
Abstract
Aims:
Neutrophil cytosolic factor 1 (NCF1) is a key regulatory component of the phagocytic NOX2 complex, which produces reactive oxygen species (ROS). Polymorphism of the Ncf1 gene is associated with increased arthritis severity. In this study, we generated targeted Ncf1 knock-in mice with inducible Ncf1 expression and determined the critical time window during which the NOX2-derived ROS protect the mice from arthritis.
Results:
Targeted Ncf1 knock-in mice lacked NOX2-derived ROS, and in vivo allelic conversion of Ncf1 by the CreERT2 recombinase led to full protein expression and ROS production within 10 days. Mice in which Ncf1 had been activated before immunization with type II collagen (CII) developed only mild clinical symptoms of collagen-induced arthritis (CIA), whereas the ROS-deficient littermates had severe arthritis. The functional Ncf1 restricted the expansion of IL-17A-producing T cells specific for the immunodominant CII peptide. When the Ncf1 gene was activated after the priming phase, Ncf1-dependent protection from autoimmune arthritis was still observed, together with a reduced number of splenic monocytes but it was not associated with alterations in peptide-specific T cell response. The Ncf1-deficient mice expressed pronounced interferon signature, which could be normalized by conditional expression of Ncf1 and was also present in the Ncf1-mutated mouse during arthritis.
Innovation and Conclusion:
Ncf1 deficiency has been known to predispose to autoimmunity in both humans and rodents. Our in vivo results point to a regulatory role of NOX2-derived ROS not only during priming but also during the effector phase of CIA, most likely via different mechanisms. Antioxid. Redox Signal. 27, 1473–1490.
Introduction
R
Ncf1 (neutrophil cytosolic factor 1) deficiency is known to predispose to autoimmunity in both humans and rodents. Our results show that Ncf1 is not needed during juvenility to protect from autoimmune arthritis later on, but conditional Ncf1 suppresses arthritis even if activated after the disease priming. The study presents in vivo evidence that Ncf1/reactive oxygen species-regulated mechanisms are different in priming than in the effector phase.
Although NOX2-dependent generation of reactive oxygen species (ROS) is believed to cause oxidative stress and tissue destruction in inflammation (38), their protective role as immunological regulatory agents was revealed by a hypothesis-free genome-wide search in animal models for arthritis (41). Subsequent studies in animal models have shown that defects in Ncf1 increase the risk to develop autoimmune disorders. These include arthritis, experimental autoimmune encephalomyelitis, autoimmune neuritis, psoriatic arthritis, gout, type 1 diabetes, and an SLE-like phenotype (21, 23, 25, 29, 31, 41, 44, 49).
Recently, a reduction in intracellular ROS due to a mutation in Ncf4, another regulatory component of the NOX2 complex, was shown to promote collagen-induced arthritis (CIA) but not the T cell-independent mannan-induced psoriasis model (57). CIA is one of the most widely used animal models of autoimmune arthritis. Ncf1 deficiency leads to more frequent and more severe CIA when compared with wild-type mice, accompanied by enhanced T cell-dependent autoimmune responses against type II collagen (CII) (25). Although ROS production by macrophages has been shown to protect from the disease (14), the exact Ncf1-regulated pathways and the time when these are critical for regulating the disease remain undetermined.
In the present study, we investigate the importance of the Ncf1-regulated pathways in detail in vivo during the development of arthritis. We generated conditional Ncf1 knock-in mice, and by switching on the Ncf1 gene in adult mice, we excluded its role in determining susceptibility to autoimmunity during the establishment of central tolerance. Newly activated Ncf1 limited expansion of autoreactive T cells and gave rise to an almost complete protection from arthritis. By in vivo gene activation after disease priming, Ncf1 still could protect against arthritis, an effect associated with a complex pattern of pathways but with interferon signature pathways as outstanding. Clearly, ROS regulates arthritis through effects on the immune system in the priming but through the innate system in the effector stage, opening a new angle for understanding the different stages for the development of RA.
Results
Ncf1 knock-ins lacked NCF1 expression
To identify the disease phase, during which Ncf1 regulates protection in CIA in mice, we generated Ncf1 knock-in mice by targeted modification into the Ncf1 locus (Fig. 1A). We aimed at generating mice in which the expression of the NCF1 protein with a loss-of-function mutation could be replaced with the wild-type protein. We utilized the natural SNP previously identified in the rat Ncf1 (41), leading to diminished capacity of activating the NOX2 complex, subsequent defects in ROS production, and, ultimately, increased susceptibility of arthritis (23) as well as several other autoimmune conditions (19).
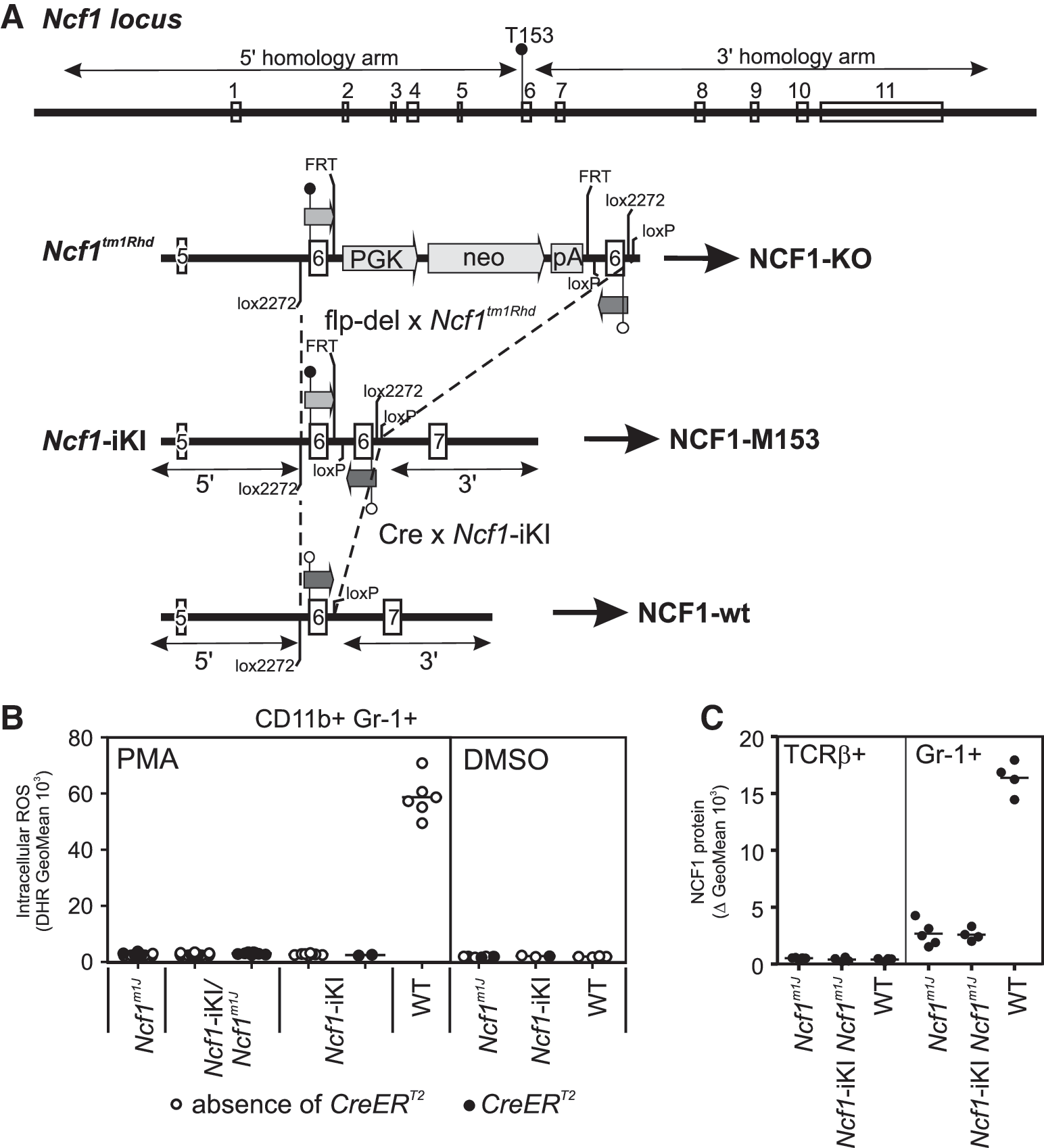
When this single amino acid change (T153 M) was introduced into mouse NCF1 protein by recombinant in vitro mutagenesis, the capacity of NCF1-M153 to activate the NOX2 complex to produce ROS was significantly reduced (unpublished observation; Sareila O., Olofsson P., Holmdahl R.), similar to the case with the rat NCF1-M153 (23). However, when this mutation was knocked into the Ncf1 locus together with the corresponding wild-type exon in reverse orientation (Ncf1-iKI in Fig. 1A), intracellular NOX2-dependent ROS production was completely abolished (Fig. 1B). The Ncf1m1J
mutation is known to be defective in activating the NOX2 complex (20, 47), and we confirmed that ROS production by Ncf1 knockout (Ncf1tm1Rhd
in Fig. 1A) cells was comparable to Ncf1m1J
cells (Supplementary Fig. S1; Supplementary Data are available online at
Similarly, NCF1 protein expression was not detected in Ncf1-iKI mice (Fig. 1C). Cells from Ncf1m1J mice served as a negative control since this mutation is known to lead to aberrant mRNA splicing and expression of trace amounts of a protein variant lacking the epitope for the monoclonal anti-NCF1 (D-10) antibody (20, 48). The absence of NCF1 protein in Ncf1-iKI mice was further confirmed by intracellular staining with a polyclonal anti-NCF1 antibody produced in-house (data not shown). We concluded that the Ncf1-iKI mice generated in this study lacked NCF1 protein and were, thus, completely defective in NOX2 complex-derived ROS, which could be caused by the palindromic exons in the locus in Ncf1-iKI mice.
As biological evidence for the absence of NCF1, we exposed the Ncf1-iKI mice to mannan-induced psoriatic arthritis known to be exacerbated in BQ.Ncf1m1J mice in comparison to the wild-type mice (31), and we found the Ncf1-iKI mice to develop remarkably more severe disease than their wild-type littermates (Supplementary Fig. S2). The disease course of Ncf1-deficient mice was similar irrespective of the presence or the absence of the inactive CreERT2 recombinase, thus proving that there is not even minor leakage from the targeted Ncf1-iKI allele before CreERT2 activation by tamoxifen administration.
Ncf1 expression can be fully activated from the ROS-deficient Ncf1-iKI locus to lead to conditional Ncf1 wild-type expression
To conditionally express NCF1, the Ncf1-iKI mice were crossed with CreERT2 mice in which the Cre recombinase may be activated by tamoxifen administration. We verified by ROS assay that the presence of the inactive CreERT2 recombinase is not able to recombine the Ncf1-iKI allele into its active form either in granulocytes (CD11b+Gr-1+) or in monocytes/macrophages (CD11b+Gr-1−) (Fig. 2A; open and closed symbols for the absence and presence of the CreERT2 allele, respectively).

One functional Ncf1 allele is enough to activate the NOX2 complex (ROS levels are typically 75–100% of those in WT) (24), and, therefore, we used Cre-activated Ncf1-iKI in combination with Ncf1m1J , to investigate the biological effects of allelic conversion of Ncf1. These mice were compared with littermate mice with one wild-type Ncf1 allele (designated WT#, Table 1). After Cre-mediated in vivo allelic conversion of Ncf1 by tamoxifen administration, the extent of intracellular ROS production, and the proportion of cells capable of producing ROS, resembled the response in WT# cells (Fig. 2B). Immense differences among the WT# mice remained undetected, and neither was ROS production affected by the presence or the absence of CreERT2 within the WT# group (Fig. 2B), which allowed us to utilize all littermate mice with one wild-type Ncf1 allele as wild-type controls.
−, absence of NOX2-derived ROS; +++, 75–100% of wild-type ROS production; ++++ wild-type ROS production.
The group WT# comprises mice with one copy of the wild-type (+) Ncf1 allele (heterozygotes), and served thus as wild-type controls to the Ncf1-iKI/Ncf1m1J CreERT2 mice in which one copy of the Ncf1 gene can be activated by tamoxifen administration.
Not only circulating cells but also tissue granulocytes expressed proper levels of NCF1 protein already 4 days after the final tamoxifen dose (Fig. 2C). ROS production enabled by the Cre-activated Ncf1-iKI allele was detected in vivo 6 weeks later (Fig. 2D), verifying the stability of the LoxP-Cre recombination event of the Ncf1-iKI allele.
Ncf1 knock-ins developed milder CIA when Ncf1 activity was restored in adulthood
To study whether Ncf1 exerts its protective role in autoimmunity during the development of central tolerance, we let the mice live without functional Ncf1 during their infancy and juvenility until Ncf1 expression from the Ncf1-iKI allele was induced by tamoxifen (average 8,1 weeks, range 5–14 weeks, median 7 weeks). Before immunization, Ncf1-iKI activation had led to almost full ROS production capacity (representative result shown in Fig. 2B). ROS-deficient Ncf1-iKI/Ncf1m1J mice developed CIA; whereas the Ncf1-iKI/Ncf1m1J CreERT2 mice developed only mild clinical symptoms (Fig. 3A) and, thus, resembled the wild-type controls. Ncf1-iKI/Ncf1m1J CreERT2 females were completely resistant to clinical symptoms of CIA (Supplementary Fig. S3). We observed a trend toward lower levels of anti-CII antibodies after the priming in ROS-sufficient mice when compared with Ncf1-deficient littermates (Fig. 3B).
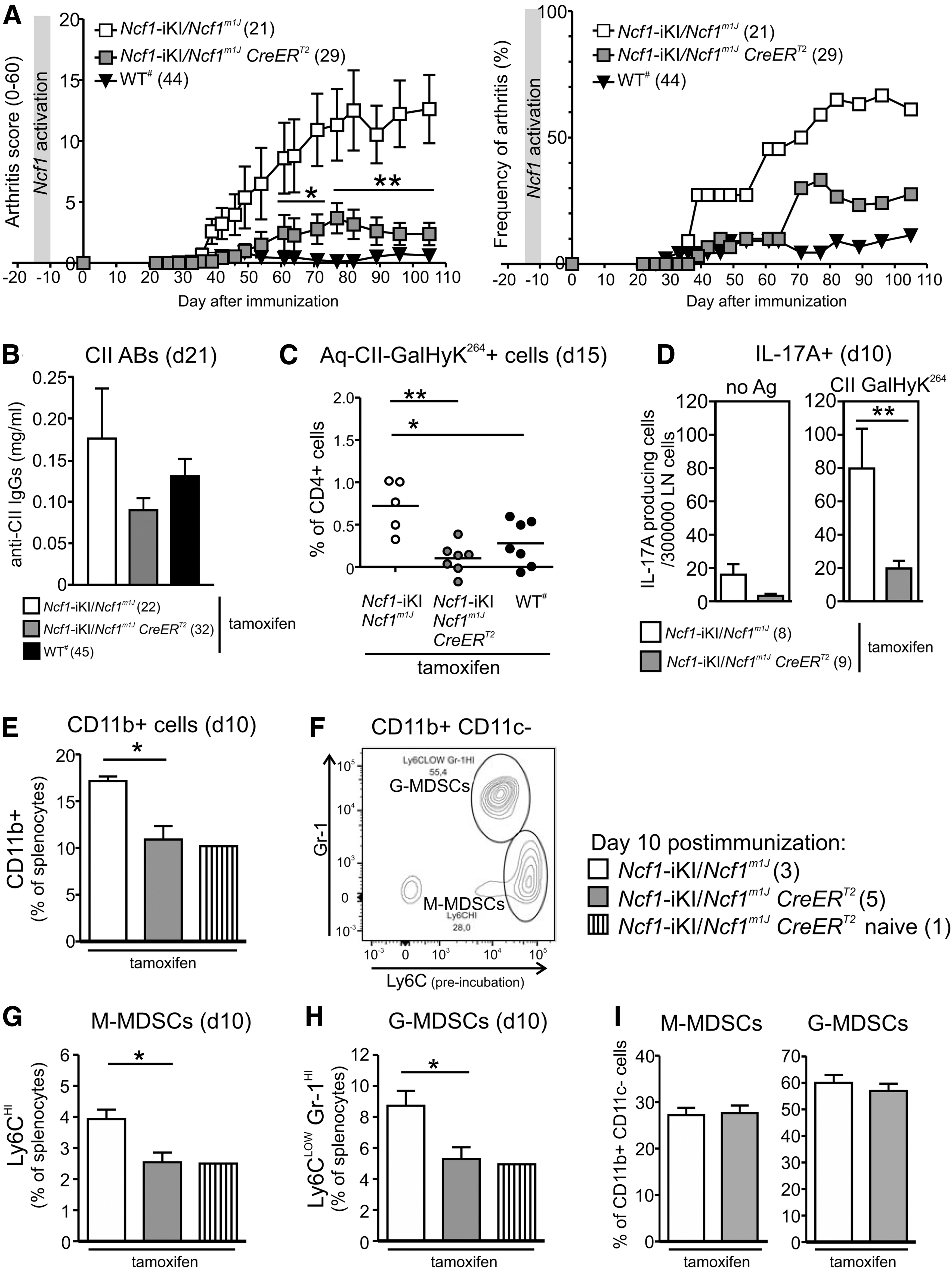
To investigate whether Ncf1-iKI activation led to alteration of the CII-specific T cell responses, we determined the number of antigen-specific T cells by using specific tetramer staining. The number of CD4+ cells recognizing the MHCII(Aq)-loaded CII-GalHyK264 peptide was significantly increased in ROS-deficient mice during priming of CIA, but Ncf1-iKI/Ncf1m1J CreERT2 mice were resistant to the oligoclonal expansion of these cells (Fig. 3C). Likewise, the number of IL-17-producing cells after the CII-GalHyK264 peptide stimulus was significantly diminished in the draining lymph nodes of mice with functional Ncf1 when compared with their Ncf1-deficient littermates (Fig. 3D).
Myeloid-derived suppressor cells (MDSCs) and their interplay with the IL-17-producing cells have recently gained attention as possible regulators of CIA (8, 12, 16, 59). MDSCs can be classified as granulocytic (G-MDSC) or monocytic (M-MDSC) based on the expression of Ly6C and Ly6G (both recognized by the Gr-1 antibody). Ncf1 deficiency was associated with accumulation of CD11b+ monocytes into the spleens during priming (Fig. 3E). The frequencies of M-MDSCs (CD11b+ CD11c− Ly6CHI) and G-MDSCs (CD11b+ CD11c− Ly6CLOW Gr-1HI) (Fig. 3F) were higher in Ncf1-deficient mice than in mice in which Ncf1 had been activated before immunization (Fig. 3G, H). A similar trend was observed in Ly6c1 gene expression in both blood and spleen of Ncf1-deficient mice during CIA (Supplementary Fig. S4). Nonetheless, the frequencies of M-MDSCs or G-MDSCs within the CD11b+CD11c− population were not affected (Fig. 3F, I).
Ncf1 exerts its protective effect also during the effector phase of arthritis
To investigate whether Ncf1 plays a role during the priming in CIA, we immunized the Ncf1-deficient (Ncf1-iKI/Ncf1m1J CreERT2 ) CIA-susceptible mice and induced Ncf1-iKI recombination with tamoxifen after the priming phase (at day 10). ROS production is not detectable in blood cells within the first 42 h after tamoxifen administration, but NCF1 expression in tissue cells reaches wild-type levels by day 9 after the start of tamoxifen administration (Fig. 2C). A CII booster immunization was given day 50 to enhance the development of arthritis. Even though the initial pathways and cellular activation during the priming had taken place without functional Ncf1, allelic conversion of Ncf1 was able to ameliorate the clinical symptoms of the disease (Fig. 4A). There was no difference in anti-CII autoantibody response between the groups before the CII re-immunization (at day 50) neither later during active disease (day 64) (Fig. 4B).
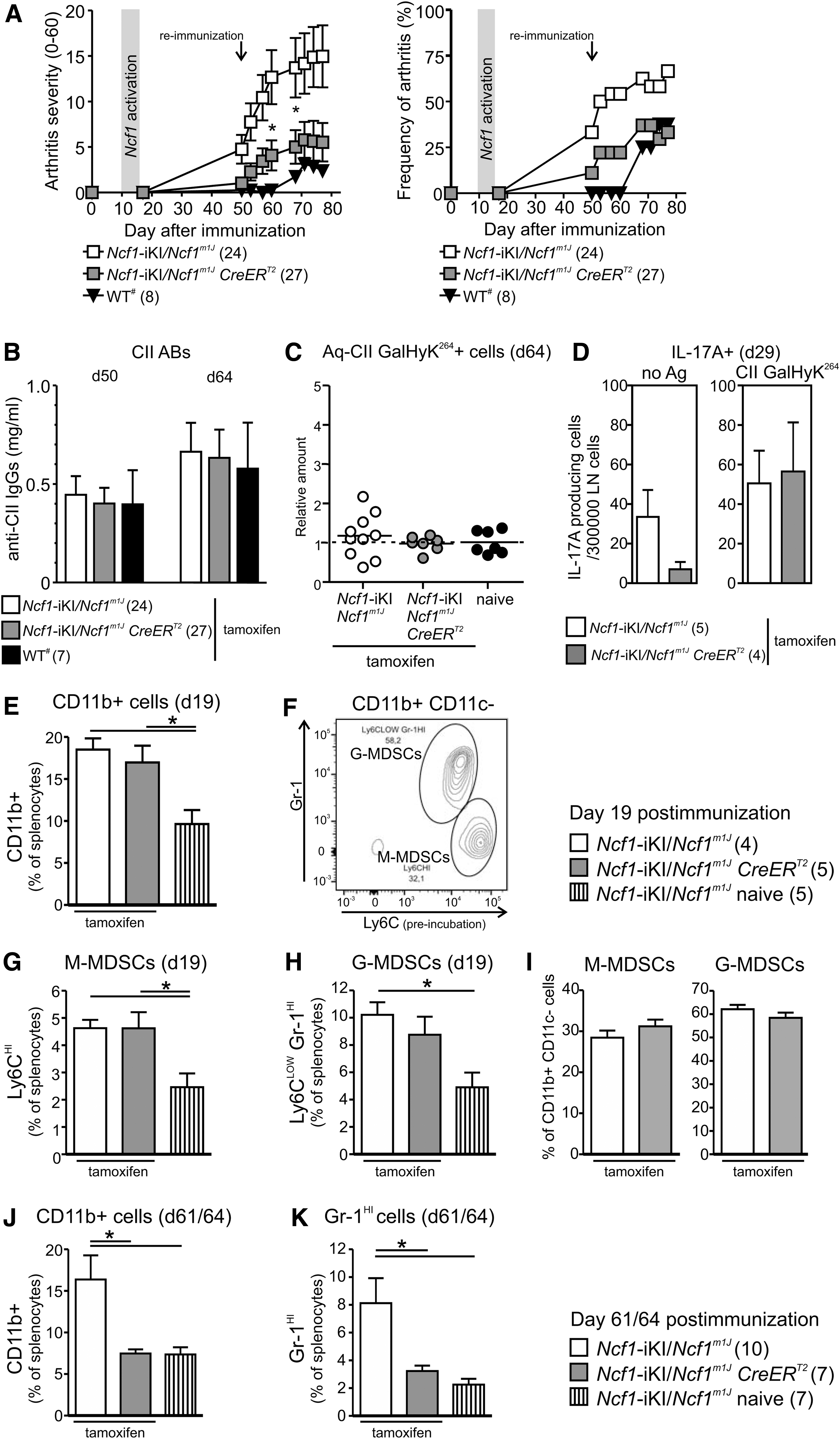
To investigate whether Ncf1-iKI activation after the priming phase of CIA is enough to alter the CII-specific T cell responses, both tetramer stainings and ELISPOT assays were performed. We could not detect consistent expansion of GalHyK264 CII peptide-specific CD4+ T cells in the spleen (Fig. 4C). Neither did the allelic conversion of Ncf1-iKI affect the number of IL-17A+ lymphocytes in the draining lymph nodes of the immunized mice when stimulated with the CII-GalHyK264 peptide (Fig. 4D).
Immunization led to an increase in CD11b+ cells in the spleens of Ncf1-deficient mice (Fig. 4E). By day 19, the recent activation of Ncf1 did not lead to a reduced frequency of CD11b+ cells in Ncf1-iKI/Ncf1m1J CreERT2 mice (Fig. 4E). Frequencies of M-MDSCs and G-MDSCs also remained unaffected (Fig. 4F–I). In course of time, functional Ncf1 diminished CD11b+ and Gr-1HI populations in the spleen when compared with Ncf1-deficient mice (Fig. 4J, K). There was a positive correlation between the clinical score of arthritis and the frequency of both CD11b+ cells (Spearman r = 0.7973, p < 0.0001) and Gr-1HI cells (Spearman r = 0.6974, p = 0.0019) in the spleens of immunized mice (n = 17) during active disease.
Ncf1-regulated gene expression changes during CIA
We have recently described that NOX2 complex deficiency due to the Ncf1m1J mutation induced significant changes in gene expression in naïve mice (29). To find the Ncf1-regulated gene expression alterations determining the susceptibility and severity of arthritis, we conducted a similar hypothesis-free genome-wide expression analysis in BQ.Ncf1m1J and BQ.Ncf1wt mice exposed to CIA. Twenty of the most significantly differentially expressed genes were confirmed by quantitative real-time PCR (Supplementary Tables S1 and S2). We also found reduced Ncf1 mRNA expression in samples from Ncf1m1J mice, known to express diminished levels of this gene (20), thus further validating the microarray data.
As described earlier (25), Ncf1m1 J mice developed more severe CIA (Fig. 5A and Supplementary Fig. S5). Flow cytometric analysis of the main cell populations revealed a statistically significant expansion of granulocytes (CD11b+Gr-1HI) in both blood and spleen of Ncf1m1J mice when analyzed after priming of CIA (Supplementary Fig. S6A, B). Similar expansion of the granulocyte pool was observed later during the disease, although the difference did not reach statistical significance in the blood samples (Supplementary Fig. S6C, D). In addition, the B cell (B220+) compartment was significantly reduced in both blood and spleen samples collected from the Ncf1m1J mice compared with samples from the wild-type mice at day 44. These results reflect changes in response to immunization, since no biologically significant differences were observed when naïve Ncf1m1J and wild-type mice were compared (29).
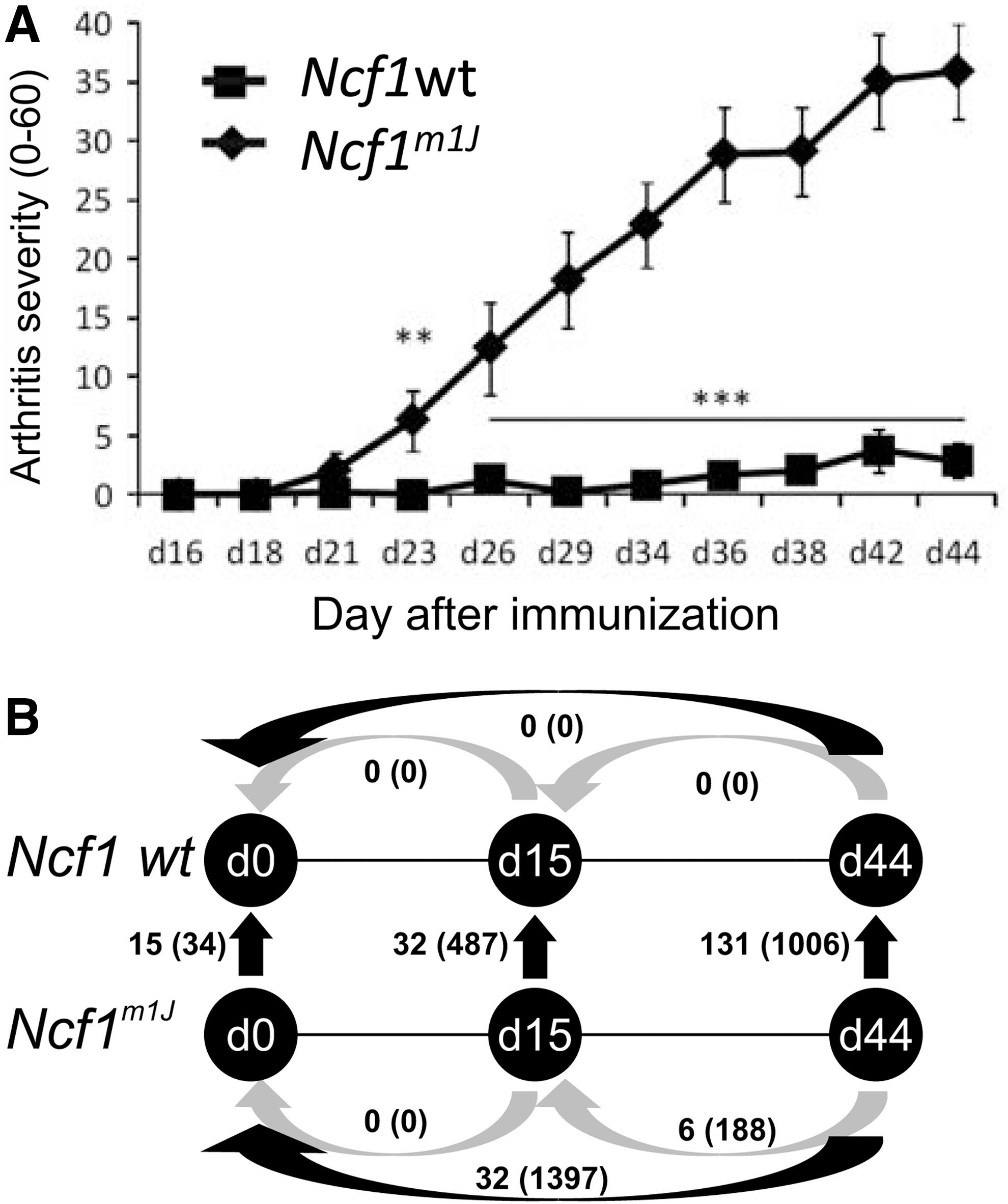
In genome-wide gene expression analysis, the number of differentially regulated genes (adjusted p-value <0.05 and FC >1.5 or FC<-1.5) was moderate (15 in blood and 34 in spleen) when naïve Ncf1m1J and wild-type mice were compared (29). Before the onset of CIA, the numbers of genes had increased to 32 in the blood and 487 in the spleen (Fig. 5B), even though no clinical signs of arthritis could be observed in either of the genotypes. Development of clinical disease further increased the number of differently expressed genes.
Ingenuity pathway analysis (IPA) was performed to gain hypothesis-free insights into the gene expression changes resulting from Ncf1 deficiency during CIA. Interferon signaling was the most significantly altered pathway in the blood (Supplementary Table S3), even though the expression of interferons themselves were not altered (Supplementary Table S4). Majority of the identified pathways were associated with immunological processes: There were very significant changes in pathways regulating antigen presentation, inflammatory disorders, and lymphocyte function as well as innate immunity. Based on this information, the most differently regulated genes (FC >2.0 or FC<−1.5, and adjusted p-value <0.05) were categorized into inflammation or lymphocyte-related genes as well as genes involved in miscellaneous processes. Because of the pronounced upregulation of interferon signaling in the absence of phagocyte ROS, we paid special attention to the genes whose expression is regulated by interferons.
Ncf1-regulated IFN signature in the blood is reversible
In naive mice, all the genes in the blood that were differently regulated in BQ.Ncf1m1J and BQ wild-type mice are known to be interferon regulated (29). Stat1 (signal transducer and activator of transcription 1) and Ifitm3 (interferon-induced transmembrane protein 3) transcripts were upregulated in the Ncf1m1J mouse profile, and upregulation of STAT1 was also shown at the protein level (29). To investigate whether the cellular phenotype is similarly Ncf1 regulated in the conditional model, we quantified STAT1 and IFITM3 protein levels in the Ncf1-iKI mice. Before the Ncf1-iKI gene activation, both STAT1 and IFITM3 were significantly over-expressed in Ncf1-deficient mice when compared with the wild-type mice (Fig. 6A, B). Already 7 days after the tamoxifen treatment, circulating cells that acquired functional Ncf1 gene had lost the interferon signature typical for Ncf1-deficient mice, and they had adopted a phenotype similar to the wild-type cells by downregulating both STAT1 and IFITM3 (Fig. 6C, D). Similar results were obtained 10–14 days later, indicating a permanent change (data not shown).
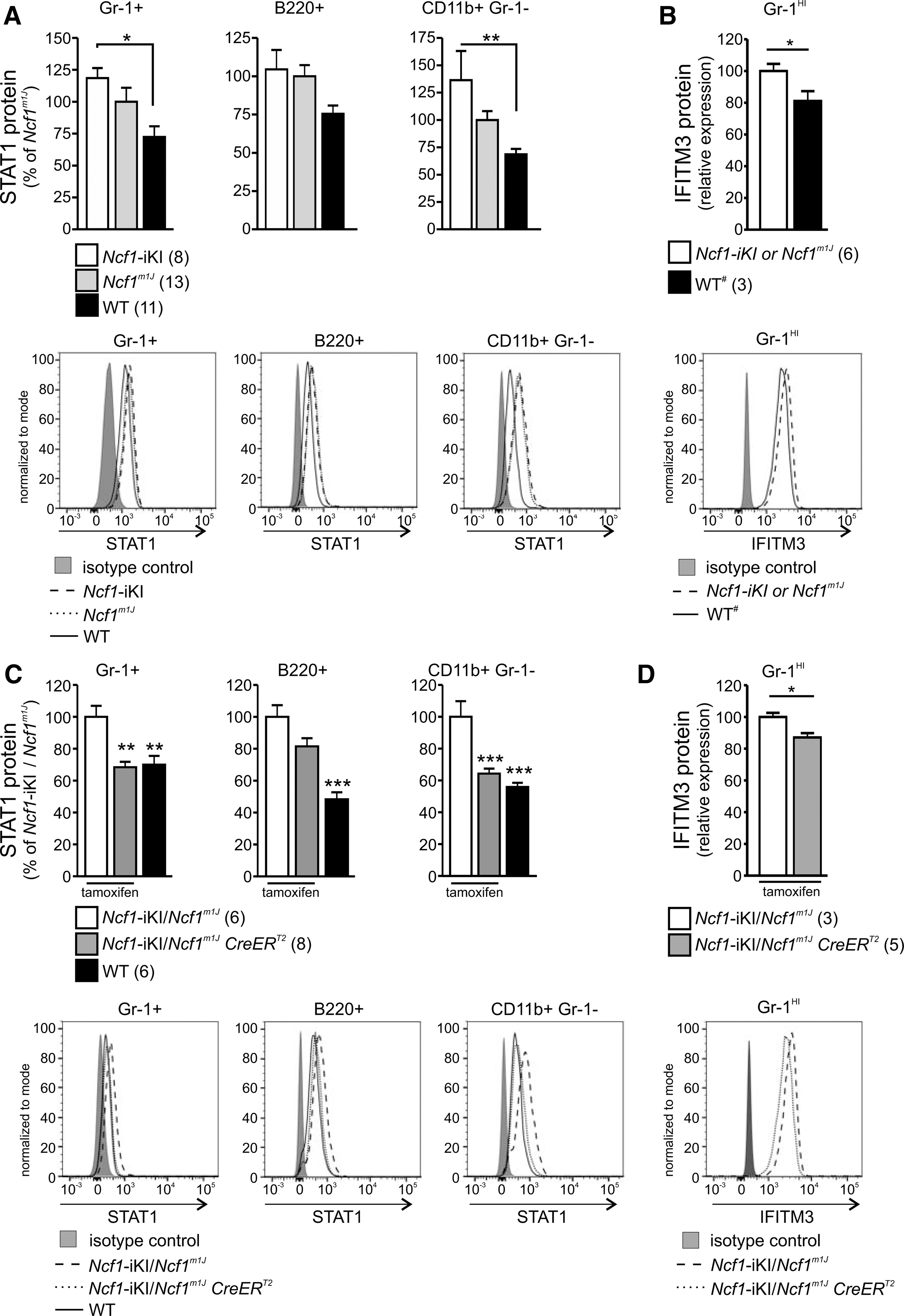
A further intensified IFN signature in BQ.Ncf1m1J mice was discovered during arthritis development in the blood (Supplementary Tables S5 and S6). Typical interferon-regulated genes (IRGs) such as Gvin1 (GTPase, very large interferon inducible 1) were remarkably upregulated in the Ncf1m1J mouse in comparison to the wild type. Majority of the inflammation or lymphocyte-related genes, in addition to genes involved in miscellaneous processes, were found to be IRGs. These include the immunity-related GTPase family M proteins (Irgm1 and Irgm2) as well as the family of interferon-induced transmembrane proteins (Ifitm2, Ifitm3 and Ifitm6), in addition to Stat1 transcription factor, some of which are already upregulated in naïve BQ.Ncf1m1J mice in comparison to wild-type mice (29).
Arthritis development was accompanied by upregulation of inflammatory transcripts
A pronounced inflammatory gene expression signature was observed in the blood and spleen samples of the Ncf1m1J mice (Supplementary Tables S5 and S6 and Fig. 7A, B). In the blood, the number of inflammation-related upregulated genes (FC >2.0, adjusted p-value <0.05) increased from one to 13 on the development of clinically apparent arthritis; whereas in the spleen samples, the increase was from 15 to 27 genes, reflecting the development of clinical arthritis.
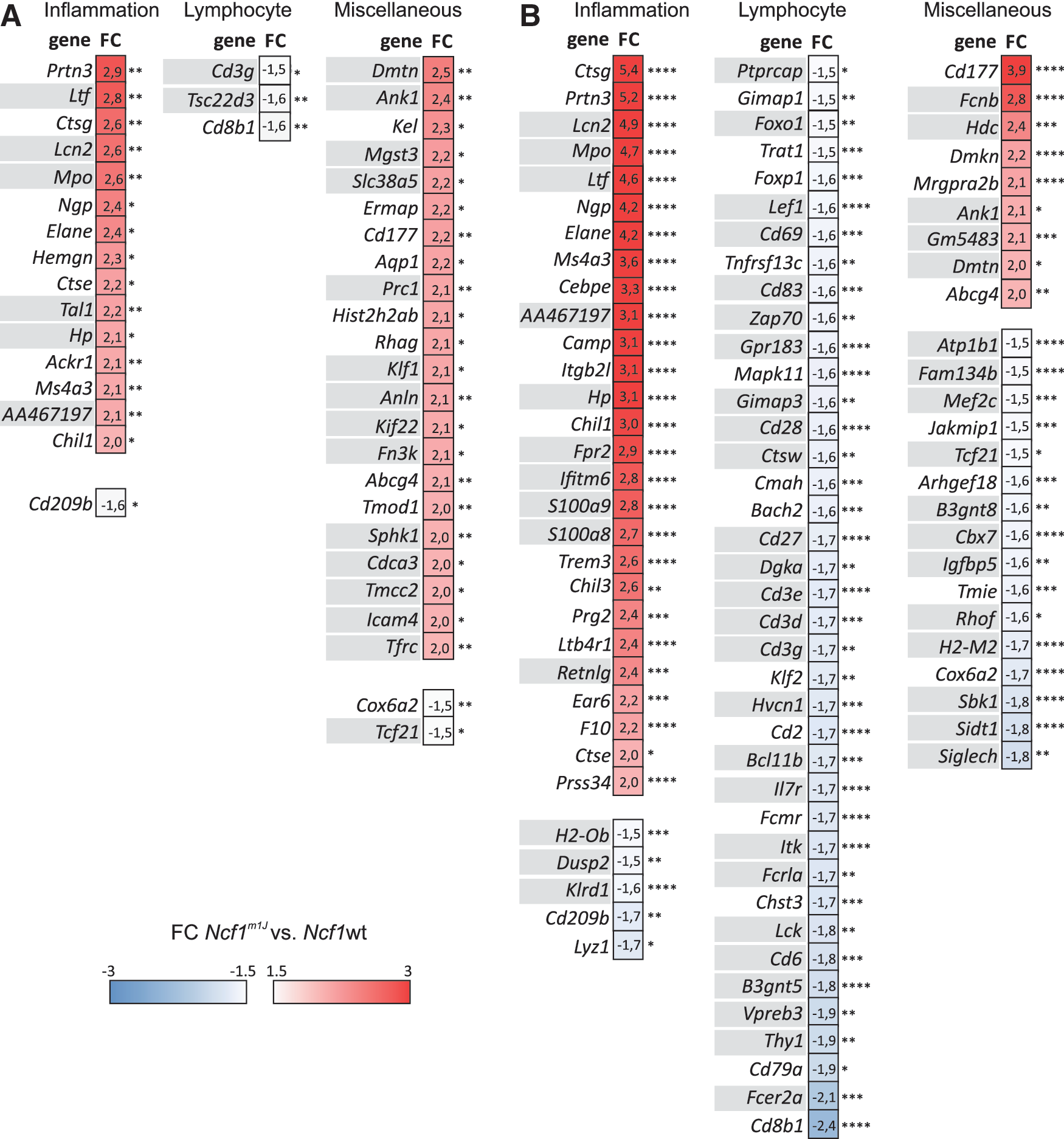
Upregulation of neutrophil-associated genes such as Ngp (neutrophil granule protein), Lcn2 (lipocalin 2), Elane (elastase, neutrophil expressed), Ltf (lactotransferrin), and Hp (haptoglobin) originated most likely from the granulocyte population expanded on arthritis development. The biologically modest increase in the granulocyte populations reached statistical significance in the blood samples after disease priming (day 15) and in both tissues collected at day 44 postimmunization (Supplementary Fig. S6A, C, D).
Remarkable downregulation of several B lymphocyte-related genes (e.g., Vpreb3, Fcrla, and Chst3) in Ncf1m1J mice during active arthritis in blood (Supplementary Table S6) and in spleen (Fig. 7B) is explained by decreased B cell compartment in both samples (Supplementary Fig. S6C, D). The arthritis-associated T lymphocyte signature in the spleen, however, could not be explained by changes in the main T lymphocyte populations (Supplementary Fig. S6D).
A significant downregulation of T cell-specific genes such as Thy1 (thymus cell antigen 1, theta), Cd6 (CD6 antigen), and Lck (lymphocyte protein tyrosine kinase) and the CD3 antigen polypeptides (CD3 g, Cd3d, Cd3e) was observed. Both conventional (CD25+FoxP3+) and unconventional (FoxP3− CD49b+) regulatory T cells are known suppressors of arthritis (6, 39), but the expression of neither Foxp3 nor Itga2 (integrin alpha 2, coding for CD49b) differed between BQ.Ncf1m1J and BQ.Ncf1wt mice during CIA (Supplementary Fig. S7). The interferon signature detected in the blood of Ncf1m1J mice (29) (Fig. 6 and Supplementary Tables S5 and S6) was obvious also in the spleen, since majority of the differently expressed genes involved in inflammation, lymphocytes, or miscellaneous processes are known to be responsive to interferons (46) (Fig. 7).
Allelic conversion of Ncf1 is able to alter gene expression after priming of CIA
To investigate whether the Ncf1-dependent gene expression changes during CIA are reversible, we studied mice that got functional Ncf1 by allelic conversion either before immunization or after the priming phase of CIA (protocols similar to those in Figs. 3A and 4A, respectively). Disease priming induced upregulation of inflammation-associated genes Ctsg, Chil1, Cd177, Hp, and Lcn2 in Ncf1-deficient (Ncf1-iKI/Ncf1m1J ) mice (Fig. 8A). Upregulation of gene expression was significantly milder if the mice expressed functional Ncf1 at the time of immunization (Fig. 8A; Ncf1-iKI/Ncf1m1J CreERT2 and WT# mice).

A similar trend was observed after re-immunization, when the expression of Ncf1 had been initiated from the Ncf1-iKI locus after the disease priming (Fig. 8B). CIA immunization decreased expression of Cd2, a representative T cell-associated transcript, in Ncf1-deficient mice (Fig. 8A). Functional Ncf1 limited extensive Cd2 downregulation during priming, and it was even able to restore a phenotype similar to naïve mice when the Ncf1 gene was activated after the disease priming phase in Ncf1-iKI/Ncf1m1J CreERT2 mice (Fig. 8B). The changes in gene expression in different genotypes are in line with the changes observed in the major leukocyte populations in the spleen samples (Supplementary Fig. S8).
Discussion
We generated conditional Ncf1 knock-in mice, which are completely deficient of NCF1 protein expression and production of NOX2-derived ROS. Ncf1 deficiency predisposed to autoimmune arthritis (CIA) and T cell-independent psoriatic arthritis (MIP) as reported earlier (25, 31). In the newly generated Ncf1 knock-ins, allelic conversion initiates physiological NCF1 expression and generation of NOX2-derived ROS, reaching levels comparable to wild type. Interestingly, it affected both T cell immune response and arthritis during the priming phase but only arthritis when exposed during the effector phase. The data provide novel information on the importance of Ncf1 not only in preventing the initial priming response to antigenic peptide but also in maintaining tolerance on re-challenge.
The development of RA may be characterized by distinct phases. Initially, an unknown environmental factor activates the immune system, leading to characteristic autoantibody production. Possibly even several years later, the joint tissue inflammation causes the first clinical symptoms to arise. Without effective medication, the disease turns into chronic relapsing arthritis that is characterized by tissue destruction and remodeling. (4, 17). Our data show that Ncf1/ROS exposure during thymus/tolerance development cannot be a major explanation for the observed regulatory effects of Ncf1 on autoimmune arthritis since newly activated Ncf1 effectively limited the number of collagen peptide-specific autoreactive T cells, thereby limiting the development of an immune-mediated arthritis.
Interestingly, however, Ncf1 activation after the symptomless priming phase of CIA resulted in milder disease. This indicates that it can reduce arthritis severity and limit inflammation even during the effector phase. Whether allelic conversion of Ncf1 during active disease would result in amelioration of the disease remains to be investigated. NOX2 agonists have been shown to ameliorate ongoing disease in animal models of autoimmune inflammation (22), thus pointing to an ROS-dependent mechanism in resolving joint inflammation.
During priming, Ncf1 suppressed the expansion of T cells specific for the immunodominant CII Gal-HyK264 peptide. The suppression is most likely due to reduced proliferation, as earlier shown by IL-2 production (24), and not due to reduced survival of the cells. Ncf1 especially limited the number of CII Gal-HyK264-specific cells secreting IL-17A.These results confirm the important role of T cells in mediating the Ncf1-dependent ROS control of arthritis (13, 24), in particular, since it is known that the Th17 cells are pathogenic in CIA (40). However, these Ncf1-dependent events during the priming of CIA do not solely determine the severity of the clinical disease later on. When Ncf1 gene was activated after the priming, no effect on T cell expansion could be observed, indicating that different ROS-regulated mechanisms operate during this phase to drive the arthritis process. Importantly, however, it should be pointed out that we were not able to measure an autoreactive T cell involvement that could play a role in the later phases of the disease. The immunodominant epitope differs at position 266 between endogenous mouse CII and the immunogenic rat CII. The peptide can still bind to the MHC molecule with a lower affinity and such autoreactive T cells, and it could trigger different sets of T cells. These are difficult to detect with tetramers or recall assays but are likely to operate in vivo.
MDSCs have been described as regulators of arthritis and the Th17 response in CIA (8, 12, 16, 59). In our studies, CIA induction was associated with an increase in both M-MDSCs and G-MDSCs in Ncf1-deficient mice, and activated Ncf1 led to a reduction of these populations in the spleen. Expansion of MDSCs was associated not only with disease severity but also with increasing number of IL-17A+ cells in an Ncf1-dependent manner. When Ncf1 was activated after the disease priming, its effects were not immediately seen in the splenic monocyte populations and detected neither in the number of autoreactive T cells nor in IL-17A+ cells. In course of time, the presence of NCF1 was associated with a reversion of the already expanded splenic monocyte population to the levels found in healthy mice.
These associations are in line with the proposed pathogenic role of MDSCs in CIA (16, 59), but we cannot exclude the possibility that these cells had expanded in response to the inflammation, and their role could, in fact, be to fight the emerging inflammatory attack as suggested by others (8, 12, 56, 60). Still, an interesting possibility is that Ncf1-dependent ROS could promote a suppressive effect of macrophages, thus explaining some of the effects mediating protection against arthritis.
Extensive studies have been conducted by others to investigate the causality between the splenic monocyte populations and CIA severity using adoptive cell transfer and in vivo cell depletion by antibodies. However, the phenotypic differences of the MDSC subpopulations due to methodological limitations such as antibody specificities and the purity of transferred cells, as well as the variation due to the genetic background of the recipients, might have contributed to the discrepancy of the conclusions. Thus, further studies are needed to investigate the role of specific MDSC populations in CIA pathogenesis, and specific Cre mice in combination with the conditional Ncf1 mouse described in the present study might be one of those tools that are used to elucidate the causative relationships between MDSCs and CIA.
ROS have previously been shown in vitro to affect both macrophages development and their polarization to type II (51, 61). Hence, there is a possibility that some of the Ncf1-regulated mechanisms involve macrophage development and/or polarization, especially considering that macrophage ROS have been shown to be critical in protecting from CIA (14).
We studied ROS-dependent genome-wide gene expression changes during arthritis development and during severe experimental arthritis in mice. Arthritis development induced changes in the expression levels of T cell transcripts as well as induced upregulation of inflammatory transcripts. Expression of some genes (10 in blood, 26 in spleen) differed in Ncf1m1J in comparison to wild type at all time points (naïve, day 15 and day 44 p.i.), indicating the presence of a stable, oxidation-modified gene expression pattern that is not affected by the induction of experimental arthritis.
Defects in Ncf1 have been found to result in type I interferon signature (and autoimmune SLE-like phenotype on the Balb/c background) in naïve Ncf1m1J mice (29). In the present study, by utilizing the Ncf1 knock-out model, we confirmed that the pronounced interferon signature is a result of Ncf1 deficiency, and we demonstrated that restoration of Ncf1 gene function by Cre-mediated recombination in vivo in adult mice normalized the interferon response.
The interferon signature that we identified in the blood samples collected from arthritic mice consisted of both type I and type II IFN responsive genes. In addition, Ncf1 deficiency led to over-expression of STAT1 and IFITM3. Recent studies show that STAT1 gain-of-function mutations underlie autoimmune diseases in humans (53). IFITM3 was upregulated in patients with primary Sjögren's syndrome, a prototypic autoimmune disease (15), and a study suggests that the haplotypes of the IFITM3 polymorphisms may be associated with a susceptibility to RA in a Korean population (35).
The co-occurrence of type I and II IFN signals during arthritis has been previously reported in RA patients' peripheral blood samples (28). Interferon-mediated immunity has also been reported as a predictor of arthritis development (36, 54), further linking interferon responsiveness and arthritis susceptibility. Our previous report (29) has shown interferon signature in the naïve mice and similarly as in humans, the present study shows that it is a good predictor of development of experimental arthritis in the mouse.
Diversification of IFN-α- or IFN-β-mediated response programs in different autoimmune diseases have been recently reported (9), and a subgroup of arthritic patients with higher interferon gene expression signature in blood neutrophils reported to respond better to TNF inhibitor therapy (58). These findings point to divergent pathogenic roles for type I interferons in different autoimmune conditions. NOX2 deficiency has been connected to an increased risk to develop autoimmunity (10), and our findings relate NOX2 deficiency with interferon signature (29).
We identified remarkable downregulation of T lymphocyte-related genes in the arthritic Ncf1m1J mouse when compared with the wild type. It is known that activated T cells downregulate CD3. T cell-specific genes (such as Thy1, Cd6, Lck, and the CD3 antigen polypeptides) are IRGs; thus, it may be a consequence of the Ncf1m1J -dependent interferon signature, differences in the cell populations, or it may partly be a more direct effect caused by the lack of ROS in Ncf1m1J mice. It is an open question as to how the downregulated lymphocyte activation pathway is related to the observed activation and expansion of antigen-specific T cells. This could be due to effects on different populations and an effect by ROS to activate regulatory T cells, as has earlier been suggested (13), even though the frequencies of regulatory T cells were not affected by Ncf1. Similarly, regulatory T cell counts were normal in NCF1/NOX2-deficient patients, but the induction of regulatory T cells by macrophages was dependent on ROS (33). We also identified a number of B lymphocyte-related genes with significantly diminished expression levels in the oxidation-deficient Ncf1m1J mouse suffering from experimental arthritis. This could, however, be explained by the reduced B cell pool in the mutated mouse, as revealed by flow cytometry. Similarly, the expanding inflammatory signature mainly consisting of gene transcripts of mostly neutrophil and macrophage origin could, at least in part, be explained by the slightly different phagocyte populations. Our results describing the inflammatory pathway are very similar with the previous findings in RA, the meta-analysis of which has been reported (52).
In conclusion, our results show that Ncf1 suppresses CIA through effects both during T cell priming and also during the effector phase when the first immune priming has already been established. Obviously, it is possible that the effects on the effector phase could be mediated by innate immune cells, as shown in the psoriasis model (26, 31), but it cannot be excluded that undetectable autoreactive T cell responses are affected in the CIA model. The situation is similar in RA in which the priming pre-RA phase is dependent on MHC II genes, whereas the association with a clinical onset and a later chronic development of the disease follows different underlying disease mechanisms (4, 17). The induced ROS response by the Ncf1/NOX2 complex is clearly instrumental in regulating both adaptive and innate immunity, and the understanding of the oxidative regulatory mechanism is also important for understanding the development of immune-mediated diseases such as RA.
Materials and Methods
Mice
Conditional Ncf1tm1Rhd mice were generated on request by Ozgene Pty Ltd. C57BL/6J BAC clones RP23-42C7 and RP23-41A15 (Roswell park cancer institute) were used to generate the targeting vector (Fig. 1A). JM8.F6 mouse embryonic stem (ES) cells (EuMMCR; European Mouse Mutant Cell Repository) derived from a C57BL/6 N mouse were electroporated with the linearized targeting vector. Neomycin-resistant colonies were picked, expanded, and screened by Southern blot to identify the ES cell clones with a properly integrated targeting vector. The targeted ES cells were injected into mouse blastocysts to obtain chimeric mice. By crossing the highly chimeric male mice with C57BL/6 J females, a germline transmission was achieved, resulting in mice that were heterozygous for the Ncf1tm1Rhd allele (C57BL/6 N-Ncf1tm1Rhd).
The FRT-site flanked Neomycin resistance cassette was removed by crossing the mice with FLP deleter mice (C57BL/6-Tg(CAG-Flpe)2Arte; originating from Taconic) that were kindly provided by Turku Center for Disease Modeling, University of Turku, Finland. The absence of the neomycin cassette in the progeny (denoted Ncf1-iKI) was verified by PCR genotyping.
The inducible CreERT2 gene originated from BQ.B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J mice from Jackson laboratories and had been backcrossed to C57BL/10.Q/rhd mice for six generations. These mice were crossed with B6 N.B10.Q.Ncf1m1J mice to generate B6 N.B10.Q.Ncf1m1J .CreERT2 mice carrying the spontaneous SNP (rs230824082, Ncf1m1 J) (20) and MHC class II Aq haplotype, making them susceptible to CIA (18). Cross between the strains B6 N.B10.Q.Ncf1m1J .CreERT2 and B6 N.Ncf1-iKI generated mice denoted Ncf1-iKI/Ncf1m1J .CreERT2 , which were thereafter intercrossed to generate the experimental mice. C57BL/10.Q/rhd mice with (denoted BQ.Ncf1 m1J ) and without (BQ.Ncf1wt) the Ncf1m1J mutation (20, 25) were used for the genome-wide gene expression studies. Sex- and age-matched littermates were used in all experiments unless otherwise stated, and the experiments started when the mice were 7–12 weeks old. The mice were euthanized with carbon dioxide, except for studies where terminal anesthesia was obtained by Mebunat vet (Orion Oyj) followed by retro-orbital i.v. injection of CD45-APC-Cy7 (BD Biosciences) antibody to label blood CD45+ cells.
Mice were housed under specific pathogen-free conditions as described earlier (47). The experiments were carried out under ethical permit numbers ESAVI-0000497/041003/2011 and ESAVI/439/04.10.07/2017. All animal experiments comply with the ARRIVE guidelines, and they were carried out in accordance with EU Directive 2010/63/EU for animal experiments.
Genotyping
Genotyping was done by PCR for the targeted Ncf1-IKI gene and by an SNP genotyping assay for the Ncf1m1J allele (Applied Biosystems) by using genomic DNA from an earpiece. Usually, the phenotype of the Ncf1-iKI/Ncf1m1J .CreERT2 mice was verified by determining the intracellular ROS production both before and after tamoxifen administration.
Reagents
Reagents were purchased from Sigma-Aldrich unless otherwise stated.
CIA and tamoxifen treatment
For CIA, the mice were immunized as described earlier (47). CIA for the genome-wide gene expression studies was induced in at least 8 week-old male mice by a single intradermal (i.d.) injection of 100 μg rat type II collagen (50) emulsified in Complete Freund's Adjuvant (CFA, Gibco, BRL, Invitrogen AB). Re-immunization (50 μg of bCII emulsified in IFA, i.d.) was given on day 21 postimmunization unless otherwise stated. Clinical symptoms were evaluated as described earlier (47). Recombination of the Ncf1-iKI allele in mice expressing the inducible CreERT2 was induced by an intraperitoneal injection of tamoxifen (Sigma-Aldrich; T5648), dissolved by sonication at +37°C into rapeseed oil with 5% ethanol, 4 mg/mouse/day, on day 0, day 1, and day 5. To exclude possible bias caused by the treatment, tamoxifen was administered to all experimental groups irrespective of their genotype, unless otherwise specified.
Mannan-induced psoriatic arthritis and in vivo imaging
Mannan from Saccharomyces cerevisiae (10 mg/mouse) was administered, and the disease followed as recently described (31). In vivo imaging of ROS production was performed as described earlier (30, 32).
Generation of MHCII(Aq)-CII peptide tetramers
The Aq/mCLIPmut molecules were designed according to a previous report (45) with modification. The native leader sequences of the a and b chains were replaced by a Drosophila BiP protein signal sequence. The extracellular domains of Aq were truncated, and the transmembrane regions were replaced by an acidic and basic zipper dimerization motif (45), respectively. A low-affinity peptide mCLIPmut (KPVSQARMATPLLMRP) followed by a thrombin cleavage site was covalently linked to the N-terminal of the b chain. In addition, at the C-terminal of the b chain, a 15-amino-acid AviTag peptide for site-specific biotinylation was inserted after the basic zipper whereas a polyhistidine tag was inserted after the acidic zipper at the C-terminal of the chain.
The genes were synthesized at Eurofins with KpnI and XhoI restriction sites at the 5′ and 3′ ends. The synthesized genes were restriction enzymes that were digested by using FastDigest™ enzymes (ThermoFisher Scientific). The digested DNA fragments were cloned into a mammalian expression vector pCEP4 (Life technologies) that was digested by using the same restriction enzymes. After sequence verification, the recombinant plasmids were co-transfected into Expi393F™ cells (Life technologies) with FectoPRO™ DNA transfection reagent (Polyplus transfection). The supernatants were harvested 6 days post-transfection. The recombinant protein was first captured by using a 5 ml HisTrap Excel (GE Healthcare Life Sciences) affinity column followed by size-exclusion chromatography on Superdex 200 pg (GE Healthcare life Sciences). The recombinant protein was purified as a single peak and was concentrated, diafiltrated into biotinylation buffer (20 mM Tris-HCl, 50 mM NaCl, pH 8.0) by using an Amicon centrifuge device with MWCO of 10 kDa.
Biotinylation using biotin-protein ligase was performed according to the manufacturer's instructions (Avidity), and the reaction was carried out at 30°C for 2 h. After biotinylation, thrombin (Novagen, 1 U thrombin to 1 mg purified protein) and the desired peptide in excess were added to replace the covalently linked mCLIPmut peptide with the desired peptide. The reaction mixture was incubated at ambient temperature overnight with slow rotation followed by incubation at 4°C for 2 days. Free biotin, cleaved peptide, and additional excess peptide were removed by size-exclusion chromatography on a Superdex 200 pg column. The Aq/peptide tetramer complexes were freshly prepared by adding PE-labeled streptavidin (Invitrogen, ThermoFisher Scientific) to the recombinant protein at a molar ratio of 1:4, and incubating at +4°C for 1 h.
Flow cytometry
Flow cytometric analyses were performed as described earlier (47), with slight modifications. Briefly, red blood cells were lysed by hypotonic buffer, Fc-receptors were blocked, and surface antigens were stained with fluorescently labeled antibodies. When antibodies against Ly6C and Gr-1 were used to stain the same sample, Gr-1 antibody was added 15 min later so as not to disturb the binding of the anti-Ly6C antibody, as they both recognize an epitope on Ly6C. FMO (fluorescence minus one) controls were used to verify specificity of signals from both antibodies. For intracellular staining, the cells were fixed and permeabilized with Cytofix/Cytoperm™ solution (BD Biosciences) according to the manufacturer's protocol. NCF1 expression was determined with either mouse monoclonal anti-NCF1 antibody (clone D-10; Santa Cruz Biotechnology) or in-house produced rabbit polyclonal anti-NCF1 (poly-B) raised against a peptide NH2-CRRNSVRFLQQRRRP-COOH. For detection, rat anti-mouse IgG1 APC or anti-rabbit IgG Alexa488 conjugates were used, respectively.
IFITM3 was stained with rabbit polyclonal anti-IFITM3 Antibody (11714-1-AP; Proteintech) and detected with Alexa Fluor 750 conjugated goat anti-rabbit IgG (H+L) secondary antibody (A-21039; Invitrogen, ThermoFisher Scientific). Rabbit polyclonal anti-human VEGF antibody A-20 (sc-152; Santa Cruz Biotechnology) was used as an isotype control. IFITM3 expression was quantified as an increase in geometric mean as compared with the isotype control. Expression in Ncf1-deficient cells was set as 100, and all values were normalized to that value.
To identify peptide-specific T lymphocytes, the cells were incubated with the Aq/peptide tetramer complexes (20 μg/ml) at +37°C for 1 h in the presence of 50 nM Dasatinib, a small-molecule protein tyrosine kinase inhibitor, whereafter they were stained for the cell surface markers. Viability staining solution (7′AAD; eBioscience) was added just before acquisition to exclude dead cells from the analysis.
The samples were acquired by using an LSR II or LSR Fortessa flow cytometer using FacsDiVa software (BD Biosciences), and the data were analyzed with either Flowing Software (Cell Imaging Core; Turku Centre for Biotechnology) or FlowJo Software (FlowJo LLC).
Measurement of intracellular and extracellular ROS
Intracellular and extracellular ROS production, as well as ELISA analysis of anti-collagen type II IgG antibody levels in the serum was determined, as recently described (47).
ELISPOT assays
Mouse/rat IL-17A ELISPOT Ready-SET-Go!® (eBioscience) was used to count the IL-17A-producing cells. Briefly, 96-Well PVDF Membrane ELISPOT Plates (Millipore, Cat. No. MAIPS4510, Merck Millipore) were coated with the capture antibody, washed, and blocked with RPMI-1640 medium containing 10% fetal calf serum.
Single-cell suspensions were obtained by mashing the inguinal lymph nodes through 70 μm cell strainers (ThermoFisher Scientific), and viable cell concentration was determined by trypan blue staining (Cellometer, Cellometer Auto 2000 Viability Counter; Nexcelom Bioscience). Lymph node cells (0.3 × 106/well) were incubated with the CII GalHyK264 peptide (CII259–273 with a β-D-galactopyranosyl residue on L-hydroxylysine at position 264) (3); 15 μg/ml or solvent control (acetic acid) at 37°C, 5% CO2 in a humidified incubator for 24 h. The spots were developed according to the manufacturer's protocol, and they were counted blindly under a dissecting microscope.
RNA extraction, genome-wide gene expression analysis, and quantitative real-time PCR
Samples for genome-wide expression analysis were collected from two independent CIA experiments with a similar experimental setup, and the results were analyzed together. RNA preservation, isolation, and purification as well as RNA amplifications, in vitro transcription, cRNA quality check, and the microarray were performed as recently described (29). The microarray data were quantile-normalized and log2-transformed. Differentially regulated genes were identified by using linear modeling (LIMMA packages, Bioconductor). For all further analyses, false discovery rate (FDR) adjusted p-values <0.05 are considered statistically significant. Microarray data have been deposited in the NCBI gene expression omnibus (GSE92530). IPA was performed as described earlier (29).
Selected transcripts passing inclusion criteria (adj. p-value <0.05 and FC >1.5 or FC < −1.5) were manually curated into functional categories based on information collected from the PubMed Gene (
For quantitative real-time PCR, splenic tissues were submerged into RNAlater® RNA Stabilization Solution (Ambion), and they were stored at −20°C. RNA was isolated with RNeasy Mini kit (Qiagen) following the manufacturer's protocol, including on-column DNAse digestion (RNAse-Free DNAse set; Qiagen). RNA quantity and quality (A260/A280 > 1.9) were assessed with a NanoDrop™ 2000 spectrophotometer (Thermo Scientific), and cDNA was synthesized with iScript cDNA Synthesis Kit (BIO-RAD). Samples were amplified by using Absolute QPCR ROX Mix (Thermo Scientific), utilizing primers and probes (Universal ProbeLibrary, Roche) listed in Supplementary Table S1. PCR cycling was performed with 7900 HT Fast Real-Time PCR System (Applied Biosystems), and relative quantitation was performed with either SDS or QuantStudio 12K Flex Software (Applied Biosystems) by normalizing to the GAPDH housekeeping gene.
Statistical analyses
Clinical arthritis scores were analyzed with the Mann–Whitney U-test, whereas cellular analyses were analyzed with the One-Way ANOVA combined with the Tukey post-test by using GraphPad Prism Software (GraphPad Software, Inc.) unless otherwise stated. In all analyses, the number of independent observations (n) refers to the number of mice used.
Footnotes
Acknowledgments
Flow cytometry was performed at the Cell Imaging Core, and the microarray was performed at the Finnish Microarray and Sequencing Centre at Turku Centre for Biotechnology, Turku, Finland. The authors thank personnel at both of these units as well as personnel at the Central Animal Laboratory of the University of Turku, Turku, Finland, for technical assistance.
Elina Wiik and Angel Yao Mattisson are acknowledged for their excellent secretarial help. The authors are thankful to Anni Kauppinen for technical assistance.
This study was funded by the Academy of Finland; Sigrid Jusélius Foundation; The Swedish Foundation for Strategic Research; The National Doctoral Programme in Informational and Structural Biology; Turku University Foundation; Knut and Alice Wallenberg Foundation; King Gustaf V 80-years Foundation; The EU Innovative Medicine initiative BeTheCure grant; The Swedish Research Council; The European Union's Seventh Framework Programme (FP7) under grant agreements Neurinox (Health-F2-2011-278611); and the Finnish Cultural Foundation, Varsinais-Suomi Regional fund. The funding sources were not involved in study design; in the collection, analysis, and interpretation of data; in the writing of this article; or in the decision to submit the article for publication.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.