
Abstract
Significance:
Lipoproteins, such as low-density lipoprotein, play a causal role in the development of atherosclerosis and coronary disease.
Recent Advances:
Lipoproteins can stimulate vascular production of reactive oxygen species, which act as important signaling molecules in the cardiovascular system contributing to the pathophysiology of endothelial dysfunction, hypertension, and atherosclerosis.
Critical Issues:
Modified lipoproteins have emerged as important regulators of redox signaling, such as oxidized or carbamylated low-density lipoprotein or modified high-density lipoproteins, that contain oxidized lipids, an altered protein cargo, and associated small molecules, such as symmetric dimethylarginine.
Future Directions:
In this review, we provide an overview on signaling pathways stimulated by modified lipoproteins in the cardiovascular system and their potential role in cardiovascular disease development. Moreover, we highlight novel aspects of how gut microbiome-related mechanisms—a growing research field—may contribute to lipoprotein modification with subsequent impact on cardiovascular redox signaling. Antioxid. Redox Signal. 29, 337–352.
Introduction
L
The physiological function of LDL is transportation of cholesterol of both exogenous and endogenous origin to peripheral cells. One of the components of LDL is the apolipoprotein (apo)B-100 (Fig. 1), which is responsible for the transportation function of LDL. Notably, LDL particles constitute about 90% of circulating apoB-containing lipoproteins in fasting blood (29).
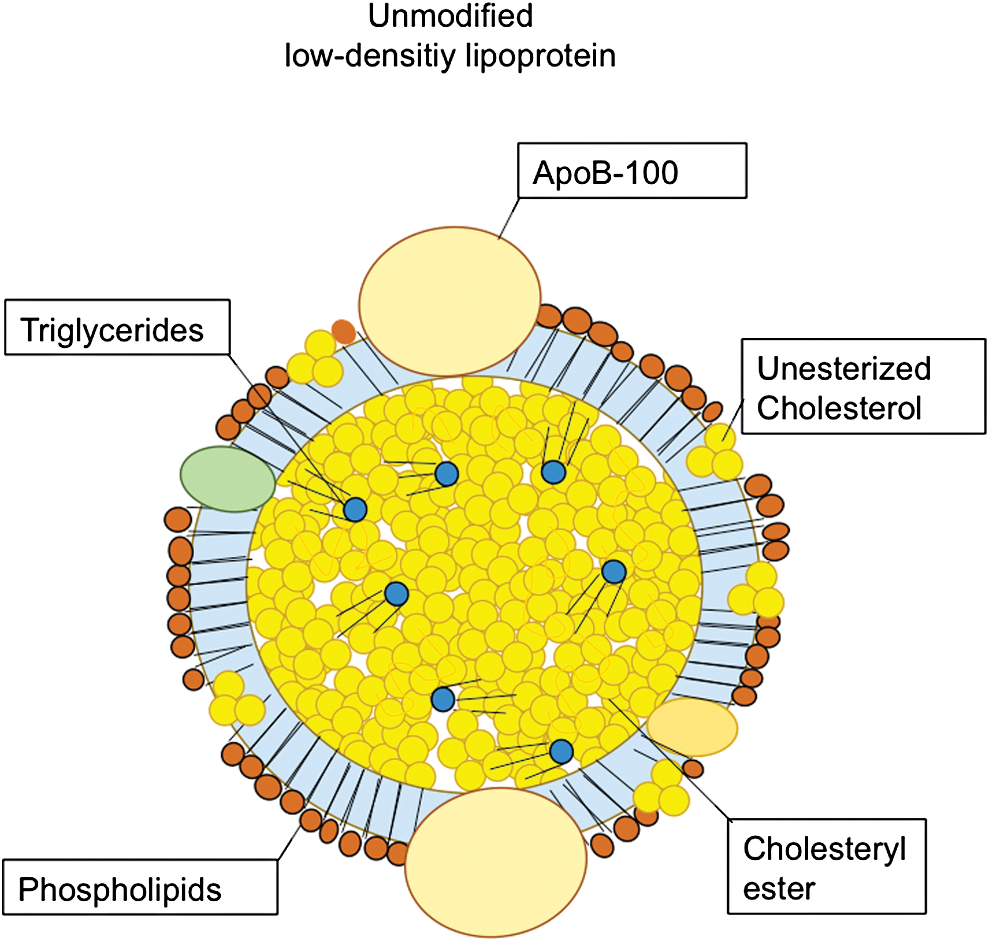
Beyond their role in transportation of lipids in the blood, biochemically modified lipoproteins mediate profound inter- and intracellular actions, in particular in vascular and immune cells, which influence the development of atherosclerosis. The major modification processes that are relevant for cardiovascular pathophysiology are acetylation, carbamylation, and oxidation (Fig. 2). Understanding detailed molecular mechanisms of how lipoprotein modification regulates oxidative events in the arterial wall and the development of atherosclerosis may help to develop novel therapeutic strategies to prevent or treat atherosclerotic vascular disease.

One of the first identified lipoprotein modifications is acetylation of LDL (42). Acetylated low-density lipoprotein (acLDL) has an increased affinity for the macrophage scavenger receptors (SRs) (82). Its activation leads to augmented uptake of acLDL by macrophages. The uptake of acLDL, in turn, contributes to increased cholesteryl ester and lipid droplet accumulation and the transformation of macrophages into foam cells (98) promoting atherosclerotic disease progression and chronic vascular inflammation (120). Oxidation of LDL (oxidized low-density lipoprotein [oxLDL]) is another mode of LDL modification that has been a subject of intensive research over the past decades (35). oxLDL exerts a wide range of effects on vascular and immune cells that can promote atherogenesis (117). Some of these effects are mediated by oxidized phospholipids within the outer shell and the cholesterol esters that are carried internally (69). These oxidized phospholipid products are also the epitopes recognized by immune cells (17), in particular monocytes and macrophages, and vascular cells, such as endothelial cells and smooth muscle cells (81). Besides lipid oxidation, protein components of LDL, in particular apoB-100, also undergo oxidation (14).
Once formed, oxLDL stimulates monocyte chemotaxis (5) and promotes foam cell formation—one of the early steps in the development of atherosclerosis (14). Moreover, oxLDL causes endothelial injury and disrupts normal functionality of the endothelium (27).
Notably, the oxidative modification of LDL occurs mainly in the vascular wall by resident vascular cells, such as endothelial cells (28) or infiltrating or local immune cells, such as macrophages (20).
The oxidation processes are mediated by both cellular and extracellular mechanisms based on enzymatic as well as non-enzymatic mechanisms. Among the enzymatic sources nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (53, 55), the xanthine oxidase (XO) (54), myeloperoxidase (116), and also the nitric oxide synthase (NOS) have been reported to mediate the oxidation pathways generating oxLDL. Importantly, these oxidation pathways can be activated by oxLDL, thereby creating a vicious cycle that potentiates LDL oxidation (90, 91).
The counterparts of the oxidants are antioxidant enzymes that are largely cell-associated proteins regulating the redox balance by removal of reactive oxygen species (ROS) of both intra- and extracellular sources. Among enzymatic antioxidants, superoxide dismutase (SOD) (36), catalase (121), glutathione peroxidases, and glutathione reductase have been suggested to control the redox state within the vascular wall (108, 109).
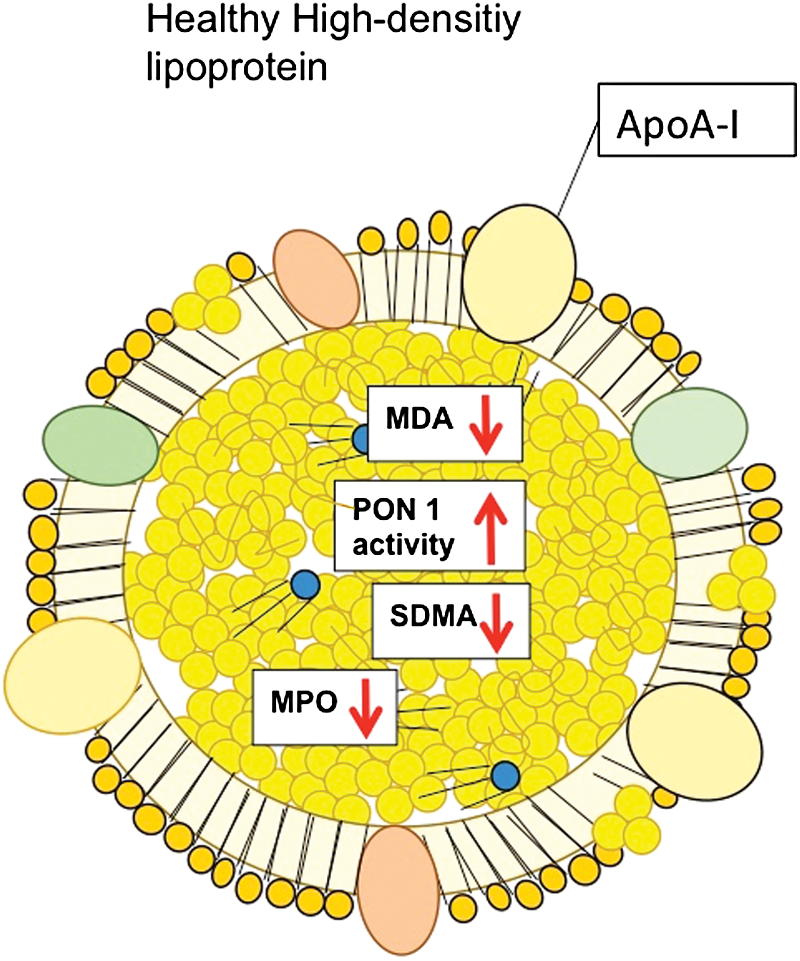
The second lipoprotein that will be highlighted in this review is high-density lipoprotein (HDL). The major physiological function of HDL is removing excess cholesterol from peripheral tissue through the ATP-binding cassette transporter A1 (ABCA1) and ATP-binding cassette transporter G1 pathways, and the reverse cholesterol transport (RCT) to the liver by its main protein constituent, apo A-I (Fig. 3) (88). Apart from the ABC transporter pathways, the cholesterol efflux from macrophages as well as the cholesterol uptake by hepatocytes are mediated by the SR-BI, which is expressed on the cell surface of hepatocytes and in the peripheral tissue, for example, on macrophages (21). Moreover, HDL has been demonstrated to also inhibit lipid oxidation, protect endothelial function, and exert anti-inflammatory and antiapoptotic actions. However, under diseased conditions, HDL may also undergo biochemical modifications (Fig. 4) that impair or reverse the atheroprotective effects of HDL, causing so-called dysfunctional HDL (88). Notably, the protective properties are mainly implemented by sphingosine-1-phosphate (S1P)-receptors and S1P-dependent signaling pathways (79, 80). Although knowledge about the receptors responsible for dysfunctional HDL is scarce, receptors of the innate immune system, such as toll-like receptor-2 (TLR-2), have been suggested to mediate, at least partly, adverse effects of dysfunctional HDL (102).

Besides modified LDL and HDL, also other lipoproteins, such as very low-density lipoprotein (VLDL) and beta-VLDL, may undergo oxidative modification (81). However, their implication for vascular redox signaling still remains largely unexplored.
Here, we summarize underlying mechanisms of how modified lipoproteins, in particular oxLDL and dysfunctional HDL, affect redox signaling in vascular cells and, consequently, impact the development of atherosclerotic vascular disease.
The primary consideration given here will be on mechanistic circuits between the lipoproteins LDL and HDL and the oxidant enzyme systems of NAD(P)H oxidases (NOXs). Moreover, we will highlight recent findings on how the gut bacterial ecosystem (gut microbiome) may regulate the lipoprotein modification with subsequent implication for the development of atherosclerotic disease—a subject that has attracted enormous interest in recent years. Finally, the potential clinical relevance of lipoprotein-related cardiovascular redox signaling is discussed, also considering results of clinical trials targeting these mechanisms.
Effects of Modified LDL—Lectin-Type oxLDL Receptor 1 Signaling on Endothelial Cell Functions: Role of the NOX
oxLDL and vascular function
Modified LDL, in particular oxLDL, plays a contributory role in the pathophysiology of endothelial cell dysfunction through a multitude of signaling pathways, some of which will be reviewed in this section.
oxLDL exerts many effects on human endothelial and smooth muscle cell physiology, particularly by activating NOX (90, 91). NOX is one of the major vascular sources of ROS (O2
−) within the arterial walls (104). For ROS generation, NOX utilizes NADH or NADPH as the electron donor for 1e− reduction of molecular oxygen (41) according to the following reaction:
Several isoforms have been identified, among which NOX2 and NOX4 are known as major endothelial isoforms (99). The NOX2 isoform consists of four subunits, the membrane-bound subunits gp91phox and p22phox, and cytosolic subunits p47phox and p67phox (39) (Fig. 5). Besides activation by oxLDL, NOX in vascular cells can also be activated by stimuli such as angiotensin II, thrombin, platelet-derived growth factor, tumor necrosis factor-α, interleukin-1, as well as mechanical forces, such as shear stress (104).

Other sources of ROS generation in the vasculature are infiltrated macrophages that are capable of ROS formation in an NOX-dependent manner, with subsequent release into the vascular wall (98).
Importantly, activated NOX signaling in various resident and infiltrated vascular cells contributes to oxidative modification of LDL, leading to increased formation of oxLDL. In turn, oxLDL constitutes a major trigger of NOX-dependent ROS generation, and thus accelerates a vicious cycle of oxLDL–NOX-mediated redox signaling and potentiation of superoxide anion release (90).
This vicious cycle of LDL–NOX-mediated redox signaling has detrimental effects on endothelial cell function (56). In particular, endothelial nitric oxide (NO) bioavailability appears to be substantially affected by increased oxidative stress (30). Beyond its major function to regulate vascular tone, NO also limits endothelial inflammatory and pro-coagulatory pathways (34). In endothelial cells, oxLDL is taken up by the oxLDL receptor 1 that is also known as lectin-type oxLDL receptor 1 (LOX-1) (94).
One of the underlying mechanisms is an aberrant function of endothelial nitric oxide synthases (eNOS) that is often referred to as “NOS uncoupling”—a state in which electron flow through the enzyme leads to a reduction of molecular oxygen at the prosthetic heme site instead of NO formation, leading to the formation of the highly reactive peroxynitrate (105) according to the following equation:
This uncoupling not only results in reduced formation of the protective molecule NO but also causes ROS accumulation, which again contributes to a condition of enhanced oxidative stress. NOS uncoupling is partly based on NOX-dependent oxidization of the NOS cofactor tetrahydrobiopterin (53).
Moreover, the condition of oxidative stress is further enhanced by suppressive effects of oxLDL on antioxidative factors, such as SOD, as shown in a recent study. In this article, it was demonstrated that oxLDL decreases extracellular SOD mRNA and protein levels in an LOX-1 dependent manner. This regulatory effect of oxLDL is mediated by a mitogen-activated protein kinase (MAPK)/extracellular-regulated protein kinase signaling pathway (64).
Enhanced expression of SOD inhibits oxLDL-induced human aortic smooth muscle cell proliferation, which may constitute a therapeutic approach to counteract oxLDL-induced oxidative stress (59).
In summary, accumulating evidence suggests a detrimental circuit consisting of oxLDL, which via the LOX-1 receptor is taken up in endothelial cells with subsequent activation of NOX. Consequently, this leads to enhanced ROS generation, which, in turn, oxidizes both LDL and the NOS cofactor tetrahydrobiopterin, leading to NOS uncoupling and generation of highly reactive peroxinitrates with a further reduction in NO bioavailability and post-transcriptional modification of cellular proteins (32, 33). All in all, this oxLDL–LOX-1–NOX circuit causes endothelial cell dysfunction, which is one of the early pathomechanistic aspects in the development of atherosclerosis (1).
Carbamylated LDL and endothelial function
Although oxLDL has attracted enormous interest in vascular research, other forms of LDL modification have been described in recent years, among which carbamylation of lipoprotein lysine residues merits further focus (101).
Carbamylation is a post-transcriptional modification process of proteins or amino acids based on the covalent adduction of isocyanic acid to specific nucleophilic functional groups (114).
One of the biochemical pathways is mediated by the non-enzymatic reaction between isocyanic acid, a decomposition product of urea, and either the N-terminus or the ɛ-amino group of lysine residues of amino acids. Another pathway is based on the oxidation of thiocyanate in the presence of hydrogen peroxide (H2O2) by the heme protein myeloperoxidase, which is one of the most abundant proteins in leukocytes, producing increased levels of isocyanate, in particular, under inflammatory conditions (118).
Importantly, elevated levels of carbamylated LDL (cLDL) have been observed in patients with coronary artery disease (CAD), in particular in those with additional chronic kidney disease (CKD) due to increased levels of urea and isocyanic acid (119). Moreover, patients with increased levels of cLDL are at higher risk for future cardiovascular events and all-cause mortality (101).
In experimental studies using organ chamber experiments on aortic rings from mice, cLDL was shown to impair acetylcholine- and calcium-ionophore induced endothelium-dependent relaxation (101), whereas non-modified LDL did not affect vascular reactivity. Interestingly, the effect of cLDL on vascular relaxation appears comparable with that of oxLDL, suggesting that different modifications of LDL lead to similar effects on vascular function and may activate similar pathways. Indeed, Speer et al. demonstrated that cLDL induces endothelial dysfunction via LOX-1 stimulation, leading to NOX activation in a p38-MAPK-dependent manner with subsequently increased endothelial ROS production and eNOS uncoupling, ultimately causing reduced NO bioavailability and impaired endothelium-dependent vasodilation (101) (Fig. 4).
In addition, cLDL directly promotes S-glutathionylation of eNOS (101), which reduces eNOS dimerization, leading to enhanced eNOS uncoupling (37). LOX-1 appears to be the predominant receptor—at least in endothelial cells—to mediate the cellular effects of cLDL. This was demonstrated in mice exhibiting endothelium-specific overexpression of LOX-1, which exhibited enhanced endothelial dysfunction in response to cLDL stimulation as compared with wild-type mice (101). Moreover, the involvement of the LOX-1 receptor was further supported by silencing ribonucleic acid-mediated silencing of LOX-1 in human aortic endothelial cells, preventing the cLDL-induced inhibition of NO production (101).
In summary, accumulating evidence suggests that cLDL induces endothelial dysfunction via activation of the endothelial LOX-1 receptor, leading to p38-dependent NOX activation, ROS production, S-glutathionylation-dependent eNOS uncoupling, and reduced NO bioavailability (101).
Vascular Effects of HDL in Healthy Subjects and in Patients with Cardiovascular Disease: Functional Shift and Altered Effects of Modified HDL on Redox Signaling and Vascular Cell Function
Protective effects of HDL in vascular cells
In this section, we will review biochemical modifications of HDL cholesterol, which occur under diseased conditions, resulting in a loss or reversal of its physiological functions.
HDL is considered a key player of the so-called RCT (89), which describes the transport of excess cholesterol from peripheral cells either to the liver for excretion into the bile or to adrenals, testes, and ovaries for steroid hormone synthesis. However, beyond its cholesterol transportation functions, direct vascular protective and potentially anti-atherogenic effects have been ascribed to HDL under physiological conditions (61). In particular, HDL from healthy subjects is known to promote NO release from endothelial cells and to increase the expression of eNOS (9). Moreover, by downregulating the expression of adhesion molecules, such as vascular cell adhesion molecule 1 (VCAM-1), HDL is capable of inhibiting the adhesion of leucocytes (78).
In addition, antithrombotic effects of HDL have been described, such as reducing tissue factor expression in endothelial cells with subsequent reduction of platelet activation (115).
Finally, HDL obtained from healthy subjects appears to improve endothelial repair mechanisms after vascular injury as demonstrated in the mouse carotid artery injury model (9) and to exert anti-apoptotic effects on endothelial cells on increased expression of anti-apoptotic signaling proteins, such as Bcl-xL (87).
Dysfunctional HDL in patients with cardiovascular disease
Unlike the protective vascular effects of HDL obtained from healthy subjects, HDL from patients with cardiovascular disease, such as CAD, acute coronary syndrome (ACS), or CKDs, loses it suppressive effect on endothelial VCAM-1 expression, resulting in increased adhesion of leucocytes to activated endothelial cells (9, 100). Similarly, anti-apoptotic effects of HDL from patients with CAD or ACS have been reported to be abolished since dysfunctional HDL fails to activate endothelial Bcl-xL, while promoting endothelial pro-apoptotic pathways, such as p38-MAPK-mediated activation of the pro-apoptotic Bcl-2 protein tBid.
Moreover, dysfunctional HDL fails to stimulate NO release and NO-associated beneficial effects in endothelial cells (9). On the molecular levels, several mechanisms have been suggested to exert detrimental effects of dysfunctional HDL, among which imbalanced redox signaling appears of high relevance for the vasculature. One of the molecular mechanisms of dysfunctional HDL in patients with CAD is reduced activity of HDL-associated paraoxanase-1 (PON-1), which under healthy conditions prevents HDL from oxidative modification (Fig. 3). Reduced activity of PON-1 leads to formation of advanced lipid oxidation products, such as malondialdehyde (MDA) (61). Consequently, reduced HDL-associated activity of PON-1 results in enhanced oxidation of HDL, in particular its apoA-I, and formation of MDA, which, in turn, activates endothelial LOX-1. Activation of endothelial LOX-1 by oxidized HDL stimulates protein-kinase C βII (PKCβII) in endothelial cells (9, 106). In turn, activation of PKCβII in endothelial cells inhibits eNOS-dependent NO production and eNOS-associated protective signaling pathways. In particular, increased endothelial PKCβII activation by oxidized HDL inhibits Akt-dependent eNOS-activating phosphorylation at Ser1177 and increases phosphorylation of eNOS at Thr495, which inhibits eNOS activity (7) (Fig. 6).

Taken together, these studies provide evidence that HDL of patients with cardiovascular diseases loses its property to stimulate endothelial eNOS-activating pathways and NO production. Consequently, this leads to defective endothelial anti-inflammatory and endothelial repair capacities of HDL. Importantly, dysfunctional HDL is characterized by stimulatory effects on endothelial LOX-1, with subsequent activation of the PKCβII signaling pathway in endothelial cells.
Dysfunctional HDL stimulates NADPH oxidases in endothelial cells: role for TLR-2
Recently, in patients with CKD-enhanced blood levels of a methylated derivative of the amino acid
Further, it was shown that this abnormal HDL activates endothelial TLR-2, increases ROS, and reduces NO production, leading to impairment of endothelial cell function, reduced endothelial repair capacity, and enhanced endothelial inflammation (102).
One of the underlying mechanisms of how HDLSDMA affects NO bioavailability and endothelial oxidative stress is TLR-2-mediated reduction in Akt phosphorylation at Ser473, with a subsequent reduction in eNOS-activating phosphorylation at Ser1177 (102) (Fig. 6).
Moreover, TLR-2 activation also stimulates NOX in endothelial cells to produce ROS—a process that has been also demonstrated in monocytes and macrophages (6). Notably, HDLSDMA-mediated activation of TLR-2 induces phosphorylation of c-Jun N-terminal kinase, which, in turn, increases NOX activity (15), leading to increased endothelial superoxide production (102). Interestingly, TLRs, in particular TLR-2 and TLR-4, are expressed on endothelial cells (24) and are activated by microbial lipoprotein patterns (108). Moreover, a potential primary role of TLR-2 for the development of atherosclerosis (75) has also been demonstrated apart from abnormal HDL signaling, underscoring its relevance for vascular biology.
Importantly, a number of studies have demonstrated elevated SDMA serum concentrations in several cardiovascular diseases, such as CAD, CKD, and pulmonary arterial hypertension (51, 83, 95), demonstrating the broad clinical implication of this bioamine. In a recent clinical study including two independent cohorts, one with 3310 subjects and one with 1424 subjects undergoing coronary angiography, the investigators observed that the combination of low SDMA levels and higher HDL cholesterol was associated with significantly lower mortality. However, high HDLSDMA levels were associated with higher mortality, confirming SDMA as a marker of HDL dysfunction (123).
In summary, these findings unveil a novel mechanism of lipoprotein modification by bioactive amino acid derivatives, such as SDMA, which is known to be elevated in several cardiovascular conditions. This modification converts HDL into a noxious particle that via TLR-2 increases NOX activity and reduces eNOS activity, ultimately leading to increased ROS and reduced NO production. It remains to be elaborated whether therapeutic strategies aiming at reducing circulating SDMA levels would improve HDL function and, thus, have beneficial effects on vascular physiology.
Relation Between Modified Lipoproteins and Endothelial Dysfunction
Impairment of endothelial function as an early functional marker of arterial disease and subclinical atherosclerosis can be measured as impaired vasodilation in response to specific endothelium-dependent and -independent stimuli within the forearm, coronary, or peripheral circulations (67). This parameter reflects functional consequences of unbalanced redox signaling in the vasculature.
One of the most frequently used method to investigate endothelial function is the assessment of flow-mediated dilation (FMD) of peripheral arteries as measured by high-resolution ultrasound (40, 113).
A negative relation between oxLDL/apoB100 ratio and FMD has been shown in a population-based cohort study including 624 subjects, even after adjustment for age, sex, glucose tolerance status, and Framingham risk score (112).
A recent study investigating circulating anti-carbamylated antibodies, which were also directed to cLDL, demonstrated a significant relation between the antibodies and endothelial dysfunction as assessed by FMD and arterial stiffness in patients with rheumatoid arthritis (103).
A similar association has been observed between dysfunctional HDL from patients with acute myocardial infarction and impaired endothelial function in these patients as evidenced by reduced FMD (19). All in all, these studies lend further support to a critical role of modified lipoproteins for vascular disease also on a functional level.
The Role of Oxidized Lipoproteins in Vascular Aging
Vascular aging is a complex process and a key factor of arterial stiffness and is strongly related to incident cardiovascular disease (CVD) (79). This process involves structural remodeling in the vascular wall marked by alteration of the extracellular matrix with fragmentation and degeneration of elastin, an increase in collagen content, arterial wall thickening, and luminal dilation. These alterations lead to arterial stiffening, with a subsequent increase in systolic and pulse pressures promoting atherosclerotic vascular disease, left ventricular hypertrophy, and dysfunction. Consequently, increased arterial stiffness is associated with increased risk for CVD, including CAD, myocardial infarction, heart failure, stroke, and cardiovascular mortality (68). Although the exact mechanisms underlying aging-related arterial stiffening are not completely understood, substantial evidence suggests an imbalance in redox hemostasis with increased oxidative stress. A major component of age-related oxidative stress is based on increased activation of the oxLDL–LOX-1–NOX signaling pathway. This assumption is supported by clinical studies demonstrating a strong association between plasma ox-LDL levels and arterial stiffness among elderly people independent of CVD risk factors (12).
In experimental settings, a crucial role for NOX2-containing NADPH oxidase activation in aging-associated damage of endothelial function was recently identified. By measuring the levels of NADPH-dependent O2 − production, the authors demonstrated a significant increase in the levels of O2 − production starting at middle age and progressing to old age in the aorta of wild type (WT) mice. This increase in O2 − production was abolished in aged NOX2 knockout mice, with improved endothelium-dependent vessel relaxation as compared with aged WT controls. Inhibition or knockout of NOX2 preserved endothelial function and delayed vascular aging (26).
Further, it has been shown that cLDL induces accelerated senescence marked by telomere shortening and β-galactosidase activity in human endothelial progenitor cells (18).
Apart from the modified LDL-mediated mechanisms, cumulative data point to aging-induced detrimental alteration in HDL composition, resulting in functional impairment in terms of reduced cellular cholesterol efflux/uptake and anti-oxidant properties. In particular, HDLs from elderly subjects appear to be more prone to lipid peroxidation and present a significant reduction of their antioxidant property, along with a decrease in PON-1 activity (47). Moreover, aging causes impairment of HDL-mediated RCT capacity, which has been attributed to a reduction in the ATP-binding cassette transporter ABCA1 pathway (8). Besides aging-induced functional alterations of HDL, aging-related changes in structure and composition of HDL have also been observed. In a recent clinical study on elderly subjects, a three-fold increase of the acute phase protein serum amyloid A, an increased content of complement C3 and proteins involved in endopeptidase/protease inhibition in HDL were identified. However, HDL from elderly subjects contained significantly less levels of apoE and cholesterol (45).
In conclusion, these studied suggest a critical involvement of modified lipoproteins in pathophysiological processes contributing to age-related vascular disease.
Current Research and Future Perspective: Interaction Between the Gut Microbiome and Host Redox Signaling
Accumulating evidence shows that the bacteria residing within the human intestinal tract, the so-called gut microbiome, influence the host risk to develop metabolic and cardiovascular diseases (110). It is currently estimated that a microbe population of up to 100 trillion cells inhabit the human body in a symbiotic manner, which constitutes 10-fold the number of the human host cells and encodes about 100-fold more genes than our own genome (58). The major proportion of the microbe community inhabits our intestines. The intestinal microbe population consists of two main bacterial phyla, Firmicutes and Bacteroidetes, and more than 1000 prevalent bacterial species (58).
Importantly, dietary habits, for example vegetarian or carnivore, are known to alter the composition of intestinal microbiota (74, 124). Metagenome-wide association studies combining clinical phenotypic data with metagenomic analysis (44, 57) have identified distinct microbiome patterns that are associated with the development of metabolic disorders such as type 2 diabetes mellitus (31) and obesity (86).
The gut microbiome has also sparked interest in the cardiovascular field due to recent studies that have demonstrated a critical involvement of the gut microbiome in cardiovascular disease development (118).
Further, it has been demonstrated that the absence of gut microbiota—as simulated in so-called germ-free mice—protects mice from angiotensin II induced arterial hypertension and vascular dysfunction. This protection appears to be mediated by reduced expression of vascular NOX2 and inducible NOS expression in infiltrated inflammatory cells (49).
Recent evidence suggested that bacterial fragments from the gut microbiota, in particular lipopolysaccharides (LPS), translocate to the host tissues and initiate metabolic inflammation and oxidative stress (13).
Studies on high fat diet (HFD)-fed mice have shown that LPS from intestinal gram-negative bacteria accumulates in the blood and establishes a state of metabolic endotoxemia on HFD (16). In a clinical study, apparently healthy individuals who were on a fat-enriched diet rather than on a carbohydrate and protein-enriched diet were characterized by higher concentrations of blood LPS (2). Moreover, individuals with type 2 diabetes and dyslipidemia also have higher concentrations of LPS (56). Importantly, LPS can also oxidize lipoproteins, leading to endothelial cell dysfunction and endothelial inflammation (70).
In view of these findings, it is conceivable that modulation of the gut microbiome may be a potential therapeutic approach to reduce the release of detrimental bacterial fragments, with a subsequent decrease of lipoprotein oxidation. Indeed, in experimental studies, modulation of gut microbiome has been tested by means of prebiotic treatment with soy products, which increased Lactobacillus spp., Bifidobacterium spp., and Enterococcus spp. and decreased the Enterobacteria population. This modulation of the gut microbiome composition was associated with a decrease of oxLDL, demonstrating the therapeutic potential of targeting the gut microbiome to improve cardiometabolic disorders and restore endothelial cell function (23).
Although gut microbiome research in the cardiovascular field is still in the early stages, it is exciting to observe the promising experimental and clinical approaches that are aimed at modulating the gut microbiome or its metabolism for therapeutic interventions.
Targeting Redox Signaling in the Clinical Arena: Current Concepts and Future Perspectives
Targeting redox signaling at different levels for therapeutic purposes has been a lingering matter of investigation (Fig. 7).
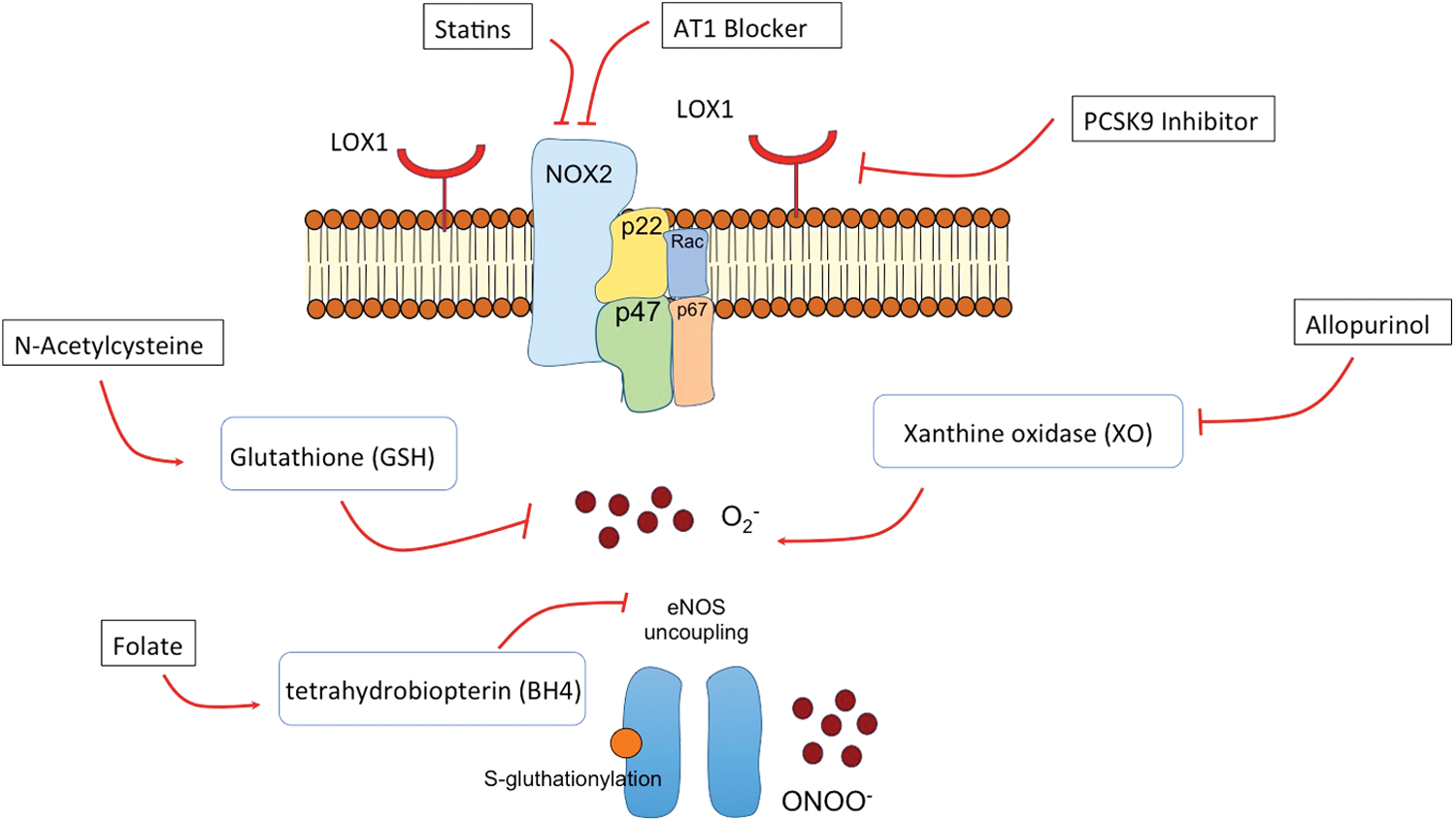
In fact, one potentially beneficial off-target effect of statins is downregulation of NOX2 or modulation of its activity (89), with an improvement of endothelial and cardiac function (46). Moreover, the regulatory effect of statins on cardiac redox signaling has been explored in a clinical study of patients undergoing cardiac surgery. In this study, myocardial superoxide anion O2 − (mainly derived from NOX) and peroxynitrite ONOO− and their enzymatic sources were quantified in samples of the right atrial appendage from patients at the time of cardiac surgery who had been randomized to receive 3-day treatment with atorvastatin 40 mg/day or placebo before surgery. The authors found reduced production of both myocardial O2 − and ONOO− by pre-operative statin treatment through an Rac1-mediated suppression of NOX activity (3).
Interestingly, recent studies suggest that the new group of LDL cholesterol-reducing drugs, the proprotein convertase subtilisin/kexin type-9 (PCSK9) inhibitors, also modulate oxLDL–LOX-1-dependent oxidative signaling since PCSK9 appears to promote the expression of LOX-1, with a subsequently increased uptake of oxLDL in vascular cells (22). This mechanism may contribute to the cardiovascular protective effects of PCSK9 inhibitors as demonstrated in a recent large outcome trial (93). Angiotensin II receptor 1 blockers (AT1 blockers) are another drug class that has been frequently demonstrated to modulate NOX-mediated oxidative stress. The regulatory property of how Ang II regulates vascular cell production of ROS has been extensively studied (77). Notably, Ang II-mediated generation of ROS is mainly driven by NOX, and it involves downstream signaling targets, including MAPKs, RhoA/Rho kinase, transcription factors, protein tyrosine phosphatases, and tyrosine kinases (77).
Interestingly, apart from AT1-mediated NOX suppressing effects, the metabolites of certain AT1 blockers, such the losartan metabolite EXP3179, exert angiotensin II-independent inhibitory effects on NOX oxidase activity by directly inhibiting protein kinase C signaling (34). This implies that losartan exerts two separate effects, which, in concert, decrease NOX-dependent ROS generation.
As far as direct modulation of the imbalance in redox hemostasis is concerned, to date, attempts to reduce oxidative stress with antioxidants have not yielded convincing results (11).
In particular, two proposed antioxidant drugs, N-acetylcysteine (NAC) and allopurinol, and several vitamins, vitamin C, E, and B, have been explored in clinical trials that merit inclusion in this review (Table 1).
CABG, coronary artery bypass grafting; CV, cardiovascular; EPIC, European prospective investigation into cancer and nutrition; GSH, glutathione; HOPE, Heart Outcomes Prevention Evaluation; i.v., intravenous; NAC, N-acetylcysteine.
N-acetylcysteine
NAC is considered to possess antioxidant properties as it serves as a substrate for the synthesis of the intracellular antioxidant glutathione (GSH) (95). GSH is a tripeptide (
In a double-blind study performed on 24 male patients with type 2 diabetes and hypertension, oral supplementation with NAC and
Allopurinol
Allopurinol has antioxidant properties by inhibiting the enzyme XO, which catalyzes the conversion of hypoxanthine to uric acid, with ROS being generated as a by-product (92). The XO-inhibiting effect of allopurinol is being currently used for treatment of gout. Several clinical studies have been performed by using intravenous or intracoronary infusion of XO inhibitors in patients with CVD. In some studies, a beneficial effect of allopurinol, in particular in patients with hypertension and heart failure, could be observed (43, 62); whereas more recent studies failed to confirm previous results questioning the therapeutic relevance of allopurinol as an antioxidant compound for patients with CVD (38). However, a multicenter, controlled, prospective, randomized trial (ALL-HEART study) (63) is currently underway that will determine whether allopurinol improves cardiovascular outcomes in patients with ischemic heart failure. The results of this trial will provide important information about the therapeutic potential of allopurinol in patients with ischemic heart disease.
Folate
Early studies focused on the effect of folate of lowering homocysteine as a potential therapeutic strategy in patients with CAD. However, a randomized clinical trial of folic acid treatment failed to improve clinical outcomes in patients with stable CAD (60), questioning the therapeutic utility of folate in patients with CAD. However, besides homocysteine-lowering effects, folate has been demonstrated to serve as an antioxidant and to have direct effects on NO-mediated endothelial function. This role of folate as an antioxidant vitamin has been comprehensively studied in the clinical setting. The findings demonstrated that folate has beneficial effects on endothelial function by decreasing superoxide and peroxynitrite production and by improving eNOS coupling as mediated by tetrahydrobiopterin availability (4). However, large-scale prospective placebo-controlled trials are required to evaluate the impact of folate on clinical outcome of patients with endothelial dysfunction.
Vitamin C
Vitamin C has long been recognized as an antioxidant with beneficial effects in a number of diseases associated with enhanced oxidative stress (1). By scavenging ROS, Vitamin C may protect DNA, proteins, and lipids against peroxidation, thereby exerting cytoprotective effects (10). In the European prospective investigation into cancer and nutrition (EPIC)-Norfolk Study, a population-based cohort study of diet and chronic disease, it was found that cardiovascular disease mortality was reduced by ∼20% through increasing plasma vitamin C by 20 μM based on increased consumption of fruits and vegetables (50). However, a meta-analysis of seven vitamin C supplement studies could not confirm a reduction of cardiovascular events by vitamin C, raising skepticism about beneficial effects of Vitamin C in cardiovascular protection.
Vitamin E
Vitamin E is one of the most intensively studied antioxidants in the clinical setting. However, the results of the trials exploring the impact of vitamin E supplementation for preventing cardiovascular disease did not show beneficial effects. One of the largest trials on vitamin E supplementation, the placebo-controlled Heart Outcomes Prevention Evaluation (HOPE) trial enrolling more than 9000 patients at high risk for cardiovascular events, showed that treatment with vitamin E for a mean of 4.5 years had no effect on cardiovascular outcomes (122). In other trials using vitamin E supplementation, an increased mortality was even reported (71). Consequently, vitamin E supplementation is no longer recommended in primary prevention of cardiovascular disease by the United States preventative task force (73).
The lack of consistency between some clinical studies (76) demonstrates the complexity of targeting redox signaling for therapeutic purposes. Moreover, the assessment of the patients redox state by means of biomarkers is often challenging (Table 2), thus, limiting the monitoring tools to evaluate the effect of antioxidant therapeutics.
CKD, chronic kidney disease; ELISA, enzyme-linked immunosorbent assay; eNOS, endothelial nitric oxide synthases; HDL, high-density lipoprotein; LDL, low-density lipoprotein; LOX-1, lectin-type oxidized LDL receptor 1; MAPK, mitogen-activated protein kinase; MPO, myeloperoxidase; NOX, NAD(P)H oxidase; ROS, reactive oxygen species; TMB, tetramethylbenzidine.
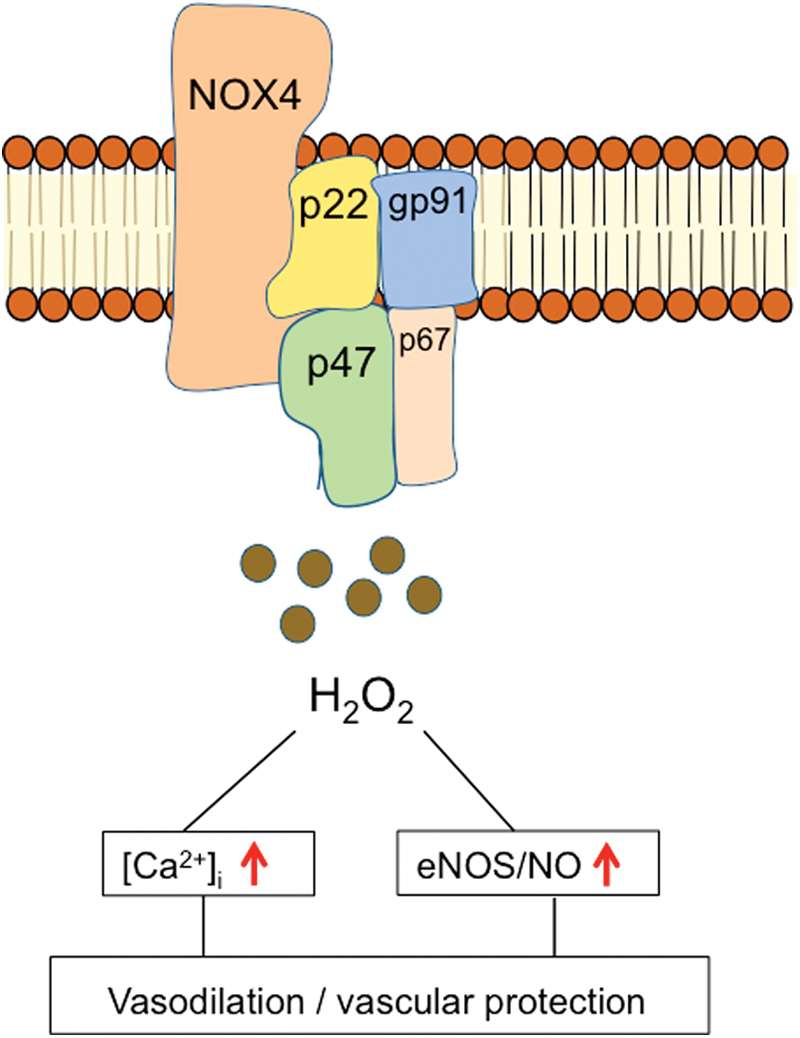
One explanation why these studies failed to show beneficial effects in the cardiovascular disease is the hypothesis that low doses of oxidants, in particular H2O2, may even exert vasoprotective effects, presumably by acting as an endothelium-derived vasodilator and improving endothelial function (96). Although the underlying mechanisms of the potential protective effects of H2O2 are not yet completely understood, altered eNOS expression/activation and activation of soluble guanylate cyclase or protein kinase G, the downstream targets of NO responsible for vasodilatation, have been suggested (96).
Another explanation is that H2O2 potentiates vasorelaxation by enhancing Ca2+ release from endothelial endoplasmic reticulum stores, which, subsequently, potentiates the opening of Ca2+-activated K+ channels (25, 97). In endothelial cells, one of the major sources of H2O2 formation is the NOX4 isoform of NAPDH oxidase (107) (Fig. 8). Numerous studies indicate that NOX4 generates predominantly H2O2 rather than superoxide. In contrast, NOX1 and NOX2 generate mainly superoxide (66). In view of the isoform specificity of NOX-generated ROS with partly opposing vascular effects (72), it is conceivable that the lack of specificity of antioxidant therapies is one reason that many therapeutic attempts were not successful. Thus, future therapeutic strategies to modulate ROS production in cardiovascular disease need to consider both potential beneficial and detrimental effects of ROS depending on the source of generation (85). Consequently, therapeutic concepts targeting ROS production require the development of more selective inhibitors of ROS-generating enzymes, for example, isoform-selective NOX inhibitors. Such inhibitors are currently under development, and future clinical studies are required to validate the therapeutic potential of selective inhibitors of ROS-generating enzymes (52).
Conclusions
During the past decades, extensive research has been devoted to exploring the role of modified lipoproteins for regulating cardiovascular redox signaling. Although atherogenic and pro-inflammatory properties of oxidatively modified LDL have been established, the role of HDL to counteract oxidative stress and vascular dysfunction and inflammation (“HDL-cholesterol hypothesis”) has been questioned in recent years, not at least because of the failure of HDL cholesterol, thus raising therapeutic strategies to improve clinical cardiovascular outcome. Experimental and clinical studies suggest that functional properties of HDL may be shifted in patients with cardiovascular disease, leading to potential pro-atherosclerotic effects on the vasculature, and, thus, HDL cholesterol is likely not a reliable surrogate marker for therapeutic interventions.
NADPH oxidases are major sources of ROS production in the vasculature and important targets of modified lipoproteins to regulate redox signaling. However, increasing evidence suggests a diverse role of distinct NOX isoforms for endothelial function, with NOX4 exerting partly protective vascular effects. Thus, therapeutic concepts targeting NOX require isoform selectivity in the vasculature. In conclusion, despite major advances in understanding underlying mechanisms of lipoprotein-mediated redox signaling, to date, therapeutic approaches to target these circuits have not been established in the clinical arena, demonstrating the complexicity of these mechanisms and the need for further investigatory effort.
Innovative concepts may arise from current research on the relationship between the gut microbiome and vascular physiology, which may open a new avenue for therapeutic strategies based on the modulation of the gut microbiome composition or metabolism for cardiovascular prevention.
Footnotes
Acknowledgment
Dr. Haghikia received a research grant from the Deutsche Stiftung für Herzforschung (F/28/16).