
Abstract
Introduction:
Glycating stress can occur together with oxidative stress during neurodegeneration and contribute to the pathogenic mechanism. Nerve growth factor (NGF) accumulates in several neurodegenerative diseases. Besides promoting survival, NGF can paradoxically induce cell death by signaling through the p75 neurotrophin receptor (p75NTR). The ability of NGF to induce cell death is increased by nitration of its tyrosine residues under conditions associated with increased peroxynitrite formation.
Aims:
Here we investigated whether glycation also changes the ability of NGF to induce cell death and assessed the ability of post-translational modified NGF to signal through the receptor for advanced glycation end products (RAGEs). We also explored the potential role of RAGE–p75NTR interaction in the motor neuron death occurring in amyotrophic lateral sclerosis (ALS) models.
Results:
Glycation promoted NGF oligomerization and ultimately allowed the modified neurotrophin to signal through RAGE and p75NTR to induce motor neuron death at low physiological concentrations. A similar mechanism was observed for nitrated NGF. We provide evidence for the interaction of RAGE with p75NTR at the cell surface. Moreover, we observed that post-translational modified NGF was present in the spinal cord of an ALS mouse model. In addition, NGF signaling through RAGE and p75NTR was involved in astrocyte-mediated motor neuron toxicity, a pathogenic feature of ALS.
Innovation:
Oxidative modifications occurring under stress conditions can enhance the ability of mature NGF to induce neuronal death at physiologically relevant concentrations, and RAGE is a new p75NTR coreceptor contributing to this pathway.
Conclusion:
Our results indicate that NGF–RAGE/p75NTR signaling may be a therapeutic target in ALS. Antioxid. Redox Signal. 28, 1587–1602.
Introduction
N
The relevance of mature nerve growth factor (NGF) inducing cell death has been disregarded because unrealistic concentrations are required in vitro. We show that post-translational modifications occurring under stress conditions confer upon mature NGF the ability to induce cell death at physiologically relevant concentrations. Post-translational modified NGF signals simultaneously through the receptor for advanced glycation end products (RAGE) and p75NTR, and we show for the first time evidence for the interaction of both receptors on the cell surface. The presence of modified NGF in amyotrophic lateral sclerosis (ALS) mice, together with the requirement of RAGE and p75NTR signaling in ALS–astrocyte-mediated neurotoxicity, suggests the therapeutic potential of targeting RAGE–p75NTR signaling in ALS.
We have previously shown that post-translational oxidative modifications regulate the ability of NGF to induce cell death. Tyrosine nitration, induced by peroxynitrite, promotes the formation of high molecular weight NGF oligomers and confers upon the mature neurotrophin the exceptional ability to induce motor neuron apoptosis at low, physiologically relevant concentrations: 10,000-fold lower than those required by native mature NGF (45). The relevance of this regulatory mechanism of NGF activity in pathological conditions is supported by the findings from two independent groups that proNGF is target of post-translational modifications in the brain of Alzheimer's disease (AD) patients and cognitive impaired aged rats (5, 6, 28). In addition to nitration (6), proNGF is also target of glycation in the brain of AD patients (28).
Protein glycation refers to the irreversible nonenzymatic modification of protein amino groups by carbonyl-containing compounds, forming adducts called advanced glycation end products (AGEs). Methylglyoxal (MG) is the most reactive glycating agent in vivo (48). It is a by-product of cellular metabolism, including glucose metabolism, ketone body metabolism, and threonine catabolism (65, 69). Glycation confers proteins the ability to signal through the receptor for advanced glycation end products (RAGEs). RAGE is a type I membrane protein that lacks catalytic activity and exerts its actions by interacting with different adaptor proteins (74). Similar to p75NTR activation, RAGE signaling can promote neuronal survival or death, depending on the cellular context and the type and concentration of the ligand (55, 64).
Although originally identified as the receptor for AGEs, RAGE is a pattern recognition receptor that is activated by an extensive pool of ligands (19). Both, p75NTR and RAGE are widely expressed in the central nervous system throughout development and their expression gradually decreases after birth. However, both receptors are re-expressed at high levels in pathological conditions associated with neuronal degeneration, including amyotrophic lateral sclerosis (ALS) (10, 26, 29, 31, 33, 56).
ALS, or Lou Gehrig's disease, is characterized by the progressive degeneration of motor neurons in the motor cortex, brain stem, and spinal cord. Most ALS cases are sporadic and only about 10% of the cases are inherited (familial ALS) (54). Studies using mutant Cu–Zn superoxide dismutase (SOD1)-linked ALS mouse models revealed that motor neuron degeneration in ALS is a noncell autonomous process that requires damage of neighboring glial cells (24). Astrocytes, the most abundant glial type in the central nervous system, adopt a reactive phenotype and play a key role in the progression of the disease (24, 75). We have shown that reactive astrocytes induce the death of cocultured motor neurons by a mechanism involving increased NGF production and p75NTR-dependent death signaling (8, 9, 43). Moreover, p75NTR signaling has been implicated in the pathology observed in mice overexpressing hSOD1G93A, the best characterized ALS mouse model (32, 57, 61, 70).
Cultured embryonic motor neurons represent an attractive model for studying NGF-mediated neuronal death. Although they express high levels of p75NTR in the absence of TrkA, embryonic motor neuron cultures are not sensitive to NGF-induced apoptosis. We showed that NGF/p75NTR-mediated motor neuron apoptosis occurs only in the presence of surrounding glial cells producing nitric oxide or other diffusible factors capable to decrease motor neuron antioxidant defenses (43, 44). In contrast, nitrated NGF induces p75NTR-dependent motor neuron apoptosis in the absence of an exogenous flux of nitric oxide (45). In this study, we show that glycated NGF also induces the death of cultured embryonic motor neurons at physiologically relevant concentrations. Moreover, although peroxynitrite (nitration) and MG (glycation) target different amino acid residues, both induce the formation of NGF oligomers and confer upon NGF the ability to signal through p75NTR and RAGE to induce motor neuron death. We also show evidence for the presence of nitrated and glycated NGF in the spinal cord of early symptomatic hSOD1G93A mice and its potential involvement in the neuronal degeneration observed in ALS cell culture models.
Results
Treatment of mature NGF with MG, the most reactive glycating agent in vivo, induces, in a dose- and time-dependent manner, the formation of high molecular weight NGF oligomers (Fig. 1A, B). Our experiments were performed using purified mature NGF from mouse submaxillary glands (NGF 2.5S; #B.5017; Harlan Bioproducts). We noticed that in some of the NGF batches used, a weak band with an apparent molecular weight corresponding to NGF dimer could be detected. This band does not correspond to an incomplete cleavage of proNGF, because its intensity increases with time after incubation at 37°C (not shown) and after MG treatment in a time- and dose-dependent manner (Fig. 1A, B). When analyzed by size-exclusion chromatography (SEC) native NGF eluted as a single peak with an apparent molecular weight corresponding to the dimer, as previously described (45). However, methylglyoxal-treated NGF (NGF-MG) eluted as three peaks with an apparent molecular weight corresponding to the dimer, tetramer, and octamer (Fig. 1C). As observed in Figure 1C, the NGF-MG dimer eluted before the native NGF dimer, suggesting conformational changes that result in a reduction of the NGF-MG dimer compactness. When aliquots from the fractions collected after SEC were analyzed by reducing SDS-PAGE and Western blot, we observed the appearance of lower order oligomers and monomeric NGF (≈13 kDa) (Fig. 1D), suggesting that NGF oligomerization involves noncovalent interactions. Nevertheless, as observed in Figure 1D, some NGF-MG oligomers are stable in reducing SDS-PAGE gels, which might indicate a nonthiol-dependent covalent crosslinking of some subunits. However, the existence of SDS-PAGE-resistant noncovalent aggregates of other proteins has been previously reported (12, 35). Remarkably, the effect of MG treatment closely resembles the effect induced by nitration of NGF after peroxynitrite or tetranitromethane treatment, which reduces NGF dimer compactness and induces the formation of noncovalent high molecular weight oligomers (45).
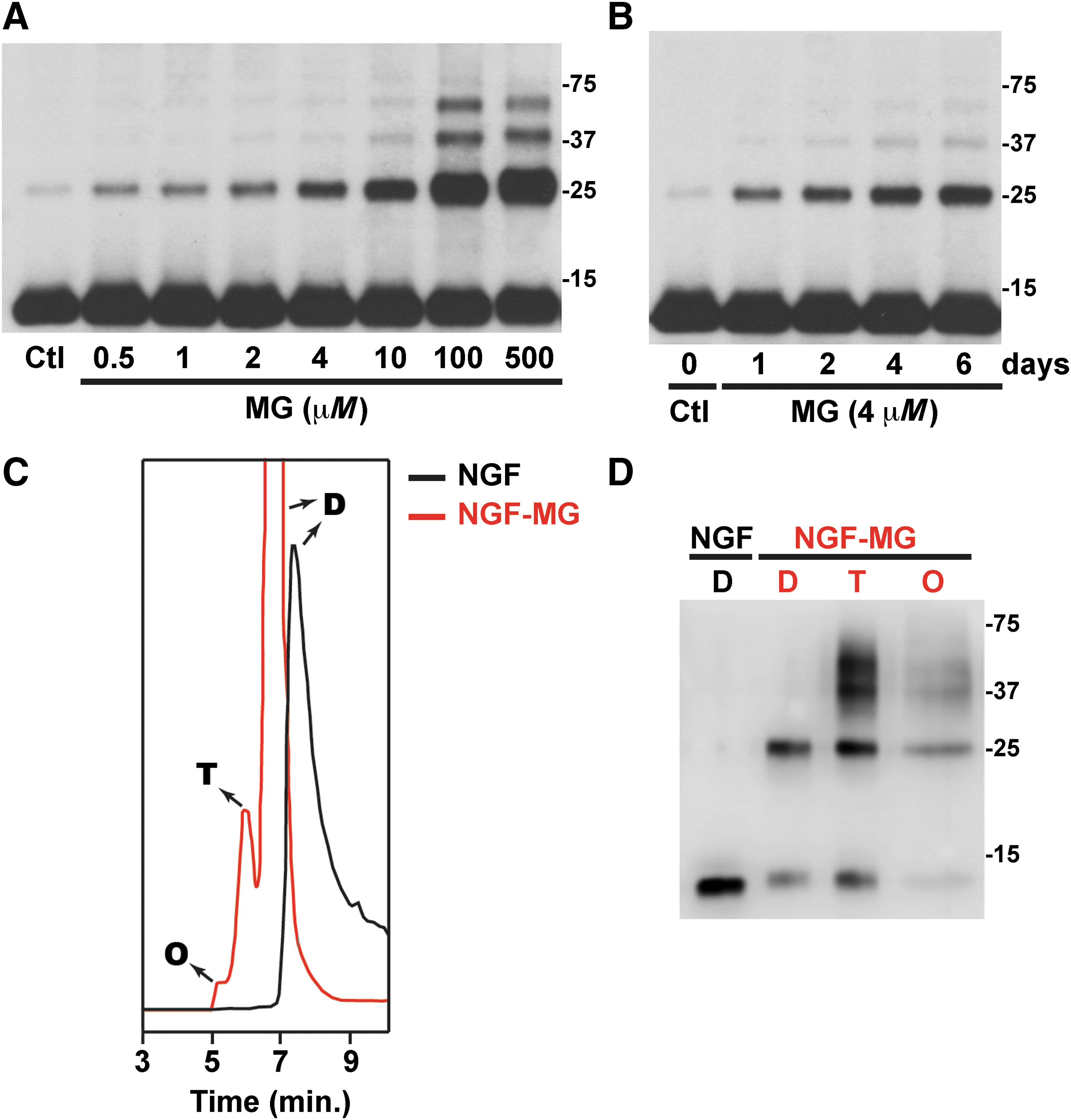
Glycation by MG is directed primarily to the guanidine group of arginine residues to initially form glycosylamine, which sequentially rearranges to form dihydroxyimidazolidine and three hydroimidazolone isomers MG-H1, MG-H2, and MG-H3 (46). MG also reacts with the ɛ-amino group of lysine residues to form Nɛ-carboxyethyl lysine (CEL) and methylglyoxal-lysine dimers (47, 65). The specific NGF residues modified by MG treatment were determined by mass spectrometry analysis. To ensure complete coverage of the NGF sequence by LC-MS/MS, multiple samples of NGF-MG were analyzed after digestion with trypsin or Glu-C.
As indicated in Table 1 and Supplementary Figure S1 (Supplementary Data are available online at
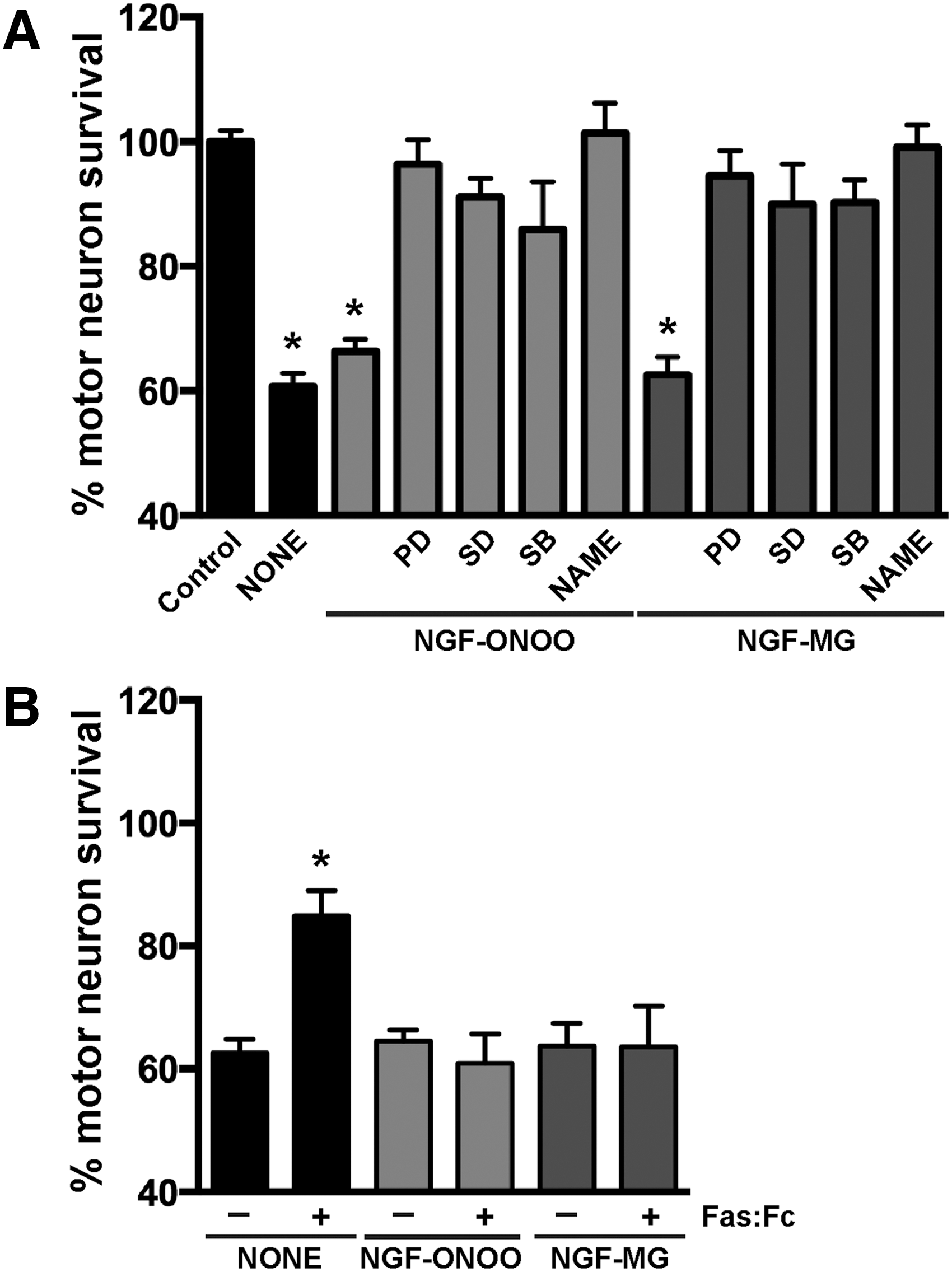
We then evaluated the biological activity of NGF-MG in primary motor neuron cultures maintained by the exogenous addition of glial cell-derived neurotrophic factor (GDNF, 1 ng/mL). Under these conditions, motor neuron cultures are not sensitive to NGF-mediated cell death (43, 73) unless high concentrations of NGF (>10 ng/mL) and an exogenous flux of nitric oxide are provided (43). In contrast, NGF-MG induced motor neuron death at subpicomolar concentrations of 10 pg/mL, and independently of the presence of exogenous nitric oxide (Fig. 2A). To perform these experiments, NGF was treated with MG (2 μM or 4 μM) for 48 h at 37°C, with residual MG eliminated by ultrafiltration. No difference in the effect was observed by doubling MG concentration from 2 μM to 4 μM. To rule out a direct toxic effect of potential residual MG, a mock sample lacking NGF was processed as mentioned and used as a control (control MG in Fig. 2A). Motor neuron survival was not affected by the addition of human glycated albumin (Fig. 2A), further supporting a specific effect of glycated NGF. In addition, motor neuron death induced by NGF-MG was prevented by the general caspase inhibitor Z-VAD-FMK and by p75NTR-blocking antibodies (Fig. 2B). These results resemble those obtained with nitrated NGF (45), suggesting that the gain in p75NTR-dependent apoptotic activity of mature NGF after glycation or nitration involves a common mechanism.
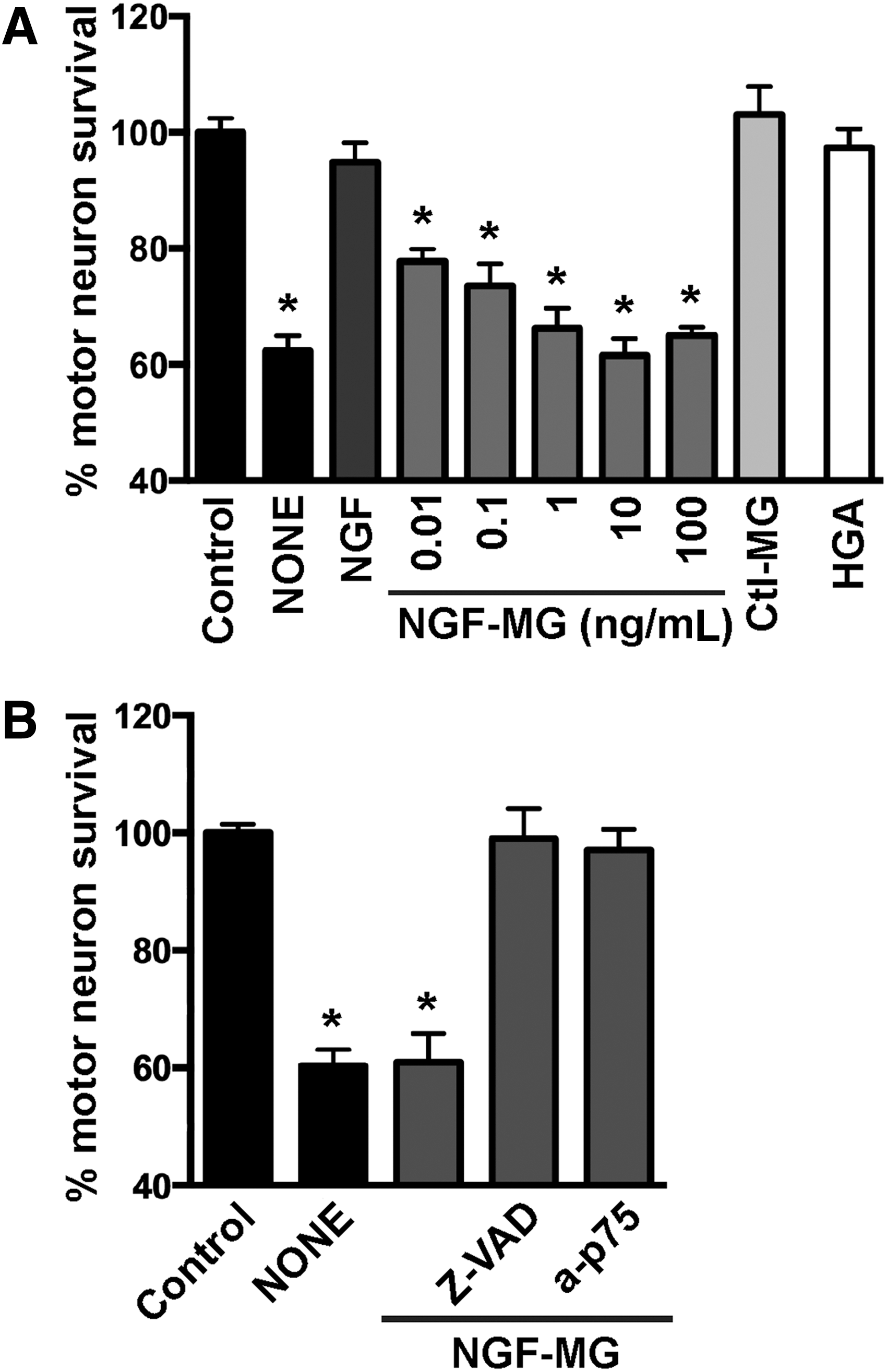
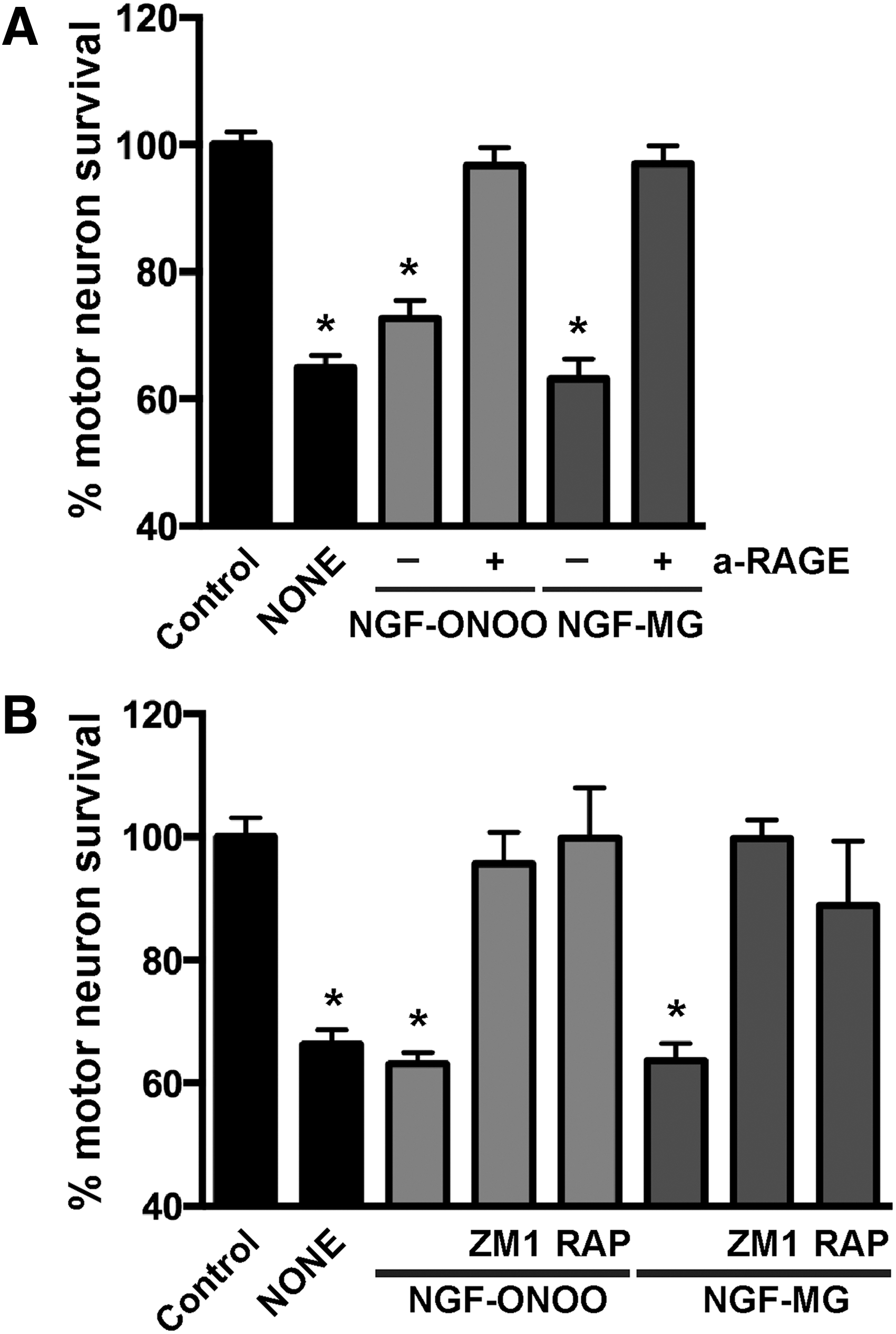
RAGE is a pattern recognition receptor able to bind multiple ligands, and most RAGE ligands are oligomeric in nature (19). Since glycation and nitration induced NGF oligomerization, we analyzed the participation of RAGE signaling in the cell death induced by post-translational modified NGF. RAGE-blocking antibodies prevented the motor neuron death induced by peroxynitrite-treated NGF or NGF-MG (Fig. 3A). A comparable protection was observed with two previously described RAGE pharmacological inhibitors, FPS-ZM1 (16) and RAP (1) (Fig. 3B).
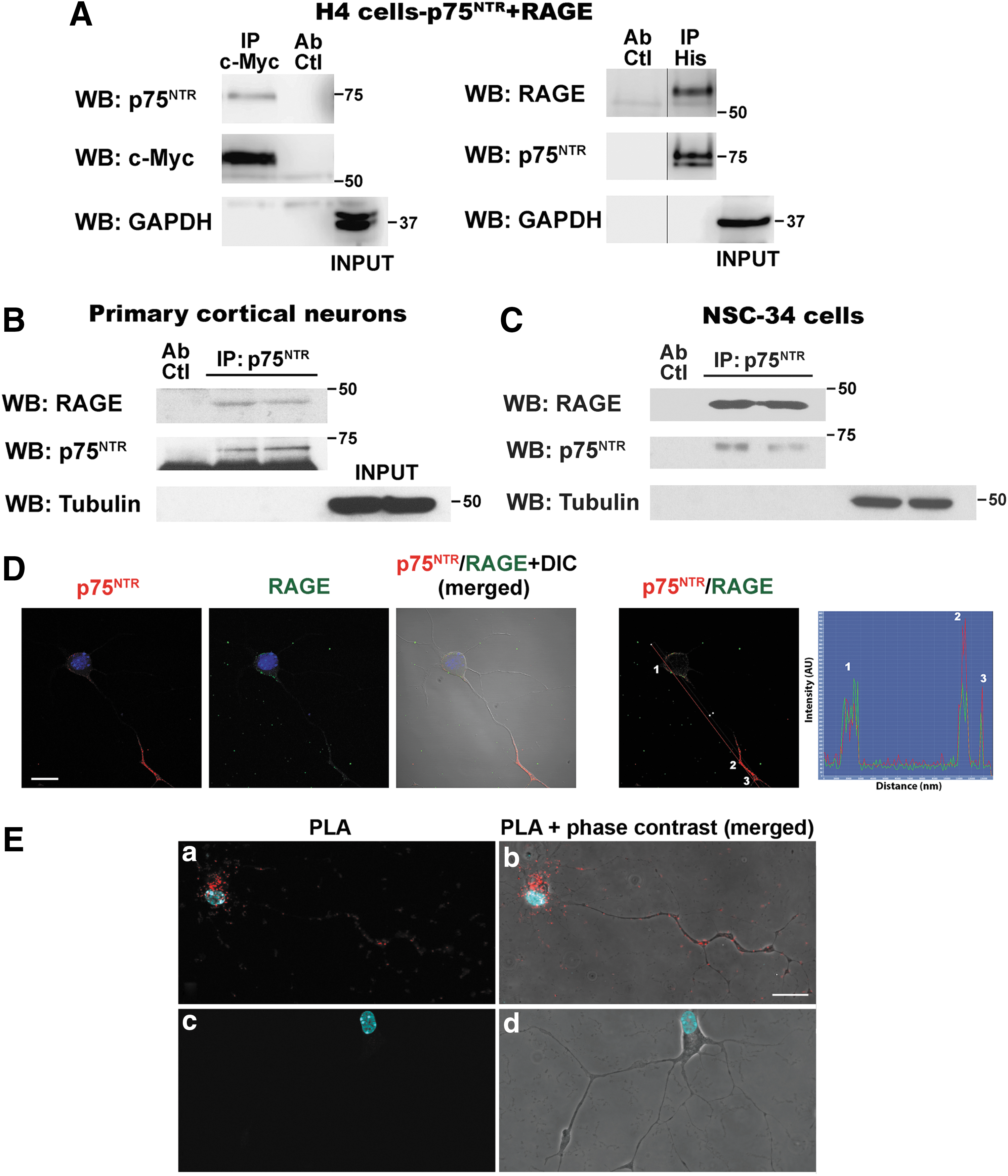
Blocking only either p75NTR or RAGE completely prevented motor neuron death induced by nitrated or glycated NGF, suggesting a functional interaction of both receptors either by independent activation by the ligand or by the simultaneous binding of post-translational modified NGF to a RAGE–p75NTR complex. This, together with the fact that p75NTR relies on its ability to interact with different receptors on the cell surface to elicit its broad array of biological functions (3), led us to evaluate the potential interaction between p75NTR and RAGE. Crosslinking and coimmunoprecipitation studies indicated that p75NTR and RAGE interact in the cell surface of H4 neuroglioma cells (Fig. 4A) or HEK293 cells (not shown) co-overexpressing a c-Myc-tagged version of RAGE and a His-tagged version of p75NTR. We also confirmed RAGE–p75NTR interaction in primary cortical neurons and NSC-34 motor neuron-like cells, expressing endogenous levels of the receptors (Fig. 4B, C). Owing to the low yield of motor neurons per spinal cord, RAGE–p75NTR interaction in primary motor neuron cultures was confirmed using an in situ proximity ligation assay (PLA), which allows to observe and determine, in fixed cultured cells, the subcellular localization of protein–protein interactions at single-molecule resolution (63). Colocalization of p75NTR and RAGE immunofluorescence was observed in cultured motor neurons (Fig. 4D). Using the same set of primary antibodies against the extracellular domain of p75NTR and RAGE, the in situ PLA detected RAGE–p75NTR interaction in the soma and neurites of cultured motor neurons (Fig. 4E).
Motor neuron death induced by different stimuli, including trophic factor deprivation and nitric oxide donors, involves activation of Fas by endogenous Fas ligand (FasL) (20, 51, 53). FasL/Fas signaling in motor neurons triggers a cell-type specific death pathway involving p38 MAP kinase activation that leads to increased nitric oxide production (52). Since RAGE activation has been shown to increase FasL expression in other cell types (67, 74), we tested the potential activation of this death pathway by post-translational modified NGF. Motor neuron death induced by nitrated or glycated NGF was prevented by the addition of three different p38 MAP kinase inhibitors and by the general nitric oxide synthase (NOS) inhibitor Nω-nitro
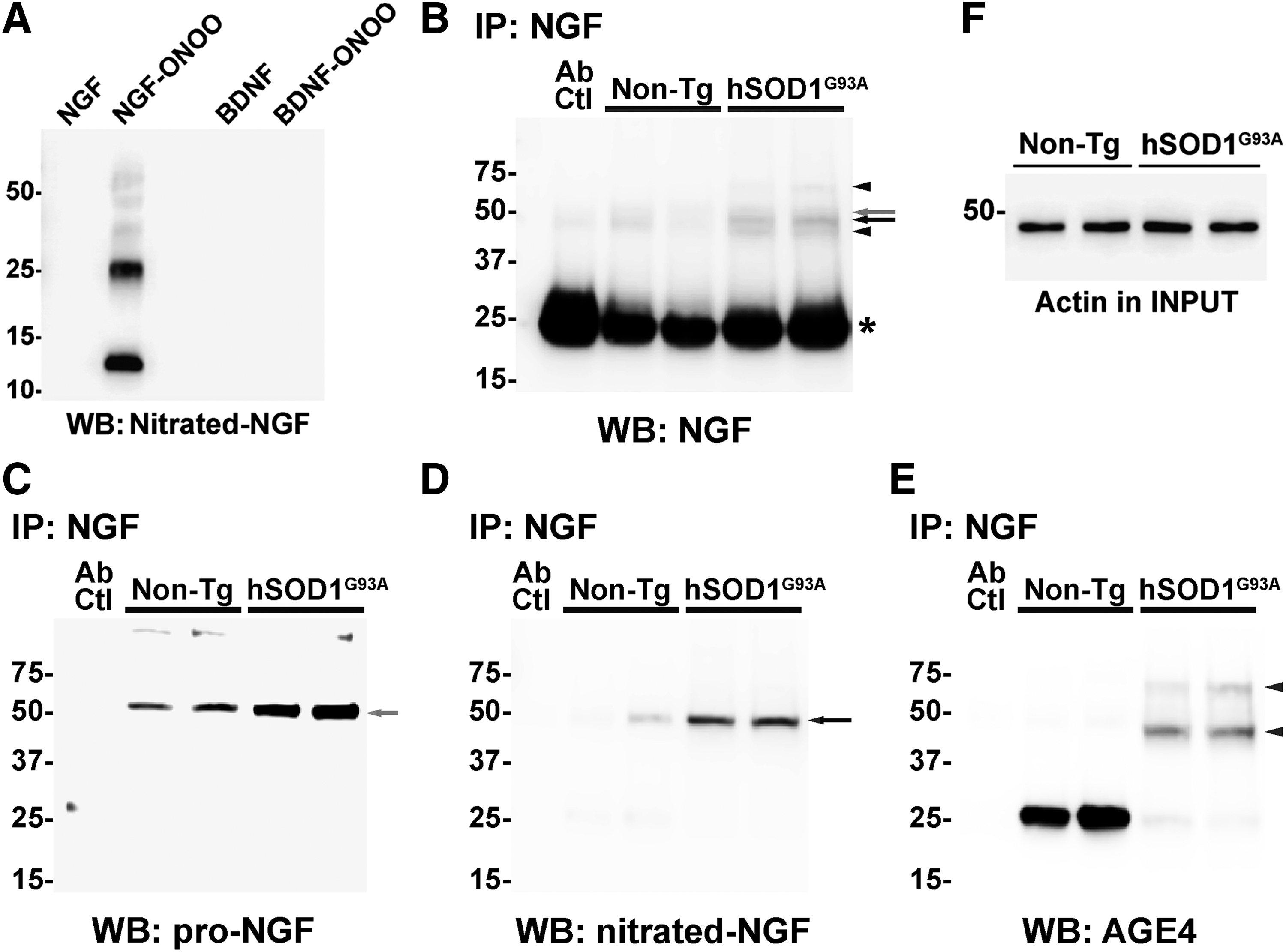
Lastly, we confirmed the presence of post-translational modified NGF in the spinal cord of early symptomatic hSOD1G93A mice using antibodies against MG-derived adducts and a specific antibody against nitrated NGF that we developed. The antibody against nitrated NGF did not recognize native NGF or nitrated BDNF, other member of the neurotrophin family (Fig. 6A). After in vitro treatment with peroxynitrite, purified NGF migrates on reducing SDS-PAGE with apparent molecular weights corresponding to the monomer, dimer, trimer, tetramer, and pentamer. The antibody against nitrated NGF detected all the oligomeric forms (Fig. 6A). After immunoprecipitation using NGF-specific antibodies, we detected the presence of high molecular weight species of NGF, whereas monomeric mature NGF (≈13 kDa) was not observed in our experimental conditions (Fig. 6B). As previously reported (43), we observed increased levels of NGF in the spinal cord of ALS mice (Fig. 6B). Moreover, using an antibody against the prodomain of NGF, we confirmed the presence of proNGF in samples from both transgenic and nontransgenic mice, which migrated as a unique band with an apparent molecular weight close to 50 kDa (Fig. 6C). This result suggests the presence of oligomeric forms of NGF, which do not correspond to proNGF, in the spinal cord of ALS mice. In spinal cord extracts from hSOD1G93A mice, four NGF immunoreactive bands were observed. Strikingly, only one band (<50 kDa; likely tetramer) was detected using the specific antibody against nitrated NGF (Fig. 6D). Remarkably, the band corresponding to proNGF does not appear immunoreactive for nitrated NGF. Because MG is the most reactive glycating agent in vivo (48, 65), we reprobed the membrane with an antibody specific to MG-derived AGE and observed that NGF is also glycated in the spinal cord of ALS mice. We detected two immunoreactive bands, different from those immunoreactive for nitrated NGF or proNGF, likely corresponding to trimers and pentamers of glycated NGF (Fig. 6E). The antibody against MG-derived adducts also detected an immunoreactive band with an apparent molecular weight close to 25 kDa. Because it migrates together with the light chain of the antibody used for the immunoprecipitation, we cannot unequivocally ascribe this band to an NGF dimer. This glycated species seems to be more abundant in samples from nontransgenic mice. However, glycated higher molecular weight species of NGF were not abundant in the spinal cord of nontransgenic mice. An experimental replicate of the immunoprecipitation experiment displaying similar results is also shown in Supplementary Figure S3. We have previously shown that the increased NGF expression in the spinal cord of hSOD1G93A mice overlaps with increased p75NTR expression (43). This occurs together with increased RAGE expression (Fig. 7A).

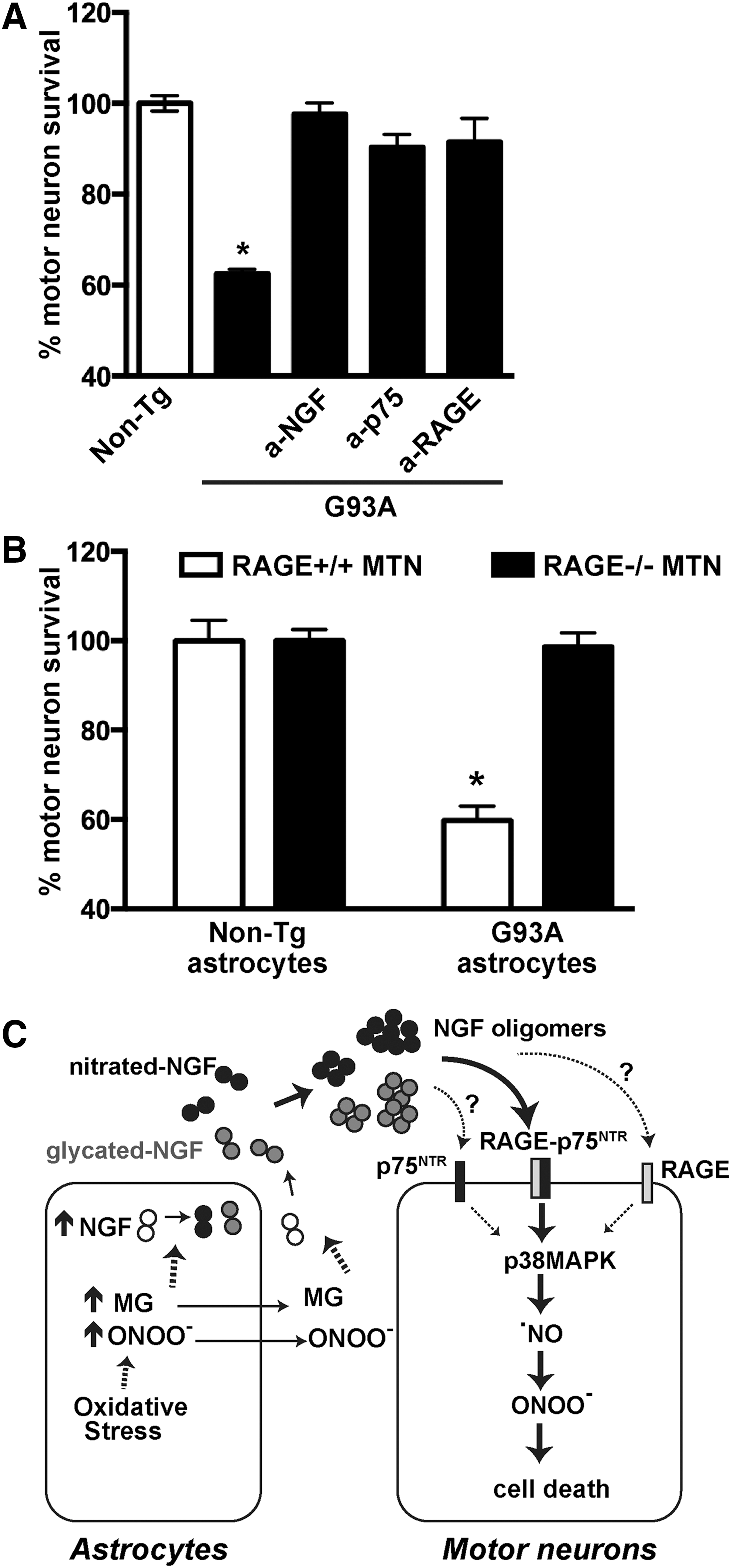
In a cell culture model, we observed that lumbar spinal cord extracts from early symptomatic ALS mice induced the death of cultured hSOD1G93A-expressing motor neurons, whereas extracts obtained from age-matched nontransgenic littermates were devoid of effect (Fig. 7B). Interestingly, motor neuron death induced by ALS extracts was completely prevented by blocking antibodies to NGF, p75NTR, or RAGE (Fig. 7B). Importantly, the NGF-blocking antibody used recognized both native and post-translational modified NGF, as evidenced by immunoprecipitation and Western blot analysis (Fig. 6). These results indicate that endogenous post-translational modified NGF present in the spinal cord of ALS mice has the ability to induce the death of cultured hSOD1G93A motor neurons by signaling through p75NTR and RAGE.
To further confirm the potential relevance of RAGE–p75NTR signaling in ALS, we employed a coculture model in which purified motor neurons are plated on top of either nontransgenic- or hSOD1G93A-astrocyte monolayers. As previously reported (39, 71), nontransgenic astrocytes support neuronal survival, whereas about 40% of motor neurons are lost when cocultured on resting astrocytes overexpressing hSOD1G93A (Fig. 8). Motor neuron death induced by hSOD1G93A astrocytes was completely prevented by the addition of either blocking antibodies that antagonize NGF, p75NTR, or RAGE signaling (Fig. 8A). The involvement of NGF and p75NTR signaling in astrocyte-mediated motor neuron loss in cocultures has been previously described (8, 18, 43). To confirm the involvement of RAGE signaling, we used motor neurons isolated from RAGE knockout mice and found that hSOD1G93A astrocytes did not induce the death of cocultured motor neurons lacking RAGE expression (Fig. 8B).
Discussion
Although the ability of mature NGF to induce cell death has been extensively reported, its relevance in vivo has been questioned because high concentrations, at least an order of magnitude higher than those required to promote cell survival, are required to induce apoptosis in vitro. Hence, the field has focused on the regulation of proneurotrophin cleavage as the switch regulating the prosurvival/prodeath signaling of neurotrophins (11, 21). Our results challenge this paradigm and indicate that the ability of NGF to induce cell death can also be regulated by post-translational modifications occurring under stress conditions, specifically nitration and glycation, which confer upon mature NGF the ability to signal simultaneously through RAGE and p75NTR.
We have previously shown that NGF is target of post-translational oxidative modifications induced by peroxynitrite (45). In our experiments, NGF was treated with peroxynitrite by 10 bolus additions of 1 μL each, to reach a final concentration of 1 mM. Thus, each bolus corresponds to a concentration of peroxynitrite in the micromolar range (100 μM to 143 μM taking into account the volume correction after each bolus addition). Accordingly, the rate of peroxynitrite production in vivo in specific compartments has been estimated to be as high as 50–100 μM per minute (68). Although acute short-lasting bolus exposure to peroxynitrite is not optimal to mimic the in vivo situation, it has allowed us to identify the potential NGF residues target of peroxynitrite modification and to characterize the changes in NGF biological activity after oxidative modification (45). Importantly, supporting the relevance of our in vitro results, the presence of nitrated NGF has been observed in the brain of AD patients and cognitive impaired aged rats (5, 6), and we have now detected it in the spinal cord of early symptomatic hSOD1G93A mice.
Nitration of the two tyrosine residues (Tyr52 and Tyr79) present in mature NGF enhances its ability to induce p75NTR-dependent motor neuron apoptosis at subnanomolar (physiological) concentrations (45). Here we show that glycation induced by MG treatment, although it targets different amino acid residues, mimics the effects mediated by tyrosine nitration. It is worth noting that the concentration of MG used to modify NGF for the cell culture experiments is in the same range as that typically found in mammalian cells (48). Typical concentrations of MG in cells and tissues are in the range of 1–4 μM, but higher levels are observed in aging and pathological conditions (48). As observed with nitrated NGF, glycated NGF induced motor neuron death at physiologically relevant concentrations (subpicomolar range). Although peroxynitrite and MG target different amino acid residues and generate different adducts, both induce NGF oligomerization and confer upon NGF the ability to signal through p75NTR and RAGE to induce motor neuron death. This suggests that oligomerization of mature NGF could be a regulatory mechanism promoting prodeath activity toward motor neurons.
A distinctive feature of RAGE ligands is their oligomeric nature (19), and both nitration and glycation induce noncovalent NGF oligomerization [(45) and Fig. 1]. Native NGF exists in solution in a self-associating dimer–tetramer equilibrium, leading to the formation of multimers ranging up to dodecamers of NGF at high concentrations of the protein (14, 40). Furthermore, a dimer of dimers arrangement was observed in the crystal structure of mouse NGF (PDB ID: 1BTG) (23). NGF has only two tyrosine residues: both are nitration targets (45). Mass spectrometry analysis indicated that glycation by MG is directed primarily to the guanidine group of five arginine residues in NGF, giving rise to the formation of the MG-H1 adduct (or its structural isomers MG-H2 and MG-H3). Nitration lowers the pK a of the tyrosine hydroxyl moiety to near physiological pH and also makes peptides more hydrophobic when the nitrotyrosine is protonated (66), whereas the formation of the MG-H1 adduct neutralizes the positive charge of the arginine residue (47). Thus, both post-translational modifications have the potential to induce structural changes. Our results indicate that they shift the NGF equilibrium in solution toward high molecular weight oligomers, suggesting the existence of structural rearrangements that facilitate the interaction.
More than 99% of the formed MG binds reversibly to protein or glutathione (GSH) cysteine thiols (hemithioacetal adduct). Since the cellular concentration of MG is approximately 0.01% of total thiol concentration, reversible MG binding does not significantly affect cellular thiol status but helps prevent the formation of irreversible glycation adducts (47). Most of the MG generated (>99%) is detoxified by the ubiquitously expressed, GSH-dependent, glyoxalase system (47). This system involves the consecutive action of two enzymes, glyoxalase I (GLO1) and glyoxalase II (48, 65, 69). GLO1 catalyzes the rate-limiting step of the pathway, and downregulation or inhibition of the enzyme leads to increased MG levels and protein glycation (49, 65). Since GLO1 activity is directly proportional to the cellular GSH concentration (49), oxidative stress can lead to decreased MG degradation and subsequent protein glycation (48). Protein quality control systems present in every cell are fundamental to prevent the accumulation of toxic protein aggregates and maintain cellular proteostasis. These quality control systems respond to different stressors with a hormetic dose response, in which low-dose stimulation by different stressors induces an adaptative response that confers resistance and enhances survival, whereas high exposure to the stressors overwhelms the defense systems and induces cellular demise (7, 15). Accumulation of oxidative-modified proteins occurs during aging and age-related neurodegenerative diseases, including ALS (2, 25). When post-translational oxidative modifications induce a gain of function in a protein, the increase in activity or the appearance of a new function could allow a small fraction of the modified protein to have a significant biological effect (66). Our results indicate that nitration and glycation induce a gain in NGF prodeath activity, conferring upon the modified neurotrophin the ability to signal through RAGE–p75NTR at picomolar concentrations. Figure 8C displays a schematic representation of our main findings and conclusions.
Blocking either p75NTR or RAGE completely prevented the neuronal death induced by modified NGF, suggesting that simultaneous binding to both receptors is essential to induce death signaling. This could be explained by the interaction of both receptors to form a signaling complex, or by functional interaction at the level of the signaling cascades activated independently by each receptor in response to post-translational modified NGF. Both mechanisms, receptor heterocomplex formation and downstream signaling pathway interaction, are not mutually exclusive and RAGE–p75NTR interaction could be occurring at both levels. Although our results do not exclude the potential interaction of both receptors at the level of the signaling cascades, using two different techniques we show evidence for the endogenous formation of a RAGE–p75NTR receptor complex in four different cellular models (Fig. 4). We observed that p75NTR and RAGE interact on the cell surface even in the absence of exogenous ligand. The preassembly of receptor complexes on the cell membrane has been observed for several receptors, including p75NTR homodimers, p75NTR–sortilin heterocomplexes, and RAGE homodimers (19, 41, 72). The binding of nitrated- or glycated-NGF oligomers to the RAGE–p75NTR complex could (i) increase the relative amount of complexes, as observed for p75NTR–sortilin complexes (41); (ii) induce a conformational change in the receptor, as observed for p75NTR homodimers (72); or (iii) induce the formation of higher order oligomerization states, as observed for RAGE homodimers (19). Further studies are required to confirm or discard these possibilities.
The presence of post-translational modified NGF has been previously observed in the brain of AD patients and cognitive impaired aged rats (5, 6, 28). In these reports, nitration and glycation were adjudicated to pro-NGF, although the potential existence of high molecular weight species of post-translational modified mature NGF was not unequivocally excluded. Owing to their close molecular weight, distinction between pro-NGF and mature NGF oligomers is not always achieved in SDS-PAGE. It was shown that mature nitrated NGF has reduced ability to activate TrkA signaling and promote neuronal survival, and its presence was linked to the cognitive decline associated with aging and AD (5, 6). Together, our results suggest the potential involvement of post-translational modified NGF and RAGE–p75NTR signaling in ALS pathology. We determined the presence of post-translational modified NGF in the spinal cord of symptomatic ALS mice (Fig. 6), which coincides with increased expression of RAGE (Fig. 7) and p75NTR (43). Moreover, motor neuron death induced by ALS astrocytes or spinal cord extracts from symptomatic ALS mice was dependent on NGF and p75NTR/RAGE signaling. Blocking either NGF, p75NTR, or RAGE independently completely prevented motor neuron death, suggesting that signaling of post-translational modified NGF through both receptors is required. Nevertheless, the potential synergistic interaction of p75NTR and RAGE at the level of the signaling cascades activated independently by different receptor-specific ligands cannot be excluded. A previous report suggested the involvement of pro-NGF in the motor neuron death induced by ALS astrocytes (18). However, in the aforementioned study, total NGF immunodepletion was performed and, therefore, the involvement of post-translational modified mature NGF was not ruled out. Our data suggest that post-translational modified NGF may have a central role in this phenomenon and our results revealed the key participation of RAGE–p75NTR signaling in the motor neuron death induced by ALS astrocytes. Both receptors are expressed by spinal cord motor neurons in ALS patients and mice (10, 18, 33, 56). Since adult healthy motor neurons lack p75NTR expression (30, 76), NGF toxicity will be directed toward stressed motor neurons that re-express p75NTR. In this context, post-translational modified NGF would propagate a vicious cycle, driving progressive motor neuron death.
Although our results were obtained using a hSOD1-linked ALS mouse model, their potential translation to familial and sporadic human ALS pathology is supported by previous reports indicating that the spinal cord of ALS patients has markers of oxidative and glycating stress (2, 13, 25, 27, 59, 60), together with increased p75NTR and RAGE expression (10, 18, 26, 33, 56), which coincides with increased levels of NGF in the cerebrospinal fluid (18). Further stressing the potential role of p75NTR-death signaling in ALS pathology, it has been shown that the urinary levels of the p75NTR extracellular domain constitute a potential biomarker of disease activity and progression in ALS patients (57, 58). Despite substantial evidence for an involvement of p75NTR signaling in ALS pathology (18, 57, 61), p75NTR ablation only modestly delayed the pathology in hSOD1G93A female mice (32, 70). This modest effect could be explained by the fact that p75NTR also promotes motor neuron survival by interacting with TrkB and TrkC (22, 73), and Schwann cell migration and myelination (31). Thus, the advance of new therapies targeting p75NTR apoptotic signaling demands a better understanding of the regulatory mechanisms defining its dual effects (36). These dual effects appear to be mediated by different ligands and by interaction of p75NTR with different coreceptors. Our results suggest that blocking the signaling through RAGE–p75NTR complex constitutes an alternative to selectively prevent p75NTR-mediated motor neuron death, without affecting its beneficial effects.
Materials and Methods
Reagents
All chemical and reagents were from Sigma-Aldrich unless otherwise specified. Culture media, serum, and supplements were obtained from Life Technologies, unless otherwise specified.
Animals
Transgenic mice overexpressing ALS-linked hSOD1G93A were obtained from The Jackson Laboratory strain B6.Cg-Tg(SOD1*G93A)1Gur/J (stock # 004435). RAGE knockout mice were previously described (38). Both transgenic lines were maintained in a C57BL/6J background. Genotype was determined by PCR. All animal procedures were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH. The Animal Care and Use Committee of MUSC (Animal Welfare Assurance number is A3428-01) approved the animal protocol pertinent to the experiments reported in this publication.
Methylglyoxal treatment
Mature NGF purified from mouse submaxillary glands (NGF 2.5S; #B.5017) was obtained from Harlan Bioproducts. NGF, 0.2 mg/mL in 50 mM sodium phosphate buffer, pH 7.4, was treated with vehicle (control) or MG (2 or 4 μM final concentration) for 48 h at 37°C, under sterile conditions. Residual MG was eliminated by ultrafiltration using Amicon Ultra-0.5 devices, after reconstituting the concentrate to the original sample volume four times with phosphate buffered saline. For the dose response experiments, samples were treated with different concentrations of MG (0.5–500 μM) as already indicated, for 72 h at 37°C. For the time response experiments, samples were treated with MG (4 μM final concentration) and incubated for 1, 2, 4, or 6 days at 37°C. After incubation, samples were diluted in reducing Laemmli sample buffer and analyzed by Western blot as described hereunder.
Peroxynitrite treatment
Peroxynitrite was synthesized as previously described (50). Peroxynitrite concentration was determined by absorbance at 302 nm in 1 M NaOH (ɛ = 1670 M −1 cm−1). Diluted stock solutions were freshly prepared in 0.01 M NaOH. NGF (0.2 mg/mL) was treated with peroxynitrite (1 mM final concentration) as previously described (45). The reaction was performed in 50 mM sodium phosphate buffer (pH 7.4) containing 20 mM sodium bicarbonate. Peroxynitrite was added to the sample in 10 bolus of 1 μL each, to reach a final concentration of 1 mM. Control experiments were performed using diluted NaOH or decomposed peroxynitrite.
Cell culture and treatment
Human neuroglioma (H4) and mouse motor neuron-like (NSC-34) cell lines were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 IU/mL), and streptomycin (100 μg/mL). Stable transfection was performed using Lipofectamine 2000 (Life Technologies), and the culture media were supplemented with 350 μg/mL G418 sulfate (Corning) and 200 μg/mL hygromycin B. All cultures were maintained at 37°C in a humidified atmosphere with 5% CO2.
Primary astrocyte cultures were prepared from the spinal cord of 1-day-old mice as we previously described (43). Pups were cold anesthetized and then euthanized by decapitation. Astrocytes were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS, HEPES (3.6 g/L), penicillin (100 IU/mL), and streptomycin (100 μg/mL). Cultures were >98% pure as determined by glial fibrillary acidic protein (GFAP, an astrocytic marker) immunoreactivity and devoid of CD11b-positive microglial cells.
Motor neuron cultures were prepared from mouse embryonic spinal cords (E12.5) as previously described (42). To prepare hSOD1G93A or RAGE knockout motor neurons, genotyping was performed by real time-PCR during dissection, and all embryos of the same genotype were pooled together for the rest of the preparation. Motor neurons were plated at a density of 500 cells/cm2 on four-well multidishes (Nunclon) precoated with polyornithine–laminin. Cultures were maintained in Neurobasal medium supplemented with 2% horse serum, 25 μM
For coculture experiments, motor neurons were plated on mouse astrocyte monolayers at a density of 300 cells/cm2 and maintained in L15 medium supplemented with 0.63 mg/mL bicarbonate, 5 μg/mL insulin, 0.1 mg/mL conalbumin, 0.1 mM putrescine, 30 nM sodium selenite, 20 nM progesterone, 20 mM glucose, 100 IU/mL penicillin, 100 μg/mL streptomycin, and 2% horse serum (43). Motor neurons were identified by immunostaining with antineurofilament 160 (mouse monoclonal [clone NN18]; #N5264; Sigma-Aldrich). Survival was determined by direct counting of all neurofilament-positive cells, as previously described (42). Counts were performed over an area of 0.90 cm2 in 24-well plates.
Primary cortical neuronal cultures were prepared from mouse embryonic cortices (E15) as we previously described (42). Cells were plated at a density of 1.5 × 105 cells/cm2 and maintained in Neurobasal medium supplemented with 1% B-27 and 0.5 mM glutamine. Cells were harvested on the 7th day after plating. Cultures were >98% pure as judge by βIII-tubulin and GFAP staining.
Plasmid constructs
The cDNA of human RAGE (NM_001136) with a C-terminal Myc-DDK tag in the expression vector pCMV6-Entry (#RC204664) was obtained from OriGene. The cDNA of human p75NTR (NM_002507) with a C-terminal His tag in the expression vector pReceiver-M77 (#EX-A0100-M77) was obtained from GeneCopoeia. The p75NTR cDNA was then cloned into the expression vector pCMV6-A-Hygro (#PS100024; OriGene).
Lumbar spinal cord extracts
Lumbar spinal cords were dissected, over ice under sterile conditions, from early symptomatic hSOD1G93A mice (≈100-day-old) or aged-matched nontransgenic littermates. One hundred milligrams of tissue was processed in 0.4 ml of ice-cold PBS containing 3 mM EGTA, 1 mM EDTA, and complete, EDTA-free, Protease Inhibitor Cocktail (Roche Applied Science). Tissue was then homogenized under sterile conditions and homogenates were centrifuged at 40,000 g for 1 h. Supernatants were collected and kept at −80°C until used. Aliquots were added to motor neuron cultures to reach a final protein concentration of 0.5 μg/mL.
Size-exclusion chromatography
Samples of NGF at a concentration of 0.2 mg/mL were treated with vehicle or MG (500 μM) for 3 days as already indicated. Aliquots containing 10 μg of vehicle-treated NGF or NGF-MG were manually injected onto a 50 μl injection loop connected to an Agilent 1100 Series pump (Agilent Technologies) equipped with a XBridge Protein BEH SEC 125 Å, 3.5 μm, 7.8 × 150 mm column (Waters). The outlet of the column was connected to a Waters 2487 Dual Absorbance UV/Visible detector. The pump was run isocratically at 0.5 ml/min at room temperature. The buffer used was 50 mM sodium phosphate/50 mM sodium sulfate, pH 7.2. Elution of NGF was determined by absorbance at λ = 220 nm. Fractions corresponding to NGF elution were manually collected at the outlet of the detector and analyzed by SDS-PAGE.
Identification of modified residues by tandem mass spectrometry
NGF at a concentration of 0.2 mg/mL was treated with MG (4 μM) for 3 days as already indicated. Samples (10 μg) were reduced in 10 mM dithiothreitol (Thermo Scientific), and alkylated in 25 mM iodoacetamide (Thermo Scientific) for 1 h at 25°C in the dark. Samples were digested with 1 μg of porcine trypsin (Sigma-Aldrich) (1:10, enzyme:protein ratio) or 1 μg of Glu-C (Thermo Scientific) (1:10, enzyme:protein ratio). Before trypsin digestion, 1 M ammonium bicarbonate was added to a 100 mM final concentration. The Glu-C digestion was done in 1 × buffer phosphate (pH 7.4). Both digestions were incubated overnight at 25°C with mild agitation. Reactions were quenched with 10% trifluoroacetic acid. Peptides were desalted using C18 ZipTip® pipette tips (Millipore). Peptides were dissolved in solvent A and loaded on a trap column (C18 pepMap 100, 300 μm × 5 mm) (Thermo Scientific) and separated on a 75 μm × 20 cm analytical column packed in house with C18 YMC ODS-AQ (Waters Corp.) using a 180 min gradient (from 5% B to 50% B). Solvent A was 2% acetonitrile (ACN)/0.2% formic acid (FA); solvent B was 98% ACN/2% FA. Eluting peptides were mass analyzed by data-dependent acquisition on the Orbitrap Elite mass spectrometer (Thermo Scientific) with Xcalibur 2.2 software. The top 20 most intense ions detected in the survey mass spectrum (Orbitrap detector at 60,000 resolution, mass range 400–1700 Da, target value of 1 × 106) were selected for fragmentation by collision-induced dissociation at 35% normalized collision energy. Fragments were detected in the ion trap. Ions with a +1 charge were excluded from selection. Dynamic exclusion was enabled with a repeat count of 2, duration of 30 s, exclusion list size of 50, and exclusion duration of 120 s.
Database searching and peptide identification
The tandem mass spectra were searched using SequestHT within Proteome Discoverer (PD) 1.4 (Thermo Scientific) against a Uni Prot (Swiss Prot + Tremble) mouse (taxonomy # 10090) protein database containing isoforms (77,220 sequences, downloaded on March 13, 2015). Parameters for peptide identification were as follows: precursor mass tolerance of 20 ppm, fragment mass tolerance of 0.8 Da, fully tryptic peptides with a maximum of three missed cleavages, and carbamidomethyl static modification of cysteine residues. Variable modifications for arginine (R) were the hydroimidazolone derivate (MG-H1/2/3; +54.011 Da), argpyrimidine (+80.026 Da), THP (+144.042 Da), and dihydroxyimidazolidine (+72.021 Da). Carboxyethyl lysine (+72.021 Da) and methionine oxidation (+15.995 Da) were also included as variable modifications. The search results were filtered to yield only high confidence peptides (1% false discovery rate) using the “target decoy validator” feature in PD 1.4. Identified peptides had mass errors <5 ppm. Fragment-labeled spectra were exported from PD 1.4; raw spectra were exported from Xcalibur 2.2. Modification sites were verified manually.
Production of monoclonal antibody against nitrated NGF
The specific mouse monoclonal antibody against nitrated NGF was produced by GenScript using as immunogen the peptide corresponding to NGF residues 48–58 with a nitrated tyrosine residue at position 52 [VFKQY(NO2)FFETKC]. The peptide was conjugated to the carrier protein keyhole limpet hemocyanin. Balb/c mice were immunized and hybridoma cell lines were generated. Antibodies were screened by ELISA for binding to the immunogen (nitrated NGF48-58 peptide) and nitrated NGF and for lack of binding to the non-nitrated NGF48-58 peptide.
Immunoprecipitation and Western blot analysis
Spinal cord protein extracts were prepared in Tris-HCl buffer pH 7.6 supplemented with 2 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.25% Nonidet P-40, and complete protease inhibitor cocktail EDTA free. After sonication, samples were centrifuged at 4°C for 10 min at 10,000 g. Protein concentration was determined by the bicinchoninic acid method (BCA protein assay; Thermo Scientific-Pierce). For immunoprecipitation, 1 mg of protein extracts in lysis buffer was incubated overnight at 4°C with rabbit polyclonal antibody anti-NGF (H-20) (#sc-548; rabbit anti- Santa Cruz). After adding BioMag Plus protein A magnetic particles (Polysciences, Inc.), the incubation was extended for 1 h. After washing three times with lysis buffer, immunoprecipitated proteins were eluted by incubation for 5 min at 95°C in 2 × concentrated reducing Laemmli sample buffer.
Immnoprecipitated proteins, protein extracts, or NGF samples were resolved on SDS-polyacrylamide gels or 4–15% Mini-PROTEAN® TGX™ gels (BioRad) and transferred to polyvinylidene fluoride membranes using the Trans-Blot Turbo transfer system (BioRad). Membranes were blocked for 1 h in Tris-buffered saline (TBS), 0.1% Tween-20, and 5% bovine serum albumin, followed by an overnight incubation at 4°C with the primary antibody diluted in the same buffer. After washing with 0.1% Tween-20 in TBS, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (GE Healthcare) for 1 h at room temperature. For immunodetection of immunoprecipitated proteins, membranes were incubated for 1 h at room temperature with clean blot IP detection reagent (HRP) (#21230; Thermo Scientific) or peroxidase-conjugated goat antimouse IgG light chain-specific antibody (#115-005-174; Jackson ImmunoResearch Laboratories, Inc.). Membranes were then washed and developed using the ECL Prime chemiluminescent detection system (GE Healthcare). Membranes were imaged and quantified using the C-DiGit Blot Scanner (LI-COR Biosciences). Protein loading was corrected by β-Actin or α-Tubulin levels.
The following primary antibodies were used for Western blot experiments: mouse monoclonal anti-β-Actin (clone AC-15; #A5441; Sigma-Aldrich); mouse monoclonal anti-AGE-4 (clone 14B5; #KAL-KG133; Cosmo Bio USA, Inc.); rabbit polyclonal anti-GAPDH (#ab9485; Abcam); mouse monoclonal anti-6-Histidine epitope tag (clone AD1.1.10; #NB100-64768; Novus Biologicals); mouse monoclonal anti-c-Myc (clone 9E10; #M4439; Sigma-Aldrich); rabbit polyclonal anti-NGF (#AB1526SP; EMD Millipore-Chemicon); rabbit polyclonal anti-NGF (#AN-240; Alomone Labs); rabbit monoclonal anti-p75 (clone EP1039Y; #NB110-58000 Novus Biologicals or #GTX61425; or GeneTex); rabbit polyclonal anti-proNGF (#ANT-005; Alomone Labs); rabbit polyclonal anti-RAGE (#R5278; Sigma-Aldrich); rabbit polyclonal anti-RAGE (#ab37647; Abcam); mouse monoclonal anti-α-Tubulin (clone DM1A; #3873; Cell Signaling Technology).
Crosslinking and coimmunoprecipitation
Cells were incubated in Hank's balanced salt solution for 30 min at room temperature with the membrane-impermeable reducible crosslinker DTSSP (5 mM; Thermo Scientific-Pierce). The reaction was stopped by addition of 1 M Tris, pH 7.5, to a final concentration of 50 mM. Cells were lysed in Tris-HCl buffer pH 7.6 supplemented with 2 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.25% Nonidet P-40, and complete protease inhibitor cocktail EDTA free. After incubation on ice, samples were centrifuged at 4°C for 10 min at 10,000 g. Immunoprecipitation was performed using the following specific antibodies: rabbit monoclonal anti-p75 (clone EP1039Y; #GTX61425), mouse monoclonal anti-6-Histidine epitope tag (clone AD1.1.10; #NB100-64768; Novus Biologicals), or the c-Myc-tagged protein mild purification kit Ver.2 (#3305; MBL International Corporation). Precipitated complexes were analyzed by Western blot as already described.
Immunofluorescence and in situ PLA
Motor neuron cultures, maintained for 48 h as already indicated, were fixed on ice with 4% paraformaldehyde plus 0.1% glutaraldehyde in PBS. Cultures were immunostained with antibodies against the extracellular domain of p75NTR (mouse monoclonal [MLR2 clone], #ab61425; Abcam) and RAGE (rabbit polyclonal, #ab37647; Abcam), and developed using (i) Alexa Fluor 594-conjugated goat antimouse (#A-11032; Life Technologies) and Alexa Fluor 488-conjugated goat antirabbit (#A-11034; Life Technologies) for immunofluorescence analysis or (ii) DuoLink In Situ PLA Probe Anti-Rabbit PLUS (#DUO92002) and Anti-Mouse MINUS (#DUO92004) with the DuoLink In Situ Detection Reagents Red (#DUO92008) (Sigma-Aldrich) for PLA. Negative controls omitting independently one or both primary antibodies were performed in each experiment. Nuclei were counterstained with DAPI (Life Technologies). The signal from each detected pair of PLA probes was observed in an Olympus Fluoview FV10i laser scanning desktop microscope. For analysis of p75NTR and RAGE colocalization by immunofluorescence, samples were imaged with a Zeiss LSM 880 NLO Axioobserver inverted laser scanning confocal/multiphoton microscope using a 63 × 1.4 N.A. Plan-Apochromat oil immersion DIC lens. Fluorescence of DAPI, Alexa Fluor 488, and Alexa Fluor 594 was excited at 405 nm, 488 nm, and 561 nm, respectively, and detected at 410–496 nm, 499–588 nm, and 600 nm, respectively at a 1-Airy-unit-diameter pinhole. The 0.38 μm thick z-stack images were processed using Zeiss Zen software.
Statistical analysis
All data are reported as mean ± SEM. Significance was determined using either Student's t-test, or for multiple group comparison one-way ANOVA with Tukey's post-test. When comparing the effect of genotype and treatments, two-way ANOVA was used followed by Bonferroni's post-test. Differences were declared statistically significant if p < 0.05. All statistical computations were performed using Prism 6.0 (GraphPad Software).
Footnotes
Acknowledgments
This work was supported by National Institutes of Health Grants NS100835, GM103542, and Shared instrumentation grants S10 D010731 and S10 OD018113. Mice were housed in a facility constructed with support from the NIH, Grant Number C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources. This work used the Cell and Molecular Imaging Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313). LC-MS/MS analysis was performed at the MUSC Mass Spectrometry Facility, supported by the Office of the Provost and the South Carolina COBRE in Oxidants, Redox Balance, and Stress Signaling.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.