
Abstract
Aims:
This study was designed to explore the neuroprotective potential of inorganic nitrite as a new therapeutic avenue in Parkinson's disease (PD).
Results:
Administration of inorganic nitrite ameliorates neuropathology in phylogenetically distinct animal models of PD. Beneficial effects are not confined to prophylactic treatment and also occur if nitrite is administered when the pathogenic cascade is already active. Mechanistically, the effect is mediated by both complex I S-nitrosation, which under nitrite administration is favored over formation of other forms of oxidation, and down-stream activation of the antioxidant Nrf2 pathway. Nitrite also rescues respiratory reserve capacity and increases proton leakage in LRRK2 PD patients' dermal fibroblasts.
Innovation:
The study proposes an unprecedented approach based on the administration of the nitrosonium donor nitrite to contrast complex I and redox anomalies in PD. Dysfunctional mitochondrial complex I propagates oxidative stress in PD, and treatments mitigating this defect may, therefore, limit disease progression. Therapeutic complex I targeting has been successfully achieved in ischemia/reperfusion by using nitrosonium donors such as nitrite to reversibly modify its subunits and protect from oxidative damage after reperfusion. This evidence led to the innovative hypothesis that nitrite could exert protective effects also in pathological conditions where complex I dysfunction occurs in normoxia, such as in PD.
Conclusions:
Overall, these results demonstrate that administration of inorganic nitrite improves mitochondrial function in PD, and it, therefore, represents an amenable intervention to hamper disease progression. Antioxid. Redox Signal. 28, 44–61.
Introduction
P
Parkinson's disease (PD) is a neurodegenerative disorder significantly affecting quality of life. Strategies to cure it, or even to slow down its progression, are not available and therapies are limited to symptoms' palliation.
We demonstrate that administration of inorganic nitrite mitigates neuropathology in multiple PD models. Mechanistically, nitrite improves mitochondrial function and activates endogenous antioxidant defenses. Nitrite also corrects impaired mitochondrial bioenergetics in patients' fibroblasts.
Our study indicates that nitrite holds clinical potential as a novel therapeutic agent for PD. Nitrite is currently tested in clinical studies to treat cardiovascular conditions, and it could, therefore, be easily repositioned for PD treatment.
Defects in mitochondrial respiratory complex I and oxidative stress have been consistently associated with PD by laboratory, clinical, and epidemiological studies, and they are mechanistically interconnected (8, 68, 69). Complex I inhibition, in fact, results in increased production of mitochondrial reactive oxygen species (ROS) (66), which, in turn, may attack complex I amino acid residues in a positive feedback loop to generate irreversible damage (56) that exacerbates deterioration. Complex I, therefore, represents a tractable target in PD.
Targeting of complex I for therapeutic purposes has been principally explored in hypoxic pathological conditions, such as cardiac ischemia/reperfusion injury (20, 67). These approaches relied on nitric oxide (NO)-mediated modification of complex I, which favors reversible over irreversible thiol oxidation and prevents its abrupt reactivation during reperfusion, limiting the burst in ROS intrinsically associated with re-oxygenation (20, 67). This strategy is, therefore, based on induction of preventive mild oxidation, emulates traditional preconditioning, and leads to significant mitigation of infarct size.
Inorganic nitrite is a biological reservoir for NO, which is produced after nitrite reduction by myoglobin, hemoglobin, neuroglobin, and/or xanthine oxidoreductase (45, 46). Nitrite has been shown to restore NO signaling and confer protection in ischemia/reperfusion via complex I reversible S-nitrosation in multiple independent studies and in several organs, including liver, heart, and brain (45, 60). NO depletion and derangement of NO-mediated signaling also occurs during aging and under oxidative stress (45). Investigations of nitrite-induced protective effects have shown that—even in normoxic conditions—nitrite increases tolerance to subsequent ischemia/reperfusion with a mechanism that modulates mitochondrial physiology and activates protein kinases (39). These findings suggest that nitrite deserves consideration also in the treatment of other conditions, particularly those where complex I defects and oxidative stress play prominent pathogenic roles. PD largely fulfills these criteria.
This study interrogated the potential of nitrite in normoxic conditions, as a neuroprotective agent in PD, in multiple and phylogenetically diverse animal models, from zebrafish to rodents. We examined the underlying mechanisms in a cellular model of DA degeneration and translated our findings to human cases by studying nitrite effects on primary dermal fibroblasts from familial PD patients with LRRK2 mutations, which are the most common variants associated with this disease (42). Collectively, our data show that nitrite slows PD progression and, therefore, holds clinical potential as a disease-modifying agent.
Results
Nitrite administration is protective in multiple preclinical animal models of PD
In a preliminary investigation, the potential of nitrite administration in PD was assessed in the small vertebrate zebrafish, which is a well-described model of PD (11, 12, 30, 50, 57). Two days treatment of larvae with the DA toxin MPP+—the active metabolite of the prototypical PD toxin 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine hydrochloride (MPTP)—results in selective degeneration of ascending DA neurons in the posterior tuberculum (PT), the homologue structure of the substantia nigra pars compacta (SNpc) in zebrafish (40, 63, 65) (Fig. 1B, C, F–G). Administration of MPP+ constitutes an appropriate experimental setting for preliminary investigation of the neuroprotective potential of nitrite because it inhibits mitochondrial complex I (61) and causes redox imbalance (59). Due to its distinctive sensitivity to MPP+, the Tupfel long fin (TL) zebrafish strain was selected for the study (13). To exclude direct pharmacological interaction between nitrite and MPP+, the former was removed from the medium before addition of the neurotoxin (Fig. 1A). DA toxicity was evaluated by counting neurons in the ventral diencephalic clusters and the associated behavioral anomalies by measuring spontaneous motor activity and swimming velocity. As expected, 1 mM MPP+ treatment for two consecutive days, starting at 5 days postfertilization (dpf), induced significant loss of DA neurons in the PT (Fig. 1C) paralleled by a sharp decrease in locomotor activity (Fig. 1L–N). Four days pretreatment with nitrite dissolved in E3 medium, starting at 1 dpf, resulted in dose-dependent neuroprotection and improvement in locomotor activity (Fig. 1C, L–N).
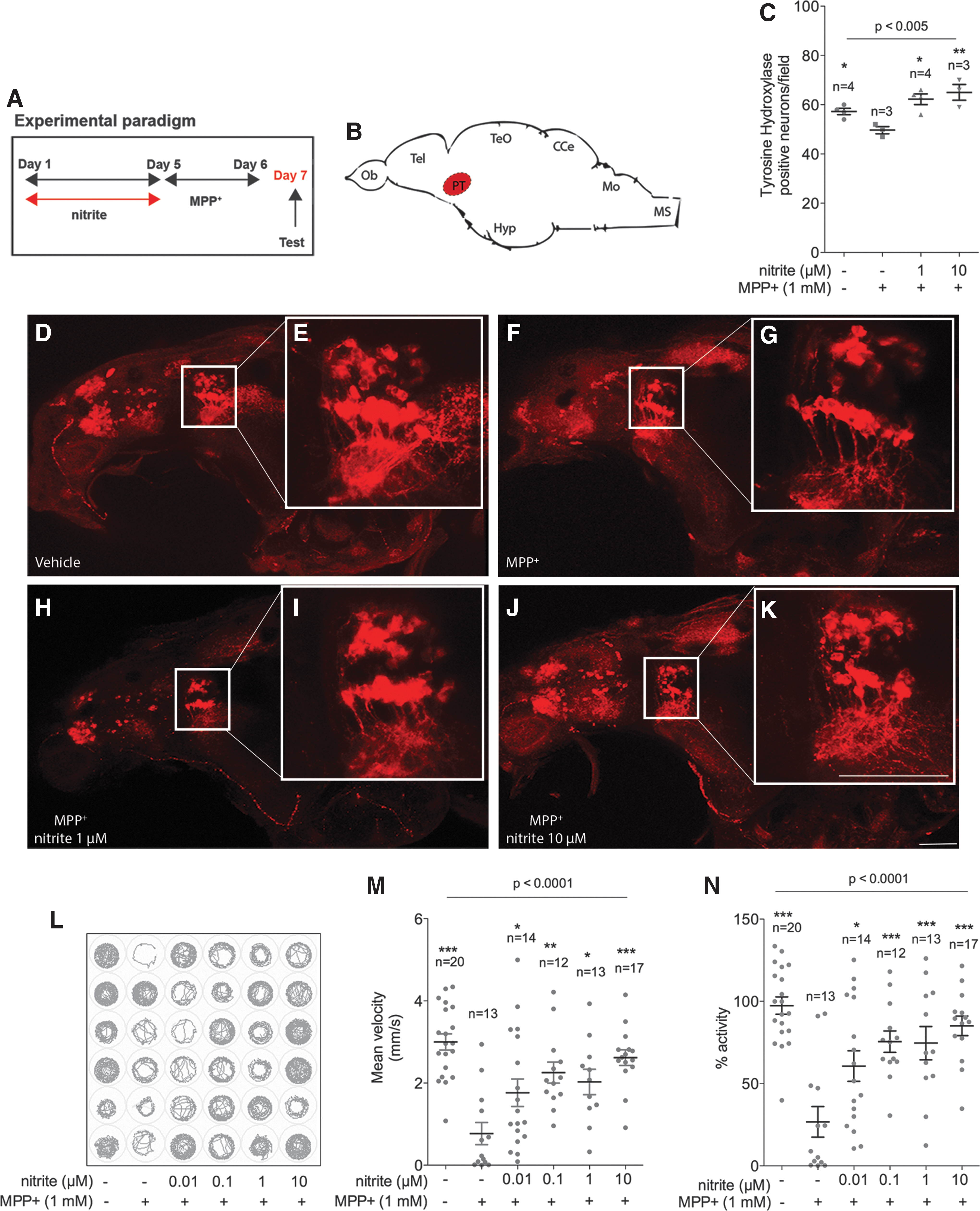
We next sought evidence for nitrite neuroprotection rodents. As expected, nitrite administration induces transient S-nitrosation of proteins extracted from the ventral mesencephalic region containing the SNpc (Supplementary Fig. S1; Supplementary Data are available online at
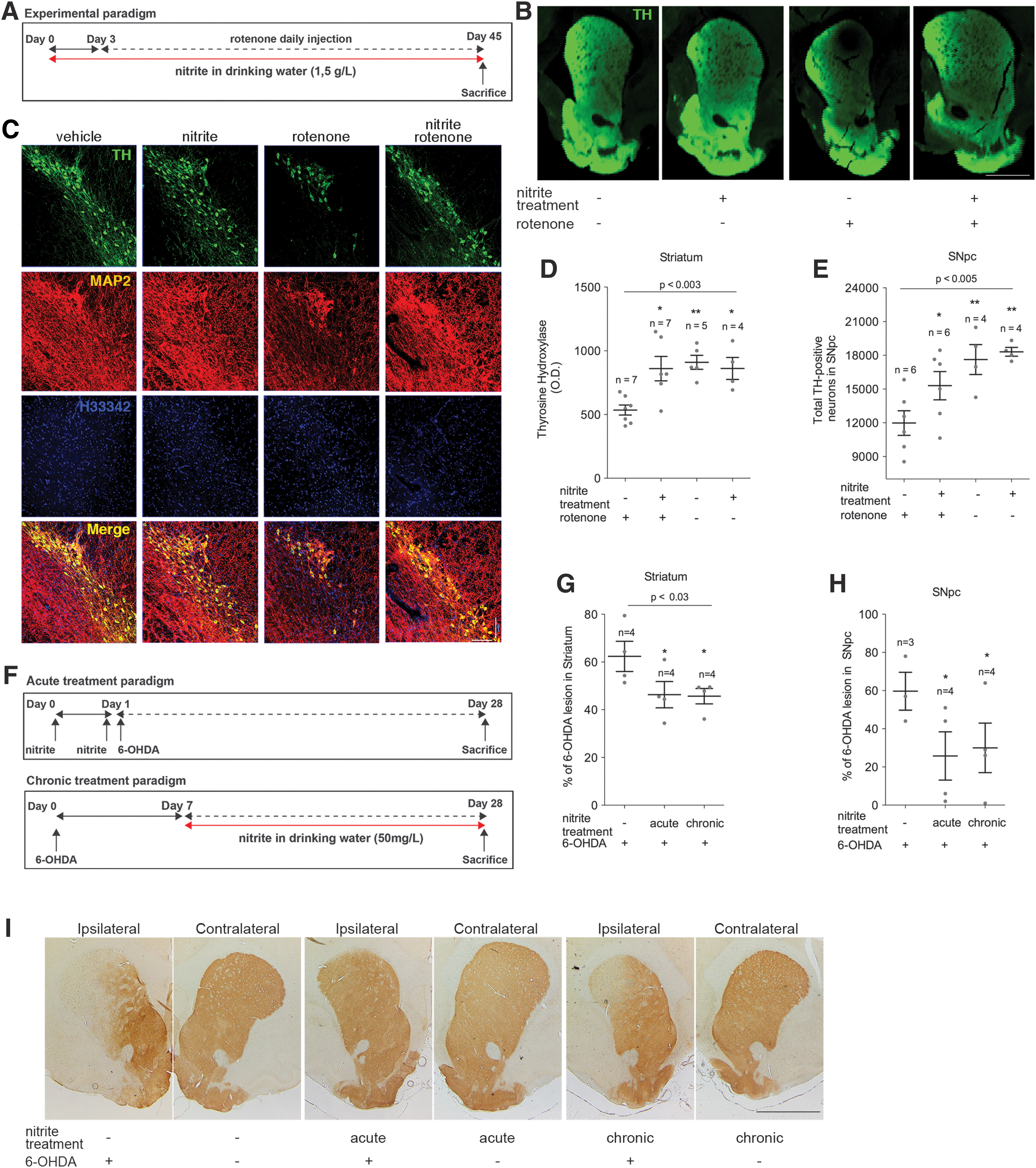
To further substantiate the neuroprotective potential of nitrite in PD, investigations were extended to the 6-hydroxydopamine (6-OHDA) rat model, in which pathology is experimentally induced by a unilateral intrastriatal injection of this toxin. This leads to progressive nigrostriatal degeneration (9), caused by oxidative stress and by inhibition of complex I dependent mitochondrial respiration (34, 35). Importantly, the 6-OHDA model has been used to screen potential neuroprotectants and all the drugs currently used in clinical practice effectively ameliorate motor symptoms in 6-OHDA-treated rats, with the only exception of antimuscarinic compounds (27). Initial experiments addressed the effects of acute administration of nitrite (2 mg/kg), which was injected in rats 24 and 1 h before stereotaxic infusion of 6-OHDA (Fig. 2F, top panel). Treatment resulted in significant reduction of the DA lesion (Fig. 2G–I, acute). Subsequent experiments evaluated the effects of chronic treatment, where nitrite was administered for 3 weeks in drinking water (50 mg/L), starting 7 days after 6-OHDA infusion (Fig. 2F, lower panel), when degeneration of the nigrostriatal pathways is already pronounced (10). Nitrite administration also successfully mitigated DA degeneration in this experimental setup (Fig. 2G–I, chronic).
Nitrite effects in human PD
We next explored whether the nitrite effects observed in PD preclinical models could be translated to human pathobiology. We investigated mitochondrial function in dermal fibroblasts obtained from biopsies of patients harboring LRRK2 mutations (Table 1) (3, 7, 23, 47), and we used a Seahorse Extracellular Flux Analyzer to characterize the bioenergetics profile of these cells (Fig. 3A). Out of the three studied PD lines, only one (ND32976) exhibited reduced basal respiration (Fig. 3B). However, rotenone-sensitive respiration reflecting complex I activity was decreased in all PD samples (Fig. 3C), substantiating the central role of mitochondrial complex I defects also in genetic PD (23). Respiratory reserve capacity, which modulates the response to stress-induced pathology (36), was also decreased in all specimens (Fig. 3D). Pretreatment with nitrite (48 h) led to significant improvement in both rotenone-sensitive respiration and reserve capacity (Fig. 3F, G) in two of the LRRK2 mutant lines, but it had no effect on cell lines from controls (Table 1). In addition, nitrite pretreatment significantly decreased basal H+ leakage in LRRK2 samples (Fig. 3E)—therefore improving mitochondrial efficiency—in agreement with previous observations (43); interestingly, nitrite augmented H+ leakage in control cells.

Effects of nitrite on MPP+-treated SH-SY5Y cells
To determine the mechanisms of action of nitrite-mediated neuroprotection observed in vivo, we performed experiments in the SH-SY5Y DA cell line, which is commonly used as an in vitro model of PD (66).
Mechanisms affecting cell survival and protein redox state
Consistent with previous data (4, 28, 66), administration of MPP+ affected cell survival, which was significantly ameliorated by nitrite (Fig. 4A–C). Treatment with nitrite alone did not induce cell death and protection was completely abolished by the NO scavenger carboxy-PTIO, thus confirming that the observed effect was mediated by nitrite-derived NO (Fig. 4B, C).

Nitrite induced an increase in protein S-nitrosation in MPP+ treated SH-SY5Y, consistent with augmented nitrite-dependent S-nitrosation (Fig. 4D, E, compare lanes 2, 3, and 5 and Supplementary Fig. S2), whereas MPP+ alone did not alter S-nitrothiol levels (Fig. 4D, E, compare lanes 1 and 2). In nitrite/MPP+ co-treated samples, administration of the NO scavenger carboxy-PTIO abolished the observed increase in S-nitrosothiol formation (Fig. 4D, E, compare lanes 3, 4, 5, and 6). Replacement of nitrite with the NO donor 3,3-Bis(aminoethyl)-1-hydroxy-2-oxo-1-triazene (NOC-18), which ensures sustained NO release because of its long half-life (21), led to comparable results and further confirmed the central role of NO in the observed effects (Fig. 4D, E, compare lanes 7 and 8).
Next, we explored whether increased reversible S-nitrosation could protect cysteine thiols from higher oxidation states (i.e., sulfinic and sulfonic acids), which can cause functional anomalies in proteins and also predispose them to aggregation (62). Protein sulfenic acid formation induced by hydrogen peroxide (H2O2) was significantly lessened by nitrite pretreatment (Fig. 4F, G), confirming that nitrite treatment favors reversible over irreversible thiol modification in proteins.
Nitrite administration did not abolish production of mitochondrial superoxide (Fig. 4H), in agreement with previous studies showing that mitochondrial ROS production is required for nitrite-mediated cytoprotection under normoxic conditions (39).
Mitochondrial respiration and complex I thiol redox status in MPP+-treated SH-SY5Y
The SH-SY5Y cell line was also used to evaluate the effects of nitrite administration on the recovery of mitochondrial respiration after transient MPP+ challenge (25). Four-hour exposure to MPP+ significantly suppressed basal mitochondrial respiration, and toxin removal was not sufficient to promote full recovery. Contrastingly, samples pretreated with nitrite successfully restored respiration to control levels (Fig. 5A).
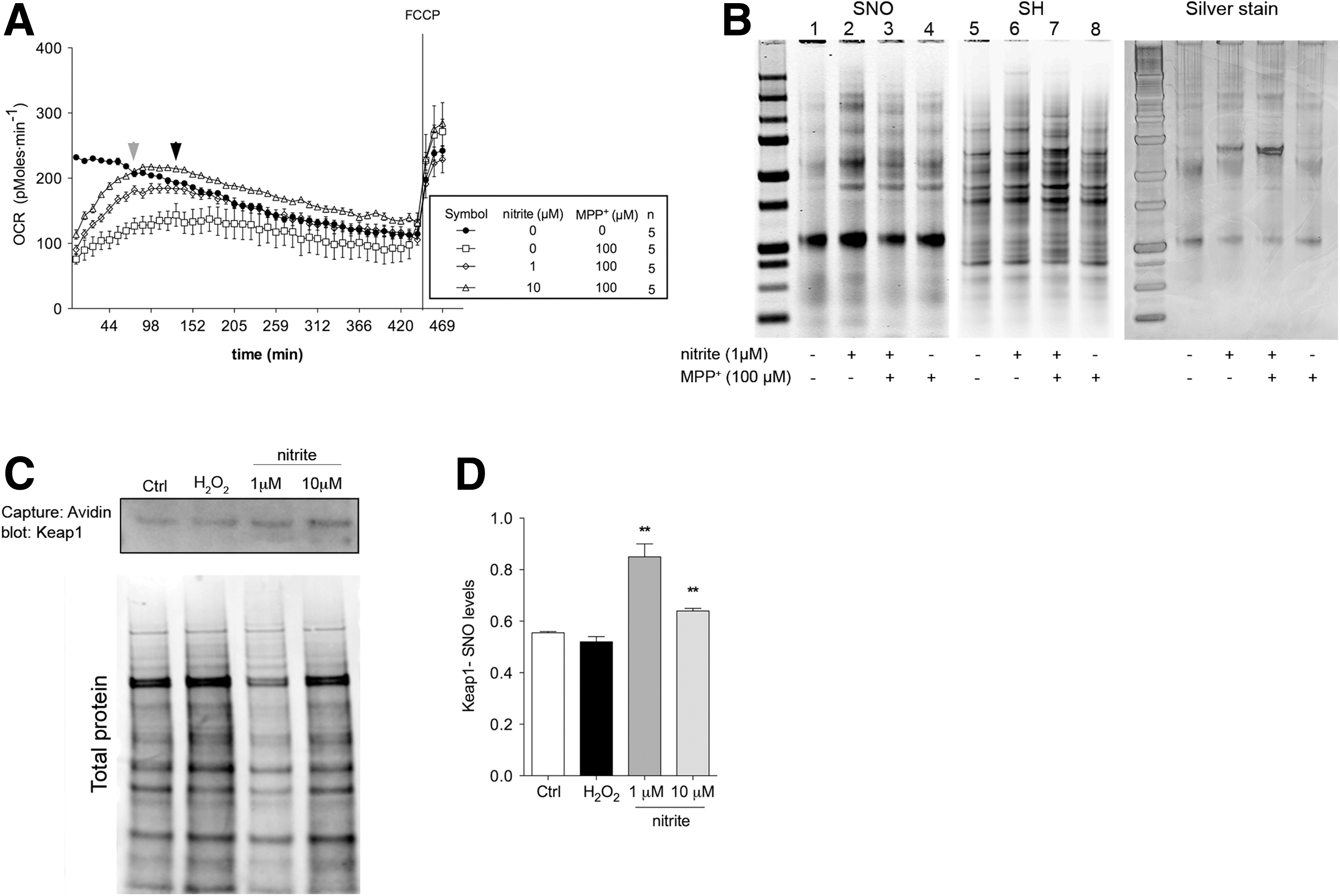
Next, the redox equilibrium between S-nitrosated and reduced cysteines was examined in immunocaptured complex I. Nitrite increased S-nitrosation without altering levels of reduced cysteines (Fig. 5B, compare lanes 2 and 6). However, when nitrite was co-administered with MPP+, S-nitrosation was paralleled by a marked increment in reduced cysteine-thiol levels (Fig. 5B, compare lanes 3 and 7), reflecting a more reduced state of complex I (Fig. 5B, lane 8). Nitrite-mediated improved respiration in MPP+-treated cells is, therefore, associated with reduction of the thiol/disulfide redox equilibrium in complex I.
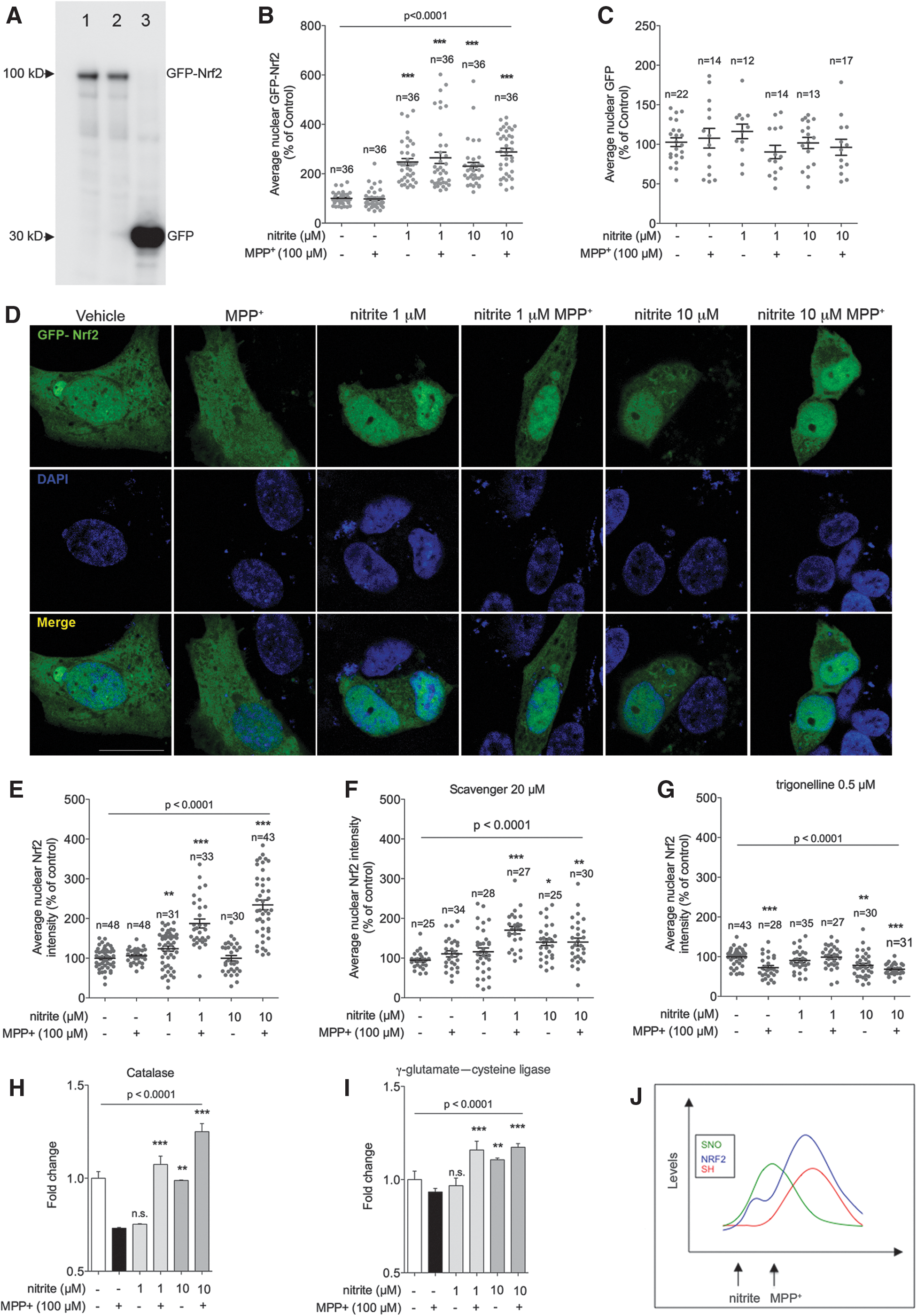
To clarify the mechanisms responsible for nitrite-mediated cysteine reduction in MPP+-treated cells, we investigated activation of the major antioxidant cellular pathway Nrf2. On augmented oxidation levels—or in the presence of NO—the Nrf2 transcription factor dissociates from its cytosolic partner Keap1, translocates to the nucleus, and promotes transcription of several genes involved in the antioxidant response (1, 16, 17, 24, 72). Nitrite pretreatment in MPP+-challenged samples—which displayed reduced cysteines in complex I (Fig. 5B)—induced S-nitrosated cysteine (SNO) modification in Keap1 (Fig. 5C, D and Supplementary Fig. S3). Nitrite administration also induced Nrf2 activation in SHSY-5Y cells overexpressing GFP-tagged Nrf2 (Fig. 6A–D). Nitrite also induced nuclear translocation of endogenous Nrf2, as evidenced by immunochemical detection in untransfected SHSY-5Y cells. Activation was also detected under physiological conditions system S-nitrosation of Keap1, as expected, and it elicited nuclear Nrf2 translocation (Fig. 6E and Supplementary Fig. S4). Collectively, these convergent data demonstrate nuclear translocation of Nrf2, which was paralleled by increased expression of known target genes (Fig. 6H, I), therefore unambiguously establishing that nitrite causes activation of the Nrf2 pathway. Activation of Nrf2 target genes was prevented, as expected, by the specific Nrf2 inhibitor trigonelline (Fig. 6G). Nrf2 nuclear translocation depended on nitrite-derived NO, as it was markedly reduced by the NO scavenger carboxy-PTIO (Fig. 6F). Nrf2 activation was not detectable in cells treated with MPP+ alone (Fig. 6B, D, E), consistently with the unaltered levels of reduced cysteines in these samples (Fig. 5B).
Discussion
This study shows that administration of nitrite—a reservoir of bioavailable NO—is neuroprotective in several preclinical models of PD and ameliorates mitochondrial efficiency in dermal fibroblasts from PD patients. Nitrite reduces DA neuron loss in multiple models of PD, and amelioration is not confined to prophylactic treatment. In fact, it is also observed when treatment is initiated at more advanced pathogenic stages, as evidenced in 6-OHDA rats treated 1 week after induction of the lesion. Nitrite treatment also corrects defective maximal respiratory capacity and increases proton leakage in LRRK2 mutant patients.
Our findings extend previous evidence in hypoxic settings demonstrating that nitrite is protective against ROS and complex I dysfunction (67) to normoxic conditions. Overall, the work emphasizes the role of NO signaling for proper biological function and confirms that reconstituting reserves of bioavailable NO preserves its integrity and protects against pathology (45). Importantly, reduced NO bioavailability characterizes conditions such as aging and oxidative stress, which constitute major risk factors for PD (22, 45, 69).
Our data are consistent with the notion that reactive species are essential signaling molecules—with effects that extend beyond mere toxicity and are instead essential for proper physiological function (5)—and are in agreement with evidence showing that NO exerts dichotomous effects depending on its concentration (45). Physiological NO levels ensure correct function and a moderate increase in NO may induce mild levels of stress, protecting against subsequent and more pronounced insults in a preconditioning-like manner (20). Consistently, we show that nitrite administration favors formation in complex I of S-nitrosothiols over sulfenic acid. Because the latter can be converted in higher and irreversible oxidation states (62), it is tempting to speculate that nitrite-induced S-nitrosation constitutes a molecular shield protecting cysteine residues against permanent damage in vivo. Further studies to test this hypothesis are warranted. Restoration of physiological NO levels can also scavenge superoxide to prevent an escalation of oxidative stress (45). Conversely, high NO levels are toxic—in fact, boosting NO production via iNOS is part of the innate immune response to kill invading pathogens—and are associated with neurodegeneration (52, 64). The key success factor is, therefore, restoration of NO bioavailability in a timely manner, before nitrosative/oxidative stress escalation.
Although nitrosative stress has previously been reported in PD (19), there is no evidence that exposure to NO-releasing drugs is causative or even constitutes a risk factor for this disorder. In fact, nitrosonium donors have been extensively used in clinical practice to treat cardiovascular diseases and membrane-permeable NO diffuses three dimensionally from the cell of origin, therefore conceivably reaching neurons (31, 51). Nonetheless, the use of this class of molecules is not associated with increased PD prevalence. Conversely, a recent and intriguing study highlighted an association between discontinuation of statin therapy and PD (44). Because improvement of NO bioavailability—also at the cerebral level (32)—is a well-described effect of lipophilic statins, it is tempting to speculate that decreased NO levels after therapy discontinuation might be detrimental for individuals at risk for PD.
In addition, our results demonstrating that nitrite restores mitochondrial reserve capacity and reduces proton leak in patients are consistent with similar, previous findings in healthy human subjects showing that nitrite improves mitochondrial bioenergetics (43).
Mechanistically, we demonstrate that, in normoxia, nitrite acts along two parallel and likely synergistic pathways involving complex I and Nrf2 activation. These findings are consistent with previous evidence showing that NO is an activator of the Nrf2 pathway (53) with a mechanism involving S-nitrosation of its cytosolic partner Keap1 (1, 16, 17, 24, 72). In addition, it has been recently demonstrated that, in normoxia, nitrite activates protein kinases (39), which, in turn, can activate Nrf2 (55). Overall, our findings are consistent with the notion that nitrite is a complex NO donor acting at multiple levels (33) and substantiate its therapeutic relevance given the attention that Nrf2 received as a target for neuroprotection (38). Future studies will be crucial in identifying the signaling cascades leading to nitrite-mediated Nrf2 activation and in determining whether other mitochondrial NO donors [e.g., mito-SNO (20)] retain the ability to activate Nrf2.
Nitrite is a source of bioavailable NO that has been successfully used in laboratory models of cardiovascular diseases and is currently being scrutinized in clinical studies (45). In addition, currently available data support the safety of nitrite use in human therapeutics. Traditionally, nitrite has been associated with two principal health-related issues: methemoglobinemia—that is, nitrite-mediated oxidation of ferrous iron in hemoglobin to the ferric state—and an alleged carcinogenic potential. None of these issues, however, constitute a threat under the pharmacological conditions envisaged for human therapeutics. In fact, nitrite EC50 to induce methemoglobinemia is ∼1 g (54), whereas the amount used in our and other studies (46) is in the order of milligrams. Under these conditions, methemoglobinemia can be safely excluded. Conversely, there is no evidence in favor of carcinogenic activity of nitrite, as demonstrated by toxicological studies prolonging administration to 2 year periods (54) and by epidemiological investigations as well (49). In summary, nitrite pharmacokinetics, safety, and dosing features are known, and the molecule is, therefore, an excellent candidate for testing in human PD.
Collectively, our study demonstrates that inorganic nitrite mitigates PD pathology through a synergistic mechanism improving mitochondrial efficiency and activating the Nrf2 antioxidant pathway, and, therefore, it represents promising therapy for PD.
Materials and Methods
Chemicals
Sodium nitrite (563218), MPP+ (D048), MPTP (M0896), Carboxy-PTIO potassium salt (NO-scavenger, C221), NOC-18 (NO-donor, A5581), 6-OHDA (H4381), oligomycin (75351), FCCP (C2920), rotenone (557368), antimycin (A8674), N-Ethylmaleimide (NEM, E3876), digitonin (D141), mannitol (M4125), glutamate (1446600), malate (4694 U), trigonelline (T5509), and 5,5-Dimethyl-1,3-cyclohexanedione (dimedone, D153303) were obtained from Sigma-Aldrich (St. Louis, MO).
Zebrafish treatment
Adult TL zebrafish were maintained at 28°C on 14/10 h light/dark cycles. Embryos were collected from natural mating and rose in E3 buffer (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4) at 28°C.
One dpf zebrafish embryos (20 embryos/plate) were treated for 4 days with sodium nitrite (0.1–10 μM) dissolved in E3 buffer and incubated at 28°C. At day 5, fish were transferred to fresh E3 buffer and treated for two consecutive days with MPP+ 1 mM as previously described (29). The solutions were daily changed. At 7dpf, zebrafish larvae were collected for behavioral testing or sacrificed to perform morphological analysis. Experiments were performed in accordance with the European Communities Council Directives (2010/63/EEC; D.L., 27.01.1992, number 116), the Dutch welfare legislation, and according to the guidelines of the Erasmus MC animal facility (EDC).
Zebrafish motor analysis
Larval motor activity was analyzed by using the Zebralab system (ViewPoint Life Sciences, Montreal, Quebec, Canada) as previously described (50). Viewpoint software was used in “tracking mode” with inactive/active and small/large thresholds, respectively, set to 0.5 and 1 mm/s. Zebrafish larvae were analyzed in a 96-well plate containing 300 μl of E3 buffer per well in a single larva/well setup. Spontaneous activity was measured at 7 dpf for hours, after 15 min of adaptation time following the transfer to the new Zebrabox setup.
Zebrafish whole-mount immunohistochemistry
Whole-mount immunohistochemistry was carried out as previously described (50). All incubation steps and washes were carried out on a shaker. Briefly, specimens were fixed in 4% PFA (from PFA 16%—28908; Thermo Fisher, Waltham, MA)-4% sucrose phosphate buffer saline solution (PBS—100110023; Thermo Fisher) overnight (ON) at 4°C. The next day, larvae were washed in PBS ON at 4°C. After washes, eyes and yolk sack were mechanically removed to improve antibody penetration in the brain tissue. Specimens were permeabilized with Proteinase K (PK—P2308; Sigma-Aldrich) 10 μg/ml for 15 min at room temperature (RT) and washed three times for 30 min with PBS-Triton X-100 0.2% (PBS-T). Larvae were then postfixed in 4% PFA in PBS-T for 20 min at RT and then washed three times for 10 min with PBS-T at RT. Zebrafish were then blocked with ON at 4°C in a solution containing DMSO 1% and normal goat serum 4% in PBS-T. Mouse monoclonal anti-tyrosine hydroxylase (TH, 1:500, MAB318; Millipore, Billerica, MA) incubation was performed with ON at 4°C in blocking buffer. Samples were washed five times for 30 min at RT with PBS-T and then incubated with Alexa 488 anti-mouse secondary antibody (A32723; Thermo Fisher) ON at 4°C. After five washes in PBS-T, larvae were transferred to an 80% glycerol solution and mounted for confocal analysis. Image acquisition was performed in an inverted Leica TCS SP5 confocal microscope (Leica Microsystems, Buffalo Grove, IL) in a z-stack mode, and the cell counting in the PT area was performed by analyzing all the consecutive layers of the stack using the ImageJ software.
Rotenone rat model
Male Lewis rats were treated with rotenone as previously described (18). Briefly, animals were treated up to 45 days (instead of 30). During the first 30 days, rats received an i.p. rotenone dose of 3 mg/kg/day and then, the dosage was increased to 3.5 mg/kg/day. Nitrite was administered in drinking water, 1.5 g/L, for three consecutive days before starting the rotenone treatment. Six- to 7-month-old male Lewis rats were used for all experiments (Hilltop Lab Animals, Inc., Scottdale, PA). The animals were maintained under standard conditions of temperature and humidity, in a 12 h light/dark cycle, with free access to water and food. The rats were adapted for 2 weeks before initiation of the experimental protocol. All studies were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh and were performed in accordance with published National Institutes of Health guidelines.
6-OHDA rat model
For the 6-OHDA model, male Sprague–Dawley rats (Charles River, Calco, LC, Italy) weighing 275–300 g were maintained under standard conditions of temperature and humidity with free access to food and water at the Centralized Animal Facility of the University of Pavia. Experiments were performed in accordance with the European Communities Council Directives (2010/63/EEC; D.L., 27.01.1992, number 116) and the guidelines for animal experimentation approved by the Animal Care Committees of the University of Pavia, Italy.
Animals were anesthetized by i.p. administration of 50 mg/kg of sodium-thiopental and placed in a stereotaxic frame (Stoelting, Wood Dale, IL). They received an injection of 6-OHDA (20 mg per 3 ml in saline containing 0.02% ascorbic acid) into the right striatum (1.0 mm anterior, 3.0 mm lateral, and 5.0 mm ventral with respect to bregma and dura) at 1 ml/min, using a Hamilton 10-AL syringe with a 26-gauge needle.
Twenty-week-old Sprague–Dawley rats subjected to the acute treatment were i.p. injected with 2 mg/kg of sodium nitrite in PBS 24 and 1 h before 6-OHDA injection. Animals underwent chronic treatment according to a previously reported administration paradigm (15), and received nitrite orally, dissolved in drinking water (concentration: 50 mg/L), for 3 weeks starting 7 days after induction of the lesion.
Cell cultures
Skin dermal fibroblasts derived from PD patients bearing the G2019S (ND33879) or the N1441G (ND32975 and ND32976) LRRK2 mutations and age-matched controls were obtained from the Coriell Biorepository of the Coriell Institute for Medical Research (Camden, NJ); a detailed description is reported in Table 1. SH-SY5Y cells were obtained from Sigma-Aldrich (94030304). Cells were cultured according to standard procedures at 37°C and 5% CO2 in DMEM medium (D6429, Sigma-Aldrich) supplemented with 10% of fetal bovine serum (F6178; Sigma-Aldrich) and 1% penicillin-streptomycin (P4333; Sigma-Aldrich). SH-SY5Y human neuroblastoma cell lines and PD-derived skin dermal fibroblast cell lines were plated and after 24 h treated for two consecutive days with sodium nitrite (1 or 10 μM) dissolved in culture medium or vehicle. For SH-SY5Y cells, on the third day, the nitrite-containing medium was removed and fresh new media containing MPP+ 100 μM or vehicle were added to the culture for 4 h. After incubation, cells were collected for analysis.
Immunohistochemistry
Immunohistological sections were processed as previously described (37). Briefly, sections were first incubated in H2O2 (Sigma-Aldrich) 3% in PBS for 30 min to block internal peroxidase activity, and subsequently in PBS-T and normal horse serum (NHS) 10% for 1 h at RT. Specimens were then incubated for 24 h at 4°C with mouse monoclonal anti-TH (1:4000, MAB318; Millipore), in PBS-T and 1.5% NHS. After several washes with PBS-T, sections were incubated with biotinylated goat anti-rabbit IgG (1:500, BA 1000; Vector Laboratories, Burlingame, CA), in PBS and 1% NHS for 1 h at RT. Immuno-complexes were revealed by Vectastain Elite ABC kit (PK 4000; Vector Laboratories), using 3,3′-diamino-benzidine (DAB Substrate kit for Peroxidase, SK 4100; Vector Laboratories). Finally, sections were dehydrated and mounted with Eukitt (Kindler GmbH & Co.). Slides were observed with an Olympus BX 51(Olympus, Parkway Center Valley, PA) microscope equipped with a Leica DFC 420 camera.
Quantification of striatal TH density
Striatal lesion in DAB-stained sections was calculated as the ratio between the lesioned area, detected by the absence of TH staining, and the area of the entire TH immunopositive striatum on the injected side (6). A total of 14 sections per animals were analyzed.
Alternatively, TH density was determined by using an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE). Four to five serial immunofluorescence-labeled sections were scanned at 800 nm at high resolution to generate the reported average striatal DA fiber intensity as previously described (70). The dorsal region of the striatum was outlined, and the average pixel intensity for each section was obtained by using the Odyssey software (3.0).
Unbiased stereological counts
In rotenone-treated rats, an automated Nikon 90i upright fluorescence microscope equipped with five fluorescent channels and a linear encoded motorized stage was used to obtain a high-resolution montage for analysis as previously reported (71). SN images were collected at 20 × (0.75 N.A.) by using a Q-imaging Retiga cooled CCD camera and the Nikon NIS-Elements software (4.2). Unbiased quantification of TH-immunoreactive cells was assessed in the SN pars compacta and reticulata from one hemisphere. All slides were scanned under the same conditions for magnification, exposure time, lamp intensity, and camera gain. After background subtraction and thresholding, quantitative analysis was determined on fluorescent images generated in three fluorescent colors (MAP2 in red; TH+ in green, and nuclei in blue) in every sixth section throughout the entire region. An average of 11–12 sections per animal was used for quantification. Stereological counts were coded and carried out by an experimenter blinded to the treatment. The coefficient of error was calculated according to West and Gundersen as described earlier, and values were <0.10 in all cases.
In rats treated with 6-OHDA, unbiased stereological estimation of the total number of the DA cells in SNpc was made by using the optical fractionator method (75) from the STEREO INVESTIGATOR program on a Neurolucida computer-controlled microscopy system (Microbrightfield, Inc., Williston, VT). The edges of the SNc in the rostro-caudal axis were defined at all levels, with reference to a coronal atlas of the mouse brain (58). TH-positive cells in the SNc of both hemispheres were counted in every three sections, on comparable sections for all the subgroups of treatment throughout the entire nucleus. Counting frames (60 × 60 μm) were placed at the intersections of a grid (frame size 120 × 120 μm) that had been randomly placed over the section. Only counting frames for which at least a part of the frame fell within the contour of the SNpc were used for counting. Cells were marked if they were TH positive and were in focus within the counting area. Guard volumes (3 μm from the top and 3 μm from the bottom of the section) were excluded from both surfaces to avoid the problem of lost caps. The reliability of the estimate was assessed by calculation of the coefficient of error according to the formulae described in West and Gundersen (74).
Bioenergetic profiling
The Seahorse XF24 Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA) was used to generate the bioenergetic profiles of human primary skin fibroblasts in real time and the neuroblastoma SH-SY5Y cell line as previously described (3). PD-LRRK2-derived fibroblasts and healthy controls were seeded on a Seahorse XF-24 plate at a density of 6 × 104 cells per well and grown overnight in DMEM (10% of FCS and 1% Pen-Strep) at 37°C in the presence of CO2. The density ensures a proportional response of FCCP with cell number (3) and resulted in confluent cultures, in which cell growth was blocked by contact inhibition. After adhesion, cells were then treated for 48 h with sodium nitrite (1 or 10 μM) or vehicle (PBS) dissolved in growth medium. Media were refreshed daily. On the experimental day, cell medium was changed to unbuffered DMEM (XF Assay Medium; Agilent Technologies) supplemented with 5 mM glucose and 1 mM sodium pyruvate, and it was incubated for 1 h at 37°C in the absence of CO2. Medium and reagents were adjusted to pH 7.4 on the day of the assay. After four baseline measurements for the oxygen consumption ratio, cells were sequentially challenged with injections of mitochondrial toxins: 0.5 μM oligomycin (ATP synthase inhibitor), 1 μM FCCP (mitochondrial respiration uncoupler), 0.5 μM rotenone (complex I inhibitor), and 0.5 μM antimycin (complex III inhibitor).
SH-SY5Y cells were seeded at a density of 5 × 104 cells per well and let to adhere overnight. On the next day, cells were treated for 48 h with sodium nitrite or vehicle at the same conditions as previously described for fibroblast lines. Culture medium was then removed, and cells were treated with MPP+ 100 μM or vehicle (PBS) for 4 h at 37°C. Medium was then changed to unbuffered DMEM supplemented with 5 mM glucose and 1 mM sodium pyruvate as described and rested for 1 h at 37°C without CO2. The recovery of mitochondrial respiration was detected for 8 h (25), followed by three measurements after the injection of 1 μM FCCP, and it was used to test cell viability at the end of the assay.
Detection of protein sulfenic acid production after H2O2 treatment in cultured cells
Confluent SH-SY5Y cells were briefly treated (10 and 30 s) with 0.5 mM H2O2 dissolved in serum-free medium and immediately suspended in RIPA buffer containing dimedone (NaCl 150 mM, NP-40 1%, DOC 0.1%, sodium dodecyl sulfate [SDS] 0.1%, Tris-HCl 50 mM, Dimedone 1 mM, and protease inhibitors) (2). After 45 min of incubation in ice, protein samples were processed, separated on a NuPAGE precast sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE, NP0321; Thermo Fisher) under reducing conditions, and transferred on a PVDF membrane. The membrane was blocked for 1 h at RT in a PBS-T solution containing bovine serum albumin (BSA) 5%, and proteins were detected through the cysteine (sulfonate) polyclonal antibody (1:1000, ON at 4°C, ADI-OSA-820-F; ENZO, Farmingdale, NY) and the goat anti-rabbit HRP-conjugated secondary antibody (1:5000, P0487; DAKO-Agilent Technologies). The signal was then revealed with the ECL detection system (GE Healthcare, Barrington, IL). After detection, the membrane was washed several times with PBS-T and incubated with the anti-actin mouse primary antibody (1:4000, ON at 4°C, MAB 1501; Chemicon, Temecula, CA) and the IRDye donkey-anti-mouse secondary antibody (1:5000; Li-COR Biosciences). The signal was finally detected by using the Odyssey Imaging System (Li-COR Biosciences).
SH-SY5Y cytotoxicity assays and superoxide production
To investigate cellular toxicity in SH-SY5Y, cells were treated with nitrite dissolved in culture medium at the described concentration. After 48 h, the solution was replaced with fresh medium containing MPP+. Cellular necrosis was evaluated after 24 h of treatment with MPP+ with Sytox green 0.5 μM (Invitrogen, Carlsbad, CA), following the manufacturer's instructions, by using a flow cytometer (FACSaria, BD Biosciences, San Jose, CA). FlowJo Software (Tree Star, Inc., Ashland, OR) was used for data analysis. Cellular cytotoxicity was further confirmed via lactate dehydrogenase release (LDH colorimetric detection kit, 630117; Clonetech, Mountain View, CA). Briefly, 6 × 104 cells were seeded in a 96-well plate, treated with nitrite dissolved in DMEM (without phenol red, 21063029; Thermo Fisher) for 48 h, and finally exposed to MPP+. After 24 h, culture medium was collected after centrifugation and then checked for lactate dehydrogenase content by following the manufacturer's guidelines.
Mitochondrial superoxide production was detected by using Mitosox Red (M36008; Invitrogen) following the manufacturer's guidelines. Briefly, after a 4 h exposure to MPP+, 106 cells were harvested, quickly washed in ice-cold HBSS without Ca++ and Mg++, and resuspendend in Hepes 0.1 M containing Mitosox 0.2 μM. Cells were incubated for 20 min at 37°C, protected from light, and finally analyzed with an FACSaria flow cytometer (BD Biosciences).
Detection of S-nitrosation levels in proteins
S-nitrosated proteins were detected as previously described, with minor modifications (73). Briefly, equal amounts of cells (5 × 105) were harvested via trypsin digestion and suspended in a lysis solution containing Tris-HCl 50 mM, SDS 1%, NEM 100 mM, and proteinase inhibitors mix (Complete mini, 11836153001; Roche, Indianapolis, IN). Lysates were incubated for 5 min at 70°C, sonicated for 15 min in a cold-water bath, and finally incubated for 30 min at RT. Protein extracts were then precipitated with a cold solution of acetone, methanol, and ethanol (50%, 25%, and 25%, respectively) suspended in the staining solution containing Tris-HCl 50 mM, SDS 1%, 20 μM fluorescent Alexa-NEM-800 dye (929-80020; LI-COR Biosciences) or EZ-Link PEG2-Biotin NEM (21901BID; Thermo Fisher), ascorbate 1 mM, and CuCl2 1 μM and they were incubated for 1 h at RT. The staining solution was removed after acetone-methanol-ethanol precipitation and proteins were finally suspended in Tris-HCl 50 mM, SDS 1%, and Alexa Fluor 680 succinimidyl ester (NHS—A20008; Thermo Fisher) 20 μM. Samples were diluted in Laemni Sample buffer containing 5% B-mercaptoethanol, and proteins were separated on a NuPAGE precast SDS PAGE gel (NP0321; Invitrogen) under reducing conditions. After electrophoresis, the gel was fixed with 50% ethanol 50% and 2.5% ortophosphoric acid overnight under mild agitation. Final images and analysis were performed with the Odyssey Imaging System (Li-COR Biosciences). Immunodetection of the mitochondrial protein Ndufa9 (1:1000, ab14713; Abcam, Cambridge, MA) in the extracts processed for the S-nitrosation assay was achieved by Western blot analysis performed according to standard procedures.
Detection of SNO modification in Keap1
SNO protein modifications were detected as described in the previous paragraph by using the EZ-Link PEG2-Biotin NEM during the staining step. Total S-nitrosylated proteins were isolated via the Streptavidin Mag Separose kit (28-9872-30 AA; GE Healthcare, Aurora, WI) according to the manufacturer's protocol. Briefly, equal amounts of biotin-SNO-modified proteins were incubated with 100 μl of streptavidin magnetic beads ON at 4°C under slow end-over end rotation. The biotin-streptavidin complex was then washed sequentially with: (1) 0.2 M borate pH 9, (2) 0.2 M sodium acetate pH 4, and (3) TBS-Urea buffer (50 mM Tris-HCl pH 7, 150 mM NaCl, and 2 M Urea) and biotin-SNO proteins were finally eluted in 100 μl of elution buffer (Tris-HCl 50 mM, 150 mM NaCl, SDS 2%, and 0.4% Urea) at 95°C for 10 min. Samples were then treated with 4 × Sample buffer, and 25 μl of the final solution was separated on the NuPAGE precast SDS PAGE gel and transferred on a PVDF membrane. The membrane was blocked for 1 h at RT in a PBS-T solution containing BSA 5%, and SNO-biotinylated proteins were detected for Keap1 (1:1000, ON at 4°C, 8046S; Cell Signaling, Danver, MA) and the goat anti-rabbit HRP-conjugated secondary antibody (1:5000, P0487; DAKO-Agilent Technologies). The signal was then revealed with the ECL detection system (GE Healthcare). As loading control, biotin-SNO samples were incubated with 20 μM Alexa Fluor 680 succinimidyl ester and equal amounts were separated on a NuPAGE precast SDS PAGE gel under reducing conditions. After electrophoresis, the gel was fixed with 50% ethanol and 2.5% ortophosphoric acid ON under mild agitation. Final images and analysis were performed with the Odyssey Imaging System (Li-COR Biosciences).
GFP-Nrf2 transient expression
Transient expression of the pcDNA3-EGFP-C4-Nrf2 vector (21549; Addgene, Cambridge, MA) or the vector alone was achieved in SH-SY5Y cells by using X-tremeGENE HP DNA transfection reagent (XTGHP-RO ROCHE; Sigma-Aldrich) according to the manufacturer's procedure. Twenty-four hours after transfection, cells were treated with nitrite/MPP+ as previously described and were then collected for analysis. For IF analysis, cells were rinsed twice with PBS, then fixed 20’ with PFA 4%, rinsed three times with PBS, covered with Vectashield containing DAPI (H1200; Vector Laboratories), and finally mounted on glass slides. Image acquisition was performed with a Zeiss LSM700 laser scanning confocal microscope. Alternatively, cells were lysed in Tris-HCl 50 mM, SDS 1%, EDTA 1 mM, and proteinase inhibitors (Complete Mini). Lysates were separated on a NuPAGE precast SDS PAGE gel and transferred on PVDF. The membrane was blocked for 1 h at RT in a PBS-T solution containing BSA 5%, and GFP-expressing proteins were detected with the anti-GFP antibody (1 h at RT, 1:1000, 11814460; Sigma-Aldrich) and the goat anti-mouse HRP-conjugated secondary antibody (1:5000, 715-035-150; Jackson Immuno Research, West Grove, PA). The signal was then revealed with the ECL detection system (GE Healthcare).
Complex I immunoprecipitation and detection of thiol and S-nitrosothiols
Mitochondria from SH-SY5Y neuroblastoma cells were isolated according to the “Mitochondria Isolation Kit for Cultured Cells” guidelines (ab110171; Abcam, Cambridge, MA), with minor modifications. Briefly, 4 × 108 confluent cells were collected by trypsin treatment and suspended in mannitol 75 mM, sucrose 25 mM, KCl 100 μM, KH2PO4, MgCl2 5 mM, Tris-HCl 20 mM, glutamate 5 mM, malate 5 mM, digitonin 0.01%, NEM 10 mM, and AlexaFluor maleimide-800 20 μM. Samples were processed three consecutive times with a homogenation step (Dounce pestle “B,” 30 strokes) and mitochondria collection by centrifugation at 12,000 g, 10 min at 4°C. Freshly isolated mitochondria were suspended in PBS, 10% maltoside, protease inhibitors, NEM 10 mM, and Alexa-NEM-800 dye (929-80020, LI-COR Biosciences) 20 μM at 5 mg/ml and mitochondrial complex I was immune-captured according to the “Complex I immunocapture kit” guidelines (ab109711; Abcam, Cambridge, MA). Isolated complex I was further processed for S-nitrosation detection assay with the addition of AlexaFluor-C2 Maleimide (NEM-680 dye, A20344; Thermo Fisher) 20 μM, ascorbate 1 mM, and CuCl 1 μM to the solution and incubated for 1 h at RT. Samples were finally diluted in Laemni Sample buffer containing 5% β-mercaptoethanol, and complex I proteins were separated on a NuPAGE precast SDS PAGE gel (NP0321; Invitrogen) under reducing conditions. After electrophoresis, the gel was fixed in ethanol in 50%, orto-phosphoric acid 2.5% ON under mild agitation. After image acquisition with an Odyssey Imaging System (Li-COR Biosciences), the gel was finally processed for silver staining, which was detected with a GS900 Calibrated Densitometer (Bio-Rad, Irvine, CA).
Nrf2 mobilization and evaluation of antioxidant gene transcription
Nrf2 activation was investigated in the SH-SY5Y neuroblastoma cell line. Overall, 105 cells were seeded on a glass coverslip, let to adhere overnight, and treated as described earlier with nitrite (48 h) and/or MPP+ (4 h). When required, NO scavenger (Carboxy-PTIO) or the Nrf2 inhibitor trigonelline was added to culture medium at the indicated concentration. Cells were then fixed and permeabilized with PFA 4% and Triton 0.5% for 20 min at RT. Subsequently, cells were blocked with BSA 3% in PBS for 1 h at RT and incubated with the anti-Nrf2-antibody (1:100, Sc-722; Santa Cruz Biotechnology, Dallas, TX) in PBS supplemented with 1.5% BSA ON at 4°C. After three washes with PBS, cells were incubated for 2 h at RT with a fluorescent secondary antibody (1:500, anti Rabbit Cy3 conjugated). Finally, coverslips were covered with Vectashield containing DAPI (H1200; Vector Laboratories) and mounted on glass slides. Image acquisition was performed with a Leica TCS SP5 laser scanning confocal microscope. Images were analyzed in a semi-automated fashion by using the Metamorph software (MolecularDevices, Sunnyvale, CA). The software automatically quantified the Nrf2 signal intensity-generating regions of interest around the nucleus based on the DAPI fluorescence.
Total RNA was isolated from SH-SY5Y cells treated with nitrite and MPP+ as described earlier. Total RNA was isolated by using the RNAqueous Kit (AM1912; Ambion, Austin, TX) according to the manufacturer's directions. cDNA was synthesized with SuperScript First-Strand cDNA Synthesis Kit (11904018; Invitrogen) from 1 μg of RNA. qPCR was performed on a C1000™ Thermal Cycler, CFX96 Real-Time System (Bio-Rad) by using SYBR Green I (S7564; Invitrogen) and Platinum Taq polymerase (10966018; Invitrogen). Primer sequences were obtained from the PrimerBank PCR Primers database for Gene Expression Detection and Quantification (
Statistical analysis
All values are expressed as mean ± standard error of the mean. Statistical significance was assessed by a two-sided Student's t-test or one-way analysis of variance followed by the Dunnett's multiple-comparison post hoc test. In all instances, a value of p < 0.05 was considered statistically different.
Footnotes
Acknowledgments
The authors are grateful to Dr. Peter Holland for providing constructive criticism on this article. Fundings: P.G.M. was supported by a grant from the Netherlands Genomics Initiative (NGI/NWO 05040202), a Marie Curie grant (IRG 247918), and the CEREBRAD grant under the EU-FP7 framework (project number 295552). The Extracellular Flux Analyzer by Seahorse Bioscience was purchased thanks to a generous donation from the “Dorpmans-Wigmans Stichting” (P.G.M.). C.M. was supported by the Ri.MED Foundation.
Dr. Gladwin receives research support from NIH grants 2R01HL098032, 1R01HL125886-01, and P01HL103455, T32 HL110849, T32 HL007563, the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania.
Author Disclosure Statement
Dr. Gladwin is listed as a co-inventor on an NIH government patent for the use of nitrite salts in cardiovascular diseases, and on provisional patents for the use of recombinant neuroglobin and heme-based molecules as antidotes for CO poisoning; the former has been licensed by United Therapeutics and the latter by Globin Solutions, Inc. Dr. Gladwin is a co-investigator in a research collaboration with Bayer Pharmaceuticals to evaluate riociguate as a treatment for patients with SCD. For all other authors, no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.