
Abstract
Significance:
Vascular oxidative stress plays a crucial role in atherogenesis and cardiovascular disease (CVD). Recent evidence suggests that vascular redox state is under the control of complex pathophysiological mechanisms, ranging from inflammation to obesity and insulin resistance (IR).
Recent Advances:
Adipose tissue (AT) is now recognized as a dynamic endocrine and paracrine organ that secretes several bioactive molecules, called adipokines. AT has recently been shown to regulate vascular redox state in both an endocrine and a paracrine manner through the secretion of adipokines, therefore providing a mechanistic link for the association between obesity, IR, inflammation, and vascular disease. Importantly, AT behaves as a sensor of cardiovascular oxidative stress, modifying its secretory profile in response to cardiovascular oxidative injury.
Critical Issues:
The present article presents an up-to-date review of the association between AT and vascular oxidative stress. We focus on the effects of individual adipokines on modulating reactive oxygen species production and scavenging in the vascular wall. In addition, we highlight how inflammation, obesity, and IR alter the biology and secretome of AT leading to a more pro-oxidant phenotype with a particular focus on the local regulatory mechanisms of perivascular AT driven by vascular oxidation.
Future Directions:
The complex and dynamic biology of AT, as well as its importance in the regulation of vascular redox state, provides numerous opportunities for the development of novel, targeted treatments in the management of CVD. Therapeutic modulation of AT biology could improve vascular redox state affecting vascular disease pathogenesis. Antioxid. Redox Signal. 29, 313–336.
Introduction
O
Oxidative stress can be defined as a shift in the physiological balance between oxidants and antioxidants within a biological system toward a more pro-oxidant phenotype and oxidizing redox state (53). In this regard, reactive oxygen species (ROS) are crucial to the dysregulation of vascular redox state. More specifically, the term “ROS” describes a range of molecules (including molecular oxygen and its by-products) that are generated in aerobic cells and can react with endogenous molecules such as proteins or deoxyribonucleic acid (DNA) resulting in their oxidation (29). Most of these ROS possess unpaired electrons and function as free radicals (e.g., superoxide anions [O2 •−]), while others (e.g., hydrogen peroxide [H2O2] and peroxynitrite [ONOO−]) have endogenous oxidizing effects.
ROS are produced by different intracellular or extracellular enzymatic systems (e.g., nicotinamide adenine dinucleotide phosphate [NADPH] oxidases, xanthine oxidase, and uncoupled endothelial nitric oxide synthases [eNOS]) (70). Interestingly, ROS are also involved in key physiological processes functioning as cell signaling molecules (153), while recent studies have revealed paradoxical, beneficial effects of endogenous ROS on vascular function (161).
However, in pathological states, excessive ROS production can overwhelm the endogenous antioxidant defense mechanisms leading to oxidative stress. A vicious circle may then ensue, which involves a series of radical chain reactions leading to the generation of more ROS and perpetuation of local oxidative stress (29). The various deleterious effects are mediated not only by damage to biological molecules at the macromolecular level but also through activation of key redox-sensitive intracellular signaling pathways that can lead to upregulation of proinflammatory and proatherogenic genes (173).
Recent studies support the importance of metabolic status and inflammation in determining vascular redox balance (61, 76, 166). In addition, adipose tissue (AT), traditionally considered a passive depot, is now widely recognized as a key regulator of cardiovascular physiology, primarily through the secretion of adipokines, bioactive molecules with endocrine and paracrine effects (13). Adipokines are a heterogeneous group of molecules that differ in their effects on vascular oxidative stress and inflammation (24). Notably, the adipokine secretome of AT is characterized by depot-specific differences and appears to be under the direct regulation of systemic inflammatory and cardiometabolic as well as local cardiovascular mechanisms (22) (Fig. 1).
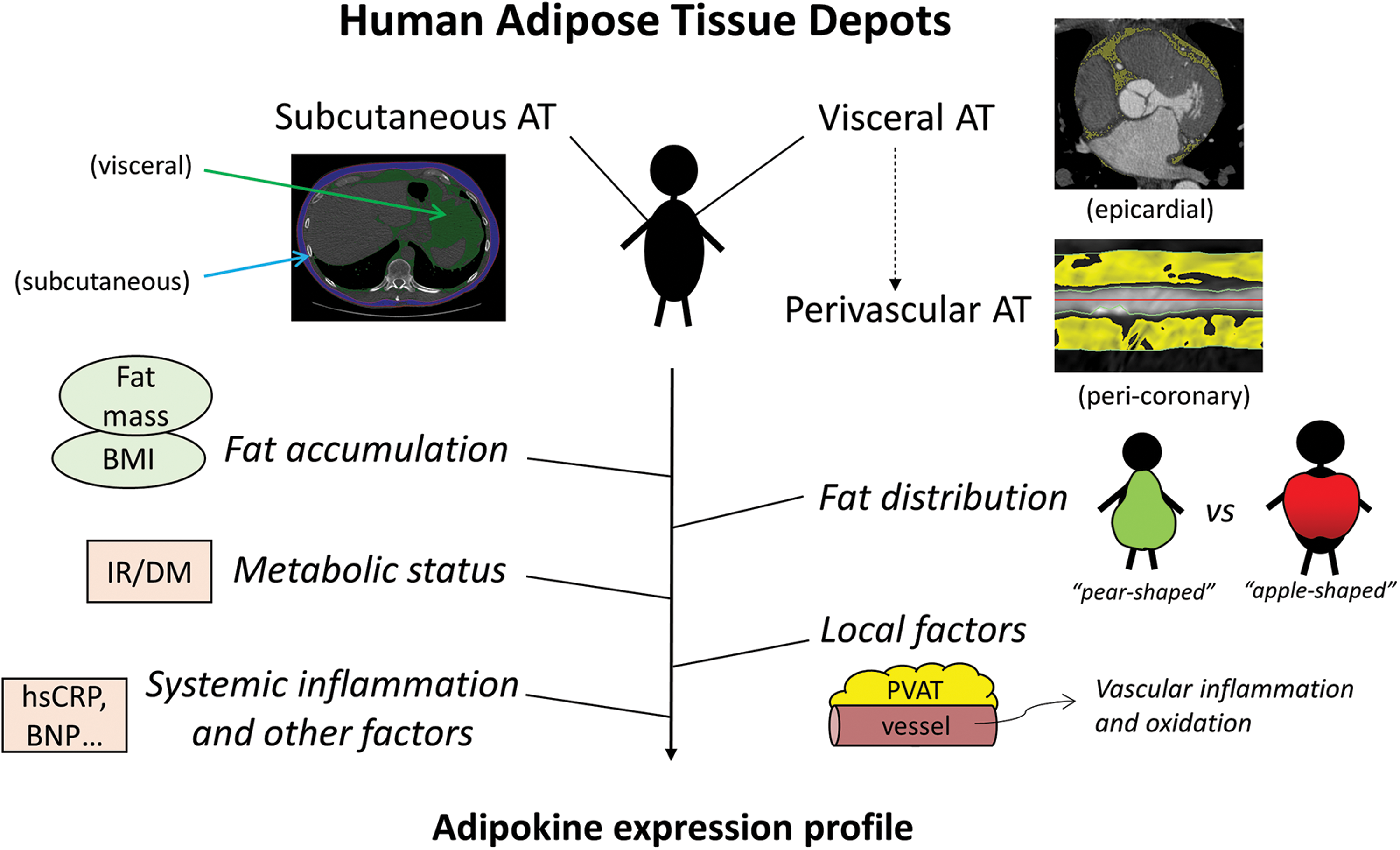
More recently, a bidirectional cross talk between cardiovascular oxidative stress and AT biology has also been shown, where AT functions as a sensor of cardiovascular oxidative damage and responds by secreting adipokines that exert antioxidant effects on the heart and vessels (11, 82, 124). Overall, it is now established that AT plays a critical role in the regulation of vascular redox state, orchestrating the complex interplay between inflammation, obesity, insulin resistance (IR), and vascular oxidative stress.
Vascular Oxidative Stress and Disease Pathogenesis
ROS as mediators of physiological signaling
ROS have been implicated in several cell processes, such as growth factor signaling, cell differentiation, proliferation, metabolism, and apoptosis (153, 175). For example, H2O2 can modify the activity of phosphatases through transient oxidation of thiol groups in specific cysteine residues (112), while O2 •− can also result in inactivation of specific proteins, leading to autophagy and cell death (38).
The mechanisms by which ROS act as signaling molecules while avoiding oxidative damage are not entirely clear. It appears that colocalization of ROS with their target proteins, careful regulation of their bioavailability in specific cellular compartments as well as their specificity for particular targets, may facilitate physiological redox signaling while limiting extensive oxidative damage (158). In addition, normal redox signaling might be confined to reversible posttranslational protein modifications (e.g., sulfenic forms), whereas subsequent nonreversible modification (e.g., sulfinic and sulfonic forms) might mediate the pathogenic effects of oxidative stress (74).
Oxidative stress and vascular disease pathogenesis
Vascular redox state is defined by the balance between ROS generation by different sources and scavenging by antioxidant defense mechanisms in the vascular wall (3, 8, 85). ROS, when in excess, adversely affect vascular physiology through multiple interrelated mechanisms that include reduced nitric oxide (NO) bioavailability, direct cytotoxic effects on vascular cells, and regulation of redox-sensitive transcriptional pathways and other biological mechanisms (29, 53, 172).
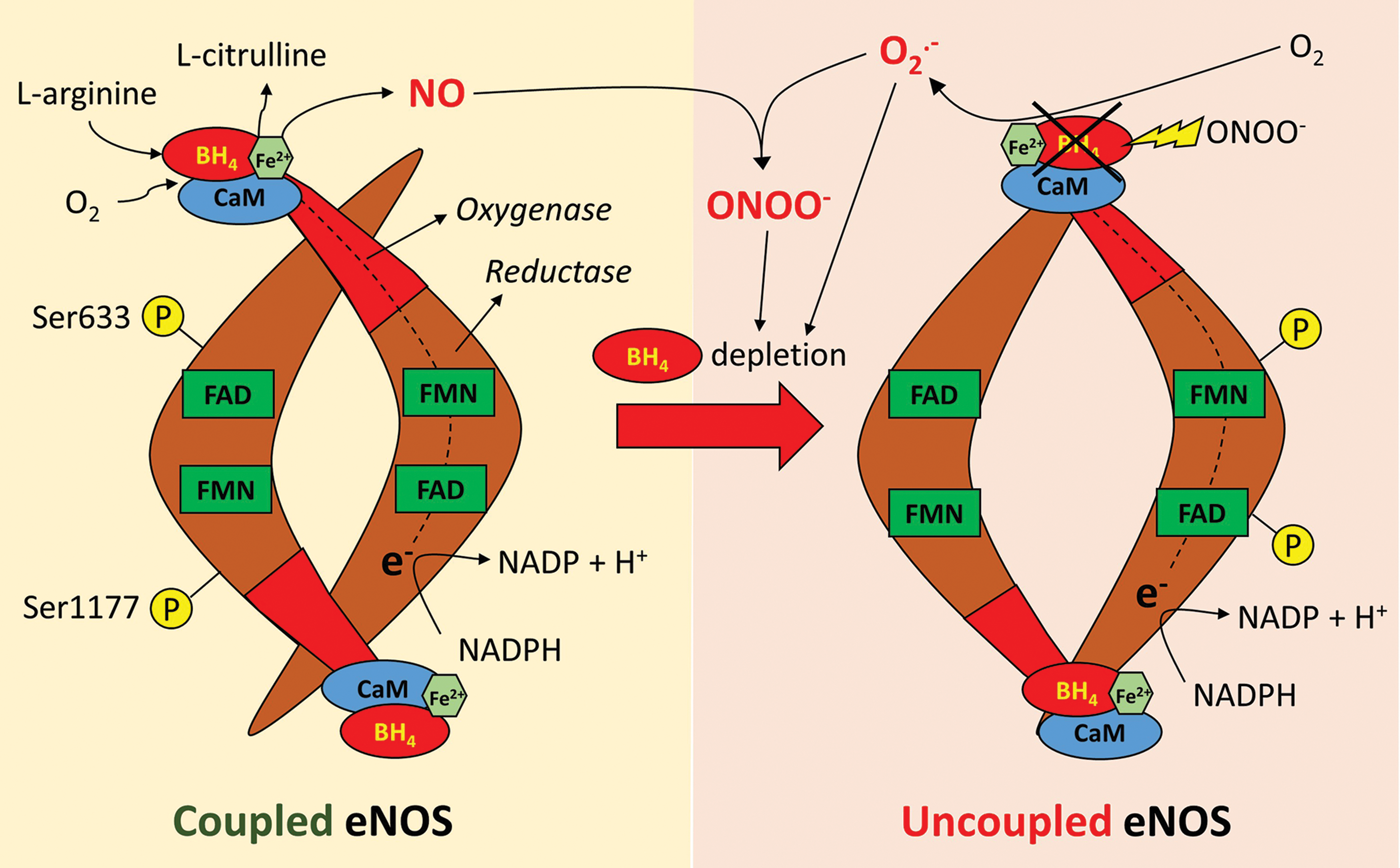
For instance, O2 •− can react with NO, decreasing its bioavailability and preventing its anti-inflammatory and antithrombotic effects (180), while also leading to the formation of ONOO−. The latter is a potent oxidant that can in turn oxidize other molecules, including antioxidants, such as glutathione reductase and superoxide dismutase (SOD), as well as dimethylarginine dimethylaminohydrolase (DDAH), an enzyme responsible for the inactivation of asymmetric dimethylarginine (ADMA), an endogenous inhibitor of eNOS (53, 188). It can also lead to depletion of tetrahydrobiopterin (BH4), a key cofactor for eNOS, leading to eNOS uncoupling and generation of O2 •− (176).
On the contrary, ROS can also activate key redox-sensitive proinflammatory transcriptional pathways, most notably the nuclear factor kappa beta (NF-κB) and activator protein 1 (AP-1) pathways, resulting in the expression of proinflammatory genes on the vascular endothelium and migration of VSMC (53, 108). Other direct effects of ROS include inflammasome activation leading to increased expression of proapoptotic (e.g., caspase-1) and proinflammatory molecules (e.g., tumor necrosis factor alpha [TNF-α]) or the oxidation of macromolecules such as DNA and proteins in the intracellular or extracellular compartment (28, 53, 127).
Sources of ROS in the vascular wall
ROS in the vascular wall are produced by different cell types, ranging from endothelial cells to VSMC and infiltrating macrophages (3, 8, 110). Similarly, a variety of different enzymatic systems are responsible for ROS scavenging, functioning as antioxidant defense mechanisms.
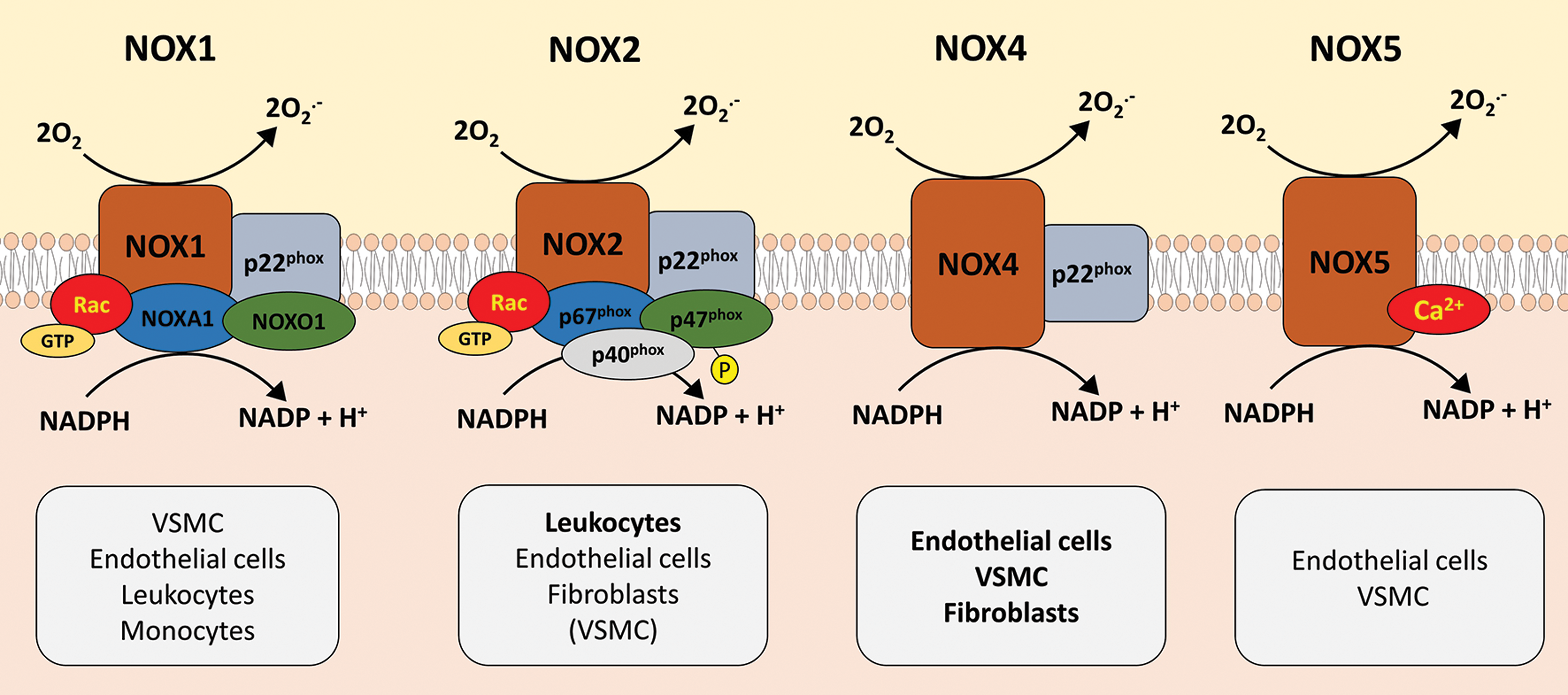
NADPH oxidases
NADPH oxidases represent a group of multisubunit isoforms that catalyze the transfer of electrons from NADPH (electron donor) to molecular oxygen (O2) to generate O2 •− (9, 103, 108). Seven isoforms have been described to date (nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1 − 5 [NOX1 − 5] and DUOX1, 2), which differ in terms of their regulatory subunits and mechanisms as well as cellular or subcellular localization within the vascular wall (Fig. 2) (19, 53, 131). For example, different NOX isoforms dominate in different anatomical regions or cellular components, with NOX4 dominating in arterial segments, primarily in the medial layers, while NOX2 is the principal isoform in veins, predominantly in the endothelium (53, 75).
Uncoupled eNOS
The importance of NO in vascular physiology is well established (180). The endothelial isoform of NOS (eNOS) represents the major enzymatic source of NO in the vascular endothelium, while the inducible isoform (inducible endothelial nitric oxide synthase [iNOS]) is mainly present in infiltrating macrophages and VSMC and is upregulated following vascular injury (34).
In its coupled state, eNOS utilizes molecular oxygen and
Other sources of ROS in the vascular wall
ROS are also produced by a wide number of other enzymatic sources, such as xanthine oxidase, cyclooxygenase, lipooxygenase, the P450 monooxygenase system, peroxidases, or the mitochondrial electron transport system (70, 178). Furthermore, hydroxyl radicals (e.g., OH·), some of the most potent ROS, can be generated from H2O2 through a metal-catalyzed (Fe2+) radical chain reaction (Fenton reaction) (158).
Antioxidant defense mechanisms
Antioxidant defense mechanisms are critical in determining the ultimate vascular redox state through continuous scavenging of ROS produced by other sources. In healthy physiological conditions, antioxidant mechanisms are able to adapt to the levels of oxidative stress to maintain an appropriate balance (166). These protective mechanisms consist of a group of enzymes, such as SOD, glutathione peroxidase, glutathione reductase, catalase, heme oxygenase, glucose-6-phosphate dehydrogenase (114, 166), as well as nonenzymatic mechanisms such as albumin and bilirubin (165, 166) or micronutrients and vitamins (e.g., vitamins A, C, and E) (119, 165). Several studies have linked the levels and activity of these enzymes to vascular disease (40, 73). Of note, increased AT accumulation is also associated with a relative depletion and impaired activity of antioxidant defense mechanisms, suggesting a possible mechanistic connection between obesity and increased vascular oxidative stress (61).
ROS and physiological signaling in the vascular wall
Interestingly, recent studies have uncovered some paradoxical, beneficial effects of ROS on vascular function. Nox5 expression increases NO release from bovine and human endothelial cells (206). Furthermore, Nox4 overexpression in mouse endothelial cells promotes angiogenesis in an eNOS-dependent manner (45), while NADPH oxidase-derived ROS are also critical in the eNOS-dependent regulation of vascular tone in coronary vessels, by activating PI3K (phosphoinositide 3-kinase)/Akt (protein kinase B) and AMPK (5′ adenosine monophosphate-activated protein kinase) (59, 160). These paradoxical effects suggest that it is not the simple presence of ROS but rather their dysregulated overproduction in conditions of oxidative stress that contributes to vascular disease pathogenesis.
Vascular Redox State and Systemic or Vascular Inflammation
Vascular redox state and vascular inflammation interact in a bidirectional loop. High levels of inflammation are known to promote increased oxidative stress through enhanced production of ROS in the vascular wall, whereas local oxidative stress also enhances inflammatory activation through redox-sensitive transcriptional pathways (82) (Fig. 4).

This “vicious circle” of vascular inflammation and vascular redox state represents a major mechanism in the regulation of atherogenesis. For example, TNF-α, a potent proinflammatory cytokine, is known to directly promote oxidative stress through upregulation of NOX1 expression (183). Activated macrophages express several ROS-producing enzymes such as myeloperoxidase (MPO) and NOX1, which produce oxidants that may also damage surrounding tissues (82, 149, 154). ROS may also promote recruitment of more inflammatory cells to the vascular wall, possibly by increasing their responsiveness to chemotactic stimuli (151). Furthermore, oxidative modification of low-density lipoprotein (LDL) particles to more proatherogenic forms, such as oxidized-LDL (oxLDL), and their subsequent uptake by vascular cells through the lectin-like oxLDL receptor 1 (LOX1) further promote atherogenesis and local inflammation (143).
Additional mechanisms may include the release of sphingomyelinase from endothelial cells and macrophages that is induced by proinflammatory molecules. This results in hydrolysis of LDL sphingomyelin to ceramide, which is known to directly activate ROS production and oxidative damage in the mitochondria (143). Angiotensin II (AngII), a molecule with well-defined proatherogenic and proinflammatory effects, is also able to stimulate O2 •− production through increased phosphorylation of NADPH oxidase and expression of the catalytic (mainly NOX1, 2, 4, and 5) and regulatory subunits of the enzyme (e.g., p22phox, p47phox, and p67phox) (47, 139).
On the contrary, it is now understood that vascular oxidative stress also has direct effects on the regulation of local vascular inflammation. In fact, various redox-sensitive transcription factors have been shown to be upregulated in response to oxidative stress (e.g., NF-κB, AP-1, hypoxia-inducible factor 1 [HIF-1]) (162). These lead to the upregulation of proinflammatory genes affecting intracellular AMPK- or calcium-dependent signaling pathways (162, 181) and expression of proinflammatory molecules, such as cytokines (e.g., TNF-α, interleukin 6 [IL-6]), cell adhesion (e.g., integrins, intracellular cell adhesion molecule 1 [ICAM-1], vascular cell adhesion molecule 1 [VCAM-1]), or other proinflammatory molecules (e.g., monocyte chemoattractant protein 1 [MCP-1], plasminogen activator inhibitor 1 [PAI-1], or platelet-derived growth factor [PDGF]) (106, 150).
In DM, hyperglycemia might further induce ROS generation, which results in increased formation of advanced glycation end products. Binding of advanced glycation end products to their receptors triggers formation of ROS, which then activate NF-κB and the expression of proinflammatory and procoagulant genes (64). The reduced bioavailability and disruption of NO signaling, a molecule with potent anti-inflammatory and antithrombotic properties, also contribute to increased local inflammation (76).
Two central signaling pathways are primarily involved in mediating the redox-induced effects on vascular inflammation. On the one hand, the NF-κB system is central in the pathogenesis of several inflammatory cellular responses (132). NF-κB is a transcription factor that is encoded by members of the Rel family and is normally present in the cytoplasm as a heterodimer (p50 and p65) bound to the inhibitory protein IκB (inhibitor of kappa beta) that blocks its translocation to the nucleus by masking its nuclear localization signal. Activation of IκB kinase by proinflammatory signals (e.g., TNF-α) leads to phosphorylation of IκB and proteolytic degradation, allowing translocation of the heterodimer to the nucleus and activation of key transcription factors (78).
Studies in aged rats have also shown that O2 •− produced in the mitochondria of endothelial cells and VSMC can be dismutated to H2O2, which can activate NF-κB in the cytoplasm of the same cells (184). Similarly, administration of salsalate (an inhibitor of NF-κB) in a double-blind placebo-controlled study in older humans resulted in upregulation of IκB and downregulation of NF-κB ιn endothelial cells, in addition to a significant reduction in the expression of the p47phox subunit of NADPH oxidase and improvement of endothelial-dependent vasorelaxation when compared to the placebo arm (150).
Contrary to the proinflammatory and pro-oxidant effects of NF-κB activation, the nuclear factor erythroid-2-related factor-2 (Nrf2) system has been linked to protective, antioxidant effects (191, 200). Indeed, Nrf2 is a redox-sensitive transcription factor that once activated, promotes the expression of several antioxidant genes that act to detoxify ROS and limit oxidative damage (115). It is evident that NF-κB and Nrf2 act in an antagonistic way to regulate the cellular response to oxidative stress.
AT in Obesity, IR, and Diabetes
AT: structure, role, and general characteristics
AT is now considered to be a key player in the regulation of cardiovascular health. While AT consists mainly of adipocytes, many other cells can be found in its stromal fraction, including inflammatory cells (e.g., lymphocytes, macrophages), preadipocytes, fibroblasts, and vascular cells. More importantly, AT is no longer regarded as a passive depot of fat and energy storage but is considered a key endocrine organ with a pivotal role in the regulation of the overall cardiometabolic health through the secretion of a wide range of adipokines (95). These regulate key processes for whole-body homeostasis, such as appetite and insulin sensitivity (95).
AT can be further separated into white and brown AT depots. White AT represents most of the AT mass found in the human body and is primarily responsible for energy storage in the form of triglycerides. On the contrary, brown AT (∼60 g in the average human adult) is well known for its thermogenic effects and might even protect against obesity (88, 89), while recent evidence suggests that, similarly to white AT, it can also produce adipokines (called “batokines”). Importantly, an inducible form of brown AT (called “beige” AT) has been recently described in humans. Beiging of white AT occurs in response to exercise or cold exposure and has been linked to improve insulin sensitivity and weight loss (88, 163).
Such differences in AT biology might explain why not all obese people develop metabolic and CVD. While the existence of a metabolically healthy versus metabolically unhealthy obese phenotype is under debate (104, 133), it is clear that differences in AT biology are at least as important as AT expansion and obesity in determining the effects of AT on cardiovascular health. IR is the most widely accepted biological feature defining the metabolic profile of obesity as “healthy” or “unhealthy”, as discussed in the section From obesity to IR and diabetes.
From obesity to IR and diabetes
Obesity, IR, and DM are all associated with changes in AT function and the adipokine profile (146) (Fig. 5). Obesity is associated with upregulation of the expression of most proinflammatory adipokines. For example, increased AT expression of leptin, resistin, lipocalin 2, chemokines such as chemokine C-C motif ligand 2 (CCL2), and other proinflammatory adipokines promotes an inflammatory state that contributes to metabolic disease (146). Several of these adipokines have been linked to AT expansion particularly in the visceral fat compartment, a depot with well-documented adverse cardiometabolic effects (13). Indeed, higher circulating levels of retinol-binding protein 4 (RBP4), visfatin, chemerin, vaspin, progranulin, and lower levels of omentin have all been linked to a higher visceral fat mass (22, 24).
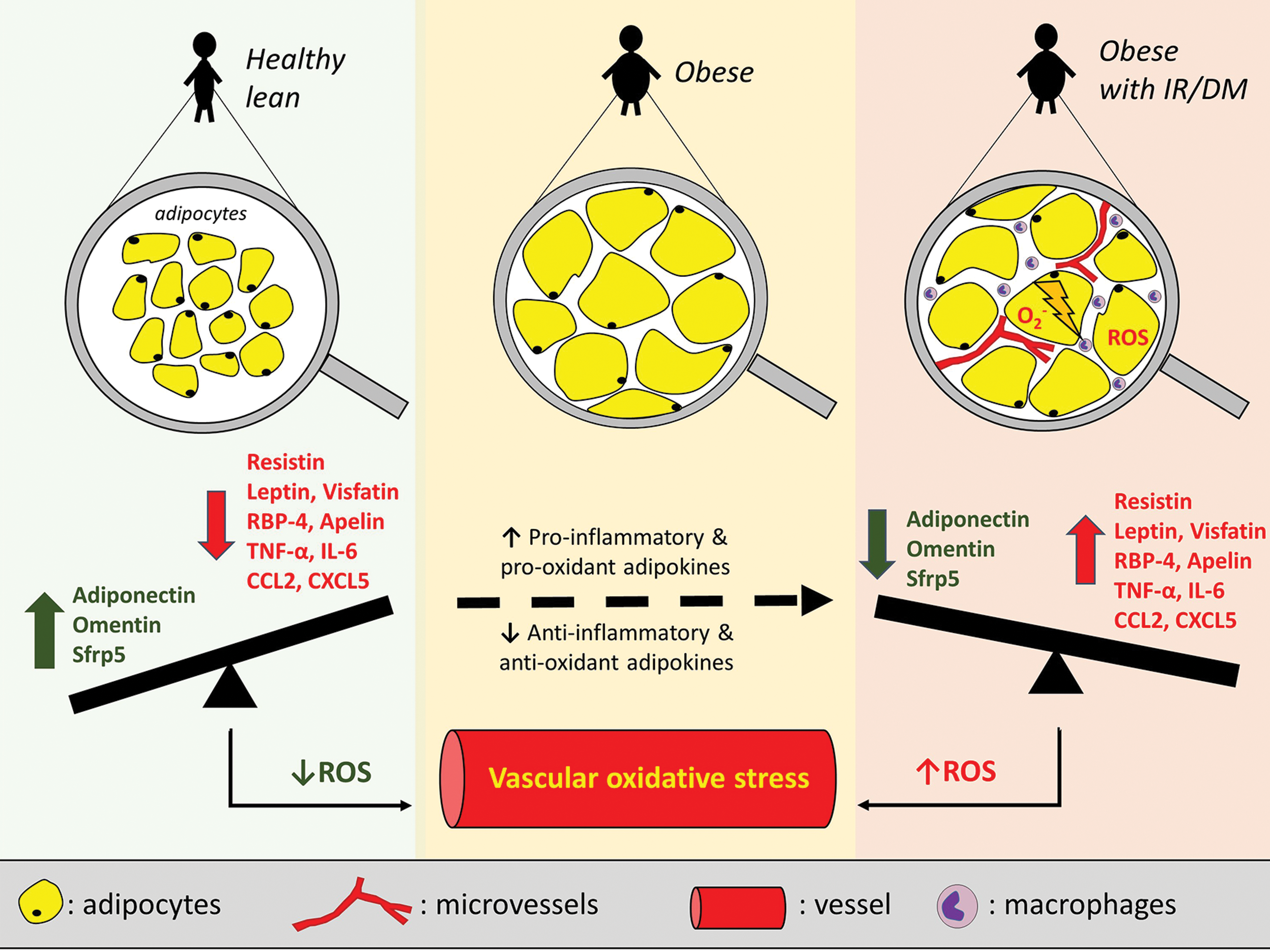
Nevertheless, even among obese individuals, AT exhibits differences in its biology and secretome depending on the underlying metabolic status. IR has been linked to higher circulating levels of progranulin, chemerin, and RBP4 and significantly lower serum levels of adiponectin independently of total body fat mass, thus defining the metabolically “unhealthy” obese phenotype (99). Several more adipokines have been shown to be differentially regulated in dysfunctional adipocytes and in states of IR. For example, dipeptidyl-peptidase 4, a recently discovered adipokine, has been proposed as a marker of visceral obesity and IR, given its higher production by visceral AT in obese and IR patients (159), while circulating glypican-4 levels correlate positively with both body mass index (BMI) and insulin sensitivity (185). Similar changes have been described for a range of adipokines, such as visfatin (32), RBP4 (204), resistin (87, 156), PAI-1 (156), and Angptl2 (angiopoietin-like protein 2) (177).
Chemokine production by adipocytes is also affected in obesity and IR. For instance, CCL2, CXCL5 (chemokine C-X-C motif ligand 5), CXCL14 (chemokine C-X-C motif ligand 14), and MCP-1 induce macrophage infiltration in the AT therefore promoting IR (35, 91, 94, 134). Moreover, proinflammatory cytokine levels that are known to be produced by adipocytes (e.g., TNF-α and IL-6) also correlate with systemic IR (17, 81). In contrast, circulating levels of anti-inflammatory adipokines such as adiponectin and secreted frizzled-related protein 5 (sfrp5) have been negatively correlated with fat accumulation and IR (51, 145).
Specific adipokine clusters have also been linked to body fat mass, inflammation, and IR (63). More specifically, ANGPTL6 (angiopoietin-like protein 6), DLK1 (delta-like protein 1), visfatin, and progranulin were found to be the strongest adipokine predictors of type 2 DM among obese individuals. However, the authors of this study acknowledge that the predictive value of this adipokine cluster was inferior to that of glycated hemoglobin (HbA1c), HOMA-IR (homeostatic model assessment—IR index), and fasting plasma glucose, questioning the potential value of these measurements in routine clinical diagnosis (63).
It should be highlighted that IR does not affect the cardiovascular system exclusively through changes in the circulating adipokine levels. Irrespective of the effects of systemic IR on the adipokine profile, vascular IR is equally important in determining vascular health and redox state. It is now suggested that vascular IR may actually precede systemic IR, while oxidative stress results in impaired insulin signaling in vascular cells (characterized by impaired PI3K/Akt signaling and enhanced activation of the MAPK (mitogen-activated protein kinase) pathway), therefore promoting endothelial dysfunction and vascular disease (54, 148) (Fig. 6).
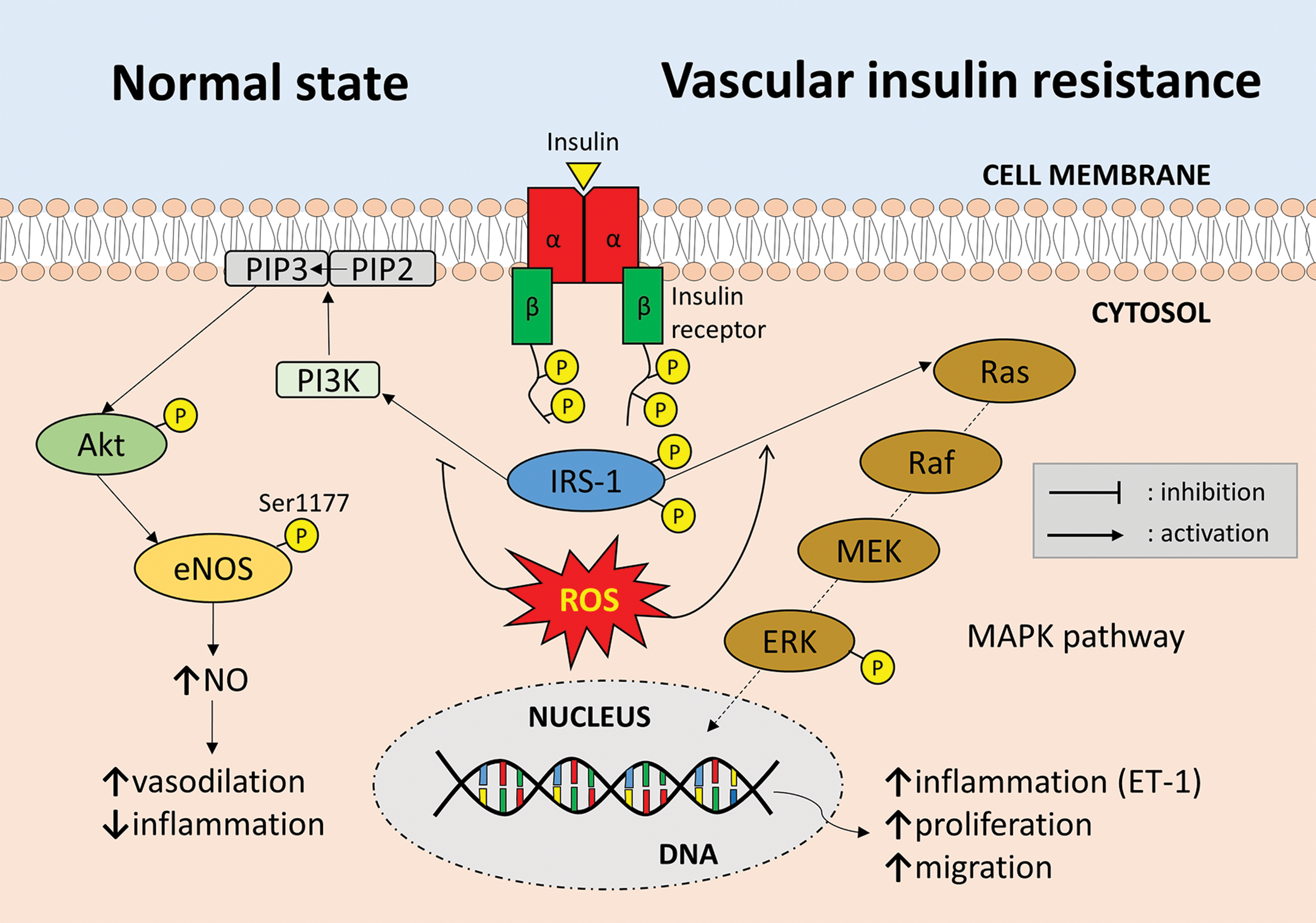
Another critical factor that regulates the effects of AT on cardiometabolic health is exercise. Experimental studies in both animal models and humans suggest that exercise induces a decrease in adipocyte size and lipid content in white AT depots, while also upregulating mitochondrial activity (170, 171). In rodent models, exercise has also been associated with browning of white AT and increased expression of uncoupling protein 1 (UCP-1), while studies in humans have provided inconsistent results (25). Interestingly, exercise-induced weight loss in humans was more effective in improving the circulating adipokine profile (increased adiponectin and decreased chemerin levels) compared to diet-induced weight loss, highlighting the importance of exercise in the regulation of AT biology (68, 96). Such observations have also highlighted skeletal muscle-derived myokines, as important regulators of AT phenotype and secretion profile (25). Finally, weight loss per se can improve glycemic and in particular GLP-1 (glucagon-like peptide 1) responses (57, 84), which may have a beneficial effect on endothelial oxidative stress (72).
AT and Vascular Redox State
While a link between AT and CVD is well established, the exact nature of this association is less clear. It is now accepted that adipokines can have direct effects on the vascular wall and are important regulators of vascular redox state. Both distant depots, such as the subcutaneous AT, and visceral depots (e.g., thoracic and epicardial AT) are potent sources of adipokines that exert direct vascular effects, ultimately promoting or protecting against oxidative stress. In this context, perivascular AT (PVAT) has emerged as an important depot in CVD pathogenesis, given its close spatial relationship to the vascular wall that allows direct diffusion of adipokines (11, 124, 197). As a general rule, adipokines not only differ in their effects on vascular oxidative stress but also exhibit pleiotropic effects, modifying vascular redox state through multiple, independent mechanisms (Fig. 7 and Table 1).
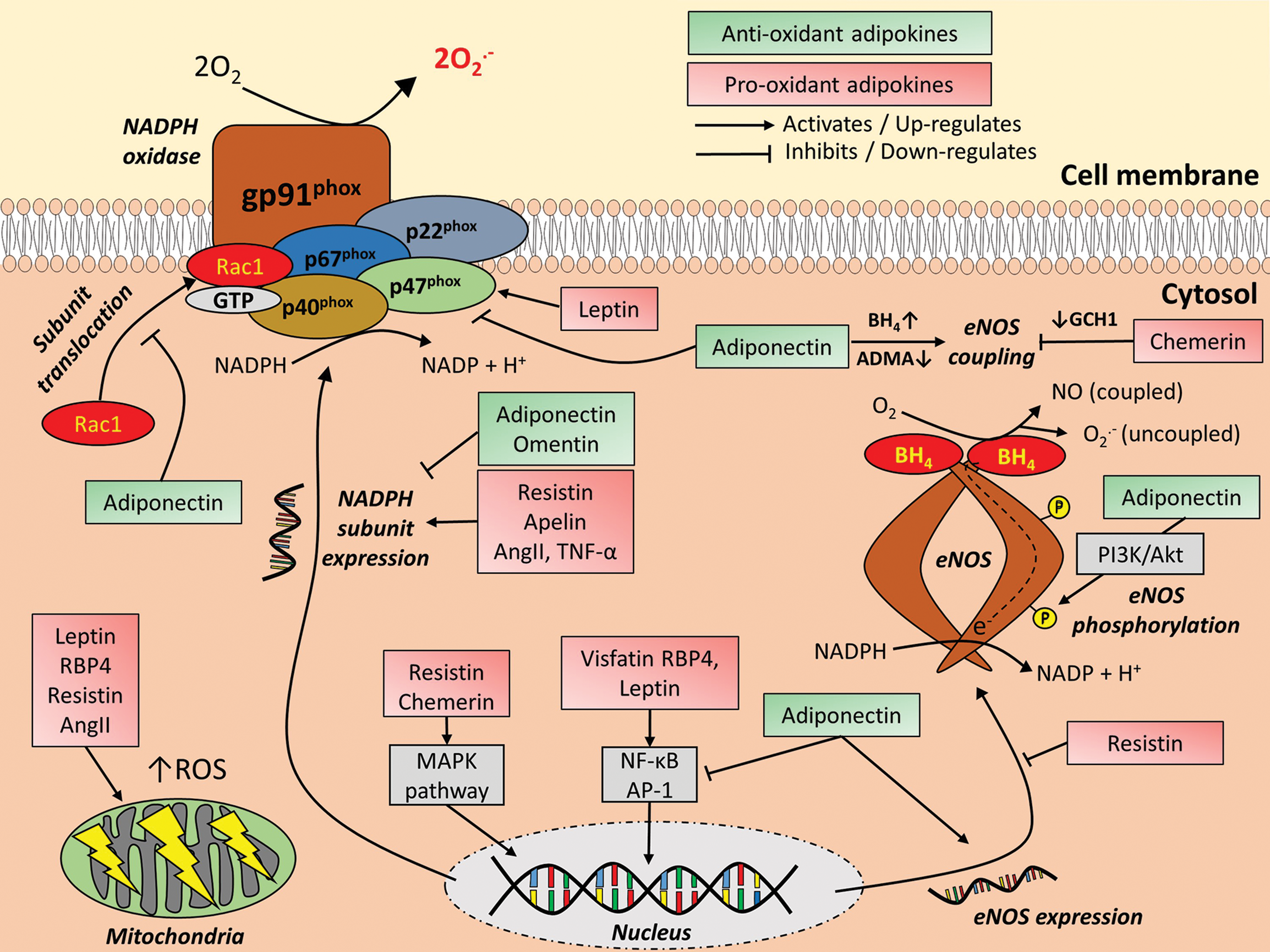
ADMA, asymmetric dimethylarginine; Akt, protein kinase B; BH4, terahydrobiopterin; DDAH, dimethylarginine dimethylaminohydrolase; DM, diabetes mellitus; EC, endothelial cells; eNOS, endothelial nitric oxide synthase; GCH1, GTP cyclohydrolase 1; GTP, guanosine-5′-triphosphate; HUVECs, human umbilical vein endothelial cells; IR, insulin resistance; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor kappa beta; NO, nitric oxide; NOX, NADPH oxidase; O2 •−, superoxide anions; PDGF, platelet-derived growth factor; PI3K, phosphoinositide 3-kinase; PKC, protein kinase C; RBP4, retinol-binding protein 4; SOD, superoxide dismutase; VSMC, vascular smooth muscle cells.
Leptin
Leptin is one of the most extensively characterized adipokines and is abundantly expressed in AT. It has a cytokine-like structure and its receptor belongs to the class I cytokine receptor superfamily (87). Proinflammatory cytokines such as TNF-α upregulate the expression of leptin in AT, while in clinical studies circulating leptin levels have been associated with C-reactive protein (CRP) levels, independently of BMI (16). Irrespective of its links to inflammation, one of the major and well-known functions of leptin is the regulation of appetite and energy homeostasis through stimulation of energy expenditure (67). Leptin improves insulin sensitivity in both skeletal muscle and liver and promotes proliferation of pancreatic beta cells (125), while its deletion in mouse models leads to obesity and type 2 DM (207).
On the contrary, studies in humans have shown that circulating leptin levels are elevated among individuals with a higher percentage of body fat, possibly due to a mechanism of leptin resistance (42, 67). In fact, IR assessed by the HOMA-IR index is positively correlated with circulating leptin levels, independently of age, gender, and body fat mass (62, 192). Despite these findings, a study of 5599 individuals (>20 years of age) from the National Health and Nutrition Examination Survey (NHANES III) did not detect an independent association between plasma leptin and DM after adjusting for BMI, a finding that was consistent in both genders (16). Therefore, it is possible that any association between circulating leptin levels and DM may be confounded by body fat (1, 16).
While most studies have focused on the effects of leptin on the regulation of metabolic status, energy homeostasis, and weight control, several studies have identified an important role for leptin in the regulation of vascular redox state. In rat models, leptin increases Akt phosphorylation and the production of O2 •− in VSMC, while both aortic leptin and O2 •− levels correlate with markers of aortic fibrosis (126). In mouse heart microvascular endothelial cells, leptin leads to increased ROS formation by promoting p47phox activation (37), while it has also been shown to increase mitochondrial O2 •− generation in bovine aortic endothelial cells through promotion of fatty acid oxidation via protein kinase A (201).
It is also known that human umbilical vein endothelial cells (HUVECs) possess leptin receptors and, in fact, leptin leads to higher production of ROS in HUVECs through activation of the NH2-terminal c-Jun kinase (JNK)/stress-activated protein kinase pathway as well as the NF-κB and AP-1 transcriptional pathways. Moreover, these effects were shown to be sensitive to the effects of N-acetylcysteine, an antioxidant treatment (26). Additional data suggest a role for leptin in the regulation of redox state in other vascular cell types, such as macrophages and VSMC. For instance, binding of leptin to receptors on the cell surface of human macrophages affects redox-sensitive transcription pathways (e.g., AP-1), resulting in the upregulation of lipoprotein lipase through redox-dependent mechanisms and activation of the PKC (protein kinase C)-dependent pathway (123).
Interestingly, leptin has been demonstrated to stimulate eNOS enzymatic activity by triggering Akt-mediated phosphorylation at its activatory site (Ser1177), while it also increases the expression of neuronal NO synthase in endothelial cells (20, 187). These studies suggest that leptin may also possess beneficial roles for the vasculature, as shown in both rat and human endothelial cells and vessels (20, 187). However, it should be highlighted that activation of NOS enzymatic activity is not always beneficial for vascular biology, given that activation of the uncoupled enzymatic form leads to increased O2 •− rather than NO, triggering redox-sensitive inflammatory pathways involved in vascular disease pathogenesis (2).
Finally, in an epidemiological study of 818 elderly participants of the Framingham Heart study, higher circulating leptin levels were linked to a higher risk of congestive heart failure and/or CVD, but the association was significantly attenuated following adjustment for BMI. This could be partly explained by the critical role of eNOS coupling status on the vascular effects of leptin, as mentioned above. Of note, the relationship between leptin levels and mortality was described as U-shaped, with higher risk shown for individuals with both high and low circulating leptin levels, reflecting the complexity of the role of leptin in CVD pathogenesis (117).
Adiponectin
Adiponectin was first described more than two decades ago as a protein that is specifically expressed by differentiated adipocytes in AT (157). It consists of 247 amino-acids and four domains; an amino-terminal signal sequence, a variable region, a collagenous domain, and a carboxy-terminal globular domain. In its circulating form it exists in a trimeric or higher order, oligomeric form. Its effects are mediated by binding to its receptors adiponectin receptor 1 (AdipoR1) and adiponectin receptor 2 (AdipoR2), which are found in a wide range of targets (e.g., liver, AT, skeletal muscle, macrophages, and pancreatic beta cells) (190).
Early studies in mice showed that adiponectin can regulate glucose metabolism by enhancing insulin sensitivity (21), while circulating adiponectin levels are negatively associated with IR in mouse models (203). More recently, it was found that adiponectin reduces local inflammation in AT and significantly alters the secretome of human adipocytes, inhibiting the release of adipokines linked to IR (50).
It should be noted that the biological effects of adiponectin may vary between its globular and full-length form. The globular form results from the proteolytic cleavage of the full-length molecule and has been shown to protect against diabetes in leptin-deficient mice as well as against atherosclerosis in ApoE (apolipoprotein-E)-deficient mice (202). In fact, despite their similarities, globular and full-length adiponectins appear to exert distinct time-dependent effects on cardiomyocyte energy metabolism (147).
In humans, adiponectin levels are negatively associated with BMI and are significantly upregulated following weight loss (121). Furthermore, circulating levels are positively correlated with insulin sensitivity independently of BMI and the degree of obesity (41, 193). The association of adiponectin with systemic inflammation, however, appears to be more complex. Recent data from our group suggest that circulating adiponectin levels are regulated by complex interactions between inflammatory cytokines and brain natriuretic peptide (BNP). While inflammatory cytokine levels (i.e., IL-6) are negatively associated with circulating adiponectin levels, BNP appears to reverse these effects resulting in increased circulating adiponectin levels. In addition, there is a regional variability in the depot-specific responses to inflammation, with femoral (but not intrathoracic or subcutaneous) fat being the only depot that appears to respond to inflammatory cytokines in an ex vivo setting through downregulation of peroxisome proliferator-activated receptor gamma (PPAR-γ) signaling and adiponectin biosynthesis (10).
Adiponectin affects ROS production through multiple mechanisms. In animal models, addition of adiponectin promotes NO production from bovine aortic endothelial cells in vitro through activation of the PI3K/Akt signaling pathway, promotion of heat shock protein 90 binding to eNOS as well as AMPK-mediated eNOS phosphorylation at its Ser1179 residue (196). Administration of adiponectin in hyperlipidemic rats did not have any effects on total NO production but was able to improve endothelial function and significantly reduce O2 •− generation through differential regulation of eNOS and iNOS activity (116).
Vascular rings from adiponectin knockout mice also demonstrate significantly higher levels of O2 •− and ONOO− generation, which are reduced following administration of exogenous adiponectin through promotion of eNOS phosphorylation (30). Adiponectin also blocks the hypertrophic effects of AngII on rat VSMC partly through a decrease in AngII-induced upregulation of ROS production and p22phox gene expression (140). Hypoadiponectinemia has also been linked to inflammasome activation in vascular tissues of diabetic mice through an increase in oxidative and nitrative stress (205).
Studies in humans show that adiponectin suppresses O2 •− generation in both neutrophils and platelets by downregulating NADPH oxidase activity through inhibition of p47phox translocation and soluble gp91phox cleavage (31, 122), while it may also prevent eNOS uncoupling in human endothelial cells by upregulating DDAH activity and decreasing ADMA accumulation in response to TNF-α (55). Adiponectin also appears to protect against palmitic acid-induced endothelial inflammation and IR, by inhibiting palmitic acid-induced activation of ROS production and the expression of inflammatory cytokines and p-IκBα in cultured HUVECs (208).
We have previously shown that circulating adiponectin levels correlate positively with higher NO bioavailability and negatively with eNOS uncoupling and O2 •− generation in vessels. In ex vivo experiments with human vessels, adiponectin was found to induce Akt-mediated phosphorylation of eNOS and increased BH4 bioavailability. These changes led to improved eNOS coupling resulting in increased NO bioavailability and reduced O2 •− generation in the human vascular endothelium (124).
In addition, we have demonstrated that circulating adiponectin levels are inversely correlated with the presence of type 2 DM and NADPH oxidase-produced O2 •− levels in internal mammary artery (IMA) segments of patients undergoing cardiac surgery. Ex vivo incubation of human IMA segments with adiponectin led to direct inhibition of NADPH oxidase activity by blocking membrane translocation of Rac1 and downregulating p22phox expression through a PI3K/Akt-mediated mechanism (11).
Irrespective of the biological effects of adiponectin, results from clinical studies exploring the predictive value of circulating adiponectin levels for mortality and cardiovascular events are controversial. In a meta-analysis of more 14,063 CVD patients, higher adiponectin levels at baseline were significantly associated with a higher risk of both all-cause and cardiovascular death (195). In accordance with these findings, in a prospective population study, lower levels of circulating adiponectin were also found to be significant and independent predictors of increased coronary heart disease risk (odds ratio [95% confidence interval] per 1 μg/mL increase: 0.78 [0.63–0.96] in males; 0.73, 95% confidence interval [0.55–0.96] in females) (43). In a separate meta-analysis of 24 prospective studies, no association was found between adiponectin levels and new-onset coronary heart disease.
However, higher adiponectin levels were a significant predictor of coronary artery disease (CAD) recurrence and all-cause as well as cardiovascular mortality (167). Similar findings were described in a study that focused on 2034 patients with type 1 DM (hazard ratio [95% confidence interval]: 1.02 [1.01–1.03] for all-cause mortality and 1.02 [1.00–1.04] for cardiovascular mortality) (66).
On the contrary, higher adiponectin serum concentrations appear to be protective against the development of type 2 DM (80, 169). In a study that included 27,548 apparently healthy individuals from the EPIC (European Prospective Investigation into Cancer and Nutrition) Potsdam Cohort, higher concentrations of adiponectin were significantly correlated with reduced risk of developing type 2 DM, even after adjusting for age, gender, smoking, and indices of obesity (waist-to-hip ratio and BMI) (169). Moreover, in the Cardiovascular Health Study, a population-based study of older adults, the association between adiponectin levels and mortality ranged from a U-shaped association in the group free of CVD to a direct positive association in the heart failure/atrial fibrillation group, while no association was found in a CVD group without heart failure or atrial fibrillation, following adjustment for potential confounders (98).
These controversial and conflicting findings have led to the notion of the “adiponectin paradox.” This could be explained by the presence of different adiponectin forms (e.g., full-length vs. globular or high-molecular- vs. low-molecular-weight forms), which have been shown to differ in their biological effects (147). However, in the Cardiovascular Health Study, no differences were observed between total and high-molecular-weight adiponectin levels in terms of all-cause and cardiovascular mortality prediction (98). Overall, it appears that when interpreting circulating adiponectin levels, the underlying disease, inflammatory and metabolic status should be taken into account, with higher adiponectin levels being protective among healthy individuals but indicating underlying cardiometabolic disease in more advanced disease states (194).
Resistin
Resistin is a 114-amino acid polypeptide that is produced by adipocytes and is secreted as a disulfide-linked homodimer. It exerts its actions by binding to its receptor (adenylyl cyclase-associated protein 1 was recently identified as a resistin receptor in human monocytes) (111) and its circulating levels are significantly upregulated in obesity, IR, DM, and inflammatory conditions. Indeed, in a cross-sectional clinical study of 79 type 2 DM patients and 30 healthy controls, serum resistin levels were linked to IR, higher in vivo lipid peroxidation, and platelet activation (155). Treatment of VSMC with resistin leads to a significant upregulation of NADPH oxidase activity (predominantly the NOX4 isoform) and ROS production through PKCɛ activation. In fact, activation of NADPH oxidase in VSMC prevented the resistin-induced proliferation and migration of VSMC as well as the expression of proinflammatory cytokines, while inhibition of PKCɛ in an ApoE-double knockout mouse model of wire injury also prevented the proatherogenic effects of resistin (152).
Experimental evidence in human coronary artery endothelial cells suggests that resistin results in downregulation of eNOS expression and NO bioavailability, impaired mitochondrial function, an increase in ROS production, and a decrease in the activity of the antioxidant defense mechanisms, catalase, and SOD. These effects appear to be mediated, at least in part, by activation of MAPK p38 and JNK and are effectively inhibited by antioxidant treatments. Moreover, resistin levels are significantly higher in atherosclerotic areas in human aortas and carotid arteries (36).
In accordance with these findings, in a recent meta-analysis, elevated circulating resistin levels in humans were associated with an increased risk of both all-cause and cardiovascular mortality (per 1 standard deviation increase: hazard ratio [95% confidence interval] 1.28 [1.07–1.54] and 1.32 [1.06–1.64], respectively) (65).
Visfatin
Visfatin is a 491-amino acid polypeptide that possesses both intracellular enzymatic activity resulting in NAD synthesis and is thought to also exert cytokine-like effects by binding its receptor (86). It was recently characterized as a visceral adipokine and has been implicated in the pathogenesis of obesity-associated CVD (97), but its role in the regulation of metabolic health and insulin sensitivity remains unclear. While some studies have shown a positive correlation between levels of visfatin gene expression in AT and IR (32, 48, 142), in other studies no significant association was observed between visfatin levels in AT or serum and glucose metabolism (186). On a separate note, visfatin is closely correlated with inflammation, with several studies demonstrating an independent association between visfatin circulating levels and markers of systemic inflammation, such as CRP and IL-6 (48, 142).
Visfatin is known to promote a proatherogenic phenotype on the vascular endothelium through upregulation of cell adhesion molecules, such as VCAM-1. Interestingly, visfatin also leads to upregulation of ROS generation in human microvascular endothelial cells through activation of the NF-κB pathway and upregulation of NADPH oxidase activity. In addition, direct ROS scavenging abolishes the effects of visfatin on the activation of the NF-κB pathway and the expression of cell adhesion molecules, suggesting that visfatin might be an important regulator of vascular redox state and may accelerate atherogenesis through the production of ROS and subsequent activation of downstream redox-sensitive transcriptional pathways (97).
In the clinical setting, higher visfatin levels have also been linked to a higher risk of future major adverse cardiovascular events (MACE) among patients presenting with ST-segment elevation myocardial infarction (STEMI) (83).
Retinol-binding protein 4
RBP4 is another recently discovered adipokine with known effects on the regulation of insulin sensitivity (204). RBP4 belongs to the family of lipocalins, is primarily synthesized in the liver, and functions as a specific transporter for vitamin A in the bloodstream. However it was recently found that it is also produced by adipocytes in the visceral depots and its production is upregulated in obesity, linking its levels with visceral fat expansion (100). RBP4 might promote IR in human adipocytes by inhibiting phosphorylation of IRS-1 (insulin receptor substrate 1) and ERK1/2 (extracellular signal-regulated kinases 1 and 2) (144). In accordance with these findings, circulating RBP4 levels in humans have been linked to the presence of obesity, IR, and DM (69), as well as to circulating levels of inflammatory biomarkers (CRP and IL-6) (15). On the contrary, other clinical studies did not identify a significant association between RBP4 levels and either obesity or IR (182).
Results from animal studies have shown that vascular oxidative damage is more pronounced in RBP4-transgenic mice compared to their wild-type counterparts (189). Similarly to visfatin, RBP4 induces proinflammatory changes in endothelial cells by upregulating the expression of proteins involved in leukocyte recruitment and endothelial adherence (e.g., VCAM-1, ICAM-1, MCP-1, and IL-6). However, these changes are independent of retinol or RBP4 membrane receptor and are in fact mediated by upregulation of NAPDH oxidase activity and activation of the NF-κB pathway (58). Furthermore, RBP4 increases O2 •− generation from mitochondrial sources in human aortic endothelial cells, while also having a negative effect on mitochondrial integrity. Overall, these results point toward a possible redox-dependent mechanism for the proatherogenic effects of RBP4.
In a recent prospective study of 468 women with CAD and 472 matched controls, higher circulating full-length and total RBP4 levels at baseline were a significant and independent predictor of CAD development during a follow-up period of up to 16 years (174).
Apelin
Apelin, a 36-amino acid peptide, was recently identified as the ligand of the apelin receptors (APJ), a G-protein-coupled receptor family (141). Apelin is upregulated in obesity and IR (198) and is considered to exert proatherogenic effects. As shown in animal studies, APJ and ApoE double knockout mice (APJ[−/−]ApoE[−/−]) have significantly less atherosclerotic lesions compared with APJ(+/+)ApoE−/−) mice, irrespective of cholesterol levels. Double knockout mice were further characterized by decreased expression of NADPH oxidase subunits (NOX1, NOX2, NOX4, and p22phox) in VSMC and decreased presence of VSMC in atherosclerotic lesions. Exposure of VSMC to apelin led to upregulation of the expression of NADPH oxidase subunits, while treatment with SOD inhibited VSMC proliferation.
Therefore, apelin appears to exert its atherogenic effects, at least in part, through local modulation of vascular redox state and upregulation of ROS generation from VSMC (77). Similarly to visfatin, circulating apelin levels can predict future MACE among STEMI patients undergoing percutaneous coronary intervention (118).
Chemerin
Chemerin is synthesized as a 163-amino acid peptide (preprochemerin) that is subsequently processed by various serine and cysteine proteases. It exerts its activities via binding to different receptors, such as G-protein-coupled receptors chemokine-like receptor 1 and C-C chemokine receptor-like 2 (CCRL2) (128). Circulating chemerin levels have also been positively associated with obesity and other cardiometabolic disorders, such as IR, hypertension, and type 2 DM (27).
Moreover, studies in rat models have shown that chemerin increases the contractile responsiveness of the vasculature and impairs both endothelium-dependent and endothelium-independent vasorelaxation mechanisms. Interestingly, these effects are reversed by BH4 administration or antioxidant interventions, such as an O2 •− scavenger or an SOD mimetic. Even though chemerin induces phosphorylation of eNOS, the protein remains mainly in the monomeric form, while the expression of GCH1, the rate-limiting enzyme in the biosynthesis of BH4, is also downregulated. This results in eNOS uncoupling, decreased NO bioavailability, and increased O2 •− production, all of which explain the overall negative effects of chemerin on endothelial function and vascular redox state (136).
Furthermore, chemerin induces the proliferation and migration of mouse VSMC resulting in increased blood pressure, an effect that is attenuated by the NADPH oxidase inhibitor gp91 ds-tat (105). Stimulation of human microvascular endothelial cells and VSMC with chemerin results in increased ROS production and phosphorylation of MAP kinases, but is reversed in the presence of Nox inhibitors and N-acetylcysteine (137).
Omentin
Omentin is a 313-amino-acid glycosylated protein, preferentially expressed in the omental/visceral AT. Contrary to visfatin, RBP4, and apelin, its levels are downregulated in obese subjects (49). In rat models, omentin exerts mainly atheroprotective effects by inhibiting the TNF-α-stimulated upregulation of NADPH oxidase activity in cultured VSMC, therefore reducing O2 •− production. The reduction in O2 •− production may then contribute to decreased TNF-α-induced VCAM-1 expression through inhibition of p38 and JNK activation (93). In addition to these anti-inflammatory effects, it was shown that omentin prevents VSMC migration and subsequent vascular remodeling by preventing PDGF-mediated activation of the NADPH oxidase and O2 •− production through inhibition of the PKC/PI3K/p47phox pathway (92).
Overall, omentin appears to protect against inflammatory activation and migration of VSMC, suggesting a protective role in hypertension and other vascular disorders. Indeed, lower omentin levels have been linked to worse outcomes in patients with heart failure (135), diabetics on hemodialysis (101), or critically ill patients (120).
Renin–angiotensin–aldosterone system
Several components of the renin–angiotensin–aldosterone system (RAAS) are produced by human adipocytes, most notably angiotensinogen (56, 90, 138). However, renin is also found in human adipocytes, while AngII might be endogenously produced by some preadipocytes (56). Overactivation of the RAAS in AT frequently occurs in states of obesity and has been implicated in the development of IR through AngII-induced activation of NADPH oxidase and NF-κB in white AT depots (90). Indeed, AngII promotes O2 •− production in endothelial cells of mice by upregulating the expression of the p47phox subunit of NADPH oxidase (107), while it may also induce production of ROS from mitochondrial sources (52).
Inflammatory chemokines and cytokines
AT is now regarded as a rich source of several proinflammatory chemokines (e.g., CCL2 and CCL5, CXCL5) and cytokines (e.g., TNF-α, IL-6) (23, 179). Even though these molecules are not exclusively produced by adipocytes, most of these have well-defined effects on vascular redox state. For example, hypertension has been linked to upregulated expression of the chemokine CCL5 (RANTES) by PVAT, which might promote leukocyte recruitment and perivascular inflammation, which in turn aggravates vascular oxidative stress and endothelial function (129). On the contrary, TNF-α is a well-known proinflammatory cytokine, whose circulating levels are increased in obesity and IR (81) promoting vascular oxidative stress through different mechanisms, including upregulation of NOX4 activity in vascular endothelial cells (18). IL-6 is another proinflammatory cytokine implicated in the pathogenesis of IR and obesity (60).
IL-6 production by adipocytes is upregulated in states of adipocyte hypertrophy (130) and correlates with the presence of IR (17). It is estimated that up to 35% of circulating IL-6 levels in humans are produced by AT (168). IL-6 has been shown to potentiate the pro-oxidant effects of AngII on VSMC of C57BL/6J mice, possibly through an indirect mechanism that includes upregulation of the AngII receptor type 1. Irrespective of the exact mechanisms involved, AT is closely associated with different inflammatory chemokines and cytokines and represents a key regulator of this complex network of molecules and their effects on vascular function and redox state.
Other adipokines
As the list of adipokines continues to expand, more adipokines are implicated in the regulation of vascular redox state. For example, serum C1q/TNF-related protein 9 (CTRP9) is a recently discovered adipokine that protects against high glucose-induced endothelial dysfunction and oxidative stress by decreasing ROS production and increasing the expression and activity of antioxidant defense mechanisms such as SOD in aortic vascular endothelial cells, possibly through upregulation of the AdipoR1 (39). On the contrary, adiporedoxin is a redox regulatory protein that is preferentially expressed in adipocytes. Adiporedoxin not only regulates the expression of adipokines, such as adiponectin, but can also be secreted and may act to inhibit inflammation-induced activation of endothelial cells possibly through its antioxidant effects (79, 199).
The Interplay Between PVAT and the Vascular Wall
PVAT describes the layer of fat that is directly adjacent to the vascular wall. Even though it comprises no more than 3% of the total AT mass in the human body (109), PVAT has been highlighted as an important AT in the regulation of cardiovascular health. Given its close spatial proximity to the vascular wall, PVAT can directly modify the biology of the adjacent vascular segment through adipokine-mediated paracrine mechanisms independently of any systemic, endocrine effects (2).
PVAT is a rich source of adipokines, which are produced either by adipocytes or other residing stromal cells, such as inflammatory macrophages and lymphocytes. Similarly to other AT depots, the secretome of PVAT is affected by both systemic, metabolic factors (e.g., obesity, IR) and local mechanisms, such as macrophage infiltration and polarization (2164). However, recent evidence suggests that changes in vascular oxidative stress and inflammation may also contribute to changes in PVAT biology, uncovering a bidirectional interplay between the two (11, 14, 124).
Inside-to-outside signaling: a paradigm shift in the study of PVAT
Until recently, scientific interest had focused exclusively on the unidirectional effects of AT biology and adipokine expression on vascular function and redox state. It is now well established that both distant and local AT depots can regulate several biological processes in atherogenesis. Koh et al. observed that eNOS(−/−) mice are characterized by reduced mitochondrial content, lower adiponectin levels, and higher levels of oxidative stress in AT, all of which were reversed by chronic administration of NO (102), suggesting that eNOS might regulate adiponectin biosynthesis.
We have recently demonstrated that, contrary to circulating adiponectin, local adiponectin levels in PVAT are positively correlated with O2 •− production and eNOS uncoupling in the underlying vessels in patients undergoing coronary artery bypass grafting surgery. In the ex vivo setting, adiponectin promoted eNOS coupling through eNOS phosphorylation and an increase in BH4 bioavailability, confirming the protective effects of adiponectin. Next, it was shown that lipid peroxidation products produced in the vascular wall as a result of vascular oxidative stress (i.e., 4-hydroxynenal) can diffuse to the neighboring PVAT and upregulate adiponectin expression, which then act to limit O2 •− generation in the underlying vessel (124).
In a subsequent study, ex vivo coculture of IMA segments with peri-IMA PVAT showed that increased vascular oxidative stress as a result of increased NADPH oxidase activity results in upregulation of adiponectin expression in the neighbouring PVAT via the release and diffusion of oxidative products and a PPAR-γ-mediated mechanism. In the same study, adiponectin was found to downregulate NADPH oxidase activity by inhibiting Rac1 translocation and p22phox expression (11). In accordance with the previous data, adiponectin expression in distant AT depots, such as the thoracic AT, was negatively correlated with vascular NADPH oxidase activity, while local adiponectin levels in peri-IMA PVAT were positively associated with the activity of the same enzyme in the underlying vascular segments.
Taken together, these findings suggest a paracrine loop between the vascular wall and PVAT, where PVAT functions as a sensor of vascular oxidation by detecting oxidation products that diffuse from the vessels to the surrounding area. PVAT then responds to these signals by upregulating the expression of protective adipokines (i.e., adiponectin), which then act on the vascular wall to reduce oxidative stress and restore a local vascular redox balance (Fig. 8). Whether a large molecule such as adiponectin can actually diffuse from PVAT to reach the inner endothelial layer is not known, however, it is likely that beneficial effects on vascular redox state in the outer vascular layers can have a secondary effect on the vascular endothelium by improving the local vascular redox state and therefore bioavailability of the important eNOS cofactor BH4 (11).

More recently, we described a similar protective mechanism for epicardial AT in the regulation of myocardial redox state. Similarly to PVAT, epicardial AT senses increased NADPH oxidase activity and O2 •− generation in the underlying myocardium through the release of oxidation products such as 4-hydroxynenal and responds through upregulation of adiponectin biosynthesis. Adiponectin then acts on the myocardial cells to block Rac1 and p47phox translocation from the cytosol to the membrane and therefore activation of NADPH oxidase (7, 12).
More recently, we found that PVAT “sensing” of vascular inflammation around the coronaries leads to changes in adipogenesis and lipolysis, changing the overall composition of PVAT surrounding inflamed areas of the human coronary tree (14). Translating this knowledge into a clinical application, we have now developed a novel noninvasive method that tracks these inflammation-induced changes of PVAT composition around the human coronary arteries noninvasively using computed tomography angiography. This method offers a powerful tool for the early detection of inflamed (“vulnerable”) atherosclerotic plaques (14), which are prone to rupture, and highlights the value of PVAT imaging in cardiovascular diagnostics.
Future Perspectives
Even though oxidative stress is recognized as a crucial factor in the regulation of atherosclerotic vascular disease, antioxidant interventions to date have failed to provide significant positive results in clinical studies (113). This has been attributed to several methodological limitations of the individual studies or therapeutic strategies used, such as the use of ROS scavengers (e.g., antioxidant vitamins), the inability to deliver the drugs in specific cellular compartments, interference with physiological redox signaling, inappropriate selection of the treatment population, and many others. This stressed the complexity of the human body, underlying the fact that redox regulation is controlled not only at a cellular level but also via endocrine and paracrine tissue–tissue or cell–cell interactions (71, 113).
As we unravel the secrets of AT and its secretome, it becomes clear that oxidative stress mediates, at least in part, the adverse vascular effects of several well-established risk factors, including immunometabolic conditions such as obesity and DM. Indeed, AT is now recognized as a major endocrine and paracrine organ, and there is growing evidence that it interacts directly with the vascular wall, affecting vascular redox state in an endocrine or paracrine way. AT secretes a wide range of adipokines with divergent roles in vascular biology. These molecules can have pro-oxidant/proinflammatory (i.e., promoting vascular disease) or antioxidant/anti-inflammatory (i.e., preventing vascular disease) roles. The balance between these molecules is largely driven by the biology of AT itself, with IR and diabetes shifting this balance toward the pro-oxidant/proinflammatory secretome.
However, recent evidence suggests that products released from the human vascular wall (such as oxidation products) act as signaling molecules driving the biology of AT and altering its secretome profile. This “inside-to-outside” signaling proves that AT and the human cardiovascular system are inter-related, affecting each other's biology, while PVAT in particular may detect changes in vascular biology. Therefore, noninvasive imaging phenotyping of PVAT may contribute to cardiovascular risk prediction, directing appropriate therapeutic interventions in primary or secondary prevention (14). In addition, immunometabolic therapeutic strategies targeting the secretory profile of AT and/or modifying the interaction signals between AT and the vascular wall may offer good alternative approaches to modify vascular redox signaling without direct ROS scavenging, and should be further explored.
Footnotes
Acknowledgments
This work was funded by the NovoNordisk Foundation through NNF15CC0018486 grant on immunometabolism, the British Heart Foundation (PG/13/56/30383 and FS/16/15/32047), and the NIHR-Oxford Biomedical Research Centre. E.K.O. is supported by an academic grant from the A.G. Leventis Foundation. C.A. received an unrestricted research grant from Sanofi.