
Abstract
Significance:
Cachexia is defined as a complex metabolic syndrome that is associated with underlying illness and a loss of muscle with or without loss of fat mass. This disease is associated with a high incidence with chronic diseases such as heart failure, cancer, chronic obstructive pulmonary disease (COPD), and acquired immunodeficiency syndrome (AIDS), among others. Since there is currently no effective treatment available, cachectic patients have a poor prognosis. Elucidation of the underlying mechanisms is, therefore, an important medical task.
Recent Advances:
There is accumulating evidence that the diseased organs such as heart, lung, kidney, or cancer tissue secrete soluble factors, including Angiotensin II, myostatin (growth differentiation factor 8 [GDF8]), GDF11, tumor growth factor beta (TGFβ), which act on skeletal muscle. There, they induce a set of genes called atrogenes, which, among others, induce the ubiquitin-proteasome system, leading to protein degradation. Moreover, elevated reactive oxygen species (ROS) levels due to modulation of NADPH oxidases (Nox) and mitochondrial function contribute to disease progression, which is characterized by loss of muscle mass, exercise resistance, and frailty.
Critical issues:
Although substantial progress was achieved to elucidate the pathophysiology of cachexia, effectice therapeutic strategies are urgently needed.
Future Directions:
With the identification of key components of the aberrant inter-organ communication leading to cachexia, studies in mice and men to inhibit ROS formation, induction of anti-oxidative superoxide dismutases, and upregulation of muscular nitric oxide (NO) formation either by pharmacological tools or by exercise are promising approaches to reduce the extent of skeletal muscle wasting. Antioxid. Redox Signal. 26, 700–717.
Introduction
B
Under many pathological conditions, a primary dysfunction in one organ may lead to aberrant inter-organ communication, causing dysfunction of a remote organ and the development of comorbidities. Examples for the coordinated development of comorbidities include cardiac cachexia, cancer cachexia, the cardio-renal syndrome, and a.o. (9, 49, 112, 141). On the other hand, short-term skeletal muscle ischemia induces a protective effect in the heart, leading to reduced infarct size, a phenomenon called remote ischemic preconditioning (46). This effect appears to be mediated by factors released from ischemic skeletal muscle, which are able to protect multiple organs from ischemic insults. Moreover, exercise causes release of trans-acting factors from skeletal muscle (58), which regulate other organs and may exert a positive effect on wasting states.
Obviously, there exists extensive organ-organ crosstalk, which substantially contributes not only to morbidity but also to adaptation under pathological conditions. In this review, we will focus primarily on skeletal muscle interaction in the context of different wasting states, including cachexia and sarcopenia, and we will underscore the impact of nitric oxide (NO)/reactive oxygen species (ROS) as mediators of organ crosstalk.
Interorgan Communication in Skeletal Muscle Wasting
Skeletal muscle wasting can be classified as sarcopenia and cachexia, respectively. Sarcopenia is the aging-associated loss of muscle mass, whereas cachexia represents a more general wasting state, which is associated with chronic disease. According to the official definition, cachexia is a “complex metabolic syndrome associated with underlying illness and a loss of muscle with or without loss of fat mass” (45). Cachexia is, therefore, the consequence of an underlying chronic disease, such as cancer, heart failure, kidney disease, chronic obstructive pulmonary disease (COPD), chronic infection, sepsis, or acquired immunodeficiency syndrome (AIDS) (Fig. 1).
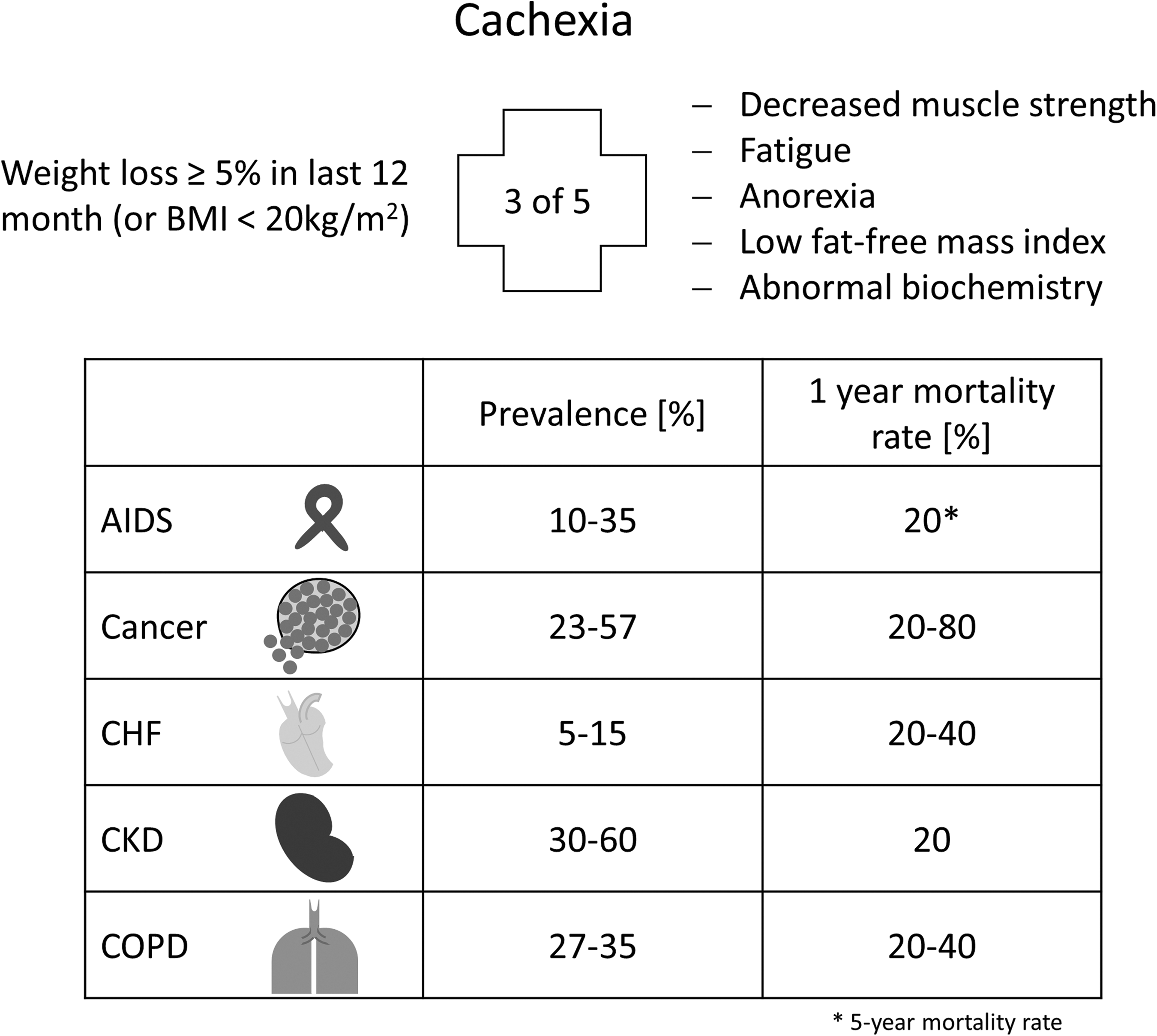
Clinical features of cachexia include weight loss, anorexia, inflammation, insulin resistance, hypogonadism, and anemia. In particular, muscle wasting plays an important role in cachectic patients, as it is mainly responsible for the weight loss, weakness, and fatigue of patients. The clinical definition of cachexia can be found elsewhere (45).
The treatment of cachexia is currently rather limited, mainly relying on treating the underlying chronic disease. Even parenteral nutrition does not prevent the ongoing loss of muscle mass; hence, once wasting is diagnosed, it has a very poor prognosis (8).
General Mechanisms in Skeletal Muscle Wasting
Loss of tissue mass in general results from an imbalance of anabolic and catabolic states due to altered substrate utilization, protein degradation, and synthesis. Although different chronic diseases may trigger cachexia via the release of a variety of different humoral factors from primary diseased organs, there seem to be some general pathogenic mechanisms underlying the cachectic phenotype (93). Until now, it remains uncertain whether loss of skeletal muscle is induced by only a primary diseased organ or via the involvement of other organs. At least, Angiotensin II (AngII), cytokines (e.g., interleukin 6 [IL6]), and growth factors of the tumor growth factor beta (TGFβ) family (e.g., myostatin) seem to be involved in the induction of muscle wasting (20, 29) (Fig. 2).

Independent of the mode of induction, it is clear that skeletal muscle wasting in cachexia is due to a disturbed protein synthesis and degradation, primarily through activation of the ubiquitin–proteasome pathway. There is also evidence for activation of the autophagy/lysosomal proteolytic pathway, but its causal role in development of muscle wasting remains controversial.
The proteasome system degrades proteins labeled by polyubiquitination (Fig. 3). Three types of enzymes E1, E2, and E3 execute different steps of the ubiquitination process. After activation of ubiquitin by E1 enzymes, activated ubiquitin is transferred to E2, which then delivers ubiquitin to E3 enzymes. The latter act as ubiquitin ligases, which covalently attach ubiquitin to the ɛ-amino groups of lysine residues of target proteins (131). Ubiquitinated proteins are then transferred to the 26S proteasome for degradation.

A prominent target of the proteasome system in muscle appears to be myosin, which was reduced in C2C12 myocytes under conditions of oxidative stress (79). Loss of myosin may explain reduced muscle mass and, on the other hand, the reduced muscle strength associated with the wasting states. More than thousand different E3 enzymes are present in the human genome, opening the way to a precise temporal and cell type-specific control of protein degradation. In skeletal muscle, the two most prominent players of the ubiquitin–proteasome pathway are the E3 ligases Muscle RING Finger 1 (MuRF1) and Muscle Atrophy F-box (MAFbx also known as Atrogin1) (111). Both these ubiquitin E3 ligases, which target many muscle proteins to degradation, are highly upregulated at the transcriptional level in nearly every model of muscle wasting (16).
Stimulation of the ubiquitin–proteasome pathway is frequently accompanied by increased oxidative stress (94). Experiments involving C2C12 myocytes have clearly shown that ROS induces E3-ubiquitin ligases (57), suggesting enhanced degradation of proteins by the 26S proteasome. However, in cachexia, the 20S proteasome is also upregulated. This proteasome subtype degrades oxidized proteins without prior modification by ubiquitination, which underscores the notion that elevated levels of ROS in cachectic muscle are involved in the pathology (96, 126).
Myocardial infarction induced atrophy of plantaris muscles in rats, which was accompanied by an increase of NADPH oxidases (Nox), activation of nuclear factor “kappa-light-chain-enhancer” of activated B-cells (NF-κB) and p38MAP kinase, and finally, elevated levels of 26S and 20S proteasomes. Almost all of these changes were reverted by apocynin, providing a clear link between elevated ROS formation and enhanced proteasomal activity in cardiac cachexia (14).
In contrast to the ubiquitin–proteasome pathway, which is responsible for clearing short-lived protein, the autophagy/lysosomal proteolytic pathway removes long-living proteins, protein aggregates, and organelles, including mitochondria (68, 89, 99) (Fig. 4).
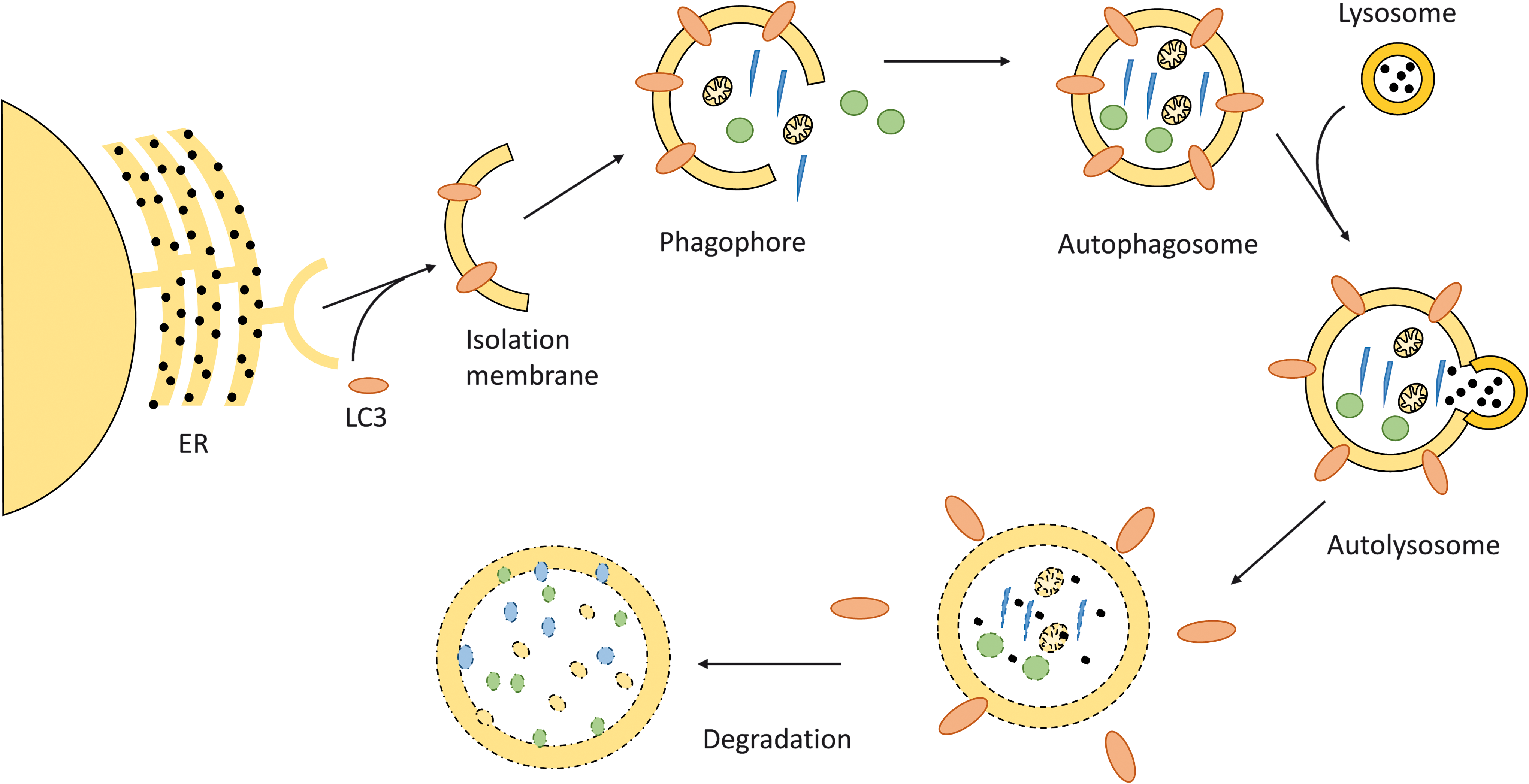
A hallmark of autophagy is the formation of a specific double membrane compartment, the autophagosome. The autophagosomal membrane originates from the endoplasmic reticulum (ER). Via elongation, the isolation membrane or phagophore grows to form the autophagosome. All of these processes are accompanied by the binding of many proteins expressed from autophagy-related genes, which not only coordinate the assembly process but also represent ligands for the recognition and internalization of cellular components destined to degradation. On fusion of the autophagosome with lysosomes, the degradation of engulfed material and recycling of its components occurs.
Experimental and clinical data demonstrate an upregulation of many genes, contributing to the regulation of autophagy and building of the autophagosomes in skeletal muscle, including mouse models of cancer cachexia as well as patients suffering from different types of cancer or heart failure (103, 104). Although there is a strong association with the loss of skeletal muscle mass, induction of autophagy may be secondary to cellular damage induced during wasting states. In this context, autophagy is important in clearing cells from damaged macromolecular complexes or organelles, which may improve the homeostasis and mitigate additional damage. For example, because damaged mitochondria may contribute to enhanced ROS production and further cell damage, mitophagy, that is, the degradation of damaged mitochondria by autophagosomes, may rather represent a protective mechanism than a cause of muscle wasting (99).
In line with this assumption, several papers describe augmented cell damage occurring with different pathological states when components of the autophagic machinery are inactivated (61, 84, 85, 97, 108).
Humoral Factors Influencing Cachexia
The induction of skeletal muscle wasting by diseased remote organs implies that circulating factors derived from the diseased tissues act on skeletal muscle to elicit catabolism in skeletal muscle.
Cytokines
Chronic diseases (e.g., COPD, heart failure, cancer, etc.) are often associated with tissue infiltration of inflammatory cells, leading to the enhanced release of pro-inflammatory cytokines by activated immune cells and/or tissue cells. These cytokines may travel through the circulation to reach remote organs, such as skeletal muscle (Fig. 5).
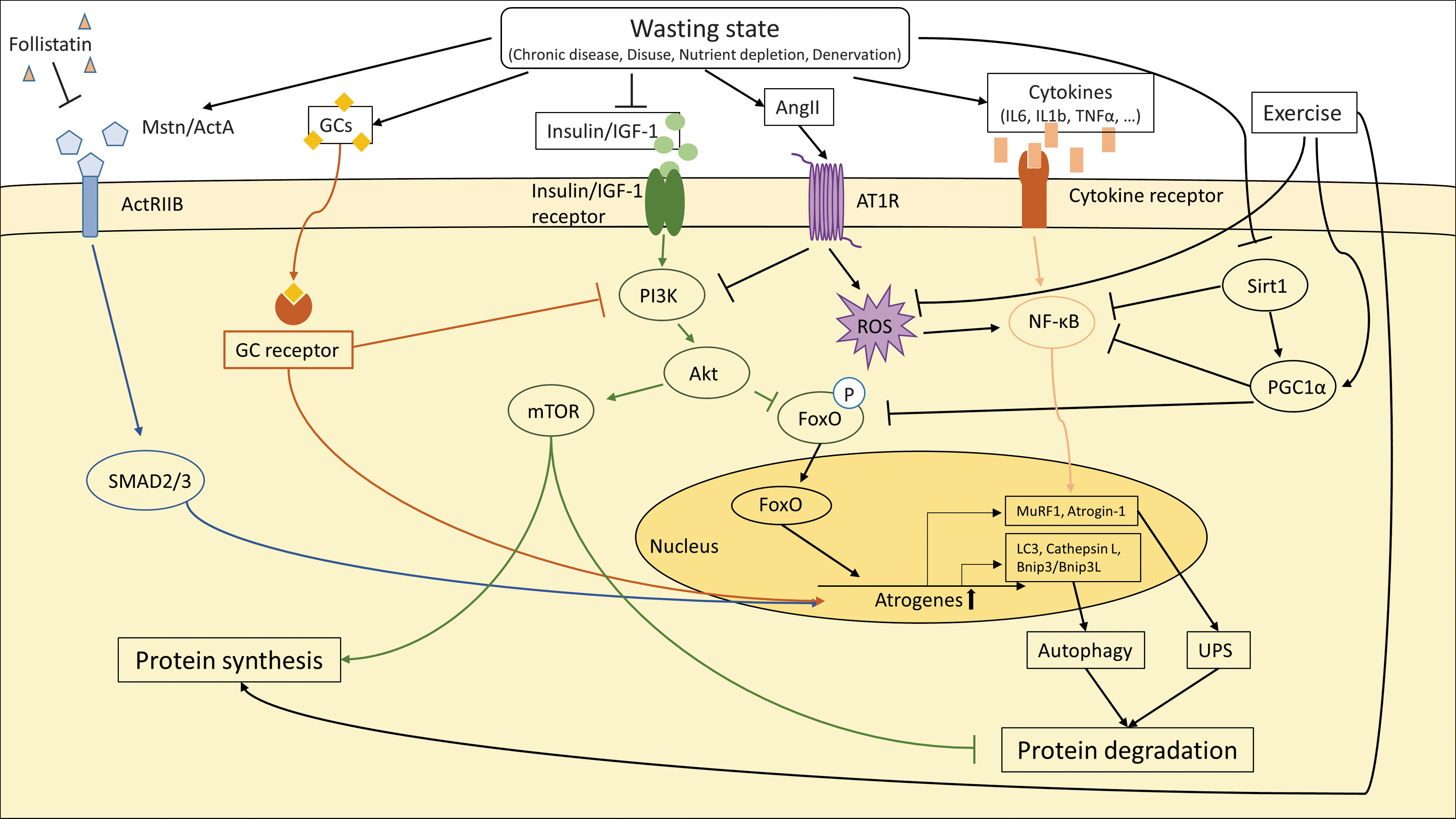
Several pro-inflammatory cytokines, including IL1, IL2, IL6, interferon γ, and tumor necrosis factor alpha (TNFα), are found to be upregulated in patients suffering from a variety of chronic diseases. Indeed, TNFα was first named cachectin, because it was identified as a factor secreted by macrophages that was able to modulate adipose cell metabolism (138). It is of note that TNFα and some other cytokines also affect muscle protein synthesis (60), the release of cortisol and catecholamines from the adrenal gland (124), affect lipolysis and lipid oxidation (114)—all leading to loss of body weight and negative energy balance.
Finally, members of the TGFβ superfamily of cytokines, including TGFβ, myostatin/growth differentiation factor 8 (GDF8), activin, and GDF11, induce the ubiquitin-proteasome system, which reduces muscle mass by increased protein degradation and promotes skeletal muscle dysfunction and weakness (144) (Fig. 5).
Also, glucocorticoids play an especially important role in sepsis as well as in cancer-induced cachexia (19, 66), characterized by an elevated transcription of ubiquitin proteasome-related genes in muscle (147), inhibition of protein synthesis, and induction of gluconeogenesis. Also, the parathyroid hormone-related peptide (PTHrP) and AngII are related to cancer and cardiac cachexia as well as to skeletal muscle wasting due to kidney failure (27, 72).
Insulin-like growth factor 1 (IGF-1) is a classic anabolic hormone that has a well-known positive effect on muscle bulk and strength (13). There is now evidence that IGF-1 plays a protective role in cachexia, and many of its actions are executed by the phosphoinositide 3-kinase (PI3K) AKT-pathway. Pro-cachectic factors such as AngII may reduce IGF-1 levels; hence, a decrease of IGF-1 levels could be an indicator of malnutrition as well as cachexia (34, 117).
AngII Induces Skeletal Muscle Wasting and Fatigue
Among the circulating factors known to induce cachexia, AngII, the active component of the renin-angiotensin-aldosterone system, is the best characterized in terms of its ability to induce skeletal muscle wasting (27, 44, 120).
Heart failure is associated with a substantial neuro-humoral activation, including a sympathetic overflow as well as stimulation of the renin angiotensin system. In the cardiovascular system, AngII may contribute to hypertension, cardiac myocyte and vascular smooth muscle cell hypertrophy, endothelial dysfunction, and insulin resistance. In skeletal muscle, AngII induces a wasting state and antagonizes the anabolic action of growth factors, such as IGF-1 (21).
As reported for many cell types, AngII induces the production of ROS in skeletal muscle (120, 132). ROS not only possess signaling functions but may also induce oxidative damage of proteins, lipids, and so on. In skeletal muscle, AngII enhances ROS generation, most likely via Nox (Fig. 6).

Wei et al. demonstrated that AngII induces an impairment of insulin sensitivity in L6 skeletal myoblasts concurrent with a reduced GLUT4 translocation to the plasma membrane (145). Small interfering RNA (siRNA), directed against the p47phox subunit of Nox, prevented this effect. Further analysis revealed that a reduced tyrosine phosphorylation of insulin receptor substrate 1 (IRS1) and the protein kinase AKT led to a concerted downregulation of the insulin signaling pathway. This was associated with a reduced translocation of GLUT4 to the plasma membrane, which may underlie the reduced glucose uptake by skeletal muscle cells. Serine phosphorylation of IRS1 also reduces its activity. This inhibitory phosphorylation could result from a feedback inhibition via activated S6 kinase or due to a direct phosphorylation by stress kinases, such as c-Jun N-terminal kinase (JNK).
Thus, reduced insulin sensitivity may contribute to the imbalance between muscle anabolic and catabolic processes in muscle wasting. In this context, it is important to note that there appears to exist a marked antagonism of AngII and IGF-1 (21, 27, 69, 127, 128).
A clinically important hallmark of cachexia is fatigue, which may be the result of degradation of muscle protein. Recent data demonstrate that AngII stimulates the Nox2 isoform via the canonical AT1-receptor—Gαq—phospholipase C (PLC)—diacylglycerol (DAG)—proteinkinase C (PKC)-dependent pathway in skeletal muscle. As a consequence, elevated ROS lead to the oxidation of critical cysteine residues of the ClC-1 chloride channel (35), which leads to reduced Cl− -current and the activation of BK potassium channels. This remodeling of resting ion currents could contribute to fatigue of skeletal muscle observed in different wasting states.
Oxidative stress in skeletal muscle induces proteolysis and, therefore, atrophy via different mechanisms. First, ROS promote transcription of the genes encoding the ubiquitin ligases MuRF1 and Atrogin-1. The basic helix-loop-helix (bHLH) transcription factor EB (TFEB) was identified as a key transcription factor in this response. TFEB binds to specific E-boxes located in the MuRF1 promoter. The transcription-enhancing properties of TFEB can be blocked by histone deacetylases 5 (HDAC5) in the nucleus. An AngII-dependent ATI receptor activation leads to induction of protein kinase D1, which phosphorylates HDAC5 and, hence, causes a cytoplasmic localization of HDAC5. As a result, derepressed TFEB activates MuRF1 expression and paves the way to muscle atrophy (44).
Moreover, activation of calcium-activated proteases, such as calpain as well as the 20S proteasome, may occur in response to AngII stimulation, favoring muscle catabolism (113, 115). Both inhibition of Nox and treatment with antioxidants prevented protein degradation in myotubes. Similarly, upregulation 20S proteasome activity and muscle wasting are sensitive to a genetic or pharmacological inhibition of Nox, indicating the importance of Nox activity in skeletal muscle wasting.
AngII also increases mitochondrial ROS production with respiratory chain complexes I and III as major sources. Other sites, including complex II, have also been identified to be the source of ROS (17). At complexes I and II, electrons are usually transferred to ubiquinone (coenzyme Q), resulting in the formation of ubiquinol, which acts as an electron donor for complex III. However, part of the electrons (0.15%–2% of total mitochondrial oxygen consumption depending on conditions) can be transferred to oxygen, resulting in the formation of O2 −· (63, 133).
A large part of O2 −· is released from complexes I-III to the mitochondrial matrix, where it is scavenged by the mitochondrial manganese superoxide dismutase (MnSOD; SOD2). This protects Fe-S clusters of aconitase, complex I, and complex II from oxidation-induced inactivation. Under ischemic conditions, complex III may release O2 −· anions to the intermembrane space (95), from where they may travel to the cytoplasm via voltage-dependent anion channels (62). This opens the possibility for mitochondria-derived O2 −· in modulating cell signaling in the cytosol.
In endothelial cells, the AngII-dependent activation of mitochondrial ROS release is triggered by an initial stimulation of Nox2 via a PKC-dependent phosphorylation of p47phox (40) and an src-mediated stimulation of Ras-related C3 (Rac1) (139). The elevated O2 −· levels act on and open mitoKATP channels and decrease mitochondrial membrane potential (mΨ), which is associated with mitochondrial permeability transition pore opening and an enhanced release of ROS to the cytoplasm. This has been shown to further increase oxidative stress, since Nox2 is among the targets for mitochondria-derived ROS, leading to a feed-forward mechanism of oxidative stress development between mitochondrial and Nox-derived ROS [for details, see the review by Schulz et al. (119)].
Overexpression of SOD2 (mitochondrial MnSOD) or the application of mitoTEMPO (a mitochondrially targeted antioxidant) could inhibit AngII-induced mitochondrial dysfunction (38), supporting the relevance of the Nox-mitochondrial crosstalk. In cardiac myocytes, a similar relationship appears to be involved in preconditioning (71). In this context, AngII via Nox stimulation and release of mitochondrial O2 −· appears to lead to a ROS-mediated activation of the apoptosis signal regulating kinase 1 (ASK1), which is well known to be regulated by the cellular redox status (18, 67).
Although these mechanisms have been studied extensively in the cardiovascular system, only few studies have addressed the possible contribution of AngII-dependent mechanism in skeletal muscle wasting.
Semprun-Prieto et al. investigated the role of Nox in AngII-mediated skeletal muscle wasting by using p47phox-deficient mice and pharmacological blockade of Nox by a specific inhibitor and antioxidant apocynin (120). They showed that AngII-induced ROS production was blocked in skeletal muscle in both models. Moreover, the elevated 20S proteasome activity and muscle proteolysis were reduced in p47 phox-/- mice treated with AngII compared with wild-type mice. In contrast to the mechanism reported for endothelial cells and cardiac myocytes, the mitochondria-targeted antioxidant Mito-TEMPO was not effective in attenuating AngII-induced muscle wasting. Therefore, Nox-dependent stimulation of mitochondrial O2 −· release appears not to be a prerequisite for AngII-induced muscle wasting (135).
Cabello-Verrugio et al. and Morales et al. demonstrated in C2C12 myoblasts that AngII-induced ROS production is mediated by an angiotensin I receptor (AT1R)-p38MAPK-Nox signaling cascade, which leads to increased TGFβ expression (26, 27, 92), suggesting a contribution of TGFβ to skeletal muscle wasting and wasting-associated fibrosis.
The effect of ROS not only depends on the expression of pro-oxidative enzymes, such as xanthine oxidase (XO) and Nox, but is also modulated by detoxifying mechanisms (SOD, catalase, and glutathione peroxidase [GPX]).
Although many studies demonstrate a connection between induction of Nox and AngII-mediated protein degradation, oxidative stress in cachexia may be the result of an imbalance between ROS-generating and -degrading systems. Sullivan-Gunn et al. demonstrated a 40% increase of superoxide levels in cachectic muscles (133). Surprisingly, Nox2, p40phox, and p67phox were downregulated in these muscles. Concomitantly, levels of antioxidant proteins, for example, SOD1, SOD2, and GPX, were also downregulated, which most likely resulted in a net increase of oxidative stress. Unfortunately, other ROS-producing systems, such as mitochondrial ROS production, were not evaluated.
Involvement of the TGFβ Superfamily in Cachexia
There exists now ample evidence that members of the TGFβ superfamily of cytokines are involved in the induction of cachexia. TGFβ and its relatives bone morphogenic proteins (BMPs), activins, and myostatin are a family of promiscuous growth factors, which share, in part, their receptors and downstream signal transducers (SMAD family of transcription factors) (118). Signaling involves the formation of protein complexes of class I and class II dimeric receptors, resulting in an active hetero-tetrameric transmembrane receptor.
Tumor growth factor beta receptor II (TGFβRII) receptors together with ALK5 type I receptors are mainly activated by TGFβ itself, leading to downstream activation of Smad2/Smad3/Smad4 complexes. BMPs signal through distinct receptor complexes activating Smad1, Smad5, and Smad8 family members together with Smad4 (11). The cytokine myostatin binds to receptor complexes consisting of the Activin Receptor IIb (ActRIIb) and ALK4 or ALK5 type I receptors, respectively. Signaling then occurs through an Smad2/Smad3 complex.
Myostatin, also known as growth/differentiation factor 8, is the first identified myokine of the TGFβ superfamily. Its role in skeletal muscle is illustrated by the fact that inactivation of the myostatin gene (knockout) results in extensive skeletal muscle hypertrophy in mice, cattle, and humans (43, 86, 87). Myostatin is one of the very prominent examples of a secreted growth factor that is involved in the development of cardiac cachexia. It is generated predominantly in muscle and acts as a major suppressor of muscle growth. Interestingly, cardiomyocyte-specific deletion of the myostatin gene in mice attenuates skeletal muscle wasting in response to pressure overload (20).
Besides myostatin, TGFβ also plays an important role in cachexia. Studies in C2C12 myocytes and in mice revealed that TGFβ-induced muscle wasting was associated with upregulation of E3 ubiquitin ligases (MuRF), and it led to reduced diameter of myotubes and myosin heavy chain degradation (2). These effects were reversible by apocynin or N-acetylcysteine, indicating the participation of ROS in the TGFβ effects. However, it was not investigated as to which Nox isoform caused myocyte shrinkage.
A recent study contributed additional important insights into the role of TGFβ in muscle wasting. Waning and Guise and Waning et al. demonstrated that TGFβ is a principal factor inducing skeletal muscle weakness in the context of cancer cachexia involving NAPDH oxidase Nox4 as a key element (143, 144). Many types of cancer are associated with bone metastases, where the activity of bone-degrading osteoclasts is elevated. Bone matrix, however, is rich with stored TGFβ, which is released during the osteolytic action of tumor cell-stimulated osteoclasts. The released TGFβ induces Smad2/3/4 signaling in muscle, which, in turn, elevates expression of Nox4. The Nox4 isoform of Nox predominantly produces H2O2 (90%) and little O2 −· (10%) (100, 121), and its activity is mainly regulated on the transcriptional level.
Nox4 activity depends on binding to the p22phox subunit, but it is independent from other “classic” Nox subunits. Nox4 is localized to cardiac and limb muscle mitochondria and to the sarcoplasmic reticulum (6, 134). Nox4-dependent oxidation of sarcoplasmic calcium release channel ryanodine receptor 1 (Ryr1) acts as an oxygen sensor in skeletal muscle. High pO2 increases oxidation of a subset of Ryr1 thiols by Nox4, leading to Ryr1 activation. Exercise induces molecular alterations of Ryr1 complex, including hyper-nitrosation and loss of calstabin binding, which is a Ryr1-stabilizing protein. The consequence is a reduced exercise tolerance due to leaky Ca2+-release channels (15). Intriguingly, Waning et al. found that this physiological control mechanism appeared to be dysregulated in cancer-associated TGFβ-induced muscle fatigue (144).
The sarcoplasmic calcium release channel Ryr1 forms a complex with Nox4, as both proteins could be co-precipitated, and Ryr1 present in these complexes was oxidized to a higher extent. Moreover, calstabin levels in these complexes were reduced in muscle from cancer-bearing mice (75, 76). Together, these changes compromised function of the Ryr1, resulting in lower muscle strength. Inhibition of Nox4 by a Nox1/Nox4 inhibitor reverted the oxidation of Ryr1, restored calstabin binding to Ryr1, and normalized strength of extensor digitorum longus (EDL) muscles. Importantly, these molecular alterations were also found in a small cohort of breast cancer patients and in a mouse model of Camurati-Engelmann disease, which is associated with increased bone degradation and elevated circulating TGFβ levels.
Although the identity of numerous circulating humoral factors that induce cachexia is known, the precise mechanisms that link the receptor activation in skeletal muscle membranes to the precise intracellular events leading to ROS activation and protein degradation are still a matter of intensive research.
Using a mouse tumor model, Fukawa et al. recently added another piece to the puzzle of “cachectic signaling” in skeletal muscle (50). A very early event identified by the use of three different tumor cell lines was a rapid induction of β-oxidation of fatty acids. In consequence, oxidative stress increased, indicated by elevated mitochondrial ROS formation and higher glutathione (GSH) levels in myotubes incubated with conditioned media from tumor cells. ROS, in turn, stimulated the activity of the stress kinase p38 MAPK, which, by itself, appears to regulate cachectic pathways.
Of note, Etomoxir, a well-known inhibitor of carnitine palmitoyltransferase I (CPT-I) and β-oxidation, efficiently blocked ROS formation in human myotubes treated with pro-cachectic supernatants of tumor cells. Moreover, systemic as well as local application of Etomoxir also reduced the oxidative stress marker 8-oxoguanidine in vivo in tumor-bearing mice and efficiently reduced the tumor-induced weight loss and p38 MAPK activation. Since cachexia, 8-oxoguanidine, and p38 MAPK activation highly correlated in human skeletal muscle biopsies from patients with gastrointestinal cancer, the axis β-oxidation, mitochondrial ROS, and p38 MAPK appear to represent a common pro-cachectic pathway that could be efficiently targeted by the established drug Etomoxir.
Fibertype-Specific Effects of Cachexia
In contrast to cardiac muscle, which is composed of homogeneous striated muscle cells, skeletal muscles are composed of two different fiber type classes—type I fibers (slow twitch fibers) and type II fibers (fast twitch fibers) (Fig. 7). Type I fibers mainly run on an oxidative profile, whereas type II fibers have to be further distinguished according to their metabolism and force generation. Type II fibers are subclassified into type IIa (oxidative), IId/x (glycolytic), and IIb (fastest glycolytic fibers—not seen in humans) fibers. In adult healthy mice, the type IIb fibers usually account for 70%–80% of total muscle fibers, whereby the exact content varies within the different skeletal muscles, depending on their rather oxidative (e.g., Musculus soleus) or glycolytic (e.g., Musculus plantaris) profile.

During wasting conditions, these fiber types are affected differentially. In cancer, sepsis, and elevated glucocorticoids, type II glycolytic fibers seem to selectively atrophy, suggesting a higher resistance of the oxidative fibers (3, 88, 137). Early work has shown that in patients with chronic heart failure (CHF), skeletal muscle fiber-type composition may shift toward type II fibers. Also, a reduced mitochondrial volume density and cytochrome c oxidase activity (41, 42) was reported. In a mouse model of cardiac cachexia, neither a switch from oxidative to glycolytic fibers nor an atrophy of oxidative fibers was observed, but cross-sectional areas of glycolytic type II fibers were substantially reduced (78).
These fiber type-specific responses to an underlying chronic disease reflect most likely a differential susceptibility of fiber types to physical inactivity as well as disease-associated factors as increased inflammatory cytokines, malnutrition, hypoxia, and oxidative stress (32, 106).
The molecular basis for the higher resistance of oxidative fibers against wasting observed in many pathological states needs to be elucidated. The transcriptional co-activator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) is a principal regulator of oxidative metabolism and mitochondrial biogenesis. Its transgenic expression in muscle leads to a shift from glycolytic to oxidative type I fibers, characterized by a switch in myosin isoform toward type I, elevated mitochondrial density, and enhanced myoglobin expression (80). Yan and coworkers analyzed skeletal muscle-specific PGC1α expression to determine whether type I fibers would be protected in the context of cardiac cachexia (51).
In transgenic mice with cardiac-specific calsequestrin overexpression, which develop heart failure and cachexia, PGC1α expression antagonized the development of muscle atrophy when compared with calsequestrin-expressing mice. This went along with attenuated FOXO3a levels, suggesting a reduced activation of the ubiquitin-proteasome system and of autophagy, because both systems are under control by FoxO3.
In cachexia-resistant type I fibers, inducible nitric oxide synthase (iNOS) and endothelial NOS (eNOS) were upregulated. Moreover, elevated levels of SOD1, SOD2, and SOD3 contributed to a lower ROS formation by type I fibers than by type II fibers. Further analyses demonstrated a pronounced protection of muscle against wasting by the presence of extracellular SOD (ecSOD, SOD3), since both transgenic mice with muscle-specific overexpression of ecSOD and in situ transfection of muscle by ecSOD expression vectors resulted in muscle protection (101).
Role of Nitric Oxide in Skeletal Muscle Wasting
As mentioned earlier, upregulation of various NOS isoforms occurs in cachexia, raising the question as to what extent NO is involved in modulating cachexia. NO has frequently been termed a double-edged sword (116), because this radical may exert both protective and pathological functions, depending on quantitative effects as well as on the spatial arrangement of NO synthases.
In skeletal muscle, the major NOS isoform is the neuronal (type I) NO synthase (nNOS). nNOS is expressed as different splice variants, giving rise to nNOS isoforms of different size. The main isoform, nNOSμ, is located at the sarcolemma, where it interacts via its N-terminal PDZ domain with α-syntrophin, a component of the dystrophin complex. Sarcolemma-associated nNOS may release NO at high rates due to activity-associated Ca2+ influx. Muscle-derived NO enhances blood flow by dilating blood vessels nearby (105, 136). Most of muscular NO is released by nNOSμ. nNOSβ is another nNOS variant that lacks the PDZ domain and is, therefore, unable to interact with the dystrophin complex. nNOSγ is associated with the Golgi apparatus, where it appears to regulate muscle fatigue (91, 105).
Yet another form of nNOS (mitochondrial NOS [mtNOS]) is claimed to reside inside the mitochondria where it could regulate respiratory chain function, although the existence of an mtNOS is still under debate (140, 151). Thus, in skeletal muscle, there is an extensive compartmentalization of NOS isoforms, and spatially defined actions of different NOS isoforms are clearly documented. Since NO is a diffusible agonist, the question remains as to how specific compartments of NO action can exist within the same cell.
Myoglobin, which is highly expressed in heart and oxidative skeletal muscle, is—when oxygenated—able to efficiently oxidize NO to nitrate. NO can efficiently inhibit mitochondrial respiration, but this function can be blocked efficiently by oxy-myoglobin (48, 55, 148). We hypothesized that myoglobin builds an efficient barrier that separates functional compartments within a muscle cell, allowing the local action of NO, for example, at the sarcolemma and the sarcoplasmic reticulum without a spillover from one compartment to another (12, 54).
To what extent the different actions of NO occur in skeletal muscle under wasting conditions remains to be investigated in detail. iNOS induction occurs in TNFα-induced cachexia in mice (23), in human skeletal muscle biopsies from patients suffering from AIDS and cancer, respectively (109), as well as in cardiac cachexia (5, 149).
Although NOS inhibition appeared to attenuate the extent of muscle wasting in one study of Buck and Choijkier (23), Yu et al. demonstrated that a robust NO-dependent, antioxidative defense may also contribute to the protection in oxidative muscles (149). Specifically, on catabolic stimuli, oxidative muscles have greater NO production and antioxidant gene expression. When the endogenous NO donor, S-Nitrosoglutathione (GSNO), was administered systemically, it mitigated dexamethasone-induced skeletal muscle wasting and attenuated MAFbx/Atrogin-1 messenger RNA (mRNA) expression (101). Moreover, GSNO injection promoted the expression of ecSOD, suggesting that ecSOD is an important player in NO-mediated protection against muscle wasting.
These findings raised the possibility of targeting the NO-dependent antioxidant defense against catabolic muscle wasting. NO could exert its protective effects via canonical cGMP-mediated signaling as well as by direct modification of proteins via nitrosylation, nitrosation, and glutathiolation (56). A more detailed analysis of NO-dependent modifications by proteomic approaches may enable the identification of novel targets in cachexia.
The precise mechanism leading to the NO-dependent modulation of skeletal muscle wasting is not fully understood. A well-defined action of NO is to inhibit the respiratory chain of isolated mitochondria at nanomolar concentrations by nitrosylation of complex IV (33). However, this may result in elevated ROS formation, because a reverse flow of electrons may occur when complex IV is blocked. On the other hand, inhibition of complex I by S-nitrosation is associated with reduced mitochondrial ROS formation. Complex I is considered to represent an “entry point” for electrons, and, therefore, a reduction of ROS formation should be the result of its inhibition. Indeed, at least in the heart, many studies have demonstrated a cardioprotective potential of complex I nitrosation, and the cardioprotection by nitrite was ascribed to this protein modification (31, 110, 125).
Besides its effects on mitochondrial respiration, NO appears to be a major regulator of glucose uptake and mitochondrial biogenesis in skeletal muscle (83), which could explain the protective effects of NO in muscle wasting. Moreover, Lira et al. presented a positive feedback loop involving NO-mediated AMP-dependent protein kinase (AMPK) stimulation, which, in turn, enhances NO formation (82) (Fig. 8).
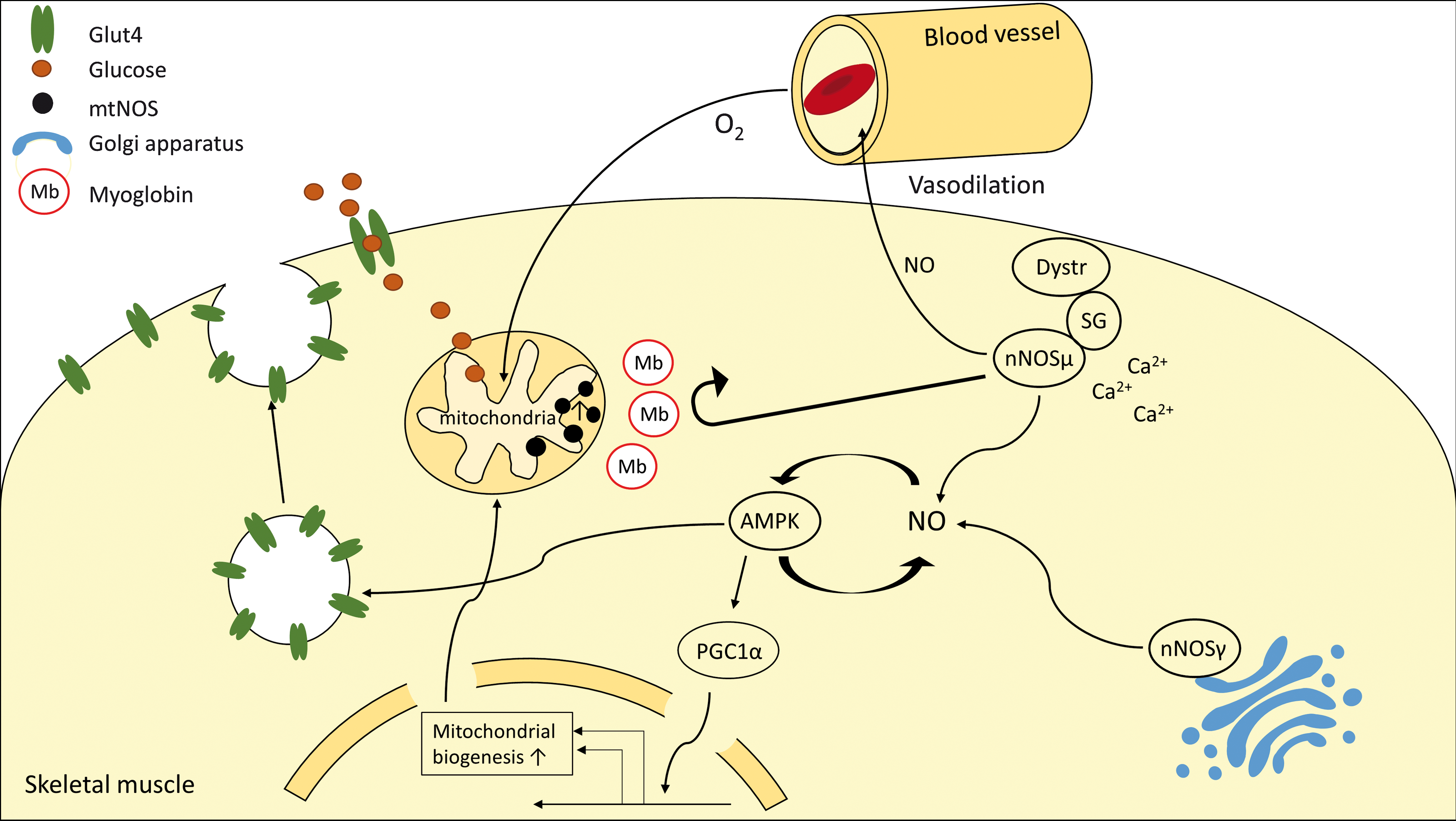
AMPK activates PGC1α, which is a master regulator of mitochondrial biogenesis by regulating peroxisome proliferator-activated receptors (PPARs) and nuclear respiratory factor 1 (NRF1)/NRF2 transcriptional activity, resulting in an enhanced oxidative metabolism. Thus, upregulation of eNOS and iNOS could protect muscle from cachexia by promoting an oxidative phenotype of muscle fibers. In particular, iNOS, which is supposed to represent a constitutively active, soluble NOS isoform, could enter the different “nNOS compartments” by diffusion and, hence, drive mitochondrial biogenesis via an AMPK-PGC1α-dependent mechanism.
Besides modulating the action of NO, myoglobin's protective effect in red muscle fibers could result from its ability to directly inactivate ROS. The pseudo-peroxidase activity of myoglobin is associated with oxidation of the central heme-bound Fe2+ to ferric- (Fe3+) or ferryl- (Fe4+) iron species, which may be involved in oxidation of biomolecules such as lipids and amino acids.
Despite its proposed function to enhance oxidative stress, for example, in myocardial infarction (7), studies in isolated hearts of myoglobin knockout mice demonstrated a higher sensitivity to H2O2 and O2 −· in the absence of myoglobin. Moreover, myoglobin-deficient hearts displayed a worse recovery from ischemia/reperfusion (47). Thus, in red skeletal muscle, this function of myoglobin may explain, at least in part, why oxidative fibers appear to be more resistant to wasting in the setting of cachexia. Experimental proof of a myoglobin-mediated protection in cachexia awaits the generation of conditional myoglobin knockout mice.
Antioxidative Therapy: A Tool to Treat Skeletal Muscle Wasting?
Current strategies for treatment of skeletal muscle wasting include anti-inflammatory and anabolic strategies. In view of the compelling evidence, that elevated ROS are important elements of the wasting pathology, a preclinical evaluation of ROS-lowering therapy was initiated.
In general, low-molecular-weight compounds with antioxidant activity, including vitamins C and E or inhibitors of ROS-generating enzymes, can be applied. In a rat cancer cachexia model that was induced by an injection of Yoshida AH-130 tumor cells, Springer et al. investigated the effects of various dosages of allopurinol and oxypurinol, inhibitors of XO, which are considered a major source of superoxide production (129). This study was triggered by the observation that uric acid, the end product of the XO reaction, is increased in many patients with heart failure and cancer (39). Interestingly, both XO inhibitors reduced uric acid levels in tumor carrying rats, demonstrating that XO was a major source of uric acid formation and of elevated ROS production in this model.
XO inhibition went along with reduced mortality, skeletal muscle wasting, ROS formation, and attenuated induction of proteasomal genes (129). In line with this pharmacologic approach, overexpression of ecSOD, as mentioned earlier, reduced oxidative stress, expression of atrogenes and attenuated skeletal muscle wasting in a transgenic mouse model of cachexia (101). In contrast to the promising results obtained by directly targeting enzymes involved in ROS formation, an antioxidant mixture containing green tea extracts, ascorbic acid, resveratrol, and a.o. failed to prevent the development of cachexia in a mouse model based on an injection of C26 tumor cells (10). Thus, a more targeted approach instead of using a broad spectrum of antioxidants appears to be a more effective strategy to inhibit ROS formation and ameliorate cachexia.
Although antioxidant therapies appear to be a logic postulate given the involvement of ROS in cachexia, trials to improve other ROS-associated diseases by antioxidants or XO inhibition have failed in many cases or yielded contradictory results. Thus, also in the case of cachexia, the development of effective therapeutic approaches based on reduction of oxidative stress will be a major challenge. For example, XO inhibition by allopurinol represents a relatively specific approach, which appeared to improve endothelial dysfunction in hypertensive patients (24) and positively influenced cardiac function in patients with idiopathic dilative cardiomyopathy (28).
However, the recently published results of the Xanthine Oxidase Inhibition for Hyperuricemic Heart Failure Patients (EXACT-HF) study demonstrated that despite a reduction of uric acid plasma levels by allopurinol, clinical status, exercise capacity, and left-ventricular ejection fraction were not improved (53). Also, vitamin E-based interventions did not yield positive results in the treatment of heart failure (1, 150).
The limited success of antioxidant therapies despite encouraging results in preclinical models may be, on the one hand, due to the choice of inappropriate antioxidants (130). On the other hand, the approach of just targeting the formation of ROS may be too simple in view of the various sources of ROS formation and mechanisms of inactivation, including mitochondria, Nox enzymes, different forms of SOD, peroxiredoxins, and so on. Under basal conditions, ROS are important parts of intracellular signaling. Thus, extensive inhibition of ROS formation may also disturb basal signal transduction, inducing unwanted side effects.
Depending on the underlying disease (cancer, heart failure, COPD, etc.) leading to cachexia, the comorbidities may even complicate the situation. ROS formation by different sources such as mitochondria, various Nox isoforms, XO, and so on most likely varies depending on the underlying disease and the type of interorgan communication. Thus, targeting may require the application of disease-specific inhibitors. In case of an Nox contribution, this may be a challenging task, since currently inhibitors with sufficient Nox and, even more problematic, sufficient Nox isoform specificity still have to be developed (30, 36, 64).
In view of the limitations of antioxidants and the lack of specific pharmacological tools to inhibit ROS-generating enzymes, the induction of endogenous antioxidant enzymes may represent an alternative therapeutic strategy. Indeed, upregulation of SOD and catalase in humans reduced lipid peroxidation (98), suggesting reduced oxidative stress. However, in a recent paper, Dey et al. investigated the contribution of mitochondrial and cytoplasmic compartments to ROS scavenging in H9c2 cardiac cells (37). It was demonstrated that the formation of NADPH, the electron donor for reduction of oxidized GSH and thioredoxin (TRX) in the cytoplasmic and mitochondrial compartments, may become a limiting factor of the ROS-scavenging capacity of the cell.
Metabolic or mitochondrial dysfunction may lead to a loss of reductive capacity, which is important to regenerate not only endogenous reductants such as TRX and GSH but also exogenous antioxidants such as MitoQ. Thus, to be effective, an antioxidative therapy may critically depend on an intact metabolic flux to sufficiently regenerate NAD(P)H, especially in the case of cachexia, which is frequently associated with mitochondrial dysfunction.
Modulation of Cachexia by Exercise
At present, there are no established pharmacological interventions that successfully treat muscle wasting in sarcopenia and cachexia. Exercise training, however, can attenuate or even reverse the process. It is well accepted that endurance exercise is the most effective (22, 142), whereas mixed results have been obtained regarding the impact of resistance exercise (73, 77). For example, a recent animal study has confirmed the effectiveness of endurance exercise with evidence of preserved muscle function and attenuation of muscle mass loss, possibly due to activation of a mammalian target of rapamycin (mTOR). In this study, resistance exercise does not seem to be beneficial, but induces genes associated with muscle damage or repair, indicative of excessive stress (70).
There are also positive findings in frail elderly humans showing that 3 months of resistance exercise significantly reduced the mRNA levels of TNFα in muscle with concomitant increases of protein levels.
Although it is well known that exercise has direct, positive impacts on muscle under the condition of cachexia, the underlying mechanism remains unclear. Exercise impacts appear to be multi-faceted. For example, endurance exercise prevents the increase in tumor necrosis factor-like weak inducer of apoptosis (TWEAK), blocking subsequent activation of NF-κB and its downstream signaling (102). As mentioned earlier, resistance exercise significantly reduced the mRNA levels of TNFα in muscle along with concomitant increases of protein levels, suggesting that exercise may attenuate muscle wasting by suppressing TNFα-mediated inflammation (59).
A recent animal study by Pigna et al. suggests the importance of autophagy in exercise-mediated protection against muscle wasting in cancer cachexia (107). These authors showed that voluntary running of mice subjected to the C26 cell-induced cancer model prevented loss of muscle mass and function along with improved autophagy flux. The increased autophagy flux may improve muscle homeostasis under the condition of cancer cachexia due to removal of, for example, damaged proteins and mitochondria.
An important mechanism by which endurance exercise provides potent protection against catabolic muscle wasting is by promoting antioxidant defense systems. As mentioned earlier, oxidative muscles, which are locomotive muscles, are more resistant to atrophy than glycolytic muscles under almost all conditions of muscle wasting except for the ones induced by reduced neuromuscular activity (3, 25, 74, 78, 90, 123). Consistently, promotion of the oxidative muscle phenotype by PGC1α overexpression in glycolytic muscles significantly attenuates catabolic muscle wasting induced by CHF (51), which is the most likely mediated by enhanced expression of iNOS and eNOS, production of NO, and expression of antioxidant enzymes, including CuZnSOD, MnSOD, ecSOD, and catalase. All of these changes could contribute to reduced oxidative stress (51).
Further supporting this is the finding that voluntary wheel running in mice (4 weeks of training) induces ecSOD and iNOS expression in skeletal muscle. Consistently, ectopic expression by somatic gene transfer and muscle-specific transgenic overexpression of ecSOD provided potent protection under the condition of glucocorticoid treatment and mitigated all aspects of cardiac cachexia, including body weight loss and exercise intolerance (101). These findings strongly support the role of NO-dependent antioxidant defense (i.e., ecSOD) in protection against catabolic muscle wasting.
Exercise is known to promote secretion of humoral factors from the contracting muscles. For example, IL6, which increases 100-fold in the circulation after prolonged exercise, was associated with muscle damage; however, IL6 also regulates the proliferation of satellite cells and promotes hypertrophic growth (122). Paradoxically, circulating IL6 levels were also associated with suppression of muscle protein synthesis and mTOR signaling in cachectic tumor-bearing mice, an effect that was attenuated by treadmill exercise training (146). Exercise also modulates the effects of myostatin. Myostatin is upregulated under conditions of muscle wasting and prevents myocyte differentiation by inhibiting the Akt/mTOR pathway. Importantly, 6 months of low-intensity endurance training in insulin-resistant adult males led to reduced myostatin (65).
The proof of beneficial effects of exercise on skeletal muscle wasting is not restricted to animal studies. A clinical study explored the impact of exercise training on the expression of antioxidant genes in patients with CHF (81). Skeletal muscle biopsies taken from healthy people and from CHF patients after completion of a 6-month training protocol or continued sedentary lifestyle demonstrated (1) a reduction of SOD, GPX, and catalase mRNA and protein levels in CHF patients compared with healthy control subjects. Moreover, this study also showed that (2) exercise training increased the activity of GPX and Cat in muscle of trained CHF patients. These changes went along with a reduction of lipid peroxides and nitrotyrosine in muscle biopsies. Thus, exercise reduces oxidative stress in skeletal muscle of CHF patients.
More recent data extended the concept of muscle protection by training. The Leipzig Catabolism Study (Leica) demonstrated that after a 4-week training protocol, MuRF1 expression, an indicator of elevated ubiquitin-proteasome-system activity, was significantly reduced in quadriceps muscles of heart failure patients (52). Also, the level of ubiquitinated proteins was lower than in biopsies from sedentary muscle. Concomitantly, IGF-1 expression increased, suggesting that exercise shifted the catabolism-anabolism ratio to a reduced muscle wasting.
Besides the positive effects of exercise training on the expression genes involved in ROS and protein degradation, a downregulation of pro oxidant genes such as Nox could also be part of the beneficial effects of training. At least in the vasculature, exercise training of coronary artery disease (CAD) patients led to reduced expression of Nox4, gp91 phox , and p22 phox (4).
Overall, exercise activates several different systems that act either independently or synergistically to reduce oxidative stress in muscle. Exercise represents a low-cost, natural, and potently effective strategy that provides protection against muscle wasting.
Footnotes
Acknowledgments
This work was supported by a grant of the Deutsche Forschungsgemeinschaft (DFG) via International Research Training Group IRTG1902 to A.G. and partially supported by the National Institutes of Health R01GM109473 to Z.Y.