
Abstract
Aims:
Living cells employ thioredoxin and glutaredoxin disulfide oxido-reductases to protect thiol groups in intracellular proteins. FrnE protein of Deinococcus radiodurans (drFrnE) is a disulfide oxido-reductase that is induced in response to Cd2+ exposure and is involved in cadmium and radiation tolerance. The aim of this study is to probe structure, function, and cellular localization of FrnE class of proteins.
Results:
Here, we show drFrnE as a novel cytoplasmic oxido-reductase that could be functional in eubacteria under conditions where thioredoxin/glutaredoxin systems are inhibited or absent. Crystal structure analysis of drFrnE reveals thioredoxin fold with an alpha helical insertion domain and a unique, flexible, and functionally important C-terminal tail. The C-tail harbors a novel 239-CX4C-244 motif that interacts with the active site 22-CXXC-25 motif. Crystal structures with different active site redox states, including mixed disulfide (Cys22-Cys244), are reported here. The biochemical data show that 239-CX4C-244 motif channels electrons to the active site cysteines. drFrnE is more stable in the oxidized form, compared with the reduced form, supporting its role as a disulfide reductase. Using bioinformatics analysis and fluorescence microscopy, we show cytoplasmic localization of drFrnE. We have found “true” orthologs of drFrnE in several eubacterial phyla and, interestingly, all these groups apparently lack a functional glutaredoxin system.
Innovation and Conclusion:
We show that drFrnE represents a new class of hitherto unknown intracellular oxido-reductases that are abundantly present in eubacteria. Unlike other well-known oxido-reductases, FrnE harbors an additional dithiol motif that acts as a conduit to channel electrons to the active site during catalytic turnover. Antioxid. Redox Signal. 28, 296–310.
drFrnE is a disulfide oxido-reductase that is over-expressed under Cd2+ stress, and its null mutant is sensitive to Cd2+ and γ-radiation. This study establishes FrnE as a new class of eubacterial intracellular oxido-reductases that could function under conditions when the other well-known intracellular disulfide oxido-reductase systems are absent or inhibited.
Introduction
D
The Trx superfamily is characterized by the presence of the Trx-fold and active site CxxC motif. The basic catalytic mechanism for Trx can be described as a bimolecular nucleophilic substitution reaction (5). The N-terminal nucleophilic cysteine of the CxxC motif forms a mixed disulfide bond between Trx and substrate protein. In the next step, C-terminal cysteine of the CxxC motif attacks the mixed disulfide, releasing a reduced substrate protein and oxidized Trx. The reduced state of the Trx active site is then regenerated by TrxR using electrons from NADPH. In case of Grx, GSH provides the necessary reducing equivalents.
Deinococcus radiodurans is a highly radiation-resistant bacterium that encodes both enzymatic and nonenzymatic components, contributing to its oxidative stress tolerance (28, 41, 42, 51). Recently, the role of the well-protected functional proteome in the recovery from radiation damage has been highlighted (20). The FrnE protein of D. radiodurans (drFrnE; NCBI Ref: WP_010887304) is induced on exposure to a sub-lethal concentration of Cd2+, and drfrnE::nptII null mutant showed reduced tolerance to cadmium and diamide (19, 32). These mutants were nearly six times less resistant to γ-radiation and displayed about twofold higher sensitivity to hydrogen peroxide (19). The molecular basis of drFrnE protecting D. radiodurans from Cd2+ metal toxicity is not yet clear. Generally, the key molecular events during oxidative stress induced by Cd2+ and Hg2+ exposure are expected to be protein S-glutathionylation, alteration in the ratio of reduced and oxidized LMW thiols, and reduced cellular accumulation of manganese and zinc ions (3, 14). For instance, bacillithiol (BSH) functions as a zinc buffer and BSH null mutants of Bacillus subtilis are found to be more sensitive to the toxic effects of Cu2+ and Cd2+ (8, 27, 40).
Here, we report the structural, biochemical, biophysical, and bioinformatics characterization of drFrnE. Structural and mutational analyses show a unique C-terminal tail harboring a novel 239-CX4C-244 motif that can channel electrons to the active site 22-CXXC-25 motif. The structural studies also provide snapshots of the reaction pathway. drFrnE is more stable in oxidized form, is located in the cytoplasm, and is functional in the presence of Cd2+. drFrnE orthologs are abundant in several eubacterial phyla and interestingly, these FrnE-positive species lack a functional glutaredoxin system. Our results suggest that FrnE is a new class of hitherto unknown cytoplasmic oxido-reductase present in many eubacteria.
Results
Crystal structures suggest involvement of C-terminus in drFrnE function
The X-ray crystal structure of drFrnE shows that it belongs to the Trx superfamily, with the core domain (residues 12–52 and 179–222) consisting of a four-stranded β-sheet surrounded by three α-helices (Fig. 1a–d). drFrnE also harbors a conserved helical insertion domain (residues 53–178) and a unique C-terminal extension (223–252) with a second 239-CX4C-244 motif. The characteristic active site 22-CPWC-25 motif in the canonical Trx fold is located on the loop connecting β-sheet 1 and α-helix 1, and it is shielded by the conserved cisPro loop (Val191-Pro192). The active site cysteines, in the crystal structures of drFrnE in the absence of reducing agent and in the presence of β-mercaptoethanol (βME), are in the oxidized form, with Cys22 forming a disulfide bond with Cys25 (Fig. 1e). In agreement with the crystal structure, drFrnE shows negligible absorbance at 412 nm in DTNB assay, suggesting that both the cysteine motifs are in oxidized form in the absence of reducing agent. The active site cysteines are in the reduced form in the crystal structure determined in the presence of nonthiol-reducing agent, tris(2-carboxyethyl)phosphine (TCEP) (Fig. 1f). Interestingly, in the crystal structure determined in the presence of 1,4-dithiothreitol (DTT), Cys22 forms a mixed intermolecular disulfide bond with Cys244 that is contributed by another drFrnE molecule (Fig. 1g).
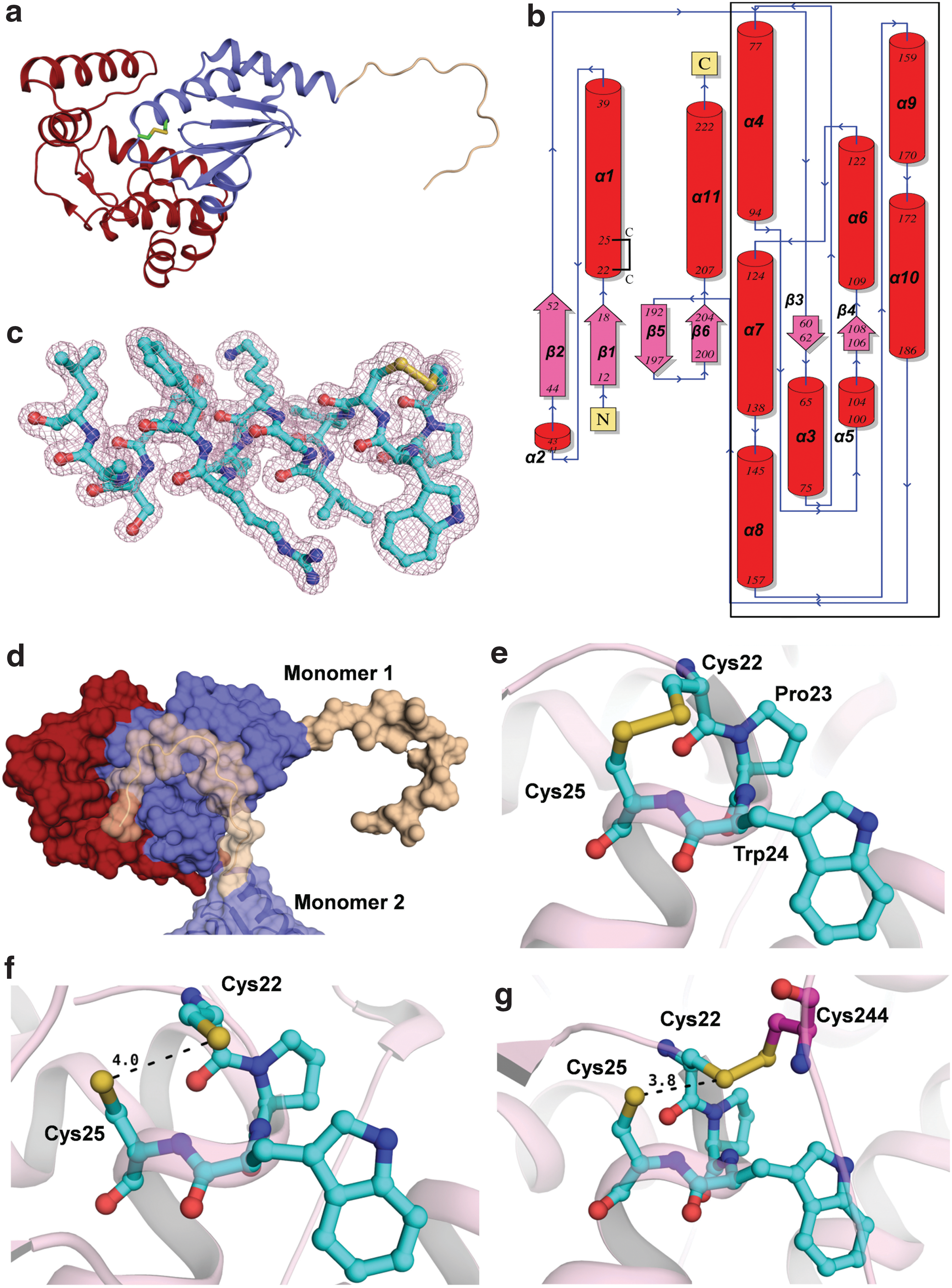
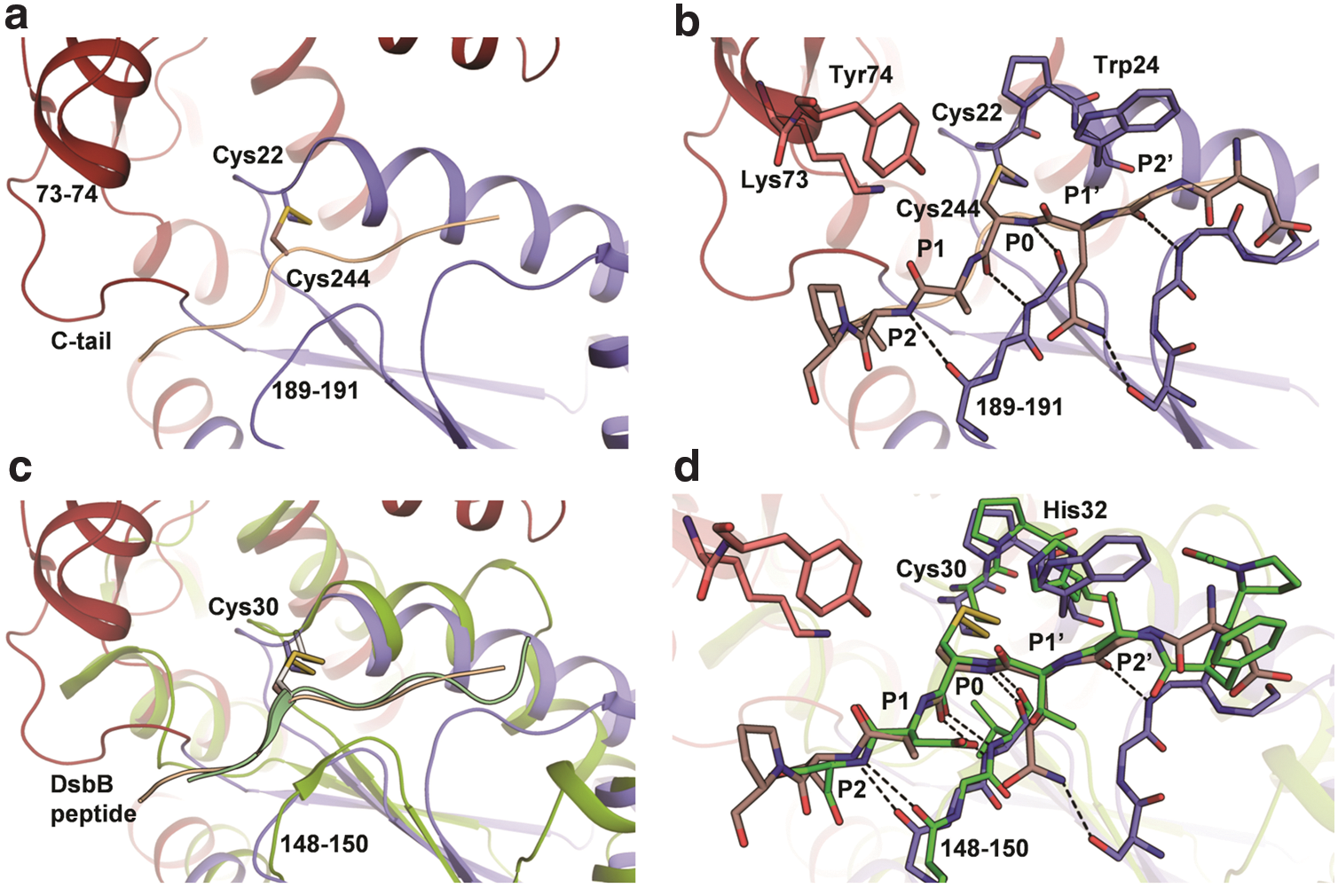
Notably, in all the structures, the C-terminal tail forms a hook-like shape and anchors itself into the active site of another drFrnE molecule with the C-terminal Cys motif located close to the active site cysteines (Fig. 1d), thus forming an extended network in the crystal lattice. The C-terminal residues displayed high flexibility (RMSF ∼1 nm) in 2 ns molecular dynamics (MD) simulations. In the crystal structures, C-tail binds to the active site in two different modes. In one of the binding poses, Glu-237 is present in the active site and in the second pose, a mixed disulfide of Cys244 with Cys22 is formed. The corresponding C-tail peptides presented in the active site are 235-PAEGC-239 and 242-GQCAV-246 (these sites can be considered as P2′-P1′-P0-P1-P2 for further discussion). These peptides bind in the cleft created by residues Lys73 and Tyr74 (from insertion domain) on one wall and residues 189–191 (from Trx domain) as the other wall of the cleft (Fig. 2). It appears that binding specificity is provided by main-chain hydrogen bonds between residues 189–191 and β-strand conformation at P0, P1, and P2 sites (Fig. 2a, b). The P2′ binding site is also characterized by the presence of the indole ring of active site Trp24. The two peptides do not share sequence identity and C-tail displays highly variable sequences in the drFrnE orthologs, except for strictly conserved C-terminal cysteines (Supplementary Fig. S1; Supplementary Data are available online at
The structures also reveal interaction of a conserved Ser18 with the resolving Cys25 (Fig. 3a), where the Oγ atom of Ser18 is H-bond donor to Cys25. The Oγ atom of Ser18 also accepts H-bond from ɛ-NH2 of Lys129, which should constrain hydrogen of Ser18 toward Cys25 (angle NH2-Oγ-Sγ ∼134°; Fig. 3a). In the reduced state, as observed in drFrnE-DTT and drFrnE-TCEP structures, Cys25 shifts toward Ser18 to form a stronger hydrogen bond that could stabilize the thiolate form of Cys25 (Fig. 3b, c). Residue Asp19 is also strictly conserved in drFrnE orthologs. Asp19 belongs to the Trx domain and forms strong H-bonds with Leu54, His113, and Thr109 residues of the insertion domain (Fig. 3d). Interestingly, these residues are also conserved in drFrnE orthologs (Supplementary Fig. S1). These interactions might be important for the structural stability of drFrnE active site and relative arrangement of Trx and insertion domain.

drFrnE exists as a dimer
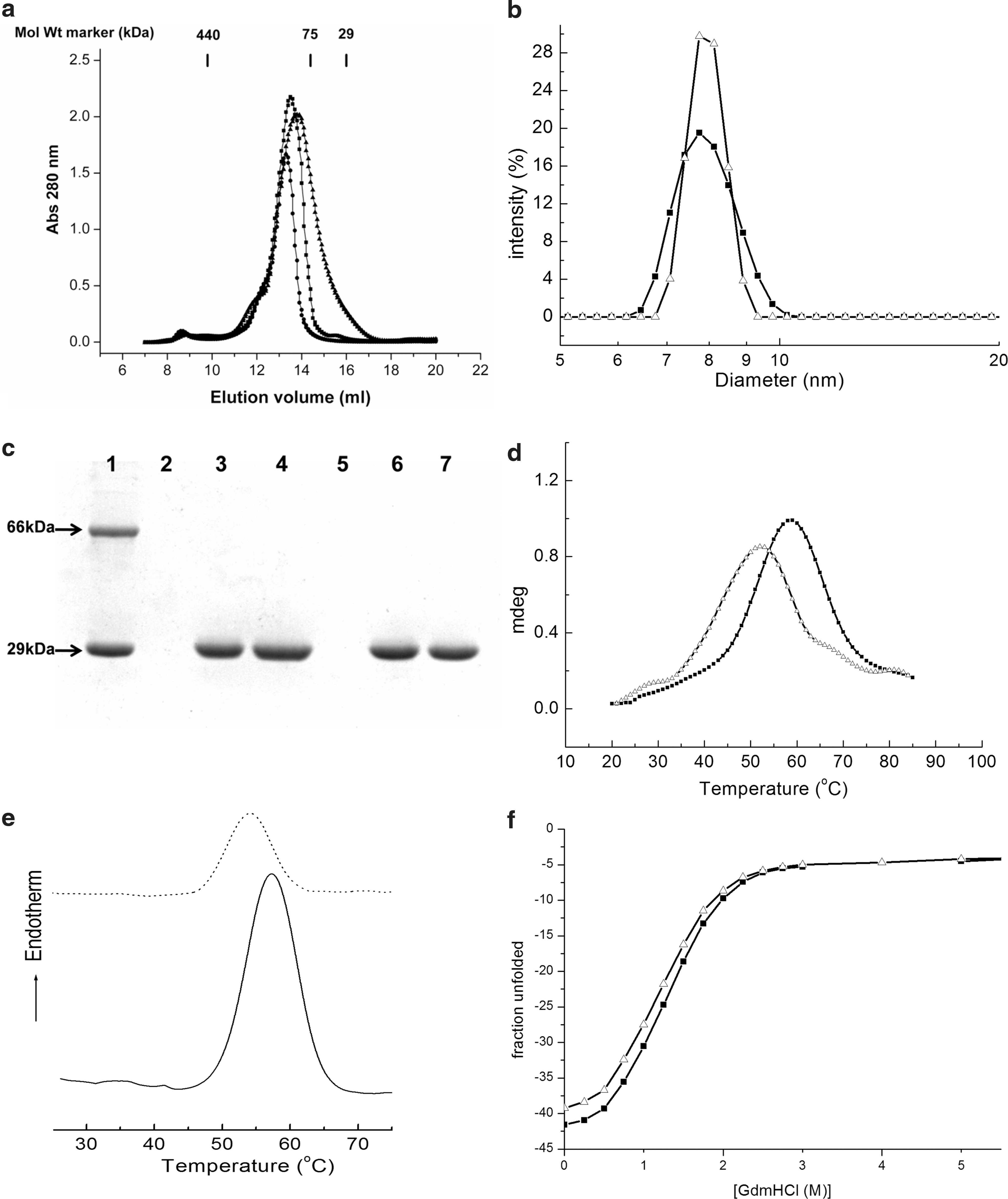
The size exclusion chromatography (SEC) analysis suggests that drFrnE exists as an oligomer in solution with an estimated Mr of ∼100 kDa (Fig. 4a). Glutaraldehyde cross-linking experiments show an additional band of ∼60 kDa corresponding to the dimer of drFrnE (Supplementary Fig. S2a). Molecular weight of drFrnE in solution estimated using average static light scattering data is ∼73 kDa (Supplementary Fig. S2b). These observations cumulatively suggest that drFrnE has a dimeric quaternary structure in solution. The Native-PAGE shows drFrnE band between bovine serum albumin (66 kDa) and chicken egg albumin (45 kDa) (Supplementary Fig. S2c). For C239S/C244S and ΔC222 truncation (residues 223–252 deleted) mutants, SEC analyses indicate similar quaternary structures (Fig. 4a). The size of drFrnE particle (7.8 nm; Fig. 4b) was not affected by the presence of 10 mM DTT, suggesting that the oligomeric status is not due to intermolecular disulfide bond, as also observed from SDS-PAGE of reduced and nonreduced drFrnE (Fig. 4c). The dimer is stable in the presence of 500 mM salt or 0.2% triton, as evident from DLS profiles (data not shown). However, the presence of 0.6% CHAPS (zwitter-ionic detergent) or 0.6% sarcosyl (anionic detergent) breaks this dimeric assembly, suggesting that both hydrophobic and ionic interactions may be involved in the dimer formation (Supplementary Fig. S3).
drFrnE is stable in oxidized form
The drFrnE protein does not show significant changes in the secondary structure on exposure to reducing agent (Supplementary Fig. S4a). However, the oxidized drFrnE (in the absence of DTT) shows a typical transition profile from native to unfolded state with T M of ∼59°C, whereas it shifts to a lower value of 52°C in the presence of DTT (Fig. 4d and Supplementary Fig. S4b). Differential scanning calorimetry (DSC) analyses shows T M of ∼57°C for drFrnE, which shifts to 52°C in the presence of DTT (Fig. 4e). Taken together, both the studies clearly suggest higher thermal stability of drFrnE in oxidized form, compared with the reduced form.
The thermodynamics of unfolding of drFrnE driven by guanidine hydrochloride (GdmHCl) reveals characteristic of the two-state model (Fig. 4f). GdmHCl unfolding of drFrnE commences at about 0.5 M, followed by a sharp transition to the unfolded end of the profile at about 2 M of GdmCl. The unfolding profiles of oxidized and reduced drFrnE were found to be similar, although with a small change in free energy values; an average of 8.6 ± 0.38 kcal/mol for oxidized drFrnE, as compared with 7.9 ± 0.23 kcal/mol for reduced drFrnE. It further supports higher stability of oxidized drFrnE.
drFrnE displays tolerance toward Cadmium (II) ions
The insulin reductase activity was estimated from the accumulated aggregates of reduced insulin, which, in turn, also depends on the nucleation-dependent process contributing toward initial lag time. Reducing agent DTT (330 μM) was used as electron donor in insulin reduction assays. drFrnE shows insulin reductase activity with a lag time of ∼25 min, which is essentially independent of enzyme concentration (Supplementary Table S1). The C-terminal Cys mutant (C239S/C244S double mutant) shows a similar lag time but a slightly higher rate of insulin precipitation (increased slope), compared with that of wild-type drFrnE. The ΔC222 truncation mutant, on the other hand, shows an increased lag phase and a significantly higher rate as compared with wild-type drFrnE (Fig. 5a). Similar activities of C239S/C244S double mutant and wild-type drFrnE suggest that C-terminal cysteines are not involved directly in catalysis. The presence of DTT in the reaction mixture, which can directly reduce active site cysteines, obviates the need of C-terminal cysteines.
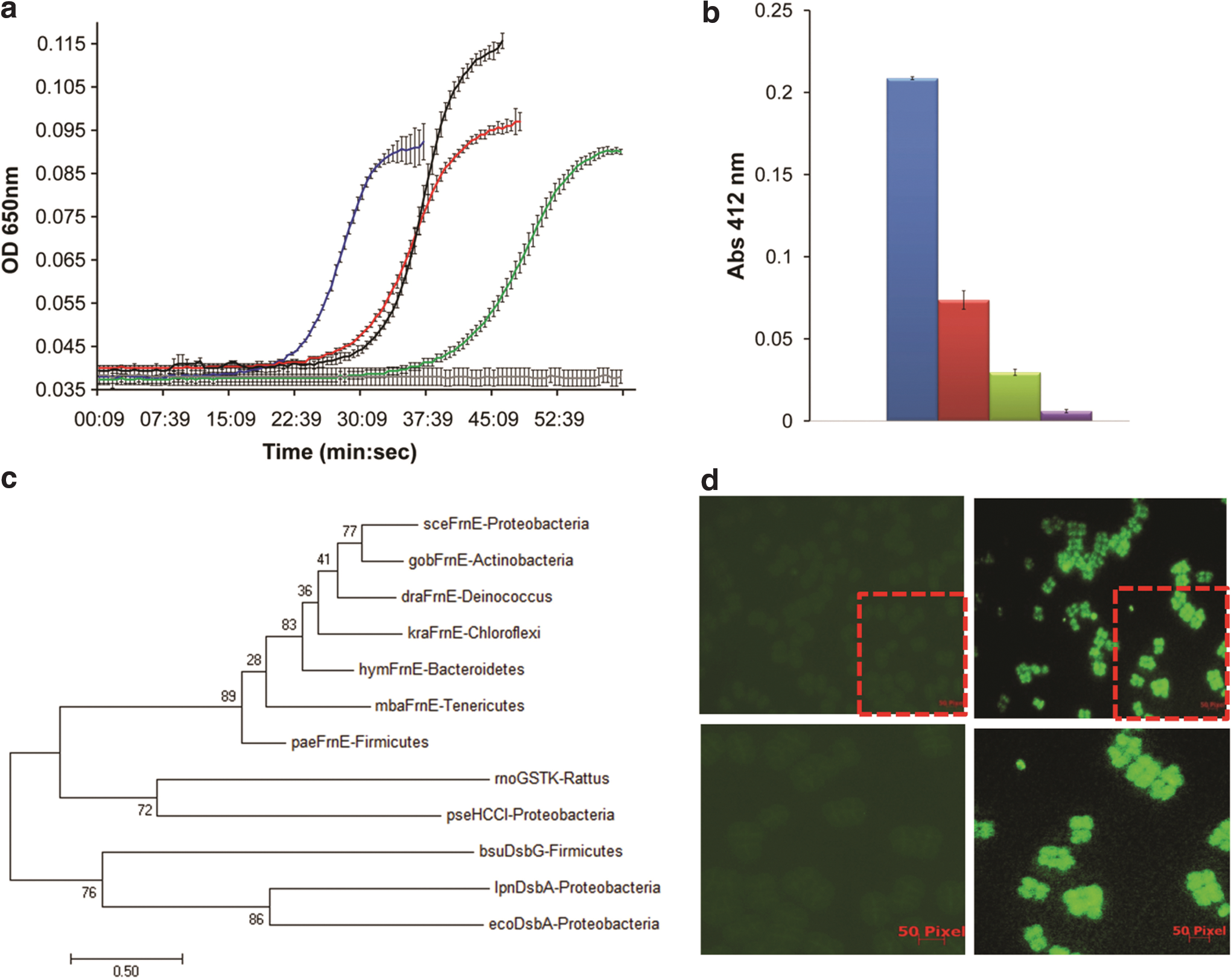
The effect of Cd2+ was probed by using 50 or 100 μM of CdCl2. The drFrnE displays maximal activity in the presence of Cd2+ in both the assays (Fig. 5a), though slope decreases and lag phase increases at a higher Cd2+ concentration (Supplementary Table S1). In comparison, recombinant Escherichia coli thioredoxin (Trx-1) was irreversibly inhibited in the presence of Cd2+ and accumulated activity did not reach the maximum (Supplementary Fig. S5).
drFrnE accepts electrons through its C-tail
The reductase activity of drFrnE and its mutants was also measured by DTNB [5,5-dithio-bis-(2-nitrobenzoic acid)] assay using E. coli thioredoxin reductase (ecTrxR) and NADPH as a coupled electron source. Significant reduction of DTNB by drFrnE suggests that drFrnE accepts electrons from NADPH/ecTrxR couple (Fig. 5b). The C239S/C244S and ΔC222 truncation mutants, however, display a marked decrease in this reduction capability by using NADPH/ecTrxR couple as electron donor (Fig. 5b).
“True” orthologs of drFrnE are abundant in many eubacterial phyla
Both the crystal structures and activity analyses suggested the functional role of C-terminal cysteine motif. Therefore, we used conserved C-terminal Cys motif as characteristic for drFrnE type oxido-reductases and used that as a probe to identify “true” orthologs of drFrnE. We find that drFrnE orthologs are abundantly present among seven eubacterial phyla. To our surprise, we did not detect the Grx system and the proteins involved in the glutathione biosynthesis (GshA, GshB, and GshF) in majority of the species carrying frnE gene (Supplementary Table S2).
In the evolutionary tree, all the “representative” FrnE sequences cluster together (Fig. 5c). The structurally related prokaryotic HCCA isomerase, mitochondrial GST class-kappa (GSTk), and bacterial Dsb proteins form independent clades. Structural similarity (based on DALI Z-score) could not discriminate the evolutionary relationship between drFrnE, GSTk, and Dsb proteins. The phylogenetic analysis, however, suggests that FrnE proteins are more closely related to mitochondrial GSTk (or bacterial HCCA isomerase) proteins than the periplasmic DsbA proteins, despite the absence of active site Cys residues in GSTk proteins. The presence of C-tail with a second Cys motif is unique to drFrnE; however, an extra helical domain insertion in the trx-fold is present in all the three proteins at the same site. Importantly, length and topology of the insertion domains in FrnE and GST class-Kappa match with each other and are different from the insertion domain of DsbA (Supplementary Fig. S6). GSTk proteins are considered to be evolved independently by convergent evolution and not to be part of the GST gene family (33). Based on their structural similarity, we hypothesize that drFrnE and GSTk proteins may share a common ancestor.
drFrnE localizes in cytoplasm
Glutathione redox couple was used to determine drFrnE-glutathione redox equilibrium by monitoring intrinsic fluorescence of Trp residues. The equilibrium constant for the drFrnE/glutathione system was found to be 2.78 × 10−4 M on nonlinear regression (Supplementary Fig. S7). The apparent redox potential value for drFrnE at 25°C and pH 7 (E o FrnE) was estimated by using the Nernst Equation to be −187 mV, which is close to cytoplasmic thiol oxidoreductase proteins.
SignalP4.1, which uses the neural network-based approach to provide reliable identification of signal peptides, did not detect signal sequence in the representative “true” FrnE sequences, except for FrnE of Deinococcus proteolyticus (Supplementary Table S3). The Cello2go server predicted with high confidence cytoplasmic localization for all the FrnE proteins and poor scores for extra-cellular and periplasmic localization. For instance, percentage probability of cytoplasmic localization for E. coli FrnE was 82.2%, as compared with extra-cellular and periplasmic probabilities of 1.8% and 5.1%, respectively (Supplementary Table S3). Thus, both the servers predicted cytoplasmic localization for FrnE orthologs with high confidence.
D. radiodurans cells harboring pFrnEGFP were induced with IPTG, and the synthesis of the drFrnE-GFP fusion protein was confirmed by immunoblotting using GFP-specific antibodies (data not shown). A microscopic examination of D. radiodurans cells expressing drFrnE-GFP fusion shows GFP fluorescence distributed throughout the cell, akin to fluorescence by GFP alone, suggesting localization of drFrnE protein in the cytoplasm of D. radiodurans (Fig. 5c and Supplementary Fig. S8).
Discussion
Thioredoxin and glutaredoxin are two well-known cytoplasmic thiol oxido-reductase systems that help to maintain intracellular redox status in eubacteria. The drFrnE belongs to the thioredoxin superfamily and displays both disulfide isomerase and insulin reductase activities (19). In Deinococcus, frnE gene is over-expressed by sevenfold on exposure to Cd2+ (19). Expectedly, we have observed purified drFrnE protein to be tolerant toward a considerably high concentration of Cd2+. Native-like insulin reductase activity of drFrnE approaching total accumulation, although with the increase in lag time, was observed even in the presence of 100 μM Cd2+ (Fig. 5a).
X-ray crystal structure of drFrnE shows a canonical 22-CPWC-25 active site motif and an additional 239-CxxxxC-244 Cys motif in a unique extended C-terminal tail (Fig. 1a, b). Crystal structure of drFrnE and DTNB assay show that both the cysteine motifs are in oxidized form in the absence of reducing agent.
The stability of oxidized vs reduced states of thiol oxido-reductases provides driving force and determines the thermodynamically favorable direction of the chemical reaction (5). Trx is stable in oxidized state and acts as a reducing agent in the bacterial cytoplasm. In comparison, DsbA is more stable in the reduced form and functions as an oxidant in the bacterial periplasm. The oxidized state of drFrnE (in the absence of reducing agents) displays higher thermal and chemical stability as observed by circular dichroism (CD) and DSC analyses (Fig. 4d–f). The apparent redox potential value for drFrnE was estimated by using the glutathione couple to be −187 mV (Supplementary Fig. S7), which is similar to that of cytoplasmic Grx 3 (−198 mV) and is significantly different from periplasmic Dsb proteins (DsbA, −122 mV; DsbC, −130 mV). These data pointed out that drFrnE is likely to be a cytoplasmic disulfide reductase. Bioinformatics analysis also suggested with high confidence that “true” FrnE orthologs may be localized in the cytoplasm (Supplementary Table S3). The localization of drFrnE in the cytoplasm was confirmed by fluorescence microscopy studies using expression of drFrnE-GFP fusion protein in Deinococcus (Fig. 5d).
We have determined drFrnE crystal structures in three different redox states. In the structures of native protein and in the presence of βME, the active site is oxidized with an intra-molecular disulfide bond between Cys22 and Cys25 residues (Fig. 1e). The active site is in reduced state in the presence of TCEP (Fig. 1f), whereas an intermolecular disulfide bond between Cys22 and Cys244 residues is observed in the presence of DTT (Fig. 1g). These structures are likely to represent snapshots of reaction pathway.
By comparison with Trx family members, Cys22 can be expected to be activity linked with lower pKa and Cys25 can be expected to be resolving residue. We have observed that mutations of these Cys residues abrogate insulin reductase activity of drFrnE. The first step of the reaction mechanism in disulfide reductases, such as Trx, is to attack the disulfide of the incoming substrate by the thiolate form of catalytic cysteine. Therefore, to function as a disulfide reductase in vivo, the active site of drFrnE needs to be in the reduced state and this pose is observed in the structure in the presence of TCEP (PDB id 5E59, Fig. 1f and Fig. 6: stage 1). In this state, reduced Cys22 is in an optimal position to react with the incoming substrate disulfide bond. Resolving Cys25 in this state forms a hydrogen bond with Ser18, and this interaction can modulate pKa of Cys25 required for its function as a resolving partner (Fig. 3b). A similar interaction of resolving cysteine with a conserved threonine in thioredoxin from Bacillus cereus was found to increase the enzymatic activity by stabilizing the resolving thiolate anion (43). Release of the substrate protein in reduced form in the next stage will leave drFrnE in oxidized form, which is represented by the structure of drFrnE in the absence of reducing agent (PDB id 5CNW) and in the presence of βME (PDB id 5COH) (Fig. 1e and 6: stage 2). The presence of C22–C25 and C239–C244 disulfides in drFrnE was confirmed by DNTB assay. The Ser18-Cys25 hydrogen bond distance increases from 3.0 to 3.5 Å in this structure. These stages are similar to the basic catalytic mechanism of the Trx superfamily.
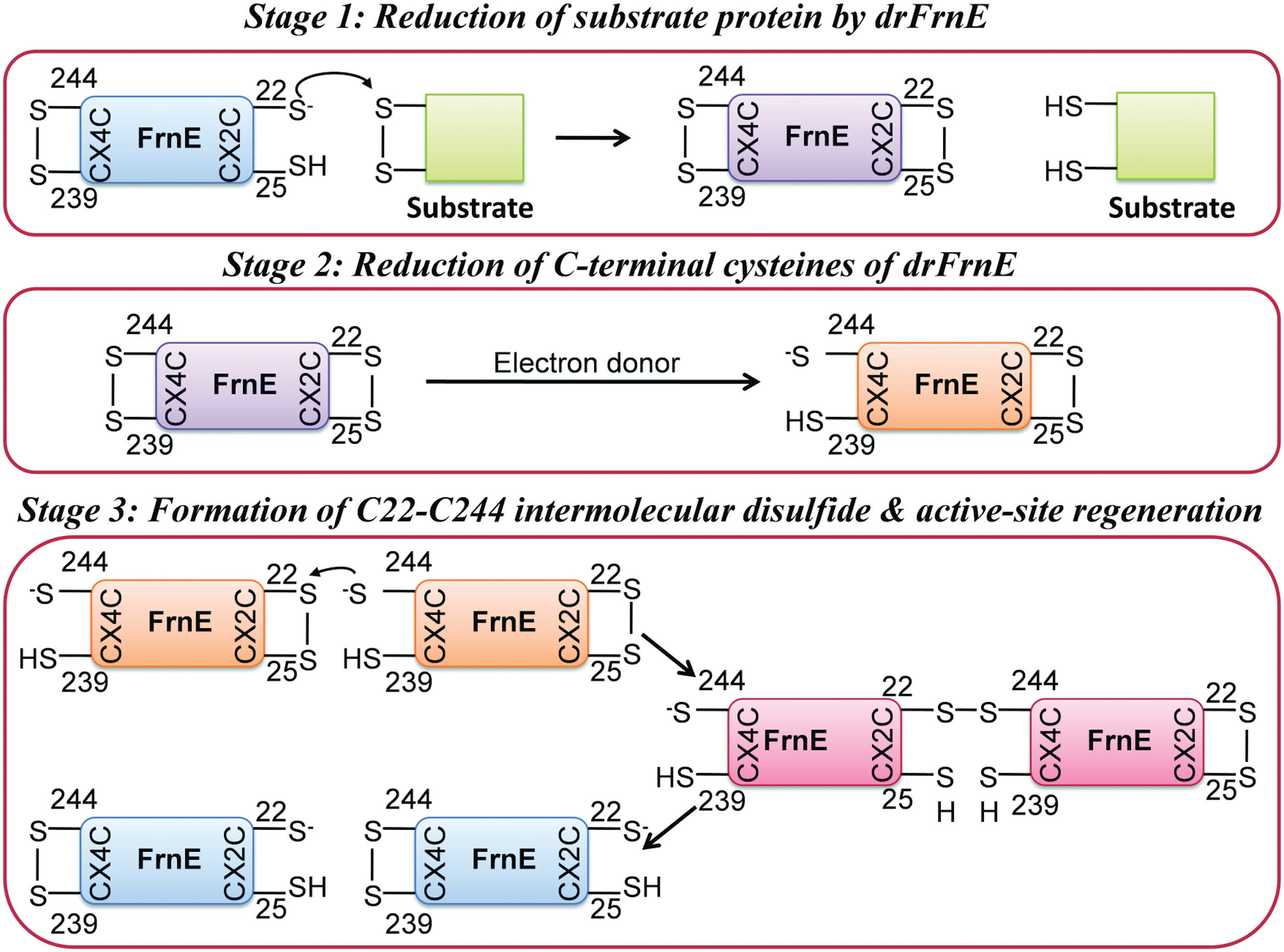
In Trx, regeneration of the reduced form of active site cysteines takes place by direct interaction with thioredoxin reductase (TrxR) through the formation of the inter-molecular Trx-TrxR disulfide bond. Analogous intermolecular disulfide bond is observed in the crystal structure of DsbA-DsbB complex. The intermolecular disulfide bond between Cys22 and Cys244 residues seen in drFrnE structure in the presence of DTT (PDB id 5CO3, Fig. 1g and 6: stage 3) suggests that the regeneration of drFrnE, however, most likely is facilitated by its C-tail, through the conserved 239-Cx4C-244 motif. The remarkable similarity in the binding modes of drFrnE-C-tail binding with DsbA-DsbB peptide complex (Fig. 2c, d) also supports the role of C-terminal cysteines in the regeneration of the active state (reduced form) of drFrnE. Similar redox behavior of CxxxxC motif and its interaction with CxxC motif have been described in several other enzymes such as eukaryotic TrxR, GR, and SBPase (25).
The role of C-terminal cysteines as a conduit to transfer electrons to the active site cysteines is evident from the loss of DTNB reduction activity in drFrnE C239S/C244S and ΔC222 truncation mutants when ecTrxR/NADPH were used as electron donor. The activity decreases to <15% in ΔC222 truncation mutant, compared with wild-type protein. The critical role of C-tail residue in electron channeling is also consistent with the observation that drFrnE alone complements the DsbCD system when exported to the periplasm (19). The C-terminal residues of drFrnE monomer display high flexibility (RMSF ∼1 nm) in 2 ns MD simulations and are expected to be easily accessible to a primary electron donor.
E. coli thioredoxin reductase is strongly inhibited in the presence of Cd2+. Proteins involved in glutathione metabolism are not found in Deinococcus (or other frnE-positive species), and, therefore, GSH is an unlikely source for reductive equivalents to drFrnE. BSH has been detected in D. radiodurans (34). Also, we could confirm the presence of BSH reductase homolog in the Deinococcus genome. It is, thus, possible that bacillithiol might act as a reducing agent for FrnE in Deinococcus.
The presence of C-terminus tail and a second CxxxxC motif is unique to drFrnE and is not found in any of the structurally or functionally related proteins. The “true” drFrnE orthologs, detected from genome databases based on the conservation of C-terminal residues, revealed the presence of FrnE protein in at least 7 out of the 27 NCBI eubacterial phyla. Interestingly, the glutaredoxin system (GR, Grx1, Grx2, Grx3) could not be detected in most of the bacterial species carrying the “true” frnE gene (Supplementary Table S2). It may, thus, be conjectured that FrnE may be the only intracellular functional oxido-reductase system in the seven identified phyla in the presence of Cd2+. Recently, the thioredoxin system has been validated as a target for development of anti-microbial compounds, especially in bacteria lacking the Grx system (23, 26). We have observed the presence of FrnE in many pathogenic bacteria, including Mycobacterium tuberculosis. Thus, due considerations for the presence of FrnE protein in target pathogens may be needed in an effective drug discovery strategy.
In conclusion, FrnE is a cytoplasmic disulfide oxido-reductase found in eubacteria that lack proteins belonging to the glutaredoxin system. Unlike thioredoxin and thioredoxin reductase, drFrnE is functional in the presence of Cd2+. Different redox states of active sites obtained under different reducing environments provide structural snapshots for different stages of disulfide reductase activity of drFrnE. It has also been shown here that essential regeneration of the active site is aided by a flexible C-tail through its novel Cys-motif. Using a combination of diverse structural and molecular biology approaches, we demonstrate that FrnE represents a novel cytoplasmic thiol-disulfide oxido-reductase system that may protect eubacterial intracellular proteome under stress.
Materials and Methods
Bacterial strains, plasmids, and materials
D. radiodurans R1 (ATCC13939) was a gift from Professor J. Ortner, Germany (46). Cloning of wild-type drFrnE and construction of C22S/C25S mutants were described earlier (19). The C239S/C244S double mutant and truncation mutant ΔC222, lacking residues 223–252, were constructed by overlap extension PCR protocol. The E. coli strain NOVABLUE was used for cloning, and the E. coli strain BL21 (DE3) was used for the expression of recombinant proteins. E. coli was grown in Luria-Bertani broth and D. radiodurans was grown in TGY medium (1% trypton, 0.1% glucose, 0.5% yeast extract) with shaking at 180 rpm at 37°C and 32°C, respectively. Recombinant E. coli harboring expression vectors and their derivatives were grown in the presence of antibiotics as required. Shuttle expression vector pVHS559 (4) and its derivatives were maintained in E. coli strain NOVABLUE in the presence of spectinomycin (40 μg/ml) or spectinomycin (75 μg/ml) in the case of D. radiodurans. All recombinant techniques were performed as previously described (44). For microscopic examination, D. radiodurans cells harboring recombinant plasmid were grown at 32°C in TGY, and they were induced with 5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). GFP antibodies were from Roche Biochemical Germany. Molecular biology-grade chemicals, antibodies, and enzymes were procured from Sigma Chemicals Company, Roche Biochemicals (Mannheim, Germany), and New England Biolabs.
Construction of pFrnEGFP expression plasmids
Genomic DNA from D. radiodurans was prepared as described earlier (2). The 750 bp frnE gene (drfrnE) was PCR amplified by using forward primer (5′CAGGAATTCACAAACCTTGCACCCG3′) and reverse primer (5′CAGGATTCCAGTTGTTAGGGCGCTG3′). The PCR product was cloned at EcoRI and BamHI sites in pDSW208 (52) to yield pDSFrnE. The 1.5 kb drFrnE-GFP was further PCR amplified by using forward primer (5′GACACGCTTAAGATGACAAACCTT GCACCCG3′) and reverse primer (5′TAACTCGAGTCAGTTGTTAGGGCGCTG3′) and cloned at AflIII and XhoI sites in pVHS559 (4), and pFrnEGFP was obtained. The pFrnEGFP was transformed into both E. coli and D. radiodurans, and the expression of drFrnE-GFP fusion protein was confirmed by immunoblotting using antibodies against GFP.
Expression and purification of drFrnE proteins
Recombinant drFrnE was purified from E. coli BL21 (DE3) to homogeneity as previously described (19). In brief, transgenic E. coli cells expressing drFrnE on recombinant plasmid were grown in Luria-Bertani medium supplemented with 100 μM ampicillin at 37°C. At an OD600 of ∼0.6, protein expression was induced with 1 mM IPTG and the cell culture was further incubated for 4 h at 37°C. However, the protein yield was higher if incubation was maintained overnight at 20°C. The cells were then harvested by centrifugation, re-suspended in cold lysis buffer (50 mM Tris, 300 mM NaCl, pH 8.0), and lysed by sonication on ice. Cell debris was removed by centrifuging the cell lysate at 11,000 g for 10 min. Clear supernatant containing soluble proteins was mixed with Ni-NTA (nitrilotriacetic acid) agarose (Qiagen, Germany) and was kept at 4°C for 2 h with gentle shaking to allow binding of His-tagged drFrnE protein. The proteins bound to the Ni-NTA resin were washed extensively with lysis buffer containing an increasing concentration of imidazole and finally eluted with elution buffer (50 mM Tris, 300 mM NaCl, 250 mM imidazole, pH 8.0). The protein fractions thus eluted were analyzed on 12% SDS-PAGE. Pure drFrnE fractions were pooled and dialyzed against 50 mM HEPES, pH 7.0. Pure dialyzed protein was concentrated to 20 mg/ml by using 10 kDa cut-off centrifugal filters.
The drFrnE mutant proteins (C239S/C244S double mutant and ΔC222 truncation mutant) were purified to homogeneity by using protocol used for purification of the wild-type protein. The Western blot of drFrnE proteins was performed by using monoclonal Anti-polyHistidine antibody (produced in mouse; Sigma), alkaline phosphatase-conjugated anti-mouse IgG (Sigma), and 5-bromo-4-chloro-3-indolyl phosphate/nitro-blue tetrazolium (BCIP/NBT) (Fig. 7).

Biophysical studies
CD spectra of the protein samples in the absence or presence of DTT (upto 5 mM) were recorded on a Jasco J-815 CD spectrometer equipped with a peltier-type thermostatic cell holder. The CD spectra of drFrnE (9 μM) in buffer A (20 mM HEPES, 20 mM NaCl, pH 7.0) in the far-UV range (200–250 nm) were recorded by using a 1 mm path length cuvette at 20°C. The thermal denaturation curves were obtained by recording changes in the dichroic intensity at 220 nm in the temperature range of 20–95°C at a heating rate of 2°C/min. Fluorescence experiments in the wavelength range 250–400 nm were performed by using Jasco fluorescence spectrometer (model: FP-8500, JASCO Inc., Japan) at 20°C. The increase in fluorescence intensity in the presence of DTT (5 mM) at 335 nm was recorded (with excitation wavelength 280 nm) by using a 1 cm path length cuvette. The concentration and time required for the complete reduction of drFrnE by DTT was optimized by using this assay.
The particle size (i.e., hydrodynamic diameter, Dh) of the protein was measured by using the Zetasizer Nano ZS (Malvern Instruments Ltd, United Kingdom) DLS instrument at 20°C at pH values of 7.0 (in HEPES buffer), and in the presence of 10 mM DTT. The oligomeric state of the protein was estimated by SEC using Superdex 200 increase 10/300 GL column (GE Healthcare). The column was pre-equilibrated with buffer A, and elution profiles of molecular weight standards (Ferritin, 440 kDa; Conalbumin, 75 kDa and Carbonic anhydrase, 29 kDa) were used to estimate the oligomeric status of drFrnE under the conditions of experiments.
The order–disorder (native–unfolded state) transition in drFrnE was measured at a heating rate of 2°C/min, in the temperature range of 20–80°C by DSC (Model: DSC 822; Mettler Toledo, Switzerland) with or without DTT (2 mM). Temperature and enthalpy calibrations of the instrument were done by using cyclohexane and indium. All samples (25 μl of 300 μM drFrnE) were hermetically sealed in aluminum crucibles.
For estimating guanidine hydrochloride (GdmCl)-induced unfolding equilibrium, drFrnE (100 μM) samples in buffer A containing increasing concentrations of GdmCl were incubated at room temperature (25°C) for 24 h. The unfolding experiments were also carried out with drFrnE in the presence of 5 mM DDT (reduced drFrnE). The GdmCl-induced unfolding curves of the native and reduced drFrnE were recorded by measuring the change in the far-UV CD signal from 200 to 250 nm at 20°C. The equilibrium unfolding curves as a function of GdmCl were obtained by plotting the CD signal at 220 nm. To generate unfolding profiles, the raw data were normalized to indicate the fraction unfolded, taking the assumption that the unfolding transition of drFrnE is complete at the highest denaturant concentration used in these experiments. Data were evaluated according to the two-state model of folding by using a six-parameter fit (29, 45).
Di-thiobis-(2-nitrobenzoic acid) (DTNB) assay
It was earlier shown that D. radiodurans Trx1 can cross-talk with E. coli thioredoxin reductase (ecTrxR) (35). DTNB assay was used to find interaction of drFrnE proteins (wild-type drFrnE, and C239S/C244S and ΔC222 truncation mutants) with ecTrxR. The assay mixture contained 50 mM phosphate buffer (pH 7.0), 200 μM NADPH, 0.1 μM ecTrxR, and 5 μM drFrnE. The reaction was started by adding DTNB (50 μM) and monitored by measuring the increase in absorbance at 412 nm by using the Shimadzu UV-1800 double-beam spectrophotometer. Fifty millimolar phosphate buffer (pH 7.0) containing 200 μM NADPH and 50 μM DTNB was used as reference in these assays. Reduction of DTNB in the presence of ecTrxR and NADPH, but without drFrnE, was used as a control. EcTrxR/NADPH by itself did not significantly reduce DTNB.
DTNB assay was also performed for quantitative estimation of the free cysteines in drFrnE and mutants. Fifty micromolar DTNB was added to the assay mixture containing 10 μM purified drFrnE proteins in 100 mM Tris (pH 8.0) and 6 M guanidine hydrochloride. Increase in the absorbance at 412 nm was monitored. An absorption coefficient of 13,700 M −1/cm was used for quantitative estimation of free cysteine groups in the protein.
Insulin reductase assay
Insulin reductase assays in the presence of DTT as electron donor were performed with wild-type and mutant drFrnE by using Synergy H1 multimode reader (BioTek). One hundred thirty micromolar insulin was mixed with 10 μM purified drFrnE in a reaction mixture containing 50 mM HEPES (pH 7.0). The reactions were initiated by adding 330 μM DTT. The rate of insulin precipitation was monitored as the increase in turbidity at 650 nm for 60 min. No protein was added to the control reaction. To understand the effect of Cd2+ on the activity of drFrnE, CdCl2 (50 and 100 μM) was added to the reaction mixture containing drFrnE and DTT before the addition of insulin. The activity of drFrnE in the presence of Cd2+ was monitored at 650 nm and was compared with wild-type drFrnE. All the reactions were performed in triplicate.
Redox potential estimation
The drFrnE sequence carries four Trp residues. The intrinsic fluorescence of the Trp was observed to increase significantly in the presence of reducing agent (5 mM DTT). This change in fluorescence intensity was used to measure the redox potential of drFrnE by measuring the equilibrium constant of oxidation of reduced drFrnE by oxidized glutathione. Oxidized drFrnE (8 μM) was equilibrated in the presence of different ratios of GSH (0–3 mM) and GSSG (0.1 mM) in sodium phosphate buffer (pH 7.0) at 25°C. After 12 h, the change in fluorescence intensity of the sample was recorded at 337 nm (excitation wavelength 280 nm), where the difference between the emission of oxidized and reduced drFrnE was maximum. The equilibrium concentrations of GSH, GSSG, and oxidized and reduced drFrnE were calculated by using the Equations (1
)–(3), where [GSHo] and [GSSHo] are the initial concentrations of GSH and GSSH, R is the relative amount of reduced drFrnE at equilibrium, [FrnEo] is the initial concentration of oxidized drFrnE, F is the measured fluorescence intensity, and F
ox and F
red are the fluorescence intensities of the completely oxidized and reduced drFrnE, respectively.
The data were fitted according to Equation (4) (12, 13), and the equilibrium constant for the drFrnE/glutathione system was found to be 2.78 × 10−4 M on nonlinear regression.
The standard redox potential of drFrnE at 25°C and pH 7.0 (E
o
FrnE) was calculated by using the Nernst Equation (5), where standard redox potential of glutathione redox couple at 25°C and pH 7 (E
o
GSH/GSSG) is −240 mv, R = 8.315 J K−1 mol−1, F = 96,485 C mol−1, T = 298 K, and n = 4.
Microscopic studies
Fluorescence microscopy studies were carried out essentially as described earlier (4). In brief, the mid-logarithmic-phase cells of D. radiodurans harboring plasmid pFrnEGFP were induced with 5 mM IPTG overnight. The expression of drFrnE-GFP was confirmed by using polyclonal antibodies against GFP. Cells expressing drFrnE-GFP were imaged by using a fluorescence microscope (Model IX83; Olympus, Japan). Image processing was done by using Image J software.
Glutaraldehyde cross-linking experiment
To determine the quaternary structure of drFrnE, glutaraldehyde cross-linking experiment was performed according to protocol described earlier (7). Briefly, 3 μl drops containing drFrnE protein (0.5 mg/ml) were placed on siliconized coverslips, inverted onto wells containing 50 μl of 25% (v/v) glutaraldehyde and the wells were sealed with vacuum grease. The plate was maintained at 30°C at Eppendorf thermomixer C. Protein drops were removed at different time intervals (10, 20, and 30 min) and were analyzed on 12% SDS-PAGE to monitor covalently linked oligomers.
Crystallographic analysis
The recombinant wild-type drFrnE was crystallized by the hanging-drop vapor-diffusion method by using 10% PEG-8000 as precipitant at pH 5.5 (36). The drFrnE was also crystallized in the presence of different reducing agents (10 mM DTT, 2 mM TCEP, or 10 mM βME) under similar conditions. The three-dimensional diffraction intensity data were acquired on the Agilent supernova X-ray diffractometer for drFrnE crystals grown in the presence of βME and TCEP, and using BL-21 beamline, INDUS-2 synchrotron (21), for all the other crystals. Crystallographic data collected on INDUS-2 synchrotron were processed by using XDS software suite (18). The data collected on the Agilent X-ray diffractometer were processed by using CrysAlisPro software suite (Agilent Technologies, Oxford, England). The highest resolution of 1.65Å was achieved for the native crystals and those grown in the presence of DTT (Table 1).
One crystal was used for each structure.
Values in parentheses are for highest-resolution shell.
βME, β-mercaptoethanol; DTT, 1,4-dithiothreitol; TCEP, tris(2-carboxyethyl)phosphine.
Initial phases for the native protein crystals were obtained by the molecular replacement method by using a homologous structure available in the PDB (PDB ID, 3GL5; Chang et al., 10.2210/pdb3gl5/pdb) as search model, which was identified based on sequence identity detected by BLAST. The truncated coordinates of the search model (coordinates of C-terminus residues removed from the list and using upto Cβ atoms for nonidentical residues) were used in the molecular replacement calculations using PHENIX (1) implementation of the PHASER (30). The molecular replacement search clearly identified unique solution with an LLG score of 143.4. Electron density maps computed with these phases and observed amplitudes were of good quality such that most of the truncated residues/atoms could be located in these. The initial model for drFrnE was subsequently refined by PHENIX using maximum-likelihood target and with intermittent model fitting using COOT (6). The refinement progressed well with the final model including all the residues, except for about 10 residues on the N-terminus and 13 residues on the C-terminus for which electron density could not be observed.
The structures of drFrnE in the presence of reducing agents were solved by using the truncated model of the refined native structure and PHASER. These structures were also refined by using PHENIX and COOT suites (Table 1). The solvents in all the structures were identified during PHENIX cycles of refinement and were validated in the electron density maps by using COOT. Also, during the final stages of refinement, the TLS model implemented in PHENIX was employed for each structure. The refined structures were validated by using PHENIX. Each structure has been refined to high precision by using all the observed data. Nearly 98% of the residues are observed in the most favored regions of the Ramachandran plot. Residues 9–239 for structures in the presence of βME and TCEP, and residues 10–247 for structure in the presence of DTT (without 232–240), were clearly visible in the electron density maps computed with 2mFo-DFc coefficients at 1.0 sigma contour levels.
MD simulations
The structural model of drFrnE revealed two independent domains and extended the conformation of C-terminals residues harboring unique 239-CXXXXC-244 motif. Two different binding modes of C-tail residues were also seen in the drFrnE crystal structures. To elucidate flexibility of these domains and the C-terminus region, MD simulations were performed by using OPLS-AA/L force field and GROMACS package (39). The monomer of drFrnE was subjected to initial energy minimization, 100 ps temperature equilibration at 298 K using Berendsen thermostat, and 100 ps pressure equilibrium at 1 atm using Berendsen barostat. The production run trajectory was simulated for 2 ns in 1,000,000 steps of 2 fs each, and structures were written every 10 ps. The MD simulation estimated very high flexibility of the C-terminus residues with an estimated RMSF of ∼1 nm.
Bioinformatics and phylogenetic analysis
A BLASTp search with drFrnE sequence was carried out against each of the 27 bacterial phylum individually to mine “true” FrnE proteins in the bacterial kingdom based on the E-value and presence of both the active site CXXC and C-tail CX4C motifs, and to probe for the global phylum-level diversity of FrnE. A small “representative” database of FrnE sequences was created by pooling a maximum of two sequences from each phylum. The selected two sequences from each phylum shared the highest and the lowest identity with drFrnE among the listed top 500 sequences (maximum) detected by BLAST search, such as to construct a diverse pool of FrnE proteins available in the bacterial kingdom.
Subcellular localization of representative sequences of FrnE protein, from each phylum, was predicted by using two independent servers, CELLO2GO (53) (
The presence of well-known cytoplasmic thiol oxido-reductase systems (Trx and Grx) and a glutathione biosynthesis system (Gsh) was probed in each of the species listed in the representative sequence database. For this purpose, ecTrx1, ecTrx2, ecGrx1, ecGrx2, ecGR, ecGshA, ecGshB, and ecGshF protein sequences were used as queries in the BLASTp analysis. The mined sequences, which shared >30% sequence identity and functional description as to be components of Trx, Grx, or Gsh systems in these species, were used against the better annotated E. coli genome to confirm the identity. Only those sequences that showed the highest identity with E. coli Trx, Grx, and Gsh system proteins were taken to be components of Trx and Grx systems in the searched bacterial species.
The structural homologs of drFrnE were searched for by using the DALI server (15) (
Phylogenetic and molecular evolutionary analyses of the representative FrnE sequences and structurally similar proteins were conducted by using MEGA version 7.0 (50). Only one FrnE sequence from each phylum in the representative database was used in the phylogenetic analysis. The tree was computed by using representative sequences from a multiple sequence alignment that was computed by using PROMALS3D web server (37) (
Accession Codes
Coordinates and structure factors for X-ray structures have been deposited in the Protein Data Bank under accession codes 5CNW, 5CO3, 5COH, and 5E59.
Footnotes
Acknowledgments
The authors are thankful to the staff of PX-BL21 beamline at Indus-synchrotron, RRCAT, for their help with diffraction data acquisition.
Authors' Contributions
The author(s) have made the following declarations about their contributions: Conceived and designed the experiments: S.C.B., L.P., Y.S.R., H.S.M., and V.K. Performed X-ray diffraction and biophysical experiments: S.C.B., L.P., and V.K. Performed biochemical experiments: S.C.B., L.P., and Y.S.R. Performed bioinformatics analysis: S.C.B. and V.K. Performed site-directed mutagenesis and fluorescence microscopy experiments: Y.S.R. Contributed to data analysis and discussion: H.S.M. and V.K. Wrote the article: S.C.B., L.P., and V.K.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.