
Abstract
Aims:
Nonalcoholic steatohepatitis (NASH) is characterized by steatosis, panlobular inflammation, liver fibrosis, and increased cardiovascular mortality. Dipeptidyl peptidase-4 inhibitors (gliptins) are indirect glucagon-like peptide 1 agonists with antidiabetic and anti-inflammatory activity, used for the treatment of type 2 diabetes. Their potential and underlying mechanisms to treat metabolic liver inflammation and fibrosis as well as the associated vascular dysfunction remain to be explored.
Results:
In the methionine/choline-deficient (MCD) diet and Mdr2−/− models of NASH and liver fibrosis, treatment with sitagliptin and linagliptin significantly decreased parameters of steatosis and inflammation, which was accompanied by suppression of hepatic transcript levels reflecting metabolic inflammation and fibrosis, including SREBP-1c, FAS, TNFα, iNOS, α-SMA, Col1α1, and MMP-12. Moreover, gliptins reduced the number of liver infiltrating CD11b+Ly6Chi proinflammatory monocytes/macrophages and liver-resident F4/80+ macrophages, with an increase of Ym1+ alternative macrophages and (anti-inflammatory) macrophage markers Arg1 and IL-10. This was paralleled by decreased hepatic and aortic reactive oxygen species (ROS) production and NOX-2 mRNA expression, a normalization of endothelial dysfunction, cardiac NADPH oxidase activity, mitochondrial ROS formation, and whole blood oxidative burst in the MCD model.
Innovation and Conclusions:
Gliptins via suppression of inflammation decrease steatosis, apoptosis, oxidative stress, and vascular dysfunction in murine models of NASH and liver fibrosis, with mild direct antifibrotic properties. They reduce the numbers of liver and vascular inflammatory monocytes/macrophages and induce their alternative polarization, with beneficial effect on NASH-associated hepatic and cardiovascular complications. Therefore, gliptins qualify as drugs for treatment of NASH and associated liver fibrosis and cardiovascular complications. Antioxid. Redox Signal. 28, 87–109.
Gliptins are antidiabetic drugs with anti-inflammatory properties. Here, gliptins normalized or significantly reduced experimental liver inflammation to attenuate steatohepatitis, hepatocyte lipoapoptosis, oxidative stress, and liver fibrosis. New is that gliptins suppressed (peripheral) vascular dysfunction and oxidative stress accompanying chronic liver injury, by suppression of NADPH oxidase and mitochondria-dependent oxidative stress. Another novelty is that our findings suggest that these effects are largely mediated through their anti-inflammatory effect on infiltrating monocytes and macrophages thereby decreasing oxidative stress and fibrosis. This makes gliptins attractive (adjunctive) drugs for the treatment of NASH, and the frequently associated insulin resistance and cardiovascular complications.
Introduction
N
Indeed, various proinflammatory mediators such as tumor necrosis factor-α (TNF-α), interleukin (IL)-6, IL-1β, and cyclooxygenase-2 are increased in the livers of NASH animal models (17, 20, 39) and secondary to this, increased formation of hepatic reactive oxygen species (ROS) was proposed as a therapeutic target in NASH (81). Similarly to NASH, an important role of immune cells is well established for the development of atherosclerosis in humans (31, 50) and also of arterial hypertension in animals (30, 87), providing another important correlation between the pathogenic mechanisms underlying the development and progression of NAFLD/NASH and cardiovascular disease. Importantly, oxidative stress has a large impact on endothelial function and is a prognostic marker for increased cardiovascular events (16, 32). Likewise, oxidative stress leads to an activation of the immune system at various levels, and activated immune cells will further increase oxidative stress, resulting in a vicious circle (8, 89).
Dipeptidyl peptidase-4 (DPP-4) inhibitors prolong the activity of the incretins gastric inhibitory polypeptide and glucagon-like peptide-1 (GLP-1) and are used to enhance glucose-induced insulin secretion (3). GLP-1 is secreted postprandially from neurons of the caudal solitary tract in the central nervous system and from intestinal L-cells, controlling food intake, stimulating insulin secretion, and inhibiting glucagon release in the pancreas, resulting in improved glucose uptake in the liver, adipose tissue, and muscle, and reducing hepatic gluconeogenesis (29). Both GLP-1 mimetics and inhibitors of DPP-4, a ubiquitously expressed cell surface enzyme that leads to proteolytic inactivation of GLP-1, are increasingly used to manage type 2 diabetes (3, 19, 78).
While animal studies demonstrated a beneficial effect of GLP-1 mimetics and DPP-4 inhibitors on the vascular system, including inhibition of atherosclerosis, myocardial and kidney fibrosis (43, 47, 72, 75), studies on experimental NAFLD and NASH are limited, demonstrating overall anti-inflammatory and antioxidant effects (42, 45, 78). The latter studies were largely limited to aspects of hepatocyte damage and steatosis, and lacked mechanistic insights into liver inflammation and fibrosis. Although a unifying immunological link between a beneficial effect on liver inflammation and the concomitant cardiovascular protection rendered by GLP-1 treatment was not provided, a few studies provided clear evidence for this connection by demonstrating synergistic actions of GLP-1 supplementation on liver inflammation and systemic atherosclerosis (85), as well as the activation of the hepatic endothelin-1 system in a model of biliary liver fibrosis (66). Besides atherosclerosis, sepsis, and NASH, the anti-inflammatory and antioxidant potential of DPP-4 inhibitors and GLP-1 analogues was demonstrated in various models of chronic inflammatory diseases such as colitis (40), asthma (68), chronic obstructive lung disease (84), and rheumatoid arthritis (9).
We therefore studied the therapeutic efficacy of two major DPP-4 inhibitors in the methionine/choline-deficient (MCD) diet model of severe lipoapoptotic liver injury and in the Mdr2−/− model of biliary fibrosis, representing key pathologies of advanced NASH, with a particular focus on the role of inflammatory monocytes and macrophages in both the liver and the vascular system. We show that pharmacological DPP-4 inhibition mitigates liver inflammation and fibrosis as well as the associated vascular oxidative stress and dysfunction, mainly via suppression of monocyte/macrophage infiltration and a beneficial shift in their polarization.
Results
Gliptins attenuate hepatic steatosis
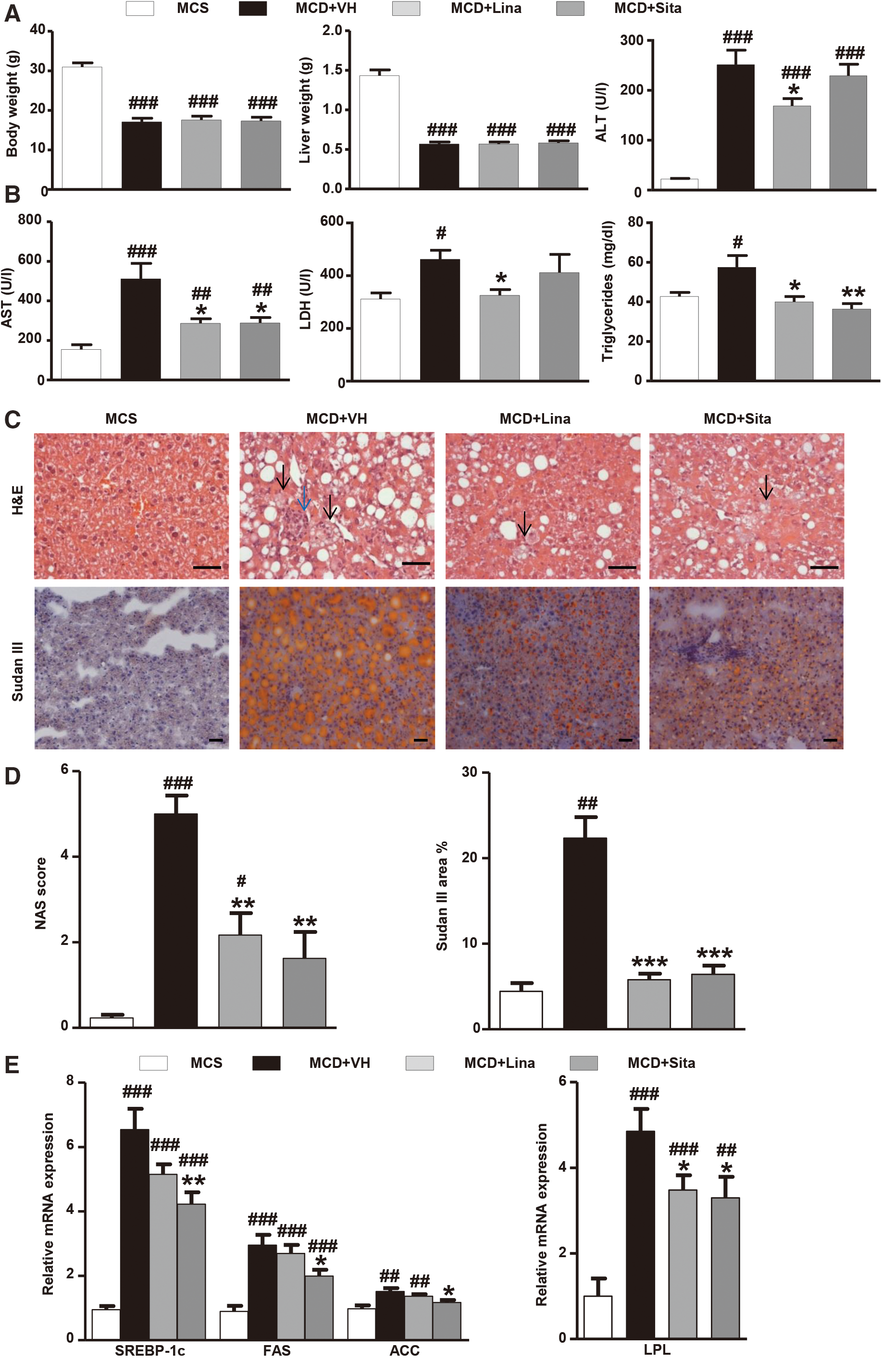
Compared to mice on the methionine and choline-sufficient (MCS) diet, mice on the MCD diet for 8–10 weeks showed a significant reduction of body and liver weight, which remained unchanged after either linagliptin or sitagliptin treatment (Fig. 1A). However, gliptin treatment normalized the elevated serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH), and triglycerides compared to vehicle-treated MCD-fed mice (Fig. 1A, B). Both gliptins decreased DPP-4 activity and mRNA levels in the serum and liver, also significantly increased plasma levels of active GLP-1 compared to vehicle treatment (Supplementary Fig. S1A, B; Supplementary Data are available online at
Effect of gliptins on hepatocyte apoptosis, liver macrophage numbers, and polarization in vivo
In mice with severe steatohepatitis induced by the MCD diet, the highly increased numbers of hepatic CD68+ and F4/80+ cells [markers for general and resident macrophages, respectively (27)], were significantly decreased, while M2 polarized and presumably beneficial Ym1+ macrophages were significantly increased in the gliptin- compared to vehicle-treated group (Fig. 2A, B). The levels of the proinflammatory cytokine IL-6 increased significantly in MCD diet-fed mice compared to the MCS diet-fed mice, reflecting the severity of inflammation seen for other parameters. Gliptin treatment normalized IL-6 levels (Supplementary Fig. S2A, B). Linagliptin and sitagliptin also reduced (hepatocyte) apoptosis, as assessed by immunohistochemistry for cleaved caspase 3, which is considered a central driver of inflammation and fibrosis in NASH (37) (Fig. 2A, B). Transcript levels related to Kupffer cell/macrophage activation and polarization were changed in line with the above data. Thus, in MCD mice, the increased expression of proinflammatory TNFα and iNOS was suppressed, and that of anti-inflammatory Arg1 and IL-10 was induced by gliptin treatment (Fig. 2C). Together, these results demonstrate a close association between DPP-4 inhibition, beneficial M2 macrophage polarization, decreased liver steatosis, inflammation, and apoptosis, and thus attenuated progression of NASH.

Gliptins suppress macrophages and other resident and recruited proinflammatory myeloid cells
The role of myeloid cells in the pathogenesis and especially treatment of NASH and liver fibrosis is little explored (33, 52, 77, 79). While the number of intrahepatic CD45+ leukocytes remained unchanged between MCD mice treated with gliptin or vehicle (Supplementary Fig. S2A), further gating based on the expression of CD11c and the macrophage marker F4/80 yielded three distinct cellular subsets: CD11c+F4/80−, CD11c−F4/80+, and CD11c+F4/80+ (Fig. 3A). In this study, the CD11c+F4/80+ M1 macrophage subset (10) was significantly increased in the MCD group, and reduced by 50% with DPP-4 inhibitor treatment (Fig. 3B). The differences were less pronounced when ratios between CD11c+F4/80− and CD11c−F4/80+ cells were calculated (Fig. 3B and Supplementary Fig. S3A).
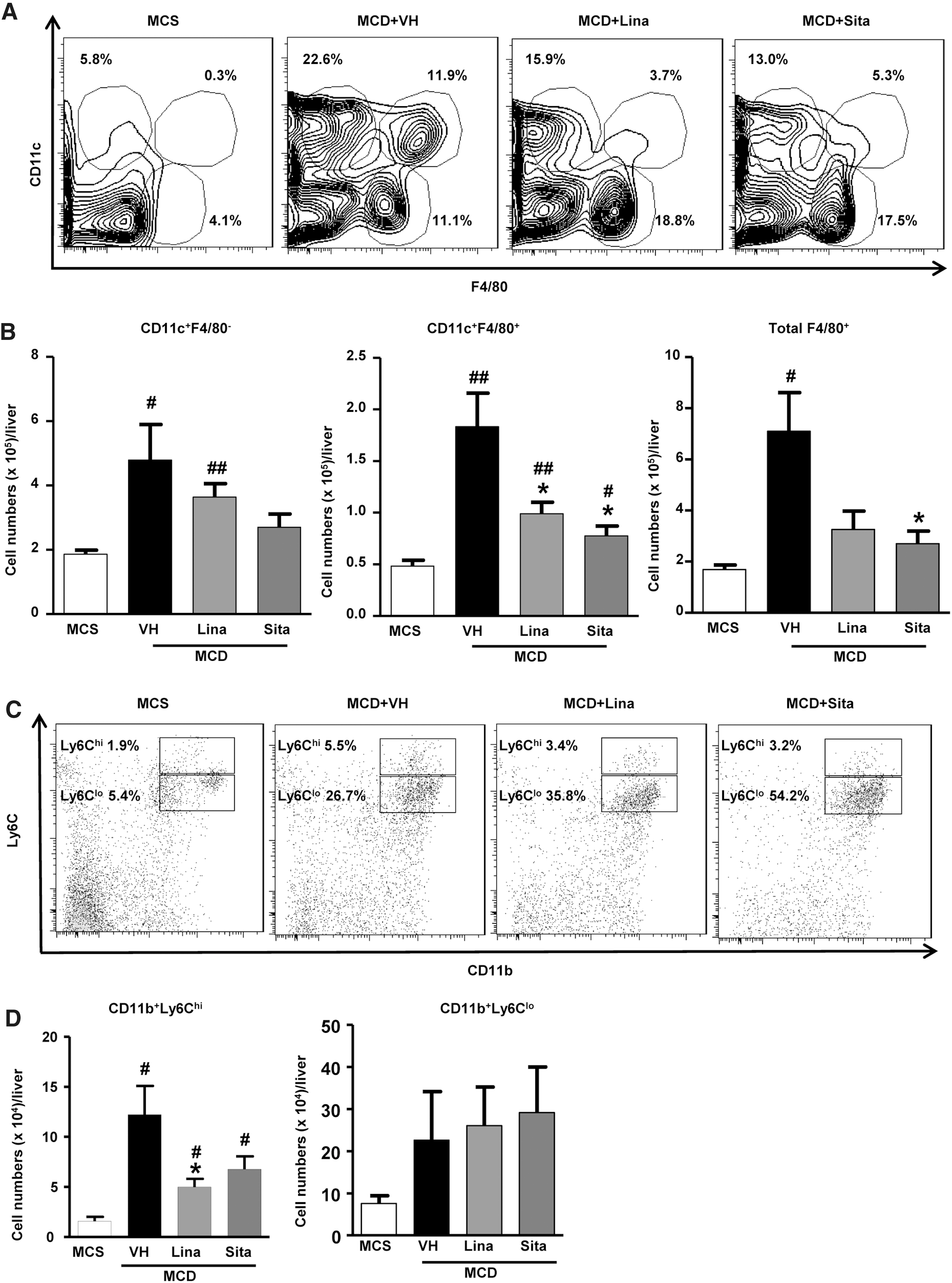
Linagliptin>sitagliptin treatment significantly reduced (by ∼40% and ∼55%, respectively) the highly (up to approximately eightfold) increased infiltrating, proinflammatory CD11b+Ly6Chi expressing monocytes/macrophages in MCD mice (Fig. 3C, D). This indicates a major suppressive effect on proinflammatory monocyte/macrophage recruitment and infiltration. Differences were even more significant when the ratio of CD11b+Ly6Chi versus CD11b+Ly6Clo per total CD45+ cells was calculated (Supplementary Fig. S3B).
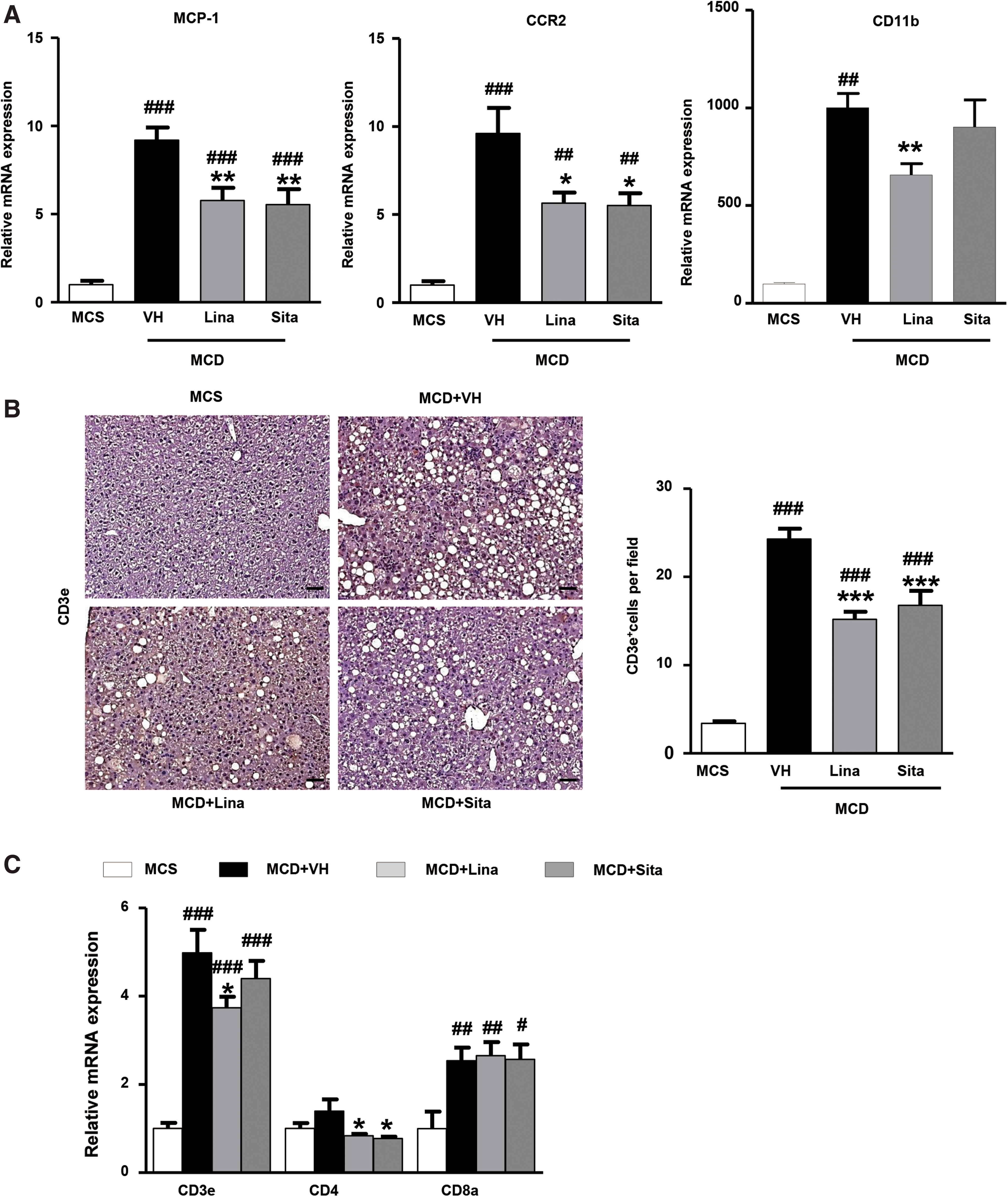
Hepatic expression of monocyte chemoattractant protein-1 (MCP-1 or CCL2, mainly expressed by Ly6Chi monocytes), C-C chemokine receptor type 2 (CCR2, a receptor for CCL2), and CD11b supported these findings (Fig. 4A). MCP-1 is a chemokine that recruits both monocytes and T lymphocytes (18). In this line, immunohistological quantification showed that CD3+ T cells were also increased eightfold in livers of MCD mice, to be significantly downregulated on gliptin treatment (Fig. 4B). This was paralleled by decreased hepatic transcript levels of CD3e and CD4 (but not CD8a), suggesting a mild secondary effect of gliptins on CD4 T cell activation (Fig. 4C). Taken together, gliptin treatment prominently mitigated the number, infiltration, and activation of myeloid cells (monocytes, macrophages, dendritic cells), and suppressed the number of freshly recruited/infiltrating proinflammatory myeloid cells in favor of anti-inflammatory (“restorative”) monocytes/macrophages, with a moderate reduction of CD4+ T cells.
Gliptins suppress diet-induced liver fibrosis
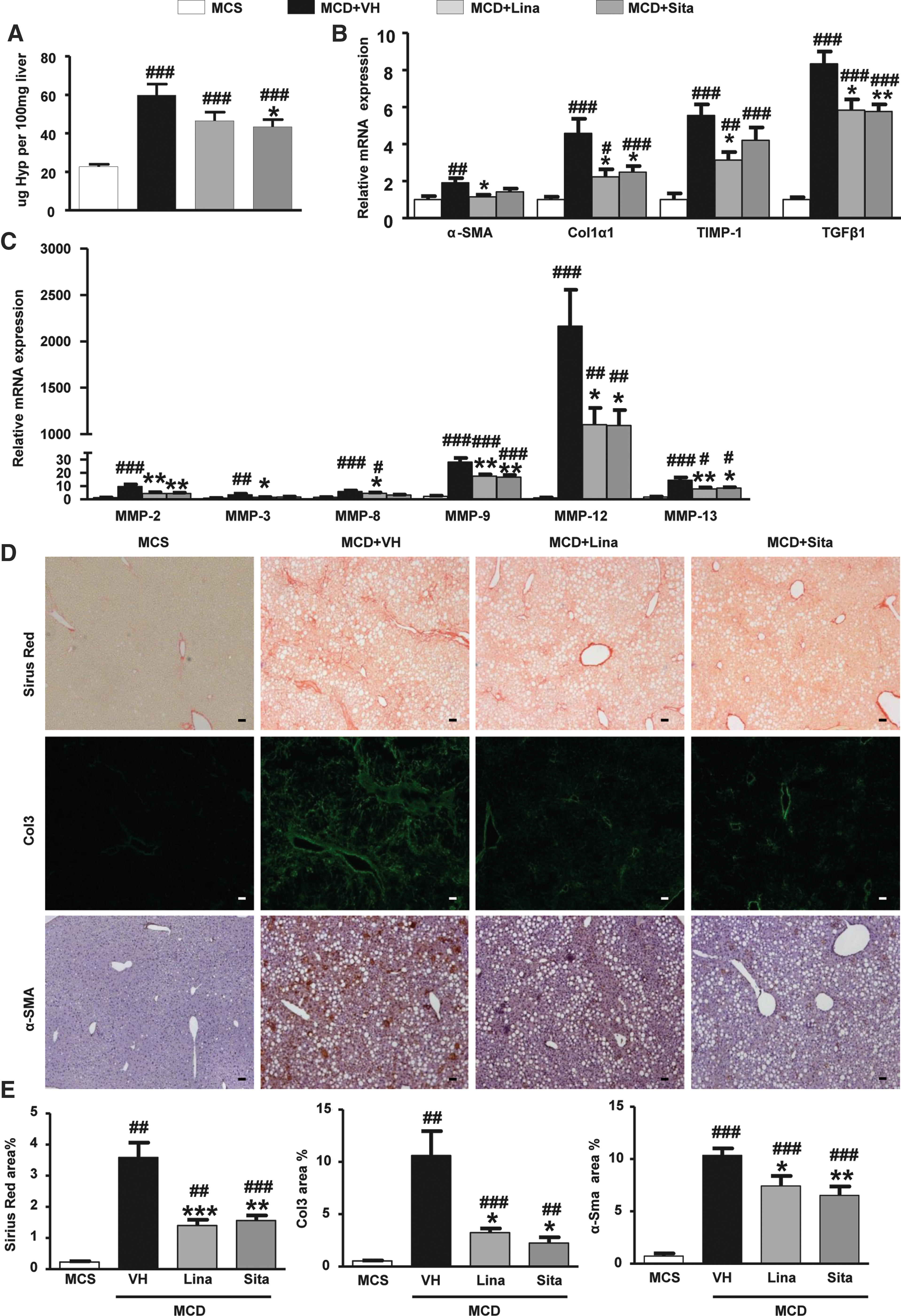
We next evaluated the development of hepatic fibrosis as a consequence of chronic liver injury. Mice on the MCD diet developed advanced hepatic fibrosis with a 2.5-fold increased relative collagen content as determined biochemically via hydroxyproline (Hyp). Sitagliptin>linagliptin reduced collagen accumulation (Fig. 5A), and both gliptins decreased hepatic transcript levels of alpha smooth muscle actin (α-SMA), procollagen α1(I) (Col1α1), tissue inhibitor of metalloproteinase-1 (TIMP-1), transforming growth factor beta 1 (TGFβ1), and MMP-2, -3, -8, -9, -12, and -13 compared to the MCD/vehicle-treated controls (Fig. 5B, C). In addition to transcript levels of TGFβ1 and MMP-12 mRNA, we also analyzed their protein levels, confirming their suppression by gliptin treatment (Supplementary Fig. S4A, B). Sirius red morphometry, which better captures parenchymal than the functionally less relevant dense portal collagen, showed an even more marked reduction in the gliptin-treated MCD mice than suggested by biochemical Hyp determination. This was paralleled by morphometry for (pro-) collagen type III (Col3), which characterizes thinner and more recently deposited collagen fibrils (Fig. 5D, E). Equally, morphometry for α-SMA protein demonstrated a reduction of activated hepatic stellate cells (HSC) and myofibroblasts in MCD mice with DPP-4 inhibition (Fig. 5D, E). These results uniformly show that both gliptins significantly attenuate MCD diet-induced hepatic fibrosis, which is accompanied by decreased expression of matrix metalloproteases (MMPs) that not only promote fibrolysis but also unfavorable extracellular matrix remodeling and that are prominently produced by activated macrophages and other myeloid cells (94).
Linagliptin attenuates liver fibrosis independent of steatohepatitis
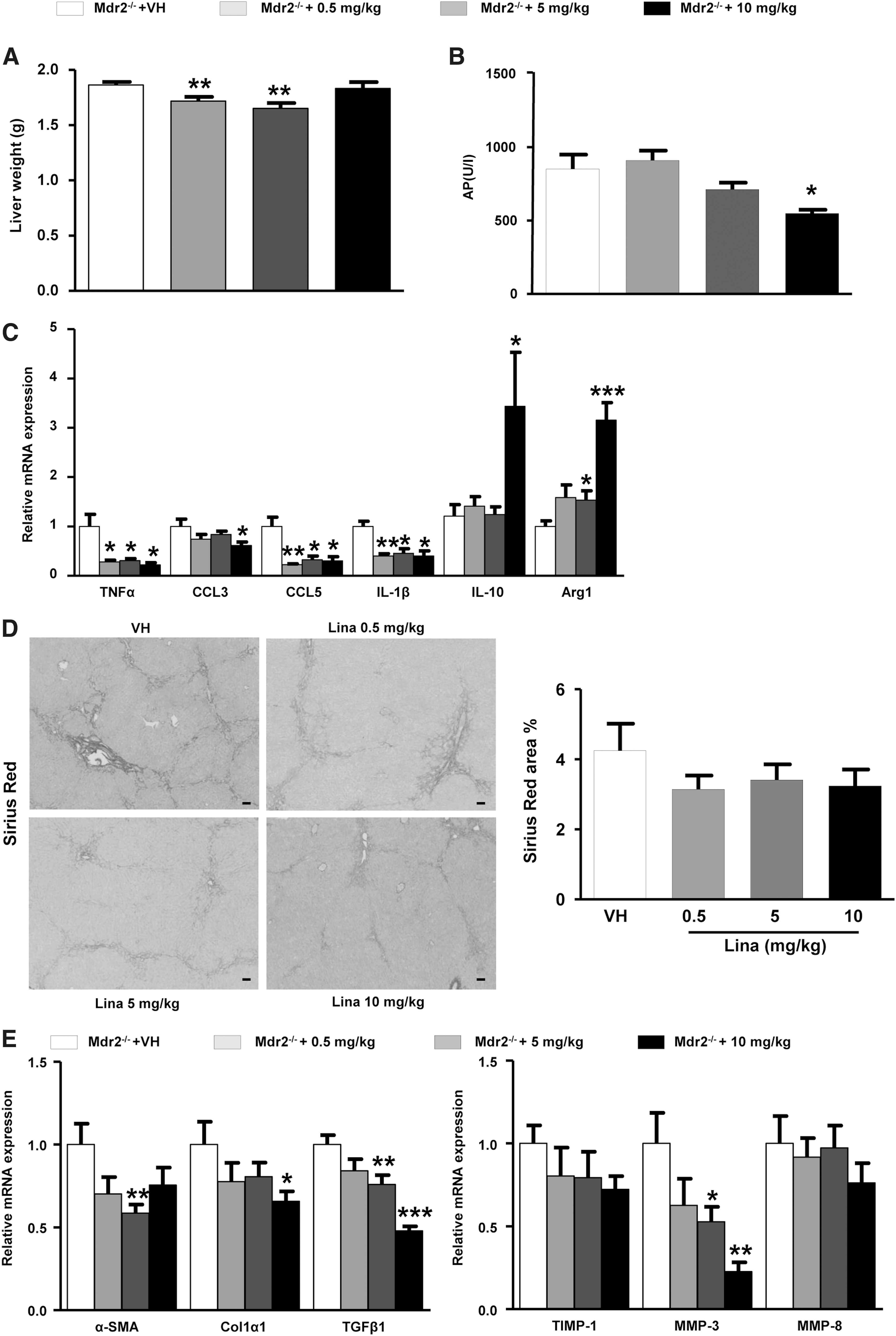
To study if and how far DPP-4-inhibitors may have a direct effect on fibrosis, irrespective of pathologic features that underlie NASH, that is, steatosis and hepatocyte lipoapoptosis that are represented in the MCD diet model, Mdr2−/− mice that spontaneously develop biliary fibrosis (63) received increasing doses of linagliptin (0.5, 5, and 10 mg/kg) for 4 weeks starting at age 7 weeks. This treatment significantly decreased liver weight at the lower doses and had no difference on body weight (Fig. 6A and Supplementary Fig. S5A). Linagliptin treatment also decreased alkaline phosphatase (ALP), a marker of biliary stasis (Fig. 6B), and ALT (Supplementary Fig. S5A). There was no effect on hepatic collagen accumulation as quantified by hepatic Hyp (Supplementary Fig. S5B), while morphometrically determined Sirius red positive collagen showed a modest decrease that failed to reach significance (Fig. 6D). However, treatment with linagliptin significantly decreased hepatic α-SMA, Col1α1, TGFβ1, and MMP-3 transcripts at higher doses, with beneficial trends for profibrogenic TIMP-1 and MMP-3 (Fig. 6E). In parallel, hepatic transcript levels of cytokines, chemokines, and Arg1 indicated a significant shift from M1 to M2 macrophage (myeloid cell) polarization, in line with mice on the MCD diet (Fig. 6C). In summary, Linagliptin modestly attenuated biliary fibrosis in Mdr2−/− mice, compatible with a primary effect on inflammatory (myeloid) cells.
Gliptins attenuate vascular dysfunction and oxidative stress in diet-induced NASH
Steatohepatitis in MCD mice was associated with severe endothelial dysfunction as shown by impaired relaxation of isolated thoracic aortic rings in response to acetylcholine. Therapy with linagliptin or sitagliptin completely normalized endothelial dysfunction (Fig. 7A). In addition, endothelium-independent vascular relaxation in response to glyceryl trinitrate (GTN), which was impaired in MCD mice, was significantly improved by DPP-4 inhibition (Fig. 7B). Likewise, in MCD mice, gliptins significantly reduced the enhanced formation of ROS from cardiac NADPH oxidase activity, mitochondrial ROS, and whole blood oxidative burst (reflecting the activation state of circulating leukocytes, especially of the myelomonocytic lineage) (Fig. 7C).

Vascular ROS formation was visualized in aortic cryosections using oxidative fluorescence microtopography. Red fluorescence indicative of ROS formation was increased in the MCD group and decreased by linagliptin therapy (Fig. 7D, E). Similarly, the highly elevated hepatic ROS formation in MCD diet-fed mice was essentially normalized by both gliptins (Fig. 7D, E).
Gliptins normalize NASH-mediated ROS formation and accumulation of NOX-2 expressing cells in the liver
The high hepatic ROS production was confirmed in liver homogenates and in hepatic mitochondria from MCD mice, which was associated with increased mRNA expression of the phagocytic NADPH oxidase isoform NOX-2 (Fig. 8A), pointing to a relevant contribution of infiltrating phagocytic cells to the overall hepatic ROS signal (Figs. 1 –3). Notably, hepatic ROS production and NOX-2 mRNA transcripts were normalized with gliptin treatment of MCD mice (Fig. 8A).
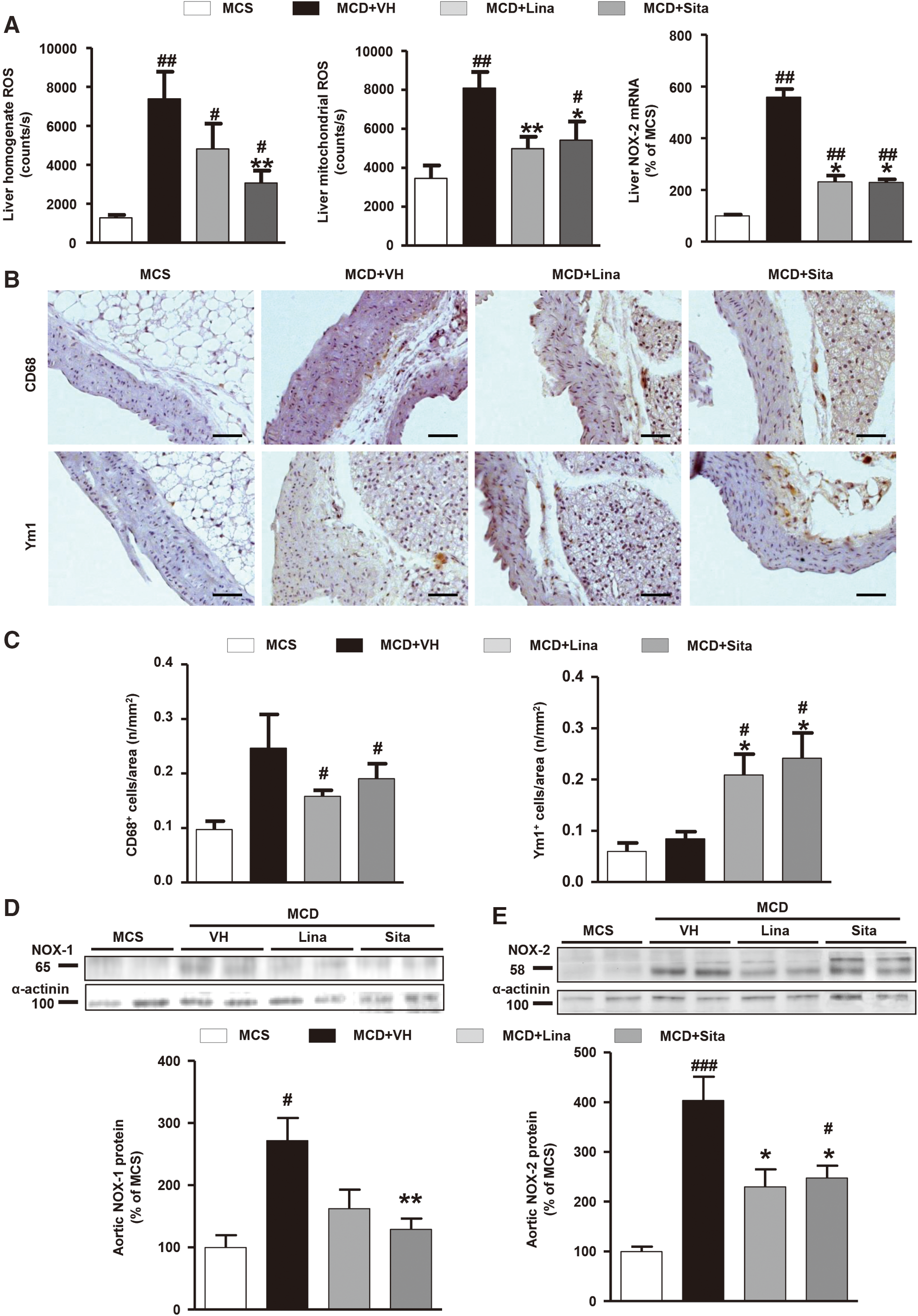
Gliptins reduce NASH-induced vascular inflammation and expression of ROS producing enzymes
Since immune cells in general and infiltrating monocytes/macrophages in particular negatively affect vascular function (30, 87), we assessed the immune cells that infiltrated the aortic smooth muscle and adventitial layer of MCD mice. Aortic CD68+ macrophages were increased compared to the MCS controls. Gliptin treatment reduced these cells significantly (Fig. 8B, C). Notably, Ym1 expressing cells that represent the anti-inflammatory M2-macrophage subset were increased in the vascular wall of gliptin-treated MCD mice (Fig. 8B, C). NOX-1, the central vascular isoform of the NADPH oxidases, was markedly increased at the protein level in vehicle-treated MCD mice and significantly suppressed by both gliptins (Fig. 8D). In accordance with the increased infiltration of immune cells into the vascular wall, the aortic phagocytic NADPH oxidase (subunit NOX-2) was increased at the protein level in MCD mice and suppressed with gliptin treatment (none).
Gliptins display only minor direct antioxidant but potent anti-inflammatory activity in vitro
Linagliptin and sitagliptin showed only marginal direct antioxidant activity, since minor suppression of peroxynitrite-mediated oxidation of phenol or dihydrorhodamine was observed when gliptins were used in excess over peroxynitrite (Supplementary Fig. S6A–D). In contrast, gliptins showed no effect at all on superoxide-dependent oxidation of dihydroethidium or hydrogen peroxide/peroxidase-mediated oxidation of Amplex Red (Supplementary Fig. S6E–G). Regarding the effective drug concentrations of gliptin therapy (plasma concentrations in the nanomolar range), it is rather unlikely that gliptins act via direct antioxidant effects in the body, even when accumulation in the lower micromolar range cannot be excluded at certain tissue sites and in specific cell types.
The most likely mechanism of how gliptins confer antioxidant effects is via their potent anti-inflammatory properties that are not only evident in vivo but also in vitro. Linagliptin and, less pronounced, sitagliptin showed a remarkable suppression of the oxidative burst in isolated human granulocytes [polymorphonuclear leukocytes (neutrophils), PMN] in response to NADPH oxidase activation by lipopolysaccharide (LPS) (Supplementary Fig. S7A, B). With increasing density of PMN, higher concentrations of the gliptins were required to suppress the oxidative burst. For 1 million cells per ml, 200 μM of the gliptins were needed, whereas with 100,000 cells per ml the effective concentrations were decreased to 2 μM for linagliptin and 20 μM for sitagliptin. Further extrapolation of this correlation most probably reaches clinically feasible gliptin concentrations. A direct suppression of the oxidative burst by scavenging of ROS by gliptins can be excluded in light of our chemical assays clearly arguing against a direct antioxidant action of gliptins (Supplementary Fig. S6).
Discussion
Macrophages are a prominent immune cell population in injured livers and fulfill critical functions by perpetuating or attenuating inflammation and fibrosis. Macrophages are highly diverse, and their functional polarization is an important determinant of liver disease progression or reversal (55, 70, 80). Our data show that in the MCD diet model, there is a marked increase in the proinflammatory monocyte and M1 macrophage population. Importantly, therapy with the DPP-4 inhibitors linagliptin or sitagliptin shifted this population toward an anti-inflammatory and antifibrogenic M2 phenotype, with partial or complete resolution of prominent features of NASH, that is, liver fat accumulation, inflammation, and hepatocyte apoptosis. Moreover, by comparing liver inflammation and fibrosis with vascular dysfunction and oxidative stress in this NASH model, we could demonstrate that monocyte/macrophage M1 polarization appears to be a common denominator of both hepatic inflammation and vascular inflammation as well as oxidative stress, and that gliptin treatment favorably addresses both compartments. AMP-activated protein kinase (AMPK) is an important regulator of macrophage polarization in adipose tissue inflammation and NASH (86) and also a suppressor of oxidative stress in the vasculature, more specifically in endothelial cells (24). Activation of AMPK via GLP-1 receptor signaling has been shown to reduce oxidative stress in cardiomyocytes and reduces activation of NADPH oxidase (4, 75).
To date, numerous investigations have focused on the effects of DPP-4 inhibitors on insulin and/or glucagon secretion, while little attention has been paid to their effect on NASH and vascular inflammation. Thus, some studies have reported either an anti-inflammatory or no direct effect of DPP-4 inhibitors on macrophages (45, 78). In this study, we clearly demonstrated a direct anti-inflammatory effect of gliptins on macrophages, as evidenced by reduced (infiltrating) macrophage numbers and a clear polarization toward an anti-inflammatory M2-phenotype, including downregulation of IL-1β, iNOS, TNFα, and MCP-1 and upregulation of Ym1 and Arg1. In the present article, these anti-inflammatory effects of gliptins were also confirmed in vitro in isolated human granulocytes by potent suppression of endotoxin-triggered oxidative burst, despite the lack of potent direct antioxidant (ROS scavenging) properties. The anti-inflammatory effects of gliptins are not restricted to macrophages and granulocytes and were also evident by inhibition of dendritic cell maturation (75), which has been recognized as an important event in adipose tissue and vascular inflammation (95, 96).
Blood-derived monocytes, which can transform into macrophages and dendritic cells, and liver-resident macrophages/Kupffer cells play a pivotal role in NASH by orchestrating local immune responses through the secretion of cytokines and chemokines (22) and via increased formation of hepatic ROS (81). Similar to NASH, these cells are also central to the development of atherosclerosis (31, 50) and arterial hypertension (30, 87), providing a potential link between the pathogenesis of NAFLD/NASH and cardiovascular disease. Importantly, the (macrophage) ROS-induced oxidative stress has a major impact on endothelial function and is a prognostic marker for increased cardiovascular events (32), and NADPH oxidase-derived ROS play a role in liver fibrosis (61). We suggest that the overall beneficial effect of gliptins in the liver as well as the periphery can largely be attributed to their repolarizing activity on hepatic and vascular monocytes/macrophages.
Apart from their immunohistochemical and transcript pattern-based characterization as M1 and M2 cells, we further defined the population of monocyte-derived macrophages by their differential expression of the myeloid activation marker Ly6C. Ly6Chi cells represent classically activated monocytes that are rapidly recruited to sites of inflammation and display an M1-like phenotype (44).These cells are distinguished by secretion of proinflammatory cytokines such as TNFα and IL-6, contribute to tissue degradation and Th1 T cell activation (26, 44), and their recruitment largely depends on CCR2/MCP-1 signaling (56). In contrast, Ly6Clo cells may serve as precursors for alternatively activated macrophages, possibly fulfilling functions in tissue repair and resident macrophage/dendritic cell turn over (26). The macrophages found in livers of gliptin treatment were predominantly polarized toward M2, as evidenced by an upregulation of the M2 markers, Arg1 and IL-10, promote tissue repair, and attenuate fatty liver inflammation (53). In this study, gliptin therapy significantly decreased hepatic CD11b+Ly6Chi cells and MCP-1 expression in MCD diet-induced steatotic livers. We also detected a significant reduction in the percentage of CD11c+F4/80+ cells (M1 macrophages) (10) and also reduced CD3e+ T cell numbers in the liver sections. These results suggest that DPP-4 inhibitors exert their major anti-inflammatory effect on myeloid cells, specifically inflammatory macrophages and M1 macrophages. Although other immune cells may also play a role in NASH pathogenesis, such as T cells and NKT cells, which are abundant in the liver (but not in the vascular system), we focused on myeloid cells that are common to both compartments and the most likely immune cells influenced by GLP-1 signaling (35). Our unpublished preliminary data also show that it is indeed the monocytes/macrophages and not these other immune cells that are prominently affected by gliptins and by GLP-1 directly (not shown).
Steatosis, hepatocyte ballooning (reflecting lipoapoptosis), and lobular inflammation are histopathological hallmarks of human NASH, as quantified by the NAS score (46). Both linagliptin and sitagliptin significantly decreased all components of the NAS score in MCD mice. This was accompanied by suppressed hepatic transcript levels of ACC and FAS, the rate-limiting enzymes of FAS in the liver, and of SREBP-1c, which controls the expression of these enzymes (64). Furthermore, the gliptins lowered hepatic LPL expression, which is implicated in hepatocyte damage due to free fatty acid release. These findings indicate that the gliptins also exert direct protective effects on hepatocytes via the reduction of fat accumulation, decreased stimulation of FAS genes, and protection from lipoapoptotic damage. However, the hepatocyte protection could also be mainly indirect, secondary to the beneficial effects on macrophages.
DPP-4 is ubiquitously expressed on cell surfaces and plays a key role in glucose metabolism and the control of glycemic state, since it degrades (among others) the hormone GLP-1 (29, 78). GLP-1 is secreted by pancreatic α- and intestinal L-cells in response to nutrient ingestion to stimulate expression of downstream hormonal cascades (including insulin signaling) via the GLP-1 receptor (3, 29). Therefore, in analogy to type 2 diabetes, inhibition of DPP-4 with a subsequent rise in GLP-1 levels and actions has been suggested but little explored as an attractive pharmacological therapy for NAFLD/NASH (19, 29, 42, 43). The beneficial effect of gliptin therapy may extend toward inflammatory and fibrotic liver diseases without features of NASH, as exemplified in our study of biliary fibrotic mice that experienced a modest reduction of fibrogenic activity. How far gliptins may also protect from the development of hepatocellular carcinoma, a complication of NASH that is associated with advanced fibrosis and upregulation of proinflammatory pathways, including IL-6, needs to be studied in long-term feeding experiments (11).
Moreover, we and others demonstrated that DPP-4 inhibition by gliptins, or genetic deletion of DPP-4, improved vascular dysfunction, oxidative stress, and survival of endotoxemic rats, which was paralleled by a reduction of CD11b-positive cells in vessel walls and reduced adhesion of human monocytes and granulocytes to cultured endothelial cells (47, 49, 75). In this study, in parallel to improvement of NASH therapy, both linagliptin or sitagliptin substantially normalized endothelial dysfunction, cardiac NADPH oxidase activity (also supported by increased aortic NOX-1 and NOX-2 protein levels), mitochondrial ROS formation, and whole blood oxidative burst (reflecting the activation state of circulating leukocytes, especially of the myelomonocytic lineage). Notably, hepatic ROS production and NOX-2 mRNA expression were also normalized in MCD mice by gliptin therapy. This can be explained by prior findings that a major (indirect) antioxidant activity of GLP-1 analogues is due to suppression of protein kinase C (PKC)/NFκB-dependent NOX activation/upregulation (6, 73), which might act in concert with the above-described AMPK activation that controls macrophage polarization and antioxidant defense (75, 86). The (indirect) antioxidant effects of GLP-1 and DPP-4 inhibitor therapy are also supported by several reports of reduced oxidative stress markers in various disease settings such as diabetes (2, 6, 59, 73), atherosclerosis (54, 72), sepsis (47, 75), cardiac ischemia/reperfusion-injury (12), chronic myocardial infarction (38), abdominal aortic aneurysm (5) and also neurological diseases (1, 67).
Since infiltrating monocytes negatively affect vascular function (30, 87), we also studied immune cells that infiltrated the aortic smooth muscle and adventitial layer of MCD mice. As observed in the liver, gliptin treatment reduced aortic CD68+ macrophage numbers and attenuated their activation, as reflected by phagocytic NOX-2 isoform reduction and an increase in the anti-inflammatory Ym1 M2-macrophage population, confirming high efficiency of DPP-4 inhibition in the prevention of oxidative stress, inflammation, and cardiovascular complications associated with this NASH model. A role of NADPH oxidase-derived ROS in liver fibrosis was previously reviewed in detail (61).
The current view on the mechanisms underlying progression of NASH favors a model in which steatosis and then steatohepatitis are induced as a result of free fatty acid overload, hepatocyte lipoapoptosis, immune infiltration with activation of (mainly myeloid) inflammatory cells, all linked to insulin resistance, and subsequent fibrogenic activation of HSC and myofibroblasts (55, 69 –71). Gliptin therapy obviously interferes with several of these underlying and associated pathologies, but prominently shifts inflammatory monocyte/macrophage infiltration toward a myeloid cell population that was previously defined as anti-inflammatory and restorative monocytes/macrophages (65).
Importantly, since the MCD model lacks the feature of insulin resistance, we may conclude that the steatotic liver inflammation itself can drive or at least favor vascular dysfunction and associated cardiovascular morbidity, a link that had been suspected but not clearly established before (21, 51). In this study, NASH models based on high-fat diet and metabolic syndrome will not permit the study of glucose-independent effects of gliptin therapy. This finding strongly supports the generally beneficial effect of gliptins on all components of NASH and the metabolic syndrome.
Limitations of the study
We selected the MCD model for NASH by purpose. This model reflects one extreme of the spectrum of NASH, that is, the typical liver damage. It is accepted in the community and frequently used to test for drugs that inhibit hepatocyte lipoapoptosis, liver inflammation, and liver fibrosis. We consider it as an advantage not to have a model that is characterized primarily by obesity and insulin resistance, such as a high-fat/high-fructose diet model, which does not lead to significant liver inflammation and fibrosis. Any intervention with drugs such as gliptins would show the expected effect of alleviating diabetes and the mild inflammation associated with it, but would not give us a clue as to its direct effect on the component of severe liver inflammation and fibrosis, which are considered the hard endpoints in drug development for NASH.
We consider our second model of secondary biliary liver fibrosis (Mdr2−/− KO), with little relation to NASH pathogenesis, a strength rather than a weakness, since it allows to separate the effects of the gliptins on the immune cells, macrophages, and fibrosis in the absence of significant oxidative stress, which is a major feature of the MCD diet model of NASH.
Sitagliptin therapy required a 10-fold higher dose, underlining that pharmacokinetics, tissue distribution, DPP-4 binding affinities, and binding regions [which may vary significantly among different gliptins (57)] might play an important role for the observed anti-inflammatory and macrophage polarizing effects. Similar drug doses of linagliptin and sitagliptin were previously used in other rodent studies (47, 75, 76).
Conclusions
In summary, we provide a direct link between severe steatohepatitis and vascular dysfunction, sharing common inflammatory (monocyte/macrophage) and oxidative stress-mediated mechanisms (see scheme in Fig. 9). In this study, the use of the MCD model, which lacks insulin resistance, an important comorbidity in human NASH, has proven advantageous in our study, since it rules out insulin resistance/type 2 diabetes as an indirect contributor to vascular inflammation and endothelial dysfunction (51), which is relevant in view of the current discussion if and how far the liver, the prediabetic/diabetic state, or both need to be addressed to treat the disease (21). Our studies provide clear evidence for a beneficial role of DPP-4 inhibitors in the treatment of key aspects of NASH (monocyte/macrophage infiltration, hepatocyte lipoapoptosis) and resultant liver fibrosis. This therapy ameliorated the associated vascular disease and exerted a modest direct antifibrotic effect, as demonstrated in the Mdr2−/− model of biliary fibrosis. We also show that the beneficial actions of gliptins are a class effect, since linagliptin and sitagliptin demonstrated similar efficacy across most parameters measured. These and related drugs that do not incur untoward side effects such as weight gain or edema and are already widely used in patients with type 2 diabetes therefore qualify as promising agents to treat NASH, the associated cardiovascular disease, and insulin resistance.

Materials and Methods
Maintenance and treatment of mice
Animal experiments were approved by the State of Rhineland-Palatinate and performed in accordance with institutional and German legal guidelines for animal protection. The MCD and the MCS (normal control) diets were purchased from Ssniff Special Diets (Germany). Eight-week-old male C57BL/6 mice were kept on the MCD or control MCS diet for 4 weeks, followed by treatment with vehicle (0.5% Natrosol 250 HX hydroxyethylcellulose), linagliptin (Lina, 5 mg/kg/d), or sitagliptin (Sita, 50 mg/kg/d) for another 4–6 weeks via daily gavage (group size: n = 10).
Mdr2 (abcb4) −/− mice that spontaneously develop progressive biliary fibrosis between 4 and 12 weeks of age were bred as described (63). They (n = 10/group) were treated with linagliptin (0.5, 5, and 10 mg/kg) from week 9 to 12. For sacrifice, mice were euthanized with vaporized isoflurane and exsanguinated via cardiac puncture. Serum was collected and frozen, dissected tissues were weighed, and aliquots flash frozen in liquid nitrogen. Isolated aortic rings, hearts, and whole blood were freshly used for in vitro determination of vascular function and ROS formation.
Serum analyses
Serum activities of ALT, AST, ALP, LDH, and serum triglycerides were determined by the Central Laboratory of the University Medical Center Mainz according to standardized and regularly validated criteria. Determination of active GLP-1 was performed with a commercial kit (K150JWC-1; Mesoscale Gaithersburg) according to the user manual. Serum DPP-4 activity was detected as recently published using a specific peptide substrate with a terminal coumarin derivative (H-Ala-Pro-7-amido-4-trifluoromethylcoumarin; Bachem, Bubendorf, Switzerland; final concentration in the assay, 100 μM) allowing quantification in a fluorescence microplate reader on cleavage by DPP-4 (83). A commercially available enzyme-linked immunosorbent assay kit was used to measure mouse GLP-1-(7 –36) amide serum levels (Linco Research, Inc., St. Charles, MO).
Dot blot analysis
For dot blot analysis, 50 μg of serum protein was spotted on PVDF membranes (Bio-Rad) blocked with TBS-T (Tris-buffered saline, 0.05% Tween 20) containing 5% nonfat dry milk for 1 h and incubated with the IL-6 antibody (1:500 in TBS-T plus 5% milk; Abcam, Cambridge, United Kingdom, Clone ab6672) overnight at 4°C, followed by horseradish peroxidase-linked anti-rabbit IgG antibody (1:10,000; Vector labs, Clone PI-1000) for 45 min at room temperature and washing in TBS-T. Signals were revealed using the ECL chemiluminescence kit (Thermo scientific) and documented on a ChemiLux ECL Imager (Intas). Dot intensities were determined using the GelPro analyzer software version 6.0 (Media Cybernetics).
Hepatic collagen content determination
Hydroxyproline (Hyp, an amino acid that is essentially unique to collagens and represents roughly 10% of most collagens) content was quantified in preweighed 200–300 mg samples from two different liver lobes per animal after hydrolysis in 6 N HCl for 16 h at 110°C as described (63) yielding the relative Hyp content. Total Hyp (mg/whole liver) was calculated based on individual liver weights and the corresponding relative Hyp content (62).
Histochemistry and immunohistochemical analyses
Formalin-fixed liver samples were deparaffinized and stained with hematoxylin and eosin for determination of the NAS (46). Collagen was stained using Sirius red (Sigma Aldrich, Steinheim, Germany). The tissues were deparaffinized and rinsed twice for 5 min in distilled water. Excess moisture was removed from the slides and these were then incubated in 0.1% Sirius red in saturated picric acid solution for 30 min. Excess dye was removed by rinsing twice in 0.05% acetic acid for 5 min. The slides were rinsed in distilled water and dehydrated by dipping several times in 70% isopropylacohol and twice in 95% and 100% isopropylacohol followed by two 5-min incubation steps for immersion in xylene. The slides were mounted with resinous medium and visualized in a Zeiss Axio Imager AX10 microscope with the appropriate filters.
Hepatic lipid was stained with Sudan III or Oil red O (Sigma Aldrich, Steinheim, Germany) on 7 μm cryosections. Tissues were dried at room temperature for 10 min, then stained in 0.03% Sudan III solution for 30 min, rinsed with deionized water, counterstained for 15 s with Mayer's hematoxylin, put under running tap water for 10 min, rinsed with deionized water again, and the slides were mounted with glycerin.
Tissues were air-dried for 5 min, dehydrated in 60% isopropylacohol by dipping them 10 times, and then stained in 0.3% Oil red O solution for 30 min. Excess dye was washed off in 60% isopropylacohol for several seconds. Next, the slides were rinsed in deionized water, counterstained for 30 s with Mayer's hematoxylin, put under running tap water for 5 min, rinsed with deionized water again, and the slides were mounted with glycerin.
Immunohistochemistry was performed on formalin-fixed sections. After rehydration of deparaffinized sections, antigens were retrieved at 95°C using citrate buffer (pH 6.0) for 30 min, followed by incubation overnight at 4°C using the following primary antibodies: CD68 (BIOZOL, Eching, Germany), F4/80 (BioLegend, Fell, Germany),Ym1 (STEMCELL, Cologne, Germany), cleaved Caspase-3 (Cell Signaling, Cambridge, United Kingdom), α-SMA (Abcam), and CD3e (BD Bioscience, Heidelberg, Germany). Biotinylated secondary antibodies were from Vector Labs (Burlingame) and incubated for 30 min at room temperature, followed by the VECTASTAIN ABC detection kit (Vector Labs) and counterstaining with hematoxylin (Merck, Darmstadt, Germany). Tissues were visualized in a Zeiss Axio Imager AX10 microscope with the appropriate filters. Representative images were taken with an AxioCamMRc5 camera. A minimum of 10 random high-power fields was quantitated for each liver. Quantitative image analysis of staining was performed using ImageJ software (National Institute of Health, Bethesda, MD).
Immunofluorescence
For immunofluorescence, frozen sections were embedded in OCT (Sakura, Japan) and fixed in 4% formaldehyde. Rabbit anti-mouse procollagen III antibodies (25) were diluted 1:800 in 1 × PBS plus 1% BSA/0.3% Triton X-100 buffer and incubated with sections overnight at 4°C. After washing three times in PBS, the sections were incubated with an Alexa Fluor 488-conjugated anti-rabbit IgG secondary antibody (1:500 diluted in PBS; Life technologies, A-11008) for 2 h at room temperature and counterstained with DAPI. In each case a minimum of 10 randomly selected fields were quantitated and quantitative morphometric analysis was performed using the ImageJ software (National Institute of Health, Bethesda, MD).
Quantitative analysis of gene expression for liver tissue
Total RNA was extracted from liver tissues with Ribozol (Amresco, Solon), and complementary DNA was synthesized from 0.5 mg total RNA by reverse transcription using a cDNA SuperMix reverse transcription kit (Quanta, Gaithersburg) with an optimized mixture of random and oligo (dT) primers. Quantitative real-time PCR (qPCR) was conducted in a StepOnePlus Real-Time PCR System using commercially available TaqMan or SYBR Green qPCR systems (Life technologies, Darmstadt, Germany) with specific primers (and probes) for hepatic expression as described in Supplementary Table S1. Expression of each target gene was quantified by transformation against a standard curve and normalized to glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) expression.
Quantitative analysis of gene expression for aortic tissue
Total RNA was isolated from aortic tissue with the RNeasy Fibrous Tissue Kit (Qiagen, Hilden, Germany) and qPCR was performed in a One Step PCR TaqMan® Assay system with specific primer/probe mixtures (Supplementary Table S1). TaqMan primers and probes for TATA box binding protein (TBP) as a housekeeping gene, CD11b (Mm00434471_g1), and NOX-2 (Mm00432775_m1) were purchased from Applied Biosystems, Foster City, CA). The expression of each target gene was quantified by transformation against a standard curve and normalized to the expression of GAPDH or TATA-box binding protein (TBP).
Immune cell isolation from livers
Mice were sacrificed, the liver removed and perfused with PBS to eliminate circulating leukocytes, then minced into small pieces and digested in a buffer containing collagenase IV (500 CDU/mL; Sigma, Steinheim, Germany) and DNaseI (150 U/mL; AppliChem, Darmstadt, Germany) for 30 min at 37°C. The liver suspension was filtered through a 100-μm cell strainer using wash buffer (PBS, 0.5% BSA, 2 mM EDTA) and the remaining suspension was collected. Hepatocytes were removed by centrifugation at 21 g for 4 min. The supernatant was collected and centrifuged at 300 g, 10 min, and red blood cells were lysed. Next, the remaining cells were counted and 106 cells were used for each staining. After using Fc block (BD Biosciences, Heidelberg, Germany) for 15 min, cells were washed and stained with antibodies CD45, CD11b, CD11c, Ly6C, or F4/80 (BioLegend, Breda, the Netherlands). After incubation for 20 min at 4°C, cells were washed and fixed with 1% PFA overnight at 4°C. After washing, samples were subjected to analysis on an FACS Canto II (BD Bioscience, Heidelberg, Germany) and analyzed using FlowJo 7.6 software (TreeStar, Ashland, OR).
Isometric tension studies
Vasodilator responses to acetylcholine (ACh) and GTN were assessed with endothelium-intact isolated mouse aortic rings (thoracic aorta, 3 mm in length) mounted for isometric tension recordings in organ chambers, as described by us (47, 75).
Detection of oxidative stress in whole blood, cardiac membrane fractions, mitochondria, aorta, and liver homogenates
Hepatic and cardiac mitochondrial ROS formation with L-012 (100 μM; Wako Pure Chemicals Industries, Osaka, Japan)-enhanced chemiluminescence (ECL) and cardiac NADPH oxidase activity by lucigenin (5 μM, Sigma-Aldrich, Steinheim, Germany)-enhanced ECL were measured as described by us in detail (47, 75). To determine whole blood leukocyte-dependent ROS formation, fresh blood (in citrate tubes) was stimulated with phorbol ester dibutyrate (PDBu, 10 μM; Sigma-Aldrich, Steinheim, Germany) and assessed in PBS containing 1 mM Ca2+/Mg2+ with L-012-enhanced ECL (47, 75). Vascular and hepatic ROS formation was determined using dihydroethidine (DHE, 1 μM)-dependent fluorescence microtopography in aortic cryosections as described (47, 75).
Western blotting
Proteins were extracted from livers in the presence of protease inhibitor cocktail (Complete EDTA-Free; Roche Applied Science, Mannheim, Germany), resolved by SDS-PAGE, and then transferred to PVDF membranes. After blocking with Tris-buffered saline containing Tween 20 (0.1%) and 3% BSA, membranes were probed with primary antibodies against NOX-1 (Abcam) or NOX-2 (BD Biosciences), TGFβ1 (Abcam), MMP-12 (Santa Cruz, Heidelberg, Germany) followed by incubation with corresponding horseradish peroxidase-conjugated secondary antibodies. For detection and quantification, an ECL imager (INTAS, Göttingen, Germany) and GelPro 3.0 analyzer software (Media Cybernetics, Rockville) were used.
Assessment of direct antioxidant properties of gliptins
Direct antioxidant effects of gliptins (100 μM) were assessed by interfering with superoxide formation from xanthine oxidase (XO, 10 and 50 mU/ml, incubated for 40 min), hypoxanthine (HX, 1 mM), peroxynitrite (authentic, synthesized by quenched-flow method (14), 10, 33, and 100 μM, incubated for 5 min) or hydrogen peroxide (100 μM, incubated for 40 min)/horseradish peroxidase (0.1 μM)-mediated 1-electron-oxidation. The oxidation reactions were detected by fluorescence using Amplex red (Ex. 530 nm/Em. 590 nm), dihydrorhodamine 123 (Ex. 488 nm/Em. 530 nm), and dihydroethidium (Ex. 480 nm/Em. 580 nm), as fluorescence probes for H2O2/peroxidase-, peroxynitrite-, or superoxide-mediated oxidations at a concentration of 50 μM on a Twinkle plate reader (Berthold Technology, Bad Wildbad, Germany) (13, 41, 90). Effects of gliptins on superoxide formation by XO/HX were also quantified by the formation of dihydroethidium oxidation products by HPLC-based quantification of 2-hydroxyethidium with PEG-SOD (100 U/ml)-dependent inhibition of the signal as a validation of the specificity of the assay (48, 92). Nitration of phenol (5 mM) by authentic peroxynitrite was quantified by HPLC-based detection of 2- and 4-nitrophenol (14, 15). All reactions were carried out at 37°C in 0.1 M potassium phosphate buffer, pH 7.4.
Measurement of anti-inflammatory properties of gliptins in isolated human granulocytes
Anti-inflammatory and indirect antioxidant effects of gliptins were measured in isolated human leukocytes in PBS (1 mM Ca2+/Mg2+) by interfering with the oxidative burst (NADPH oxidase activation) induced by the endotoxin LPS (50 μg/ml). PMN were isolated by sedimentation of red blood cells with dextran and centrifugation on Ficoll as described previously (13). Total blood cell count and the purity of the fractions were evaluated using an automated approach using a hematology analyzer KX-21 N (Sysmex Europe GmbH, Norderstedt, Germany). The typical constitution of the blood cell fractions obtained by this method was recently published (91). Activation of PMN was quantified by ROS formation during oxidative burst in response to LPS using L-012 (100 μM, 60 min)- or luminol (100 μM, 30 min)/horseradish peroxide (0.1 μM)-enhanced ECL on a Centro plate reader (Berthold Technology, Bad Wildbad, Germany). Gliptins were incubated at concentrations of 2, 20, and 200 μM. All reactions were carried out at 37°C in PBS (1 mM Ca2+/Mg2+).
Statistical analyses
Data were analyzed using GraphPad Prism 5.0 (GraphPad software, La Jolla). Statistical analyses used one-way or two-way ANOVA with Bonferroni's correction. Data are expressed as mean and standard error mean.
Footnotes
Acknowledgments
The technical assistance of Jessica Rudolph, Jörg Schreiner, Angelica Karpi, and Michael Mader is gratefully acknowledged. The study was supported by an ERC Advanced Grant and an unrestricted grant from Boehringer-Ingelheim Pharma to DS, and by an internal research grant of the University Medical Center Mainz to AD. SS holds a Virchow-Fellowship from the Center of Thrombosis and Hemostasis (Mainz, Germany) funded by the Federal Ministry of Education and Research (BMBF 01EO1003). We also thank M. Neuser for graphic arts.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.