
Abstract
Aims:
Asymmetric dimethylarginine (ADMA), an endogenous inhibitor of nitric oxide synthase, is mainly degraded by dimethylarginine dimethylaminohydrolase (DDAH). Emerging evidence suggests that plasma ADMA accumulation and DDAH1 activity/expression reduction are linked to chronic kidney disease (CKD) pathology, but the mechanisms remain largely unknown. Here, we examined the role of ADMA/DDAH1 in the epithelial–mesenchymal transition (EMT) of tubular epithelial cells (TECs), an important mechanism for the pathogenesis of renal fibrosis.
Results:
In HK-2 cells, DDAH1 expression was reduced by aldosterone treatment, and overexpression of DDAH1 significantly attenuated aldosterone-induced EMT. More interestingly, DDAH1 deficiency resulted in EMT-related changes in primary TECs via increasing oxidative stress, impairing adenosine monophosphate-activated kinase (AMPK) signaling, and downregulating of peroxiredoxin 5 (Prdx5). However, those effects could not be mimicked by increasing the ADMA concentration. After regular feeding for 24 months or inducing type 2 diabetes, Ddah1 −/− mice had higher serum creatinine levels than wild-type (WT) mice. In the kidneys of the aged or diabetic mice, loss of DDAH1 resulted in more interstitial fibrosis, more collagen deposition, and greater induction of EMT-related changes and oxidative stress than in the WT kidneys.
Innovation and Conclusion:
Our results provide the first direct evidence that the DDAH1 has a marked effect on kidney fibrosis and oxidative stress induced by aging or diabetes. Our findings suggest that strategies to increase DDAH1 activity in TECs may provide a novel approach to attenuate CKD development. Antioxid. Redox Signal. 27, 1347–1360.
Introduction
K
Plasma asymmetric dimethylarginine (ADMA) accumulation is linked to kidney disease pathology, but the mechanisms remain largely unknown. The results of this study highlight the importance of dimethylarginine dimethylaminohydrolase (DDAH1), an enzyme degrading ADMA, in the development of chronic kidney disease. We demonstrated that DDAH1 deficiency promotes the epithelial to mesenchymal transition in renal proximal tubular epithelial cells and causes fibrosis, and oxidative stress in aging and diabetic kidneys. Thus, strategies to increase DDAH1 activity could be a novel approach to attenuate progressive kidney damage.
Asymmetric dimethylarginine (ADMA) is an endogenous inhibitor of nitric oxide (NO) synthase that blocks NO production (42). The plasma ADMA levels are increased in patients with end-stage renal failure (42, 53) and observational studies in CKD have shown an inverse association between plasma ADMA and kidney function, as measured by the estimated glomerular filtration rate (5, 32). Chronic ADMA administration in mice results in increased blood pressure, renal oxidative stress, and interstitial and glomerular fibrosis (29). ADMA may also be involved in peritubular capillary loss and tubulointerstitial fibrosis (28, 41), contributing to the progression of CKD. ADMA is metabolized to citrulline by dimethylarginine dimethylaminohydrolase (DDAH), which has two isoforms, DDAH1 and DDAH2 (40). In rodent CKD model, reduced DDAH expression was associated with ADMA accumulation and subsequent elevation in blood pressure (27). Conversely, overexpression of DDAH1 can lead to the amelioration of renal fibrosis and kidney injury (15, 28, 30, 41). While studies have clearly demonstrated that DDAH1 plays a critical role in renal failure, the effect of DDAH1 on the EMT is not recognized. To address the issues, we first examined the effects of ADMA and DDAH1 on the EMT process in HK-2 [an immortalized human proximal TEC line (34)] and primary TECs. Then, we investigated the role of DDAH1 in renal fibrosis using global DDAH1 knockout (KO) mice and wild-type (WT) littermates.
Results
DDAH1 is downregulated by aldosterone in HK-2 cells and renal cortex
To induce the EMT, we treated HK-2 cells with aldosterone (Aldo), a well-known EMT inducer (19, 43, 47, 50), for 24 h and observed that E-cadherin was downregulated, while alpha smooth muscle actin (α-SMA) was upregulated in Aldo-treated cells in a dose-dependent manner. Aldo treatment dose dependently reduced DDAH1 protein expression with a noticeable effect at 50 nM and maximal effect at 200 nM (Fig. 1A). Similar results were also obtained in primary renal proximal TECs (Supplementary Fig. S1; Supplementary Data are available online at
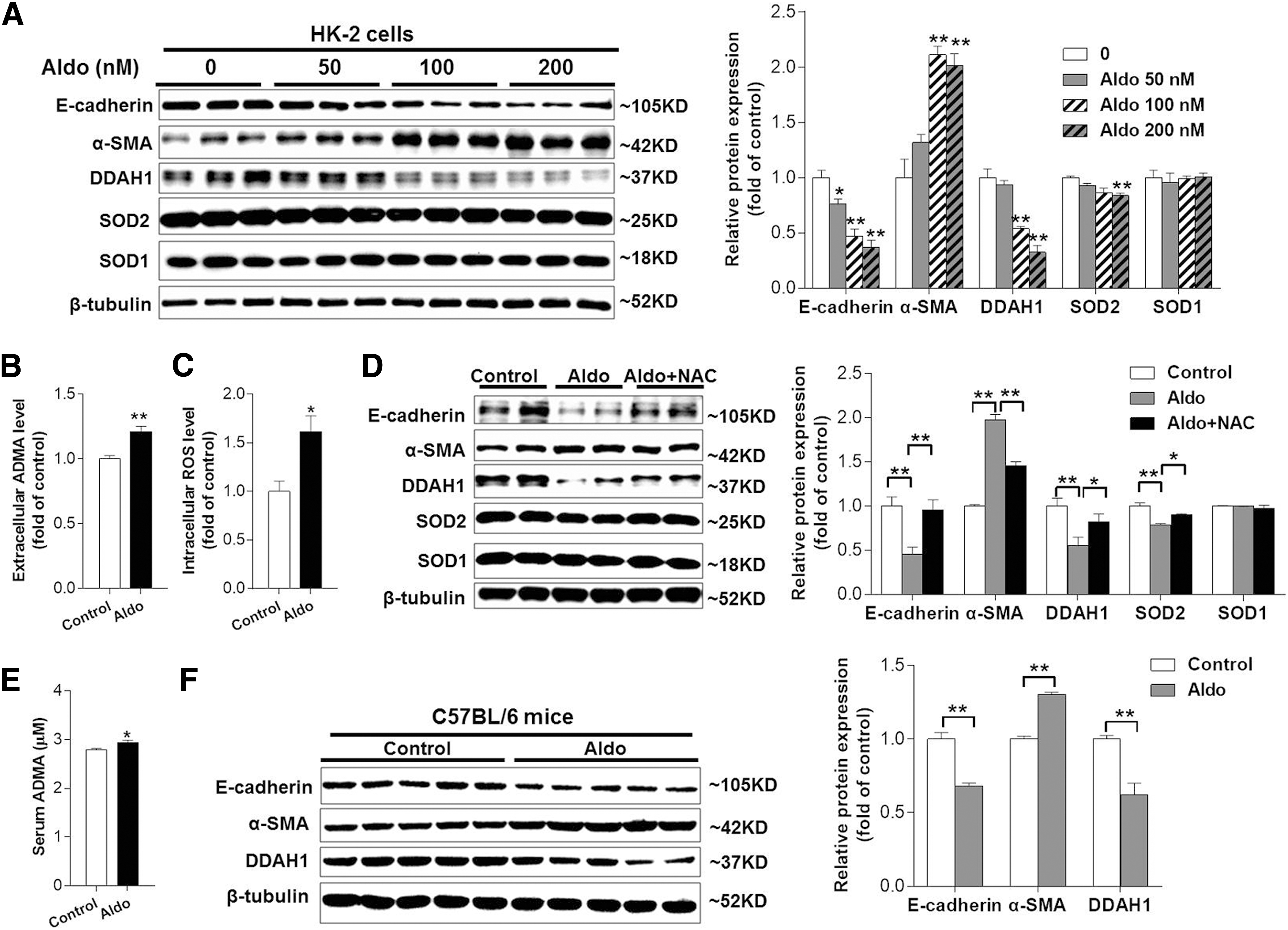
Next, we hypodermically injected C57BL/6J male mice with Aldo (1 mg/[kg·day]). After 7 days, we found that mice with Aldo administration exhibited elevated levels of serum ADMA (2.79 ± 0.03 vs. 2.93 ± 0.06, p < 0.05) (Fig. 1E). Western blotting of renal cortex homogenate showed that E-cadherin and DDAH1 were downregulated, while α-SMA was upregulated in Aldo-treated mice (Fig. 1F).
Overexpression of DDAH1 attenuates the Aldo-induced EMT and oxidative stress
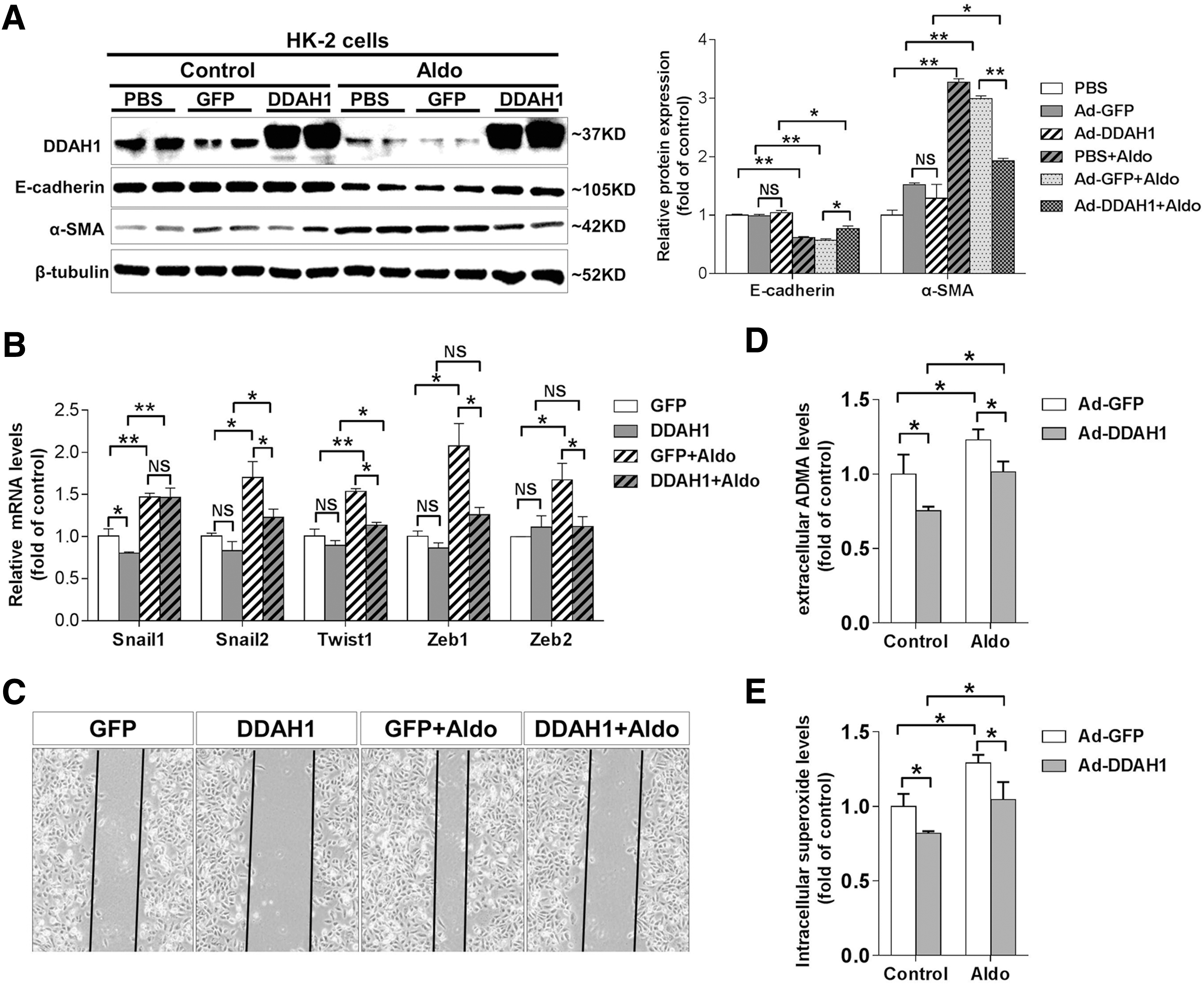
To verify whether DDAH1 overexpression could affect the Aldo-induced EMT, we overexpressed DDAH1 in HK-2 cells by adenovirus transfection. Phosphate-buffered saline (PBS) solution was used as control of adenovirus. Compared to GFP controls, DDAH1 overexpression significantly attenuated the Aldo-induced downregulation of E-cadherin and upregulation of α-SMA in HK-2 cells (Fig. 2A). It has been well established that several transcription factors, including Snail1, Snail2, Twist1, Zeb1, and Zeb2, function as molecular switches for the EMT program (18). Using reverse transcription and quantitative real-time polymerase chain reaction (qRT-PCR), we observed that DDAH1 overexpression significantly attenuated Aldo-induced upregulation of Snail2, Twist1, Zeb1, and Zeb2 in HK-2 cells (Fig. 2B). Furthermore, cell migration that had been enhanced by Aldo treatment (200 nM, 48 h) was dramatically suppressed by DDAH1 overexpression (Fig. 2C). DDAH1 overexpression also significantly decreased the levels of extracellular ADMA and intracellular superoxide in both control and Aldo-exposed cells (Fig. 2D, E).
DDAH1 deficiency induces the EMT in primary proximal TECs in an ROS-dependent manner
To understand the role of DDAH1 in the EMT, we isolated primary renal proximal TECs from WT and Ddah1 KO mice and compared the mRNA levels of EMT-related genes with qRT-PCR. Compared to WT cells, Ddah1 KO TECs exhibited lower mRNA levels of E-cadherin and higher mRNA levels of α-SMA, N-cadherin, Snail1, Snail2, Twist1, Zeb1, and Zeb2 (Fig. 3A). The downregulation of E-cadherin and upregulation of α-SMA in Ddah1 KO TECs were confirmed by Western blot (Fig. 3B). As a critical enzyme for ADMA degradation, DDAH1 deficiency increased the extracellular and intracellular ADMA levels by 12% and 20%, respectively (Supplementary Fig. S3). To determine whether ADMA accumulation was responsible for EMT induction in Ddah1 −/− TECs, we treated WT TECs and HK-2 cells with different concentrations of ADMA (20–60 μM) for 24 h. Western blot results showed that the protein expression levels of E-cadherin and α-SMA were not affected by exogenous ADMA treatment (Supplementary Fig. S4), indicating that increased ADMA levels could not induce the EMT in Ddah1 −/− TECs. Then, we examined the levels of intracellular NO, ROS, and mitochondrial O2•− in WT and Ddah1 −/− TECs using 3-amino,4-aminomethyl-2′,7′-difluorescein, diacetate (DAF-FM DA), 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), and MitoSOX, respectively. Compared with WT cells, Ddah1 −/− TECs exhibited lower levels of intracellular NO (1.00 ± 0.02 vs. 0.58 ± 0.02) (Fig. 3C) and higher levels of intracellular ROS (1.00 ± 0.06 vs. 1.30 ± 0.05) (Fig. 3D) and mitochondrial O2•− (1.00 ± 0.09 vs. 1.61 ± 0.06) (Fig. 3E). Then, we treated Ddah1 −/− TECs with 1 mM NAC, 10 μM tempol (an SOD mimetic), MnTBAP (a cell-permeable SOD mimetic and peroxynitrite scavenger), or NO donor sodium nitroprussiate (SNP) for 24 h and found that E-cadherin was upregulated, while α-SMA was downregulated in these antioxidant or SNP-treated cells (Fig. 3F), indicating that the EMT in Ddah1 −/− TECs could be attenuated by antioxidants and NO donors. Scratch assays also showed that DDAH1 deficiency enhanced cell migration and this effect was attenuated by treatment with 10 μM tempol or SNP (Fig. 3G).

Activation of the phosphoinositide 3-kinase and mitogen-activated protein kinase (MAPK) signaling pathways has been reported to promote the EMT in TECs (20). However, DDAH1 deficiency has no obvious effect on phosphorylation of AKT (Ser473), GSK-3β (Ser9), ERK (Thr202/Tyr204), and p38 MAPK (Thr180/Tyr182). Inhibition of these kinases by specific inhibitors did not affect mRNA levels of E-cadherin and α-SMA in Ddah1 −/− TECs (Supplementary Fig. S5). We also found that the mRNA levels of transforming growth factor β (TGF-β), a well-known EMT inducer (48), were similar in WT and Ddah1 −/− TECs (Supplementary Fig. S6), suggesting that the TGF-β pathway may not be involved in EMT induction in Ddah1 −/− TECs.
DDAH1 regulates AMPKα signaling and peroxiredoxin 5 expression in primary TECs
To verify whether DDAH1 deficiency regulates the redox state in primary TECs through modulating mitochondrial antioxidant enzymes, we measured the mRNA levels of Sod2, thioredoxin 2 (Trx2), peroxiredoxin 3 (Prdx3), and peroxiredoxin 5 (Prdx5) in WT and Ddah1 −/− TECs. It was interesting that only Prdx5 was significantly downregulated in Ddah1 −/− TECs (Supplementary Fig. S6). The reduction of Prdx5 expression in Ddah1 −/− TECs was confirmed by Western blot (Fig. 4A). Consistent with our previous finding that DDAH1 deficiency impaired adenosine monophosphate-activated kinase (AMPK) signaling in hepatocytes (22), we also found DDAH1 deletion decreased phosphorylation of AMPKα at Thr172 in primary TECs (Fig. 4A). By contrast, overexpression of DDAH1 in WT TECs significantly increased Prdx5 protein expression and phosphorylation of AMPKα (Fig. 4B). We also confirmed the regulatory effect of DDAH1 on Prdx5 and AMPK signaling in HK-2 cells by inhibiting DDAH1 activity with PD 404182, a tightly bound, covalent, and irreversible inhibitor of DDAH1 (6) (Supplementary Fig. S7).
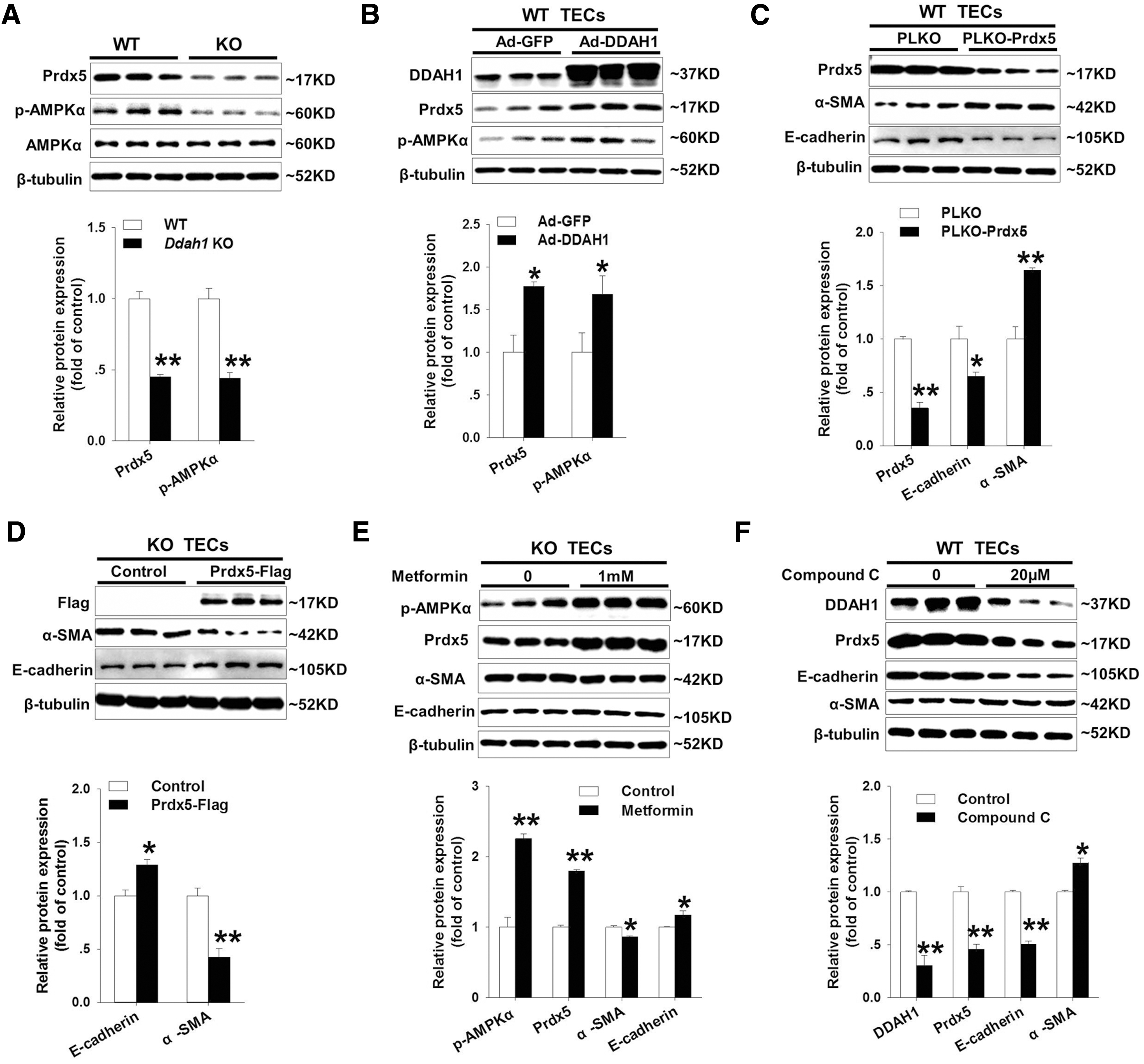
To investigate whether Prdx5 reduction contributed to EMT in Ddah1 −/− TECs, we transfected WT TECs with PLKO.1-scramble shRNA (PLKO) or Prdx5-specific shRNA plasmid (PLKO-Prdx5), which resulted in an ∼65% reduction in Prdx5 protein expression (Fig. 4C). Compared with control, Prdx5-depleted TECs exhibited an ∼35% reduction in E-cadherin protein expression and ∼64% increase in α-SMA expression (Fig. 4C). Depletion of Prdx5 in TECs also enhanced cell migration (Supplementary Fig. S8A). Conversely, overexpression of Prdx5 using PCMV-N-Flag-Prdx5 plasmid significantly increased E-cadherin and decreased α-SMA expression in Ddah1 −/− TECs (Fig. 4D), which was consistent with the inhibition of cell migration (Supplementary Fig. S8B).
Activation of AMPK has been found to inhibit profibrotic cytokine-induced EMTs in HK-2 cells (19). Therefore, we treated Ddah1 −/− TECs with 1 mM metformin for 24 h and observed that activation of AMPKα by metformin suppressed the EMT program and increased Prdx5 expression (Fig. 4E). On the contrary, inhibiting AMPKα activity by compound C significantly decreased the expression of DDAH1 and Prdx5 as well as stimulated the EMT in WT TECs and HK-2 cells (Fig. 4F and Supplementary Fig. S9). The mitochondrial complex I inhibitor rotenone could also activate AMPK signaling and stimulate glucose transport in rat skeletal muscle (8). To determine whether rotenone exhibits similar effects as metformin, we treated Ddah1 −/− TECs with 1 μm rotenone for 24 h. However, rotenone did not replicate metformin actions on EMT repression and Prdx5 upregulation (Supplementary Fig. S10). Together, these results indicated that DDAH1 deficiency induces the EMT program, at least in part, by impairing the AMPK-Prdx5 pathway in TECs.
DDAH1 deficiency induces fibrosis, oxidative stress, and the EMT program in kidneys of aged mice
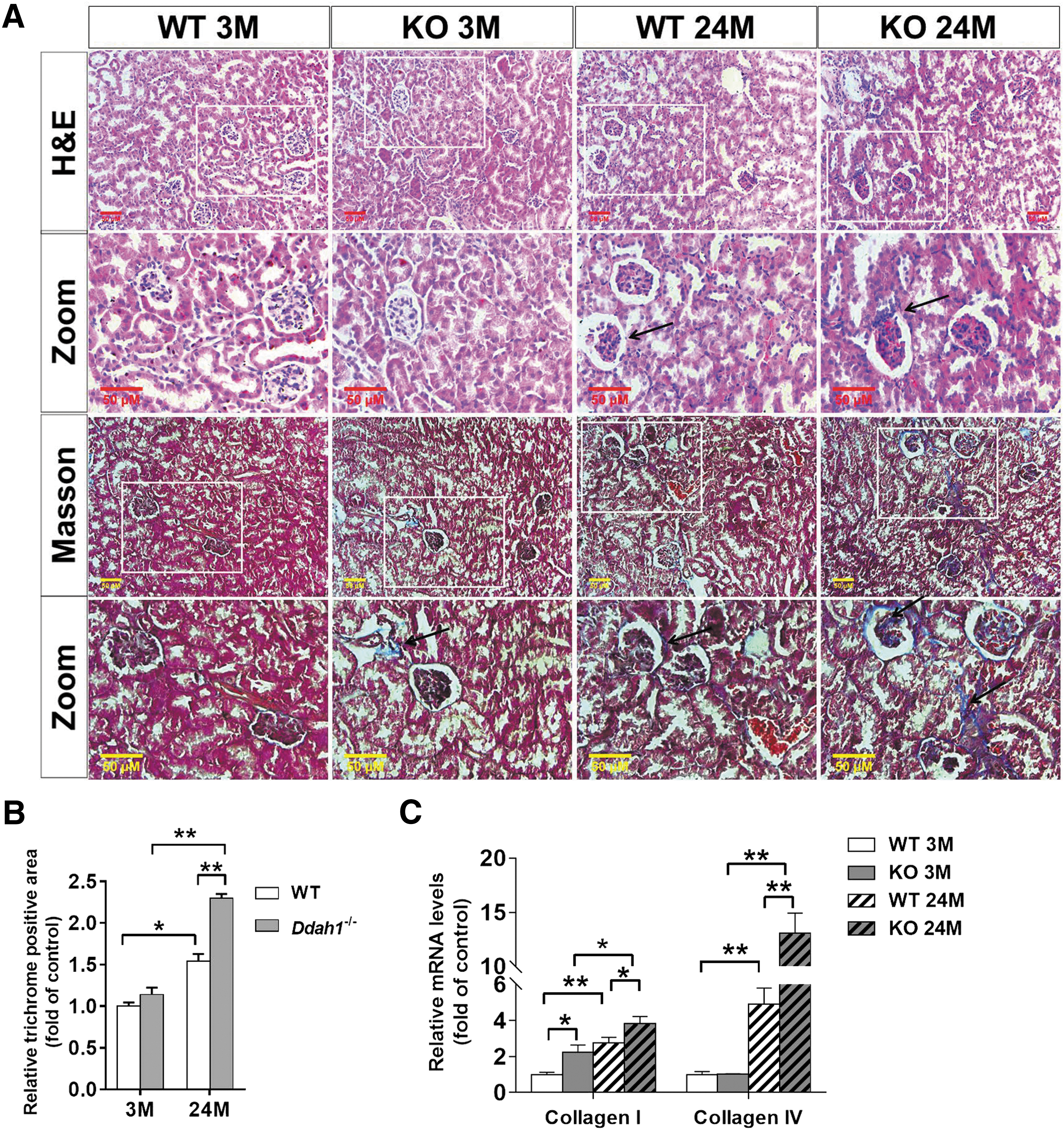
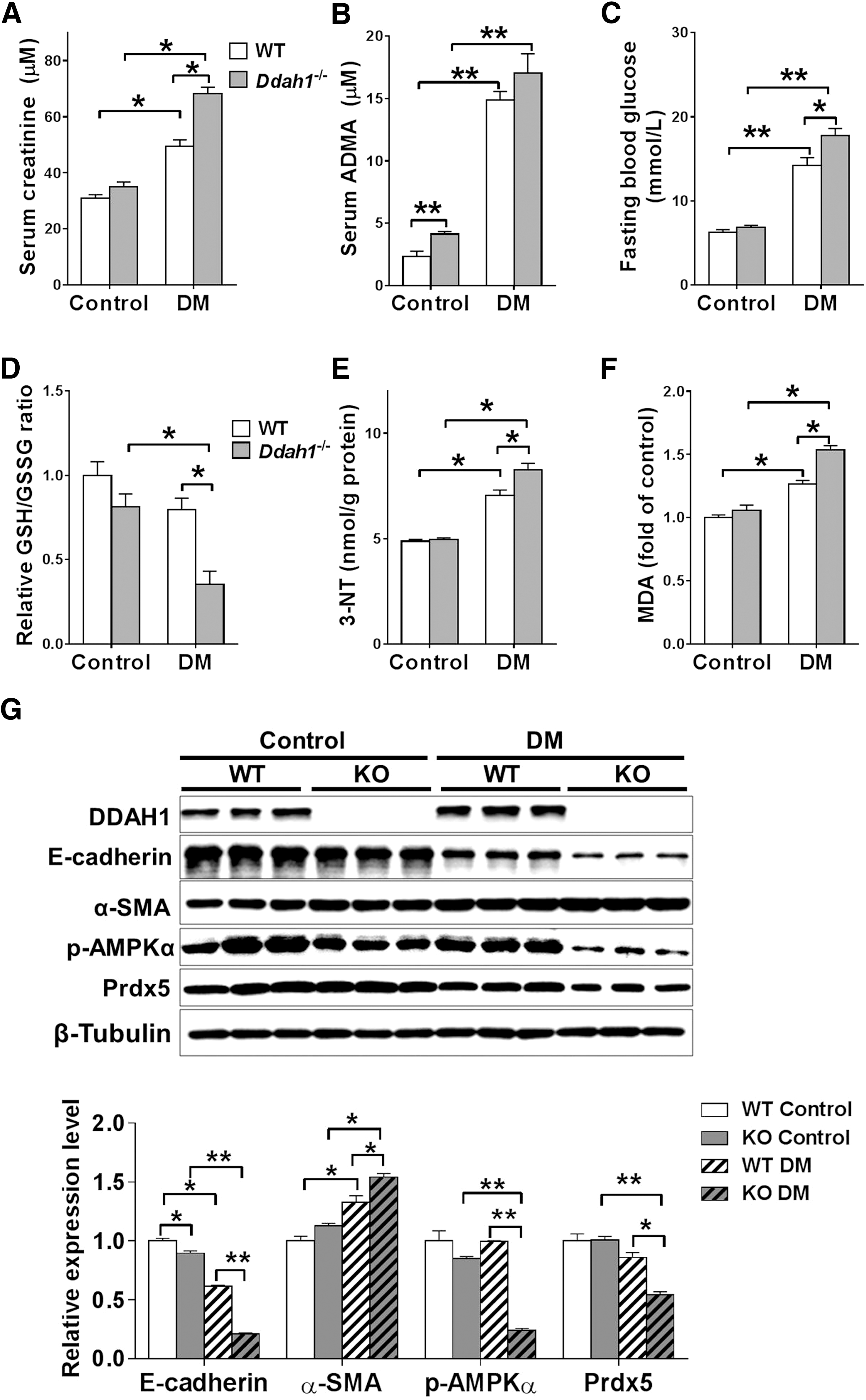
To explore the in vivo effect of DDAH1 on EMT, we compared the difference between WT and Ddah1 −/− mice at different ages. The serum ADMA levels were significantly higher in Ddah1 −/− mice than in WT mice at both 3 and 24 months of age (Fig. 5A). At 3 months of age, the levels of serum creatinine, proteinuria, and renal cortex 3-nitrotyrosine (3-NT), and the ratio of reduced glutathione (GSH)-to-oxidized glutathione (GSSG) in the renal cortex were similar between the WT and Ddah1−/− mice. At 24 months of age, the serum creatinine, proteinuria, and renal cortex 3-NT levels were significantly increased in both groups. However, elderly Ddah1−/− mice had higher levels of serum creatinine, proteinuria, and renal 3-NT, while they had a lower GSH/GSSG ratio than elderly WT mice (Fig. 5B–D and Supplementary Fig. S11). As anticipated, DDAH1 was undetectable in the KO mice. In the renal cortex from 3-month-old Ddah1 −/−, E-cadherin was significantly decreased, while α-SMA, p-AMPKα, and Prdx5 were unchanged. At 24 months of age, there was a significant increase in α-SMA and reduction in E-cadherin, p-AMPKα, and Prdx5 in both groups, while the reduction in E-cadherin, p-AMPKα, and Prdx5 expression was significantly exacerbated in Ddah1−/− kidneys (Fig. 5E). Aging also increased the mRNA levels of Snail1, Snail2, Twist1, Zeb1, and Zeb2 in both genotypes, and DDAH1 deficiency significantly enhanced the upregulation of those EMT-related transcription factors (Fig. 5F).

Histopathological analysis using hematoxylin and eosin (H&E) and Masson's trichrome staining demonstrated that the kidney harvested from 24-month-old Ddah1−/− mice displayed more interstitial fibrosis and collagen deposition than age-matched WT mice (Fig. 6A, B). Consistent with this finding, the mRNA levels of collagen type I and IV were significantly higher in Ddah1−/− kidneys at 24 months of age (Fig. 6C).
DDAH1 deficiency induces fibrosis, oxidative stress, and the EMT program in the kidneys of diabetic mice
Considering the critical role of EMT in initiating and promoting kidney fibrosis in diabetic nephropathy, we used the widely accepted high-fat diet (HFD) plus low dose of streptozotocin (STZ) method to induce type 2 diabetes in WT and Ddah1 −/− mice. The levels of serum creatinine, ADMA, fasting blood glucose, renal 3-NT, and malondialdehyde (MDA) were increased, while the GSH/GSSG ratio was decreased in diabetic mice (DM); however, diabetic Ddah1 −/− mice had higher levels of serum creatinine, fasting blood glucose, 3-NT, and MDA, as well as lower GSH/GSSG ratio than diabetic WT mice (Fig. 7A–F). Similar to the results from 24-month-old mice, the renal cortex from diabetic Ddah1−/− mice exhibited more interstitial fibrosis and collagen deposition than diabetic WT mice (Supplementary Figs. 12 and 13). DDAH1 deficiency also significantly enhanced the EMT program and impaired AMPK-Prdx5 signaling in the renal cortex of DM (Fig. 7G and Supplementary Fig. S13).
Discussion
Emerging evidence suggests that the plasma ADMA levels are elevated in CKD patients and higher plasma ADMA concentration is thought to contribute to CKD development through decreasing vascular NO production and increasing oxidative stress. Therefore, DDAH1 may exert a protective effect against CKD via regulating the cellular concentration of ADMA. Genetic DDAH1 overexpression decreases the circulating ADMA and protects against renal fibrosis in angiotensin (15) and surgical nephron reduction models of CKD (28). Overexpression of DDAH1 also attenuates the ischemia/reperfusion (30) and hypertensive (37) injuries in the kidney. In agreement with those findings, we found that aged or diabetic Ddah1 −/− mice displayed higher serum creatinine levels as well as more renal fibrosis and oxidative stress, suggesting that DDAH1 is an important regulator of kidney function and redox balance during the development of CKD.
The finding that DDAH1 deletion induces EMT in the primary proximal TECs suggested a novel mechanism for the protective effect of DDAH1 in kidney fibrosis. Clinical studies utilizing human kidney biopsies demonstrated that tubular expression of mesenchymal markers, such as vimentin and fibroblast-specific protein-1 (FSP-1), could be found in various progressive kidney diseases (9, 31, 33, 36). Furthermore, the expression of these transitional proteins in TECs was well correlated with declining renal function (9, 31, 44), indicating that EMT may be involved in the pathogenesis of CKD. It was originally suggested that renal TECs can convert into matrix-producing fibroblasts under pathologic conditions and participate in the pathogenesis of CKD through an EMT-like program (14). Although such a hypothesis has been challenged by subsequent studies (13), the two latest studies demonstrated that TECs acquire a partial EMT program during which they remain associated with their basement membrane and express makers of both epithelial and mesenchymal cells in renal fibrosis models induced by unilateral ureteral obstruction or folic acid (7, 26). Inhibition of the partial EMT program through targeting Snail1 or Twist1 in TECs during chronic renal injury could ameliorate established renal fibrosis and, therefore, represents a potential antifibrosis therapy (7, 26). In HK-2 cells, ours and a previous study (23) demonstrated that silencing DDAH1 expression could effectively induce the EMT, while overexpression of DDAH1 has the opposite effect on the EMT. Deletion of DDAH1 also enhanced the EMT program in primary proximal TECs, as evidenced by the upregulation of α-SMA- and EMT-related transcription factors as well as the reduction in E-cadherin. More importantly, the enhanced EMT program in Ddah1 −/− TECs was associated with higher levels of serum creatinine and more collagen deposition in aged or diabetic Ddah1 −/− mice, indicating DDAH1 regulates kidney function and renal fibrosis at least partially through inhibiting the EMT in TECs.
While our data identify a novel role for DDAH1 in the EMT of TECs, the mechanism by which DDAH1 regulates the EMT remains unclear. It has been consistently demonstrated that the EMT induced by a number of profibrotic cytokines/growth factors, including TGF-β (46), Aldo (47, 50), high glucose (20), and angiotensin II (2), is mediated through ROS generation (1, 45). In agreement with those findings, we found that deletion of DDAH1 increased the intracellular ROS levels and treatment with antioxidants such as NAC, tempol, and MnTBAP inhibited the EMT in Ddah1 −/− TECs. DDAH1 deficiency also exacerbated renal oxidative stress in aged or DM. Since exogenous ADMA treatment did not affect the EMT in HK-2 cells and primary TECs, it seems that ADMA accumulation in Ddah1 −/− TECs was not sufficient to induce the EMT. Prdx5 is an antioxidant enzyme that reduces oxidative stress by catalyzing intramolecular disulfide bonds (16, 17), and it can protect against TGF-β-induced fibrosis in rat kidney interstitial fibroblast cells (3). Here we found that DDAH1 deficiency significantly decreased Prdx5 expression in primary TECs and kidneys from aged or DM. By contrast, overexpression of DDAH1 increased Prdx5 protein expression. Furthermore, knockdown of Prdx5 directly caused changes in EMT-related genes, while overexpression of Prdx5 attenuated DDAH1 deficiency-induced EMT in primary TECs. Accordingly, the reduction of Prdx5 provides an alternate important mechanism for ROS production and EMT induction in Ddah1 −/− TECs.
AMPKα has been found to regulate antioxidant enzymes in cardiac myocytes (11) and endothelial cells (54). In the present study, DDAH1 deficiency significantly decreased AMPKα phosphorylation in primary TECs and the cortex from aged or DM, while DDAH1 overexpression activated AMPK signaling. More interestingly, an AMPK activator, metformin, increased Prdx5 expression in Ddah1 −/− TECs, while an AMPK inhibitor, compound C, decreased DDAH1 and Prdx5 expression in HK-2 cells. Thus, DDAH1 may regulate Prdx5 through AMPK-dependent pathway.
Although the concept that DDAH1 is protective in kidney fibrosis has been supported by previous studies (15, 28, 29), our findings differ from a previous report that proximal tubule-specific Ddah1 KO (Ddah1 PT−/−) mice were protected from reduced kidney tissue mass, collagen deposition, and profibrotic cytokine expression in two independent renal injury models (39). As deletion of a gene may produce effects on other nearby genes and thus cause additional phenotypic changes, these discrepancies may be the result of technical differences between the studies. For example, deletion of exon1 of mouse Ddah1 leads to embryonic lethality (21), while deletion of exon 4 has no obvious effect on embryonic development (10). There were also significant differences between two vascular endothelial-specific Ddah1 KO strains (4, 12). As a result, more careful studies are still needed to address these discrepancies.
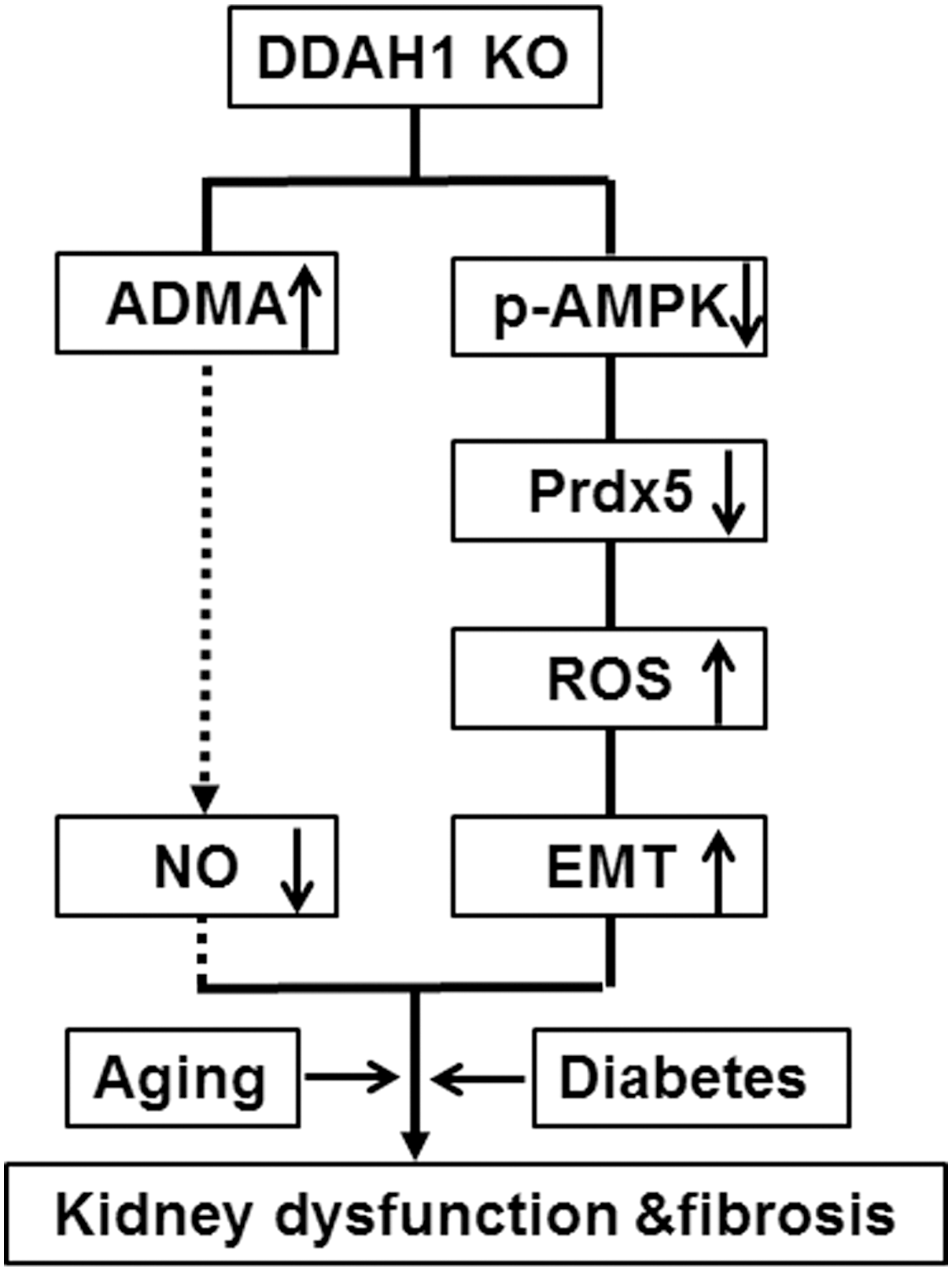
As summarized in Figure 8, our study provides the first direct evidence that DDAH1 protects against renal fibrosis via inhibiting EMT program, maintaining AMPK-Prdx5 signaling, and attenuating ADMA accumulation and oxidative stress. Our results suggest that increasing DDAH1 activity in TECs is a new strategy to treat CKD.
Materials and Methods
Antibodies and reagents
Antibodies against β-tubulin, DDAH1, α-SMA, and Prdx5 were from Abcam PLC (Cambridge, UK); E-cadherin, AMPKα, phospho-AMPKαThr172, phospho-p38Thr180/Tyr182, phospho-AKTser473, phospho-ERKThr202/Tyr204, and phospho-GSK-3βSer9 antibodies were from Cell Signaling Technology (Danvers, MA). Dulbecco's modified Eagle's medium: nutrient mixture F-12 (DMEM/F-12) and fetal bovine serum (FBS) were purchased from Gibco BRL (Grand Island, NY). MnTBAP [Mn(III)tetrakis (4-benzoic acid) porphyrin chloride] and tempol (4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy) were purchased from Cayman Chemical (Ann Arbor, MI) and Selleckchem (Houston, TX), respectively. STZ, ADMA, DAF-FM DA, SNP, metformin, DCFH-DA, NAC, PD 404182 (6H-6-imino-(2,3,4,5-tetrahydropyrimido)[1,2-c]-[1,3]benzothiazine), and dihydroethidium (DHE) were purchased from Sigma (St. Louise, MO). PCMV-N-Flag and PCMV-N-Flag-Prdx5 plasmids were purchased from Biogot (Nanjing, China). The pLKO.1–TRC Cloning Vector (Addgene Plasmid 10878) was used to construct clone shRNA against mouse Prdx5 (PLKO-Prdx5, target sequence is TTGGTGTCTCTCTTTGGGAAT). All other chemicals made in China were analytical grade.
Experimental animals
The DDAH1 global KO mice were kindly provided by Dr. Yingjie Chen from the University of Minnesota. Animal studies were performed in accordance with the principles of laboratory animal care and with approval by the University of Chinese Academy of Science Animal Care and Use Committee. For Aldo infusion experiment, C57BL/6J male mice (10–12 weeks) were hypodermically injected with PBS or Aldo (1 mg/[kg·day]) for 7 days. For the type 2 diabetes murine model, mice were fed an HFD (60% fat; purchased from HFK Bioscience Co., Beijing, China) for 8 weeks; then, a single low dose of STZ (120 mg/kg intraperitoneal, formulated in 0.1 M citrate buffer, pH 4.5) was given after a 6-h fast to induce diabetes. Four weeks after STZ injection, the 6-h fasting blood glucose level of the HFD/STZ mice was measured to confirm hyperglycemia.
Cell isolation and culture
Primary renal proximal TECs from murine kidneys were harvested according to the previous report (38) with minor modifications. Briefly, kidneys were surgically removed from anesthetized WT and Ddah1 −/− male mice. The renal cortices were sliced, minced, and digested in PBS containing 1 mg/ml collagenase type I. Highly purified proximal tubules were isolated through a 45% Percoll density gradient centrifugation. The HK-2 cell line was obtained from the Cell Bank of the Institute of Biochemistry and Cell Biology (Shanghai, China). Freshly isolated tubules or HK-2 cells were grown in DMEM/F-12 supplemented with 10% (v/v) FBS and 1% penicillin and streptomycin at 37°C with 5% CO2.
Measurement of ROS, superoxide, and NO
The levels of intracellular ROS, NO, and superoxide were determined by spectrophotometry using DCFH-DA, DAF-FM DA, and DHE, respectively. The cells were washed with PBS and incubated with 5 μM fluorescence dyes (final concentration) at 37°C according to the manufacturer's instructions. Then, cells were washed three times and the fluorescence intensity was determined using a Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek). The following wavelength settings were used to monitor the oxidized fluorescent metabolite of each probe: H2DCF-DA and DAF-DA, excitation 485 nm/emission 530 nm; DHE, excitation 530 nm/emission 610 nm.
In vitro scratch assay
Cells were seeded in 12-well culture plates and cultivated until subconfluence. Subsequently, the cell monolayer was scraped with a 200 μl pipette tip held at an angle of 45°. Culture plates were then washed twice with FCS-free DMEM/F-12, and 500 μl of this medium was then added per well. Following this procedure, the first image of each well was captured. Cells were treated with or without aldosterone, tempol, or SNP in starving medium and were incubated for 48 h. Following this incubation, the second images were taken from the exact same location as the first picture for each well.
Measurement of serum and kidney biochemical markers
Blood glucose levels were measured using an Accu-Chek® glucometer (Roche Diagnostics, Indianapolis, IN). Serum ADMA levels were determined using an ADMA ELISA kit (DLD Diagnostika GmbH, Hamburg, Germany). The mouse 3-NT ELISA kit and kits for MDA and glutathione measurement were obtained from the Abcam and Beyotime Institute of Biotechnology (Shanghai, China), respectively. At least five mice per group were used for each assay.
Tissue processing
Murine kidney tissues were harvested, washed, and then fixed with formalin and embedded in paraffin. Kidney sections (5 μm) were stained with H&E or trichrome stain kit (modified Masson's; ScyTek Laboratories, Inc., UT) to assess fibrosis. At least five mice per group were used for these experiments. The interstitial fibrosis quantification was performed as previously reported (35).
Western blots
Renal cortex was isolated from the kidney and pulverized or cells were scraped. Then, proteins were extracted using buffer (50 mM Tris-Cl, 150 mM NaCl, 100 μg/ml phenylmethylsulfonyl fluoride, protease and phosphatase inhibitor cocktail, and 1% Triton X-100) on ice for 30 min. After centrifugation at 12,000 g and 4°C for 20 min, the supernatant was used for Western blot analysis.
Quantitative real-time PCR
Total RNA was extracted with TRIzol reagent (Invitrogen) and cDNA was synthesized using the PrimeScript RT Reagent Kit (TaKaRa) according to the manufacturer's instructions. Two microliters of cDNA was used to determine the mRNA expression by quantitative PCR using the SYBR® Premix Ex Taq™ II Kit (TaKaRa). The cycling conditions were as follows: initial denaturation at 95°C for 30 s, followed by 40 cycles of 5 s at 95°C, 30 s at 60°C, and 30 s at 72°C. Primers are listed in Supplementary Table S1. The results were normalized to Gapdh.
Data and statistical analysis
All values are expressed as mean ± standard error. Statistical significance was defined as p < 0.05. One- or two-way analysis of variance (ANOVA) was used to test each variable for differences among the treatment groups with StatView (SAS Institute, Inc.). If ANOVA demonstrated a significant effect, pairwise post hoc comparisons were made with the Fisher's least significant difference test.
Footnotes
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81270319, 81470520, and 91643206) and the Chinese Academy of Sciences (KJRH2015-005, Hundred Talents Program, and CAS/SAFEA International Partnership Program for Creative Research Teams).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.