lead to nitrosothiol formation, which decompose to generate NO. Last, NO can also generate reactive nitrogen species (RNS) through a diffusion-limited reaction with superoxide to form peroxynitrite (ONOO−).
Carbon monoxide
What reminds most people when a fire alarm sets off or whenever an automobile puffs out smoke? Carbon monoxide. CO is an odorless gas and is released during incomplete combustion of fuels and is biologically poisonous. According to the Centers for Disease Control, every year many people die or have severe complications from environmental or industrial CO poisoning. However, its role as a physiological mediator is less well understood both by the public and within the scientific community.
In 1870, decades after its discovery by Joseph Priestley, sometime between 1772 and 1799, Claude Bernard identified the reaction of CO with hemoglobin. CO reacts with hemoglobin, forming carboxyhemoglobin (COHb), which disrupts transport of O2 from lungs to tissues and results in death due to carboxyhemoglobinemia (88, 151). In 1951, Torgny Sjöstrand discovered that this poisonous gas is in fact a normal product of the body's metabolism (222). However, beneficial physiological effects of CO have come to light via its endogenous production.
CO is endogenously produced in the body by the heme oxygenase (HO) enzymes—inducible heme oxygenase-1 (HO-1), constitutive heme oxygenase-2 (HO-2), and heme oxygenase-3 (HO-3)—in low nanomolar concentrations (270). These enzymes are responsible for the rate-limiting step in the degradation of heme to CO, ferrous iron, and biliverdin (BV) in equimolar concentrations (180, 235). They all equally have cofactor requirements of NADPH and O2. CO and BV were initially thought to be waste products through HO activity (85), but are now known for their regulatory roles in redox and stress response (11, 12). Last, CO-releasing donors called CORMs, metal carbonyl complexes, have been used to confirm and induce biological effects of CO in various models (81).
Unlike other members of the gasotransmitter family, much less is known regarding the metabolism of CO. As per the prevailing thought in the literature, excess CO is simply exhaled through the lungs (193). However, CO has a strong affinity for iron (Fe), forming COHb. CO has 200 times more affinity for heme than O2 in the blood and 60 times more in tissues (85). CO replacement of O2 in the blood reduces its oxygen-carrying capacity to <50% (45). CO also interacts with other Fe-containing molecules, notably the detoxifying enzymes, cytochrome P450 and cytochrome A3. Together, these create a hypoxic environment within tissues resulting in compensatory responses (including increased cardiac output) that when coupled with binding of CO to myoglobin result in cardiac failure (169, 197). It is ironic that HO enzyme expression is induced in stress responses such as ischemia and protects tissues from endotoxic shock, yet this concept illustrates the principle of Paracelsus that dose makes the poison (203).
Hydrogen sulfide
H2S is a recent addition to the gasotransmitter family, where it plays a role in several physiological functions. Similar to NO and CO, H2S is also an environmental pollutant and hazardous gas with great biological toxicity. However, recent work on H2S has identified it as an important cell signaling molecule, much like NO and CO. Endogenous production of H2S has been well documented in the literature (255). It is known for some time now that H2S formation occurs as a product of the transsulfuration pathway. Enzymatically, H2S is synthesized by cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE) and 3-mercaptopyruvate (3-MP) sulfurtransferase, and cysteine aminotransferase (CAT). Both CBS and CSE are heme proteins and pyridoxal 5′-phosphate (PLP)-dependent enzymes located in the cytoplasm, whereas 3-mercaptopyruvate sulfurtransferase (3-MST) is zinc dependent and found in the mitochondria.
CBS is primarily localized in the brain and the nervous system and liver tissues (60, 199). It is a homotetramer with NH2 terminal containing binding sites for both PLP and heme and a COOH terminal containing a regulatory domain with an autoinhibitory role. A CaM binding consensus sequence has also been identified at the carboxy terminus that is responsive to Ca2+-mediated activation of this enzyme (9, 116). CBS facilitates the first reaction in the transsulfuration pathway through condensation of homocysteine, a sulfur-containing amino acid intermediate involved in methionine metabolism, and serine to form
In addition to enzymatic production, H2S can also be generated through liberation from persulfide and polysulfides, via different mechanisms, including glycolysis, phosphogluconate, and intracellular reductants. Apart from this, H2S may also be released from other storage forms of sulfide such as bound sulfane sulfurs under alkaline conditions (131). H2S is typically metabolized into thiocyanate, methanethiol, and thiosulfate that may also be recycled to H2S via intracellular reductants. 3-MST desulfurates 3-MP to generate thiosulfate, which can be reduced to generate H2S (124). However, thiosulfate may undergo further oxidation to form sulfite and sulfate with diminishing ability to recycle H2S (131). Last, similar to other gasotransmitters, H2S can either covalently bind to the porphyrin of hemoglobin to form sulfhemoglobin or bind with myoglobin, forming sulfmyoglobin with reduced oxygen binding affinity (132).
In aggregate, all gasotransmitter molecules readily undergo oxidation. The degree to which this occurs for individual gasotransmitters varies and is reflective of its chemical properties. Table 2 identifies primary metabolic oxidation products of CO, NO, and H2S along with their oxidation states. It is clear that CO has one main oxidation product, whereas NO and H2S have 5 and 10, respectively. While NO is a free radical that can engage in numerous chemical reactions with various molecules, its oxidation products act in different ways such as a biochemical reservoir for non-NOS nitric oxide generation (NO2 -/NO3 -) or oxidizing (NO2) and nitrosating agents (ONOO−).
ROS, reactive oxygen species.
Conversely, H2S being a diprotic acid, exhibits a reactivity profile similar to cysteine amino acid. Sulfide can be oxidized to sulfenic acid (HSOH). HSOH is a highly RSS and can be oxidized rapidly to sulfinic acid (HSO2H) and sulfonic acid (HSO3H). Sulfenic acid may also be reduced to a disulfide by intra- and intermolecular thiols. HSOH also can react with sulfide, resulting in the formation of a complex mixture of polysulfides (H2Sn, n = 0–6). The biological relevance and implications of these oxidation products remain poorly understood at present and are important areas for future study.
Biological measurement of gasotransmitters
A common feature among the various members of the gasotransmitter family is their ephemeral nature. Numerous reports have documented trials and tribulations of measuring NO, H2S, and CO levels in biological specimens, leading to important insight regarding how these molecules react and behave. While a comprehensive discussion regarding measurement of gasotransmitter molecules is beyond the scope of this review, it is useful to identify established aspects of their measurement so as to better appreciate experimental findings.
Sample preparation
Arguably, the single most important aspect for determining gasotransmitter bioavailability is sample preparation. Proper and efficient preparation of samples for analysis is critical and requires that investigators understand the best way to prepare and preserve their specimens for downstream measurements. Numerous studies have established solutions, buffers, and processing conditions that help preserve various gasotransmitter metabolites. For example, control of sample pH, metal and thiol binding, presence of oxygen, and processing time are profoundly important for optimal sample preparation. Unfortunately, a universal sample preparation method does not exist to preserve all gasotransmitters in a single sample as optimal preservation methods for each of them are different. Additional detailed information and guidance on sample preparation can be found elsewhere (131, 281).
CO measurement
Several methods are used for CO measurement, including simple microdiffusion into gas detectors or colorimetric, infrared absorption, and spectrophotometric methods, as well as indirect methods (36). However, gas chromatography (GC) is universally viewed as a standard reference method due to its sensitive detection and specificity. Detection of CO is based on thermal conductivity detection (TCD) or flame ionization detection (FID) after methane conversion by a nickel catalyst (80, 112). However, a sample preparatory phase is needed especially if the specimen is blood (245).
Enzymatic activity assays are also important tools to study the production of CO indirectly via functional activity of HO enzymes. This can be performed either by a conventional spectrophotometric method or by high-performance liquid chromatography (HPLC). These methods are based on the biological principle of HO metabolism of heme, resulting in CO production. The components in this process are NADPH:cytochrome P-450 reductase, which oxidizes heme to BV and CO, and biliverdin reductase (BVR) that mediates heme degradation by reducing BV to bilirubin (BR). Conventional detection of HO activity involves spectrophotometric detection of BR (205, 236). This direct measurement of BR in protein samples can be complicated by spectral interference and differences in molar extinction coefficients of BR and the protein extract. Kutty and Maines have reported an alternative method based on the aqueous phase using chloroform (138). In this method, the concentration of BR is calculated from the A464 to A530 of the chloroform extract and the blank where the yield is near the lower limit of detection compared with the spectrophotometric method. Applying these techniques, HO enzyme activity has been reported in many in vivo models such as rats, monkeys, and mice under various conditions (200, 226, 244, 246). However, the HPLC detection method has an advantage over the conventional assay where the HO activity can be calculated even under limited BVR activity.
Last, measurements of CO can also be carried out in biological gases such as exhaled breath with a semiportable analyzer with activated carbon filter and an electrochemical (EC) sensor developed by Stanford University (247). The sensor is structured as a liquid electrolyte system with three electrodes, including a counter electrode, all consisting of platinum surface deposited on a porous Teflon membrane. Upon diffusion of CO through the membrane, it oxidizes on the platinum surface. The transfer of electrons accompanied by the redox reaction flows from the working electrode through an external circuit and constitutes the output signal of the sensor. Studies demonstrate that CO levels from breath of human subjects are comparable with the results obtained by GC (247).
NO measurement
Measurement of NO bioavailability is challenging and subject to significant sources of variability and potential error. Historically, initial NO measurements were performed using a gas detection probe that was reasonably sensitive, but often variable over time (281). Another popular method to measure NO metabolites, nitrite and nitrate, is the Griess reaction assay with spectrophotometry (281). This method has become one of the most popular assays for determining NO bioavailability; however, there are several issues that must be considered, including (i) this is an indirect assay of NO levels via measurement of its oxidation products, which may be altered independent of NO production (i.e., plasma levels could be influenced by renal clearance rates of nitrite/nitrate); (ii) the spectrophotometric plate reader-based assay is limited in its detection sensitivity; however, recent refinements have been made that employ Griess-based chemical derivatization, followed by HPLC detection, leading to significantly enhanced detection; and (iii) simultaneous measurement of nitrite and nitrate often overestimates the amount of NO produced based on the fact that nitrate metabolism can be independent of NOS activity.
Other methods to measure NO from biological samples include electron spin resonance (ESR) (83, 90, 157), fluorescent dye indicators (129, 260) and chemiluminescence detection (158, 201), GC-mass spectrometry (MS), and HPLC (217, 268). These assays are advantageous in their selectivity and sensitivity. Taking advantage of the free radical nature of NO with spin trap agents enables one to clearly and cleanly measure NO by ESR (14). However, limitations of this method exist with regard to quantitation and the ability to directly measure NO generation in real time. Several fluorescent dyes have been generated that react with NO or its metabolites that can be assayed using fluorescent spectroscopy, microscopy, or HPLC (132). Diaminofluorescein and its derivatives (DAF-FM and DAF-2DA) have been widely used to evaluate NO production in cells and tissues using fluorescent spectroscopy and microscopy (194). While these dyes are easy to use in various formats, it is important to keep in mind that they do not directly react with NO itself, but do react with NO metabolites (e.g., N2O3 and others) (260). 2,3-Diaminonapthalene (DAN) can also be used with biological specimens to measure nitrite levels via formation of the fluorescent product 2,3-naphthotriazole (NAT) measured by reverse-phase HPLC (142, 249). Recently, a novel organelle-targetable fluorescent probe called HTDAF-2DA to detect NO in live cells needs further validation (252). However, one limitation of fluorescent methods is their unreliable detection sensitivity beyond low micromolar concentrations without using analytical methods. On the other hand, tri-iodide-based NO chemiluminescence detection methods provide significant flexibility and sensitivity in measuring NO and other nitrosated species, including S-nitrosothiols (SNOs) and nitrosohemoglobin (SNOHb) (55, 78). This assay takes advantage of NO reaction with ozone to generate photons within a reaction chamber, enabling highly sensitive detection of bioavailable NO. A reaction sparger is coupled to the detector that allows experimental flexibility to measure bonafide NO levels itself, the amount of NO contained within specific metabolites upon chemical liberation, or rates of NO production from chemical and cell models. However, all these assays have their own strengths and weaknesses and it is important to understand them as well as which approaches are best suited to accomplish the desired task.
NO production can also be measured indirectly by assays of NOS activity. NOS activity is most accurately measured using radiolabeled
H2S measurement
Measurement of H2S has proven to be particularly challenging for investigators. Similar to NO, H2S detection can be accomplished using a gas detection probe or an ion-selective electrode (14, 184). Both operational and chemical limitations exist with these methods due to the need for individual sample measurements or that a certain pH is required that can influence sulfur release from other biochemical sources. Similar to the Griess reaction assay for NO measurement, a simple spectrophotometric-based assay employs zinc acetate to trap sulfide that is subsequently reacted with N,N-dimethyl-p-phenylene diamine under acidic conditions, resulting in the formation of methylene blue. This assay was first described by Fisher in 1883 and redefined by Cline in 1969 to measure H2S in natural waters (34). However, studies from several groups, including ours, have revealed that use of this methylene blue assay is not compatible with biological samples for several reasons, including its insensitive nature, harsh chemical treatments, and inability to obey Beer's law, leading to exceedingly high measurements of biological sulfide (14, 184, 215).
Significant progress and reliability have been made regarding accurate H2S detection using analytical approaches similar to those used for NO. The fluorophore monobromobimane has been shown to react with sulfide-generating sulfide dibimane (SDB), which is quantified using reverse-phase HPLC with fluorescent detection (215, 266). This assay has been refined by different groups to also measure different biochemical forms of sulfide, including acid-labile and bound sulfane sulfur (including polysulfides), enabling researchers to study H2S metabolism in a comprehensive manner (102, 216). Similar to NO, chemiluminescence-based methods can also measure H2S through selective liberation of H2S gas that is separated by GC and introduced into a reaction chamber containing ozone (242). Results from this detection method are sensitive and similar to results using the monobromobimane (MBB) HPLC method. Together, these analytical methods provide useful and accurate ways to measure H2S metabolite bioavailability.
H2S production can also be measured indirectly through the use of transsulfuration enzyme activity assays. This approach is similar to those of CO and NO and employs the use of pyridoxal 5′-phosphate with cystathionine or cysteine substrate, followed by ninhydrin spectrophotometric detection. Caveats exist with this assay, in that it employs nonrate-limiting amounts of substrate that do not reflect what exists in intact cells or tissues. Thus, appropriate interpretation of resulting data is very important.
Biochemical molecular modifications facilitating heterocellular communication
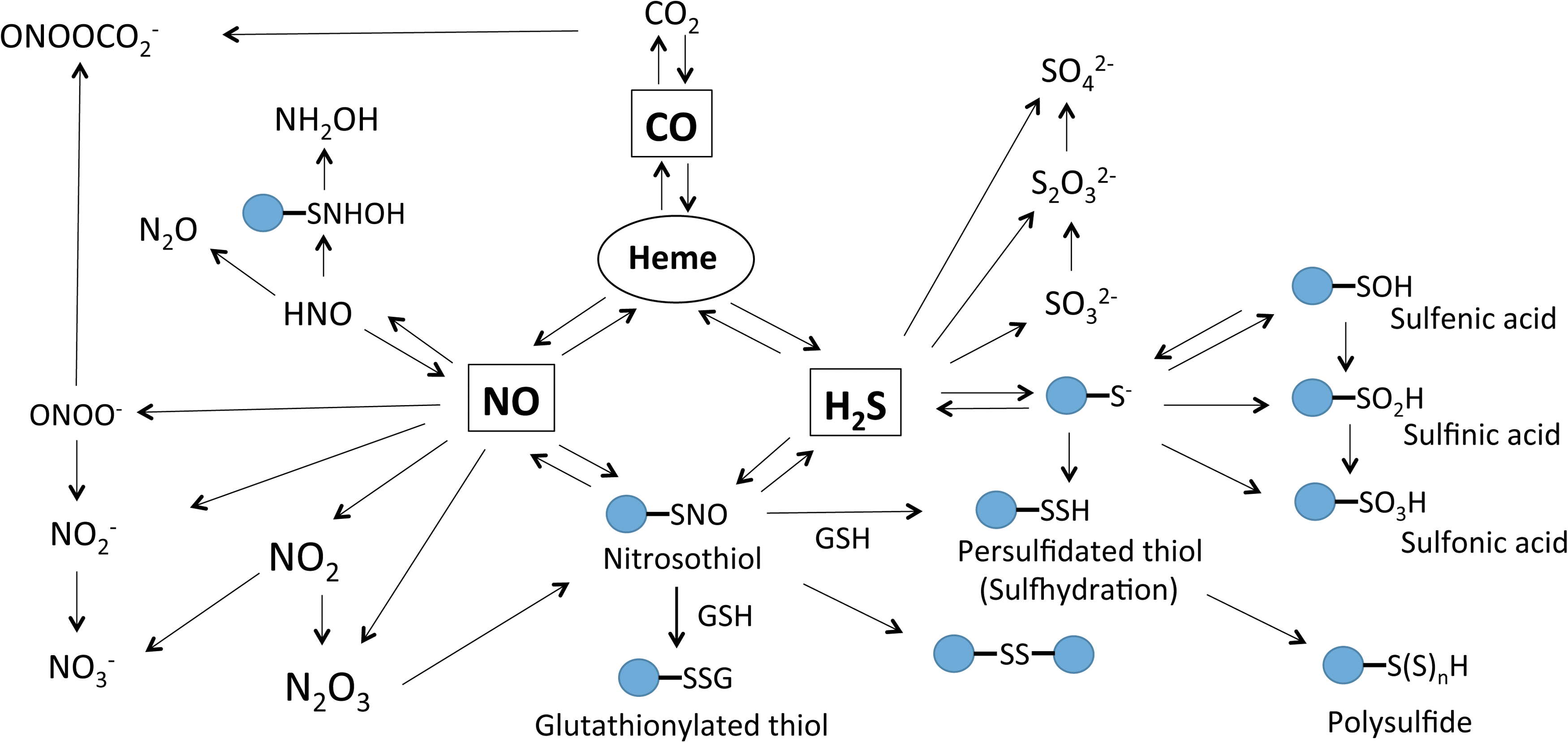
Major biochemical reactions of NO include its oxidation to nitrite and nitrate as well as conversion to peroxynitrite in the presence of superoxide (6). Target proteins of peroxynitrite include lipids and protein thiols, resulting in nitration of tyrosine, protein carbonylation, and lipid peroxidation (133, 181, 183). NO metabolites such as HNO or N2O3 can react with glutathione (GSH) and protein thiol to form S-nitrosoglutathione (GSNO) and protein S-nitrosothiols (RSNOs) (99). Overall, interactions between NO and protein typically include the following: (i) Binding to metal ions (such as copper and iron) of metalloproteins, such as soluble guanylate cyclase (sGC), cytochrome c oxidase (COX), and others. Upon NO-heme ligand binding, sGC becomes activated in a specific and reversible manner (103). COX is the terminal complex (Complex IV) of the electron transport chain and includes a copper–copper dimer, heme A, and a binuclear heme–copper coupled center. Importantly, NO binding to COX results in inhibition of electron transfer (38, 118). (ii) As mentioned above, NO binding to protein thiols through an indirect reaction forming protein SNOs (223), which is called S-nitrosation popularly called S-nitrosylation, plays important roles in physiological signaling processes (224). GSNO, formed by S-nitrosation of GSH, is likely the most abundant intracellular low-molecular-weight S-nitrosothiol and is able to transfer its NO group to other neighboring thiol groups (91). (iii) Protein tyrosine nitration is a covalent interaction of NO and protein, resulting in addition of a nitro group onto tyrosine residues. Aside from peroxynitrite, many enzymes are capable of nitrating tyrosine residues (20, 239, 271), such as myeloperoxidase, eosinophil peroxidase, myoglobin, and cytochrome P-450s. Last, peroxynitrite can also directly react with carbon dioxide (oxidation product of CO) and facilitate tyrosine nitration (141).
H2S can react with oxidants to form various sulfur compounds (polysulfide, polythionates, elemental sulfur, thiosulfate, sulfite, sulfate), sulfide radical, and disulfide. Under neutral pH, sulfides can form complexes with divalent metals and proteins with metal centers (e.g., heme) (69, 144). However, another way H2S affects protein function is through protein sulfhydration (also named protein persulfidation), which is the product of protein cysteine residues and sulfide. In this reaction, the sulfhydryl group of cysteine is modified to −SSH. Under physiological conditions, many cysteine residues exist as thiolate anions, which interact with ROS to generate sulfenic acid (−SOH) that can be further oxidized to sulfinic acid or sulfonic acid (127, 195). However, sulfenic acid can facilitate thiol sulfhydration through its reaction with sulfide (59). S-sulhydration can be reversed by thioredoxin (136) and this post-translational modification is known to modulate enzyme activity. For example, sulfhydration of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) at cys150 results in significant increase in glycolytic activity (172).
CO plays an important role in both cellular antioxidant defense and vascular protection. For example, CO avidly binds to heme groups, in particular sGC, but is 30–100 times less potent than NO in inducing sGC activity (270). CO activation of sGC and cGMP formation can blunt NADPH oxidase-dependent ROS formation, indicating a biological role for this interaction (31, 171). CO is also known to blunt O2 consumption by inhibiting mitochondrial COX activity. Thus, inhibition of COX by CO suppresses oxidative phosphorylation and reduces adenosine 5′-triphosphate (ATP) production. However, specific molecular mechanisms responsible for these effects and their overall importance in relation to other gasotransmitter effects on the same pathway are not well understood. Figure 3 summarizes the major gasotransmitter metabolic products and target molecules discussed in this section.
Discrete gasotransmitter formation in tissues and cells
A systematic network is key for efficient cellular communication. This is necessary for signaling to integrate cellular and tissue responses regulating physiological functions. Importantly, gasotransmitter molecules can exhibit their effect in different ways, including autocrine, paracrine, and endocrine responses, which are influenced by their discrete production. These pathways are complex with a range of mechanisms controlling formation and metabolism such as enzyme expression and/or activation (all gasotransmitters), microbial contributions (H2S and NO), and metabolic recycling. Importantly, nearly all of these responses contain feedback loop control mechanisms to limit production of one or more gasotransmitters.
To understand how discrete formation and signaling of NO, CO, and H2S could occur, several concepts influence this response: (i) Overlapping functions—one or more gasotransmitters may influence similar physiological functions in an overlapping manner. (ii) Intra and extracellular environment—gasotransmitter formation is clearly impacted by intra and extracellular conditions such as substrate availability, pH, presence of regulatory proteins or cofactors, and oxygen levels. (iii) Janus-faced properties—all gasotransmitters are notorious for being environmental pollutants and very toxic at higher concentrations, yet potentially beneficial at much lower concentrations. These properties clearly contribute to discrete formation and metabolism.
How tissues and organ systems coordinate the production and metabolism of all these three gases with similar physiological targets and biological implications remains unclear. However, one important and likely dominant mediator of this response involves specialized regulation of gasotransmitter formation in different cell types such as endothelial cells, smooth muscle cells (SMCs), fibroblasts, neurons, astrocytes, microglia, intestinal epithelial cells, skeletal and cardiac myocytes, and hematopoietic cells such as leukocytes, monocytes, macrophages, platelets, and many more. Moreover, the relative health state of these cells, along with other communication and signaling responses during health versus disease, has a significant impact on gasotransmitter formation. While it is not possible to provide a comprehensive survey of this topic in this review, this section discusses some of the major ways in which cellular gasotransmitter production occurs.
NO production
The orchestration of variant NOS isoforms to produce NO is complicated by their distinct structure and expression pattern and also their differential subcellular localization. The compartmentalization of eNOS at the plasma membrane and the Golgi apparatus has been well documented. This process requires myristoylation of Gly2 and dual palmitoylation of Cys15/Cys26 (182). While myristoylation of eNOS is irreversible and required for general membranous association, palmitoylation may target eNOS to the plasma membrane (74, 149). Functionally, eNOS localized at the plasma membrane and the Golgi complex has the highest activity. A small amount of eNOS can also be found in the cytosol, mitochondria, and even the nucleus. However, eNOS in those compartments only exhibits minute NO-producing ability. This difference in eNOS activity is thought to involve Ca2+ gradient, which is highest at the submembranous region (108). In addition, since the sensitivity of eNOS to Ca2+/CaM is increased by phosphorylation at multiple serine residues, the subcellular location of specific protein kinases, such as Akt, also plays a role in the spatial regulation of eNOS activity (70, 71). On the other hand, eNOS partially localizes in caveolae, where caveolin-1 associates and inhibits eNOS activity (73). Similarly, palmitoylation of Cys3 residue is required for intracellular trafficking of inducible NOS (iNOS) (179). The iNOS mutant lacking Cys3 palmitoylation is nonfunctional and accumulated in the Golgi network (179). However, intact iNOS targeted to different subcellular compartments displays the same NO-producing capacity (108). This could be due to the higher affinity of iNOS to Ca2+/CaM. Moreover, the interaction between iNOS and caveolin-1 results in iNOS degradation and reduced NO production (56). Unlike eNOS and iNOS, nNOS compartmentalization is not mediated by lipid modification. Instead, nNOS has a unique N-terminal PDZ domain that mediates its interactions with other PDZ domain-containing proteins such as postsynaptic density-95 (PSD-95), C-terminal PDZ ligand of nNOS (CAPON), and α1-syntrophin. With their help, nNOS is directly or indirectly tethered to the synaptic membrane, synaptic vesicles, and sarcolemma, respectively (33, 92, 106). In short, all NOS isoforms are routed to certain subcellular compartments where NO production is needed.
H2S production
Spatial regulation of H2S generation remains poorly understood. However, it is reasonable to speculate that H2S production should be compartmentalized for it to serve as a signaling molecule. Although CBS and CSE were first recognized as cytosolic enzymes, they have been found to be present in mitochondria in specific tissues under normal and stress conditions. In the liver, CBS is expressed in mitochondria and associated with mitochondrial Hsp70, which may mediate the shuttling of CBS from the cytosol to the mitochondria (150, 234). Moreover, CBS translocation also involves its N-terminal domain since a CBS mutant lacking amino acids 1–70 is favorably translocated to mitochondria over the cytosol (234). Interestingly, these residues form the N-terminal heme-binding motif that is recognized and degraded by the mitochondrial Lon protease under normoxic conditions (163, 234). In SMCs, CSE is not detectable in the mitochondria. However, CSE is translocated to mitochondria with a sustained increase in intracellular Ca2+, which may facilitate the interaction between CSE and translocase of the outer membrane 20 (Tom20) (68). It is worth noting that the compartmentalization of CSE, CBS, and 3-MST not only regulates the availability of H2S but also its metabolism. Since the mitochondrion is the main organelle where H2S is oxidized, CSE, CBS, and 3-MST localized in mitochondria can be a major source of RSS.
CO production
HO-1 is a major endoplasmic reticulum (ER) protein. It is anchored to the ER membrane by the C-terminus whereby the C-terminus is inserted into the ER lumen with the N-terminus facing the cytosol (19, 82). However, HO-1 can also be found in other compartments such as the nucleus, mitochondria, and caveolae (10, 15, 37, 258). Although the detailed mechanism of HO-1 translocation remains to be investigated, it is thought to require the cleavage of the C-terminal anchor by signal peptide peptidase (SPP) (19, 146). Additionally, a nuclear shuttling sequence (amino acids 207–221) mediates the localization of HO-1 to the nucleus (146). By contrast, the first 33 amino acids from the N-terminus have been shown to signal HO-1 for mitochondrial translocation (10). In addition, as a heme protein, HO-1 is also subject to the degradation by Lon protease in mitochondria under normoxia (234). Functionally, nuclear localization results in reduced HO-1 activity, while mitochondrial HO-1 retains its activity (15, 146).
Molecular Signaling Targets of Heterocellular Communication
Redox regulation of molecular targets
Gasotransmitter molecules can influence cellular redox potential through numerous biochemical, metabolic, and signaling pathways. RNS and RSS engage in various oxidation/reduction reactions that may influence ROS availability. Moreover, both RNS and RSS can exist as free radicals that effectively mitigate oxidative challenges. The ways in which this may occur remain poorly understood and are just beginning to be examined.
ROS are generated through numerous mechanisms; however, membrane-bound NADPH-dependent enzyme oxidases (NOX enzymes) are predominate sources of ROS formation within cells (109). Under normal conditions, eNOS catalyzes the generation of NO from
H2S has been shown to be a reductant that can exert antioxidant effect through direct and indirect actions. Direct antioxidant effects of H2S have been reported; however, the antioxidant strength of H2S itself is rather weak when compared with other intracellular antioxidants. Rather, H2S likely exerts its effect through modification of protein thiols via conversion of cysteinyl thiolates to cysteinyl persulfide. Due to the lower pKa of persulfides compared with corresponding thiols, the majority of persulfide would be in the deprotonated form at physiological conditions, suggesting that it is more nucleophilic. Enzymatic generation of persulfides may also occur via CBS, CSE, 3-MST, and sulfide–quinone oxidoreductase (SQR) (102, 145, 275). Alternative nonenzymatic generation of persulfides by H2S can occur via protein S-sulfenylation (29, 284) and S-nitrosation (230), whereby reaction with H2S produces persulfide. H2S may also react with disulfides leading to persulfide formation; however, Zhang et al. did not observe S-sulfhydration as a consequence of the reaction of H2S with disulfides of BSA (284). Last, H2S can guard against redox stress indirectly by increasing the GSH level (125) or by facilitating S-sulfhydration of kelch-like ECH-associated protein 1 (Keap1) and nuclear factor erythroid 2-related factor 2 (Nrf2) (26, 61).
Ion channels
Ion channels are one of the common molecular targets regulated by gasotransmitters. This is due, in part, to the fact that they act as an interface between the cells and their microenvironment. Moreover, ion channels regulate membrane potentials that impact fundamental cell physiology processes, including neuron depolarization, myocyte contraction, epithelial barrier function, and numerous other responses (66). Ion channels may be broadly classification based on sodium (Na+), potassium (K+), calcium (Ca2+), chloride (Cl−), and unspecific cation channels. The impact of gasotransmitters on ion channels is reflected by changes in cell–cell interactions, electrolyte homeostasis, and communication, thereby influencing normal morphology and function such as intercellular permeability, proliferation, and survival. Many diseases have physiological dysfunction resulting from mutations involving voltage-gated channels, including Na+, K+, Ca2+, and Cl-, and (Ach)-gated channels (50). With the increasing attention on gasotransmitter roles as potent regulatory molecules affecting ion channels, research has diversified in this area with studies elucidating their properties on channel expression and function.
Nitric oxide
NO regulation of ion channel function is well established (42). NO effects on ion channels may elicit various responses through either cGMP-dependent or -independent pathways (1, 269). Activation of cGMP-dependent kinases and direct ion channel activation are critical for NO regulation of vascular tone. NO is known to regulate ion channels, including Ca+2-dependent K+ channel (KCa) and ATP-sensitive K+ (KATP) channels, across different cell types such as endothelial, SMCs, epithelial, and neuronal cells, as well as immune cell types such as neutrophils, macrophages, and eosinophils (3, 8, 58).
NO mediates SMC relaxation through the sGC/cGMP-dependent pathway via KCa activation (24240871, 7519783, PMC507785). However, NO also mediates cGMP-independent relaxation by directly activating calcium-dependent potassium channels (18). NO also invokes changes in the behavior of mitochondrial KATP (mtKATP) channels (204). mtKATP is influenced by modulators such as ATP, 5-HD, and Ca2+, where NO regulates both its activation state and kinetics. However, Brookes et al. also reported that NO exerts bidirectional effects on opening the mitochondrial permeability transition pore and inhibition of cytochrome C release (21). Moreover, NO regulation of ion channel function also controls intracellular calcium levels by influencing T-type and L-type Ca2+ channels (89).
NO can also modulate synaptic transmission involving ion channels that are important for neurodegenerative diseases (202, 225). NO modulates several neurological ion channel activities through cyclic GMP-independent molecular mechanisms. The neuronal N-methyl-D-aspartate receptor (NMDAR) has crucial functions in the central nervous system such as development, learning, and memory that are mediated via postsynaptic Ca2+ influx and nNOS. However, NMDAR activity and function and consequent neurotoxicity are attenuated through S-nitrosation of cysteine residue 399 (Cys399) under physiological conditions (32, 147). NO also induces Ca2+-activated K+ (BKca) channels (139) and activation of ryanodine receptors (RyRs) through S-nitrosation that mediate Ca2+ release at the neuromuscular junction involved in muscle contraction (274).
Carbon monoxide
Studies reporting a role for CO in both physiology and disease continue to grow. Some of them include a role for CO in regulating ion channels, such as calcium-activated K+ (BKCa), voltage-activated K+ (Kv), and Ca2+ channel (L-type) families, epithelial Na+ channel, and tandem P domain K+ (TREK1) channels. However, the mechanisms by which CO regulates these ion channels remain unclear and somewhat controversial. CO regulates [Ca2+]i that is critically involved in cardiac and vascular smooth muscle function (206). CO can also mediate electron leak from mitochondria via complex III (189) and inhibit BKca channels by acting on cysteine residues, involving rapid ROS formation (41, 206). On the contrary, CO also induces cardioprotection via reduced Ca2+ influx via L-type Ca2+ channels. However, a majority of these findings regarding CO were demonstrated through the use of CO-releasing molecules (CORMs).
Several research groups have studied the regulation of high-conductance, Ca2+-dependent K+ channels [Slo1] (KCNMA1), variously termed KCa1, BKCa, or maxiK channels, by CO (43, 53, 95, 107). cGMP-activated K+ currents are the major ion channels regulated by CO found in jejunal smooth muscle and epithelial cells (53, 198). However, CO did not directly impact cGMP production to regulate these channels. Recent reports indicate that CO conveys this regulation via direct interactions or indirectly through heme binding and redox regulation (43, 107). Further studies are clearly needed in relation to CO-mediated regulation of cGMP-dependent ion channels.
P2X2 receptors are found throughout the nervous system, including sensory neurons. These are the only ligand-gated ion channels modulated by CO reported thus far. Studies have shown that CO selectively modulates P2X2 receptors using the CO donor CORM-2 (265). Studies using CORM-2 also reveal CO regulation of TREK1 channels, which are involved in neuronal activity (44, 190). As stated above, CO can activate K+ channel activity that is typically dependent upon sGC/cGMP. Conversely, Kv2.1 is inhibited by CO through a PKG-dependent pathway and elicits antiapoptotic neuroprotective effects (43). However, this is controversial as a separate study could not confirm a role of CO in this mechanism (110). Taken together, CO regulation of ion channels does occur, yet associated mechanisms are substantially unclear that require further investigation.
Hydrogen sulfide
It is well established that H2S is involved in inflammation, regulation of blood pressure and vascular tone, neuromodulatory, and cytoprotective functions. H2S also modulates synaptic activities, ion channels, and transporters to mediate biological functions in relation to the vasculature and brain (120, 231, 261, 285). H2S interacts with ion channels, including KATP channels, and transient receptor potential (TRP) channels, which they typically activate while inhibiting L-type calcium (Ca2+) usually in cardiomyocytes, T-type Ca2+ in the viscera, and chloride (Cl−) channels in the heart, facilitating neurotransmitter activity (120, 143). This may possibly be aided by H2S-mediated release of antioxidants that exhibit neuroprotection, via its interaction with the transporter systems mentioned above (143, 152). H2S has also been shown to influence mitochondria in relation to ion channels in a dose-dependent manner. H2S donor NaHS was shown to induce mitochondria swelling of rat hearts at 10−12 to 10−4 mol/L, suggesting activation of KATP channels (209).
Vasorelaxant effects of H2S have been attributed to small-conductance Kca channels. H2S may mediate changes in membrane potential in SMCs through low electrical resistance (254). H2S-mediated vasorelaxation occurs through KATP channel activation involving the cGMP/PKG pathway (23, 285). Work by Mustafa et al. in isolated rat mesenteric arteries suggests that H2S induced voltage-mediated hyperpolarization upon Ach stimulation independent of NO (173). This was substantiated with CSE knockout mice, where hyperpolarization effects were significantly reduced. This group and others have also demonstrated that H2S hyperpolarization of endothelial cells occurs via S-sulfhydration of Cys 43 of Kir6.1 subunit of KATP channels and interaction with Cys 6/Cys 26 of SUR1 (111, 173). Together, these findings illustrate the importance of H2S on ion channel function that requires further investigation under pathophysiological conditions.
Intracellular organelles
Eukaryotic cells are compartmentalized by cellular membranous organelles, including nucleus, ER, Golgi complex, endosomes, lysosomes, and plasma membrane. Vesicles bud off and fuse in these organelles, delivering structural and signaling molecules. This transportation between different compartments makes it possible for the exocytosis of neurotransmitters in neural synapses, the degranulation in leukocytes and endocrine cells, the release of adhesion molecules from endothelial cells, and the endocytosis of surface proteins. Therefore, vesicular trafficking plays critical roles in intercellular cell communication. This is a complex and well-orchestrated process involving cytoskeleton, vesicle coats, cargo adaptor proteins, Rabs, soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNAREs), and proteins facilitating membrane fusion. Gasotransmitters have been shown to regulate intracellular organelle responses by directly targeting these molecules or indirectly by altering upstream signaling pathways.
Nitric oxide
In isolated synapses, S-nitrosation has been found on a large spectrum of proteins, including syntaxin-1, synaptosomal-associated protein 25 (SNAP-25), and synaptobrevin of the SNARE family (196). There are two subtypes of SNAREs, known as vesicular SNAREs (v-SNAREs) and target SNAREs (t-SNAREs), which are located on the synaptic vesicles and plasma membranes. The coupling of v-SNAREs and t-SNAREs forms a SNARE complex, which pulls and fuses the membranes together. S-nitrosation of SNAREs may interfere in the process of exocytosis. Indeed, S-nitrosation of syntaxin-1 at Cys145 obliterates the binding of Munc-18, a chaperone which stabilizes the closed conformation of syntaxin-1 (187). Furthermore, this cysteine residue is highly conserved in all neuronal syntaxins and similar regulation is also found in islet β-cells (S-nitrosation at Cys141 of syntaxin-4) that increase insulin secretion (267).
Dynamin is a large GTPase assembling into helical polymers at the neck of budding vesicles to scissor them off, which is crucial in endocytosis. Dynamin binds to eNOS (28). Both endogenous and exogenous NO have been shown to S-nitrosylate dynamin at Cys86 and Cys607, resulting in increased dynamin assembly and activity, increased internalization of β2 adrenergic and epidermal growth factor receptors, and increased microbial invasion (251, 259). NSF is another central component in vesicular trafficking. It interacts with SNARE molecules via soluble NSF attachment protein (α-SNAP). Although the mechanism is not fully understood, α-SNAP interferes with SNARE protein-mediated membrane fusion, which is inhibited with NSF binding (188). However, NSF is S-nitrosylated resulting in reduced exocytosis (161).
Heterocellular communication also depends on microdomains where signaling molecules accumulate in a context. There are numerous scaffolding proteins and cofactors on the membrane to facilitate the establishment of such microdomains. Postsynaptic receptors, such as NMDAR and γ-aminobutyric acid receptor (GABAR), are located at a region called PSD. S-nitrosation of scaffolding protein PSD-95 and gephyrin inhibits their clustering and NMDAR and GABAR synaptic transmission, respectively (46, 93). On the other hand, S-nitrosation of stargazin and NSF increases α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) surface expression (101, 207).
Moreover, NO may also modify target protein by forming 3-nitrotyrosine. Dynamin can undergo nitration at Tyr354, which impairs its self-assembly and association with an integral membrane protein, synaptophysin (155, 156). Synaptophysin is abundant in vesicles and is involved in the formation of synapses and exocytosis/endocytosis. Synaptophysin is nitrated at Tyr250 by peroxynitrite, resulting in the disassembly of dynamin/synaptophysin complex (155). Therefore, nitration of dynamin and synaptophysin may negatively regulate endo/exocytosis. Members of SNAREs, including SNAP-25, Munc-18, and synaptotagmins, also undergo 3-nitrotyrosine formation that may interfere with the docking/fusing step of exocytosis (48, 248).
Hydrogen sulfide
H2S induces protein post-translational modification by forming per/polysulfide on cysteine residues. Recently, H2Sn formed through oxidation of H2S has been identified as a potential signaling molecule (122). H2Sn has physiological implications, including cardioprotection, vascular tone regulation, barrier functions, and tumor suppression (121, 123, 280). Sulfane sulfur has been implicated to mediate beneficial effects of H2S signaling by inducing conformational changes in cysteine residues of enzymes and receptors via a process termed sulfhydration/sulfuration/persulfidation (172, 237). Some recent studies have attributed the biological effects of H2S to H2Sn (128, 136, 173, 177).
One of the first described targets was GAPDH (172). Such modification on Cys105 of GAPDH favors its association with Siah, an E3 ligase, which promotes the ubiquitination of PSD-95 (166). In addition, H2S mitigates the decreased expression of PSD-95, SNAP-25, and synaptophysin in the brain induced by homocysteine treatment (115), although it is not clear whether this protective effect is due to H2S-induced modification on these proteins.
On the other hand, H2S also affects vesicular trafficking by regulating upstream pathways. NaHS decreases the exocytosis of secretory granules both during resting conditions in GH3 pituitary tumor cells and in response to depolarization, which depends on Ca2+ (174). NaHS also decreases the cAMP level by inhibiting adenylate cyclase and stimulating phosphodiesterase, which decreased renin degranulation in As4.1 and rat renin-rich kidney cells (153). Moreover, NaHS targets EGFR (Cys797/Cys798) and activates the EGFR/gab1/PI3K/Akt pathway to increase Na+/K+ ATPase endocytosis in renal tubular epithelial cells (75).
Carbon monoxide
Early studies show both endogenous and exogenous CO increases presynaptic neurotransmitter release and end plate current, which is not blunted by sGC inhibition, suggesting that this effect is independent of NOS inhibition (220). However, CO-enhanced synaptic activity may partially result from reduced dopamine and glutamate uptake in the synaptosome, rather than altered intracellular trafficking (233). Recent evidence indicates that CO inhibits antigen presenting from dendritic cells to T cells by decreasing endosome–lysosome fusion (232). In SMCs, dynamin-2 polymerization is increased by CO, which mediates the internalization of TGF-β receptor 1 (97). On the other hand, CO inhibits SNARE complex formation in LPS-stimulated platelet, which involves decreased expression of SNARE proteins, including STX-2, SNAP-23, and VAMP-8, as well as the upstream TLR4/PKCθ/STXBP-1 pathway (148). Additionally, CO increases adenylate cyclase/cAMP increasing Ach release in neuromuscular junction and insulin secretion in islet (170, 221).
Gasotransmitter Heterocellular Signaling in Health and Disease
While numerous reports have shown that gasotransmitters exert profound influence over normal and pathological tissue responses, this section addresses specific aspects involved in signal mechanisms as well as areas where gasotransmitters overlap influencing pathophysiological responses. The discussion below represents distinct areas of investigation rather than a litany of all gasotransmitter reports for a given organ system.
NO–H2S share a unique and complex relationship in the biological systems. Both of these gasotransmitters have an influence on each other, including their production, signaling mechanisms, and unique chemical product formation. Early report from Kimura's group suggests that H2S and NO work synergistically to enhance vascular relaxation (94). Works from Moore and coworkers have identified the interactions of NO and H2S as they reported the antioxidant properties of H2S mediated through inhibition of peroxynitrite (263). Subsequent works by this group suggest that NO and H2S interact to form a novel intermediate nitrosothiol or thionitrous acid (SNO−), which has vasorelaxant activity (264). However, recent studies by Feelisch and coworkers suggest the role of nitrosopersulfide (SSNO−), a bioactive product of the chemical interaction of NO or nitrosothiols with sulfide, instead of SNO− to have physiological implications such as vasoregulation and redox signaling (39). Further research in this direction is needed to identify the physiological relevance using animal models and in humans. Figure 4 highlights concepts of H2S and NO gasotransmitter reaction products that may participate in heterocellular communication and cooperative pathophysiological interactions.

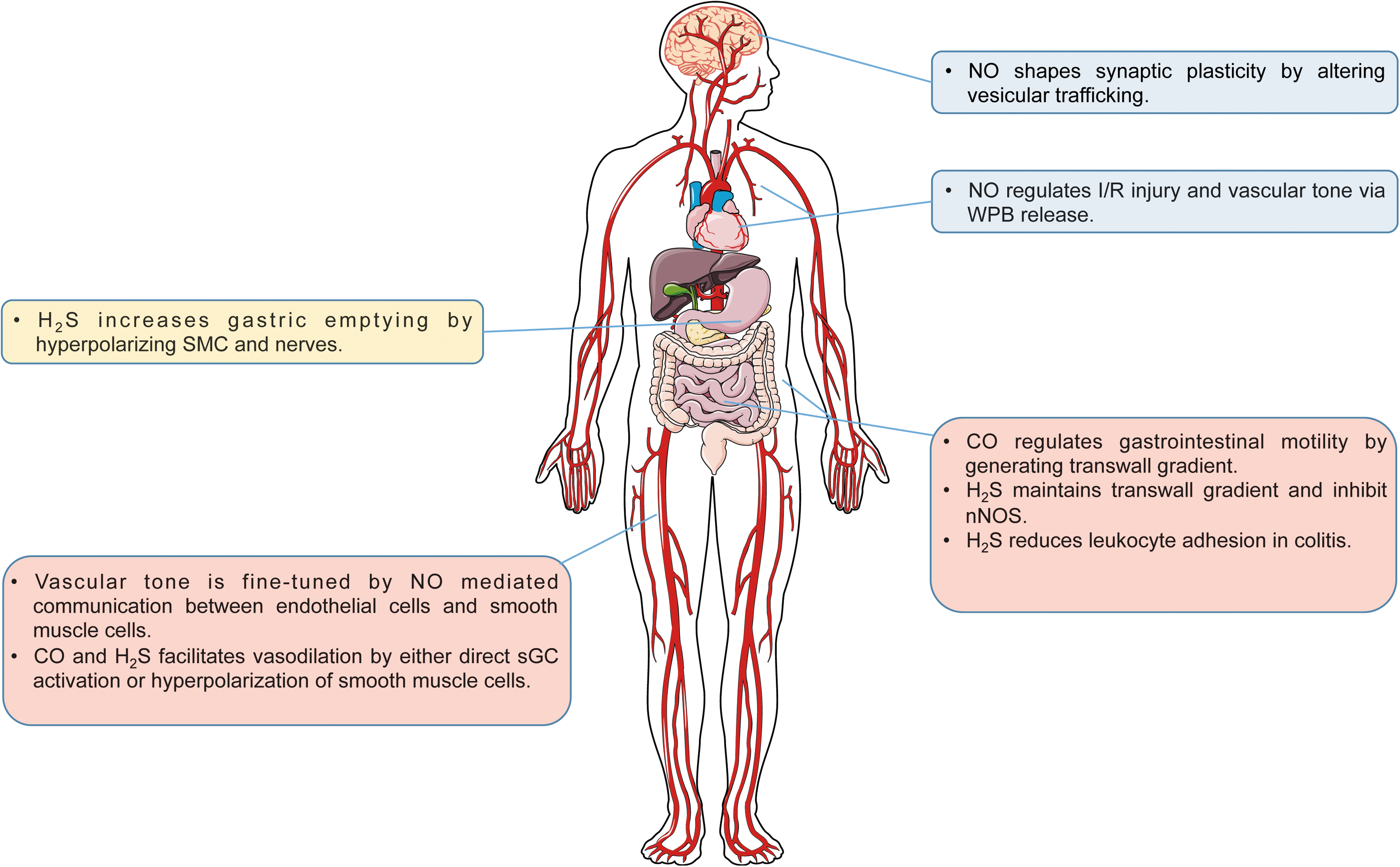
Cardiovascular system
In the cardiovascular system, vascular tone is regulated by Ca2+ and NO communication between endothelial cells and SMCs. Connexin 43-based gap junctions are localized at the myoendothelial junction. Constitutive S-nitrosation of Cys271 is required for the open channel conformation (227). In small resistance arteries, α1-adrenergic signaling results in increased inositol triphosphate (IP3), Ca2+ release from SMCs, and consequent vasoconstriction. IP3 and Ca2+ diffuse to endothelial cells via gap junctions. In the endothelial cells, IP3 activates IP3 receptor type 1 (IP3R1) on ER to further increase cytosolic Ca2+, which promotes eNOS activity (104, 105). To maintain the endothelial Ca2+ concentration, S-nitrosation of Cx43 is removed by S-nitrosoglutathione reductase (GSNOR). As a result, NO diffuses back to SMCs causing vasodilation (227). Moreover, it is known that NO-regulated vasodilatory response is more prominent in conduit arteries, but not resistant arteries (24). A possible explanation is hemoglobin α (Hbα), which has been found to be expressed in small resistance arteries and large mesenteric arteries, mediated NO sequestration at the myoendothelial junction (228). It is worth noting that cytosolic Ca2+/CaM also activates HO-2, which contributes to endothelium-dependent vasodilation (17, 282). Although via different mechanisms, H2S is also considered as an endothelium-derived relaxing factor. Increased Ca2+ in endothelial cells activates CSE (276). H2S activates SKCa on endothelial cells and KATP channels in SMCs (173). The outward flow of K+ causes hyperpolarization and vasodilation. However, it is not clear whether HO-2 and CSE are subject to a similar regulatory mechanism as eNOS at myoendothelial junctions.
In endothelial cells, Weibel-Palade bodies (WPBs) in endothelial cells, containing P-selectin, von Willebrand factor (vWF) tissue plasminogen activator (t-PA), interleukin-8 (IL-8), and endothelin-1, mediate inflammation thrombosis and vasoconstriction. Increased WPB exocytosis is related to vasoconstriction of aged arteries and myocardial ischemia/reperfusion injury (27, 79). NO can regulate WPB exocytosis by interfering in multiple stages of vesicular trafficking. For example, hypertensive stretch induces WPB release via the VEGFR2/PLCγ1/Ca2+ pathway, while the VEGFR2/Akt/NO pathway serves as a negative pathway involving S-nitrosation of NSF (272). Similarly, NO inhibits platelet degranulation by targeting NSF (168). Additionally, syntaxin-4, an essential part of SNARE complex in endothelial cells, platelets, and cardiomyocytes, can also be S-nitrosylated to inhibit exocytosis (57, 67, 267).
Cooperative action among gasotransmitters is evident within cardiovascular tissues. While it is clear that endogenous H2S and NO generation regulate blood pressure and cytoprotection responses during ischemia (16, 51, 100, 126, 130, 214, 278), recent reports reveal cooperative functions between these molecules. Investigation into the pathophysiological importance of both exogenous and endogenous sulfide during tissue ischemia–reperfusion injury has shown that eNOS-dependent NO generation is important for cytoprotection responses. Minamishima et al. found that exogenous sulfide therapy could not effectively mediate cytoprotection against experimental cardiopulmonary arrest in eNOS-deficient mice (165). King et al. further reported that exacerbation of myocardial ischemia–reperfusion injury in CSE/H2S-deficient mice was due to reduced eNOS phosphorylation and NO bioavailability (126). Moreover, Kondo et al. found that exogenous H2S therapy protected against murine heart failure in an eNOS/NO-dependent manner (134). These findings are in agreement with a study by Coletta et al. showing that exogenous H2S treatment of cultured endothelial cells increased eNOS phosphorylation and that exogenous NO could also augment cellular H2S formation (35). This is perhaps due to the fact that H2S can stimulate eNOS S-sulfhydration that facilitates its phosphorylation and prevents its S-nitrosation to stabilize its enzymatic function (5).
Similar cooperative behavior between NO and H2S is observed during chronic tissue ischemia-dependent vascular remodeling. As mentioned above, Coletta et al. revealed that each gasotransmitter reciprocally influenced the other and facilitated endothelial cell proliferation under normoxic cell culture conditions (35). Conversely, a study from Bir et al. exploring ischemic vascular remodeling in the hind limb ischemia model found that exogenous H2S therapy increased NO generation from both NOS and nitrite reduction pathways (16). This relationship was further clarified by Kolluru et al. demonstrating that CSE/H2S deficiency blunted ischemic vascular remodeling in an NO-dependent manner that could be rescued by nitrite/NO prodrug-based therapy (130). Importantly, this study revealed a unique temporal relationship of gasotransmitter induction, in that plasma H2S bioavailability rapidly increased upon hind limb ischemia preceding an increase in plasma NO bioavailability, and that as plasma NO bioavailability increased, plasma H2S levels decreased back down to baseline levels. These findings clearly illustrate cooperative relationships that work to restore tissue homeostasis.
Oxygen sensing of arterial blood by the carotid body also entails gasotransmitter cooperation. Peng et al. reported that carotid body glomus cells express CSE and produce H2S, which are important in modulating ventilation rates during hypoxia (192). In a subsequent study, these same authors reported that CO production via HO-2 in the carotid body is important for regulating H2S production in a spontaneously hypertensive (SH) rat model that was found to be important for controlling hypertension (191). Most recently, Yuan et al. demonstrated that HO-2-dependent CO formation regulated PKG activity that leads to CSE Ser377 inhibiting H2S production (279). Interestingly, in HO-2-deficient mice, nNOS/NO compensated for a loss of CO in oxygen sensing in a PKG-dependent manner. These reports further illustrate complementary and overlapping functions of gasotransmitters, which require further study in different organs and model systems.
Nervous system
In the brain, NO evokes presynaptic vesicular release through cGMP signaling and S-nitrosation of synaptic proteins, which enhances presynaptic syntaxin binding with VAMP and SNAP-25 (30, 187). NO may also recruit resting vesicles into recycling pools to potentiate NMDAR-dependent synaptic transmission. Moreover, S-nitrosation of RyR channel also increases channel opening and Ca2+-mediated vesicular release (113). At the PSD, nNOS is tethered to PSD-95, where NMDARs are accumulated. Presynaptic neural transmitters, glutamate and glycine, stimulate NMDARs, causing Ca2+ influx and nNOS activation. AMPARs are the predominant ion channels for fast excitatory transmission in central synapses. The expression and conductance of AMPARs in the PSD determine synaptic plasticity, which is the basis of learning and memory. NMDAR activation regulates synaptic plasticity that is believed to be mediated by NO through various mechanisms. S-nitrosation of GluA1 subunit at Cys875 facilitates its phosphorylation at Ser831 by CaMKII (208). Phosphorylated AMPAR is more permeable to Ca2+. Moreover, S-nitrosation of GluA1 also favors its binding to AP2 and increases GluA1 endocytosis. In contrast, S-nitrosation of stargazin and NSF enhances the recruitment of GluA1 and GluA2 respectively, regulating AMPAR subunit composition (101, 207). However, NMDAR activation also S-nitrosylates PSD-95, weakening its interaction with AMPAR and NMDAR. Therefore, NO signaling plays complex roles in regulating synaptic transmission. Additionally, neurotoxins, such as Aβ oligomers and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), can also increase iNOS expression in astrocytes, macrophages, and microglia cells. The elevated NOS activity increases NO production and excessive S-nitrosation of a large spectrum of proteins in the brain, which contributes to neurological disorders (178). Alzheimer's disease, specifically, is characterized by Aβ plaques and synapse loss via AMPAR internalization (98). The roles of S-nitrosation-mediated change of synaptic plasticity in neurological pathogenesis remain to be investigated.
All three H2S-producing enzymes are expressed in the brain tissue, but compartmentalized in different cell types. CBS, the predominant source of H2S in the brain, is mostly found in the Bergmann glia and astrocytes (52, 167). Physiologically, CO produced by HO-2 in neurons and endothelial cells inhibits CBS activity. However, hypoxia reduces CO production, which releases CBS from inhibition. As a result, CBS-derived H2S mediates vasodilation to increase blood supply (167). In comparison, CSE resides in vascular SMCs and neurons. The lack of CSE does not affect hypoxia-induced vasodilation in the cerebral cortex (167). Therefore, the site and the source of H2S production are crucial for H2S-mediated heterocellular signaling in the brain. 3-MST not only localizes in neurons in the brain and spinal cord but also concentrates in the synaptosomes isolated from the brain tissue, which suggests important roles of 3-MST in synaptic communication (219). Additionally, H2S increases intracellular Ca2+ in astrocytes and microglia, also implying that H2S mediates signaling between astrocytes, neurons, and microglia (140, 175). Moreover, H2S potentiates NMDAR activity and enhances long-term potentiation induction, which may be mediated by cAMP/PKA and postulated modification of NMDAR cysteine residues (2, 114, 119). H2S can also protect the brain from oxidative stress by increasing glutamate uptake, enhancing GSH synthesis, and possibly inhibiting NADPH oxidase (125, 152, 263, 277).
Gastrointestinal tract
Gasotransmitters regulate the gastrointestinal tract on many different aspects. H2S donors accelerate gastric emptying of liquid in awake mice by relaxing pyloric sphincter muscles via activation of KATP channels in SMCs and TRPV1 receptors in afferent nerves (162). Similarly, NaHS has also been shown to inhibit smooth muscle contraction and motility in the small intestine and the colon of mice, rats, and guinea pigs (72, 76, 176). Endogenous H2S derived from CSE and CBS involved also inhibits colonic motility (76). Moreover, the regulation of gastrointestinal motility by the central nervous system is modulated by CSE-derived H2S (212). In addition, there is a transwall gradient of resting membrane potential across the circular muscle of the gastrointestinal tract, which regulates circular muscle contractility (229). This transwall gradient depends on endogenous CO production (54, 210, 211, 229). Endogenously generated H2S physiologically potentiates resting membrane potential hyperpolarization, which is regulated by its inhibition of nNOS (213).
The effect of H2S on ion channels also regulates visceral pain. On the one hand, H2S acts on Cav3 channels to enhance nociceptive pain in the mouse (160), on the other hand, H2S also has an antinociceptive effect mediated by KATP channels and NO (49). CBS-derived H2S also contributes to chronic visceral hyperalgesia, which may be meditated by TRPV1 (137, 273).
H2S is an important regulator of inflammatory bowel disease. Both endogenous and exogenous H2S protects rats against colitis (62, 250). In dinitrobenzene sulfonic acid (DNBS)-induced colitis in a rat colitis model, CSE is acutely upregulated in colonic ulcerative sites, but diminished after 14 days (62, 63). This may be partially explained by the regulatory role of H2S in leukocyte adhesion. Under physiological conditions, endogenous H2S inhibits leukocyte adhesion on endothelial cells via activation of KATP channels, while the lack of H2S increases surface expression of adhesion molecules (P-selectin on endothelial cells and CD11/CD18 on leukocytes) (283). However, it is not yet clear whether deficiency of CSE affects the development of experimental colitis.
Remaining Questions and Future Directions
It is without question that our understanding of gasotransmitter chemical biology, biochemistry, and biological functions remains largely unknown. The evolutionary role of these molecules for the development of life on this planet illustrates the overall importance of them and is still shrouded in mystery (185, 186). While significant information is known about toxicological properties of CO, NO, and H2S, researchers have just begun to scratch the surface of the incredibly complex and rich chemistry between these molecules during normal physiological responses. The following issues below as well as others beyond the scope of this review are very important areas that will require careful consideration and future study.
Gasotransmitter reciprocal regulation of formation and signaling
As discussed throughout this review, one gasotransmitter molecule may act to limit or compliment the production of another. However, the mechanisms and conditions by which these events occur are highly opaque at the present time. Gasotransmitter regulation of signaling responses may also be complimentary or limiting, yet insight into how these reactions could occur is also unclear. Future studies aimed at delineating specific mechanisms whereby gasotransmitters influence formation and metabolism of one another under a broad set of conditions will provide substantial understanding to seemingly contradictory reports found throughout the literature.
Spatial and temporal relationships of gasotransmitter production and metabolism
The chemical nature of gasotransmitters coupled with mechanisms of production suggests that we know very little regarding how gasotransmitter molecules are made within cells and tissues. With an exception of a few reports, our understanding of the temporal nature of gasotransmitter formation is not well understood. Figure 5 illustrates some pathophysiological features involving cooperative gasotransmitter interactions. However, future studies are clearly needed to delineate gasotransmitter formation and metabolism in a temporal manner that will provide important insight into cellular and tissue responses.
Biological function of specific gasotransmitter reaction products
While many studies have reported beneficial effects of exogenous NO, H2S, or CO administration, it is unknown whether reaction products of these gasotransmitters are the actual mediators of beneficial effects. This is especially important given that the use of gasotransmitter donors varies substantially between studies and likely reflects biological effects of metabolic products. An excellent example of this is where many studies have used high micromolar concentrations of H2S donors, which likely involve a prominent role of polysulfide formation (84). However, much less is known regarding biochemical and biological effects of these reaction products. Thus, studies in this area are essential for a clear understanding of which molecules are the most biologically active.
Therapeutic potential of gasotransmitter metabolic products
A holy grail of biomedical research is aimed toward therapeutic uses or some other direct application. This is certainly a laudatory and important goal. However, given the complexity of gasotransmitter reaction products, future therapeutic studies must also consider the impact of specific metabolites on tissue function and disease models. In summary, it is now clear that gasotransmitter biochemistry and signaling serve prominent roles in normal physiology and disease with many more discoveries yet to come.
Footnotes
Acknowledgments
This work was supported by an NIH grant HL113303 and ADA grant 1-15-TS-18 to C.G.K. S.Y. was funded by a fellowship from the Malcolm Feist Cardiovascular Research Endowment, LSU Health Sciences Center—Shreveport.
Author Disclosure Statement
C.G.K. has intellectual property rights regarding NO/nitrite and H2S chemistry and is a cofounder of Theravasc, Inc., and Innolyzer LLC. X.S. has intellectual property rights regarding H2S chemistry and is an advisor for Innolyzer LLC. All other authors have no competing financial interests.