
Abstract
Significance:
Eicosanoids are endogenous lipid mediators that play important roles in brain function and disease. Acute brain injury such as that which occurs in stroke and traumatic brain injury increases the formation of eicosanoids, which, in turn, exacerbate or diminish injury. In chronic neurodegenerative diseases such as Alzheimer's disease and vascular dementia (VD), eicosanoid synthetic and metabolizing enzymes are altered, disrupting the balance between neuroprotective and neurotoxic eicosanoids.
Recent Advances:
Human and experimental studies have established the opposing roles of hydroxy- and epoxyeicosanoids and their potential utility as diagnostic biomarkers and therapeutic targets in neural injury.
Critical Issues:
A gap in knowledge remains in understanding the cellular and molecular mechanisms underlying the neurovascular actions of specific eicosanoids, such as specific isomers of epoxyeicosatrienoic (EETs) and hydroxyeicosatetraenoic acids (HETEs).
Future Directions:
EETs and HETEs exert their actions on brain cells by targeting multiple mechanisms, which include surface G-protein coupled receptors. The identification of high-affinity receptors for EETs and HETEs and their cellular localization in the brain will be a breakthrough in our understanding of these eicosanoids as mediators of cell-cell communications and contributors to brain development, function, and disease. Antioxid. Redox Signal. 28, 987–1007.
Introduction
E
Recognition of the biological importance of eicosanoids has evolved over the past several decades. The importance of prostaglandins in various physiological functions has been recognized and extensively studied since the mid-1930s (184); the physiological and pathophysiological roles of leukotrienes and lipoxins were introduced in the late 1970s (152, 160); whereas the roles of EETs and HETEs were not recognized until the early 1980s (34). More recently, non-enzymatically formed isoprostanes and non-AA products of COX, LOX, and P450 metabolites, such as resolvins, protectins, and maresins, were discovered and implicated in tissue injury and resolution of inflammation (124, 159).
Prostanoids and leukotrienes are involved in a multitude of normal and pathological conditions, such as vascular reactivity, platelet aggregation, cell maturation, and inflammation (45). The diverse roles of these prostanoids and leukotrienes are mediated, in part, by a family of membrane-bound G-protein coupled receptors (GPCRs) that can activate distinct intracellular signaling pathways that are triggered by ligand binding to the receptors (45). Extensive data in the literature suggest that EETs and HETEs may also exert their effects via membrane-bound receptors (38, 79, 185, 192, 198), although their corresponding putative receptors are largely unknown.
There is a duality and reciprocity to the roles that P450 eicosanoids play in brain injury. As a general rule, epoxygenase products are neuroprotective, whereas hydroxylase products are neurotoxic. Furthermore, the conversion of AA to eicosanoids by COX-, LOX-, and P450 enzyme-catalyzed reactions generates reactive oxygen species (ROS) such as superoxide anion (O•−) and hydroxyl radical (OH•) (63, 64). Under normal physiological conditions, the generation of free radicals is balanced by the capacity of cellular anti-oxidant systems (63, 64). The low level of ROS serves as an intracellular signal and can be beneficial, but at high concentrations, ROS causes lipid peroxidation, DNA damage, and protein modifications (111). As summarized in Figure 2, in general, the level of cellular ROS is determined by the balance between its production and neutralization.

ROS is produced by the mitochondrial electron transport chain and enzymatic reactions utilizing oxygen, whereas they are neutralized by intracellular scavengers such as antioxidant enzymes and antioxidants (111). The redox balance in cells may be disturbed due to a rapid increase in prooxidant mechanisms and a limited antioxidant capacity of cells, resulting in a lag in the antioxidant response (63). In disease states, such as ischemia and inflammation, inflammatory cytokines and ROS increase the activity of phospholipase A2 (PLA2), leading to a higher PLA2-catalyzed release of AA from cell membranes (102).
As mentioned earlier, at a higher rate of synthesis, AA metabolizers, in turn, generate ROS, worsening the redox environment and exacerbating tissue injury (82, 146, 189). Furthermore, as discussed later, eicosanoids themselves can augment or suppress oxidative stress. These observations demonstrate the reciprocal relationship between eicosanoids and ROS. The current review summarizes recent data on the interplay between P450 eicosanoids and ROS in the context of brain injury caused by stroke, traumatic brain injury (TBI), and neurodegenerative diseases such as Alzheimer's disease (AD) and aging-related vascular cognitive impairment (VCI). We synthesize these findings to highlight the dual role of P450 enzymes and their metabolites in brain disease as pro- and anti-inflammatory, and pro- and anti-oxidant and neuroprotection.
Selectivity of regio- and stereoisomers
As mentioned earlier, EETs and HETEs act in opposite manners under normal physiological conditions and disease states. For example, HETEs tend to induce vasoconstriction whereas EETs tend to be vasodilators (57, 69, 131, 158, 197, 206, 217). HETEs augment ROS production, whereas EETs reduce ROS production (82, 146, 189). HETEs promote inflammation, whereas EETs suppress inflammation (41, 48, 97, 129). However, these generalizations ignore the unique and tissue-specific roles that different regio- and stereoisomers of EETs and HETEs play under different conditions (61, 73, 129, 137, 167, 176). For example, levels of 5-, 8-, 12-, and 15-HETE and 5,6- and 8,9-EET but not 11,12- or 14,15-EET increase after intestinal ischemia induced by superior mesenteric artery occlusion (73). In a vascular inflammation model, 11,12-EET inhibited the expression of vascular cell adhesion molecule-1 (VCAM-1) induced by tumor necrosis factor-a (TNF-a), in contrast to 14,15-EET, which had no effect on VCAM-1 expression (129).
Evidence suggests that different HETEs regio- and stereoisomers also have different efficacies, and their effects are tissue- and organ specific. For example, 12(S)-HETE induced a more profound constriction than 12(R)-HETE and 15-HETE in isolated rat kidney afferent arterioles (206), whereas 15-HETE induced vasoconstriction in rat internal carotid arteries (217); 20-HETE has been recognized as a potent vasoconstrictor in multiple vascular beds (69, 70, 207). In rat cerebral arteries, 20-HETE formation correlated with an increasing intraluminal pressure, and an increasing 20-HETE concentration dose dependently reduced the diameter of isolated rat cerebral arteries (69). This pressure-dependent vasoconstriction, known as myogenic vasoconstriction, is the hallmark of autoregulation of blood flow in the brain, heart, and kidney (69). Nevertheless, even for 20-HETE, its effect seems to be tissue specific. For example, in rat basilar arteries, 20-HETE produces dilation that is inhibited by indomethacin, suggesting the conversion of 20-HETE by COX to secondary products (58). In isolated mouse aortic rings, 19-HETE induces vasodilation by activating the prostacyclin receptor (179). Later, we discuss possible explanations for the tissue selectivity of different EETs and HETEs and their regio- and stereoisomer-specific effects.
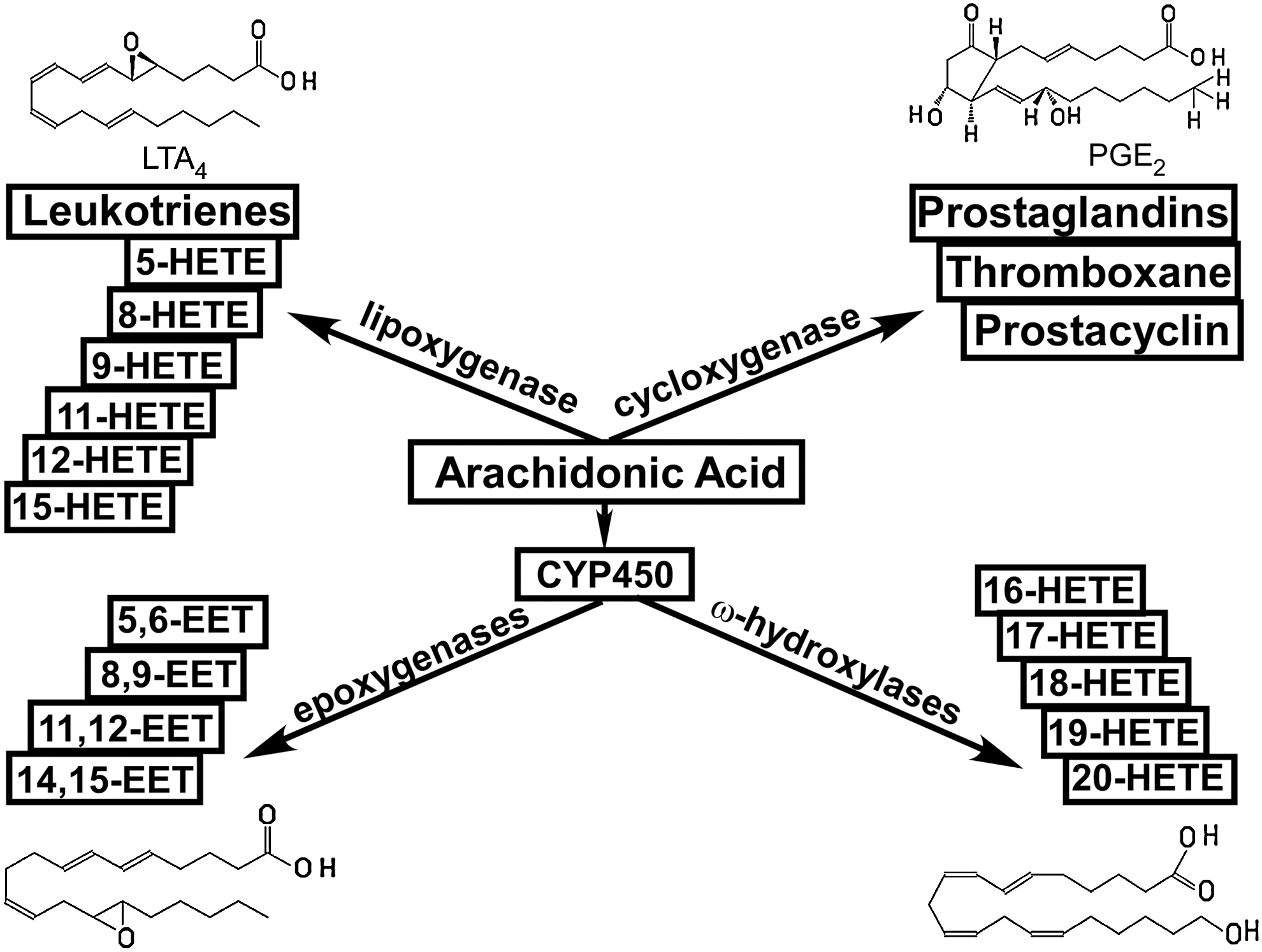
Synthetic enzymes
In addition to LOX-catalyzed production of midchain HETEs (35, 55, 135), HETEs and EETs are produced by members of the cytochrome 450 enzymes superfamily (Fig. 3) (8, 29, 34, 40, 46, 55, 72, 96, 108, 148, 210). Among the panel of enzymes discovered, several are selective for ω-terminal HETEs regioisomers (labeled in red). Rat CYP2C11 (r-CYP2C11) selectively produces EETs (labeled in green). In addition, the activation of several enzymes (labeled in blue) catalyzes the production of both HETEs and EETs, although the efficiencies for HETEs versus EETs production by these enzymes differ. For example, human CYP1A1 (h-CYP1A1) is more efficient in synthesizing terminal HETEs than midchain HETEs and EETs; h-CYP1A2 is more efficient in synthesizing EETs; h-CYP1B1 is more efficient in synthesizing midchain HETEs; and mouse CYP1B1 (m-CYP1B1) is more efficient in synthesizing EETs (40).

Local concentrations of HETEs and EETs
In addition to synthetic enzyme activation, local concentrations of HETEs and EETs are affected by additional factors. First, differential incorporation of HETEs and EETs into phospholipids can affect their bioavailability (20, 24, 59, 98, 165, 183). For example, the level of lipid-bound 14,15-EET in rat plasma is more than three times higher than the level of 8,9-EET and approximately 20% more than 11,12-EET (98). In human umbilical artery smooth muscle cells, 70% of (3H)5-HETE was incorporated into cell lipids, whereas only 12% of 12-HETE and 8% of 15-HETE were incorporated into smooth muscle cell lipids over 20 h of incubation (24).
Second, the conversion of HETEs and EETs into secondary products could change their bioavailability. For example, EETs are converted into dihydroxyeicosatrienoic acids (DHETs) by soluble epoxide hydrolases (sEH) in vascular cells (59, 121, 183, 211), and HETEs can be converted into secondary products such as 20-hydroxyendoperoxides, 20-COOH-AA, and 20-OH-PGH2, which are products of 20-HETE (44, 58, 158). The secondary products may sometimes be active and exert additional effects that may differ from those induced by the primary products. For example, in pre-constricted bovine coronary artery rings, 14,15-DHET induced vasorelaxation of the artery rings, although with less potency than 14,15-EET (31). The vasodilatory effects of other DHETs such as 8,9-, 11,12-, and 14,15-DHET have also been observed in isolated human coronary arterioles where they induce vasodilation by activating BKCa channels (103). The secondary products produced from 20-HETE such as 20-hydroxyendoperoxides, 20-COOH-AA, and 20-OH-PGH2 displayed vasoconstrictive properties (158). Interestingly, as mentioned earlier, in mouse basilar arteries, both 20-COOH-AA and 20-HETE induced vasodilation (58).
Third, cell-specific release of HETEs and EETs could, in addition, modulate the local concentration and bioavailability of these compounds. Earlier studies indicated that HETEs were produced in platelets and neutrophils (149, 180). However, later studies demonstrated that they are also produced by vascular smooth muscle cells (VSMCs) (70, 125, 157, 219); 14,15-EET is produced by endothelial cells (ECs) and astrocytes (8, 32, 68, 128) but exerts its effect on VSMCs, inflammatory cells, and neurons (57, 93, 131, 197).
Direct modulation of membrane protein functions
Incorporation of HETEs and EETs into phospholipids (20, 24, 59, 98, 165, 183) suggests that they may modify the function of membrane proteins directly in the lipid bilayer. For example, in HEK cells expressing the transient receptor potential cation channel subfamily V member 4 (TRPV4) channel, which is a Ca2+-permeable non-selective cation channel (143), 500 nM 5,6-EET opens TRPV4 channels directly as demonstrated by both whole-cell and inside-out single-channel recordings (186). To a lesser extent, 8,9-EET also increased intracellular Ca2+ in HEK cells expressing TRPV4, whereas 11,12- and 14,15-EET had no effect on TRPV4 channel function (186); 5,6-EET increased intracellular Ca2+ in ECs isolated from mouse aorta (186). In contrast, both 5,6-EET and 14,15-EET were shown to increase endothelial permeability in rat and mouse lungs where the expression of TRPV4 was confirmed in bronchiolar epithelium as well as in smooth muscle cells (SMCs) in human and rat extra-alveolar vessels (10). In this case, however, it is not clear whether the effects of 5,6-EET and 14,15-EET were due to direct modulation of channel activity since their effects were measured 45 min after treatment (10). In another study, 3 μM 11,12-EET induced TRPV4-like cation current in VSMCs as well as vasodilation in mesenteric arteries of the wild-type (WT) mouse. These effects were absent from TRPV4 knockout mice and the effect of 11,12-EET was fully reversed by the potent, non-specific TRPV4 blocker, ruthenium red (54).
Several other channels are also modulated by EETs and HETEs. For example, 20-HETE induced dose-dependent increases in blood pressure, coronary perfusion pressure (isolated Langendorff), and pressure-induced constriction of resistance arteries and these effects were attenuated in TRPV1 knockout mice or after treatment with the TRPV1 antagonist RP67580 (25). In cultured rat brain astrocytes, exogenously applied 20-HETE (100–300 nM) significantly reduced the activity of two types of Ca2+-activated potassium channels (KCa), with unitary conductances of 71 pS and 161 pS (72). Also, 20–125 nM EETs decreased the open probability of porcine cardiac L-type Ca2+ channels that were reconstituted in lipid bilayers (36). On the other hand, 3 μM 14,15-EET increased the opening probability but not single-channel conductance of large-conductance Ca2+-activated K+ (BKCa) channels that were assayed in inside-out patches obtained from rat pituitary GH(3) cells (194). In freshly isolated vascular muscle cells from cat cerebral arteries, 5 μM 8,9-EET or 11,12-EET increased the open probability of a 98-pS K+ channel that was recorded in the cell-attached mode (71).
Receptor targets for HETEs and EETs
High-affinity receptors
Besides the impact of local production, their incorporation into membrane lipids, and their secondary metabolism, the effects of HETEs and EETs also depend on the availability of their putative membrane receptors as well as on modes of ligand-receptor interaction and the signal transduction pathways activated by receptor-ligand engagement. Due to the low level of EETs and HETEs produced endogenously (6, 76, 163), their effects are likely to be mediated by high-affinity receptors under normal physiological conditions. Multiple ligand binding studies suggest the existence of a high-affinity receptor for 14,15-EET (37, 192, 198), and this putative 14,15-EET receptor exerts its vasodilator effect via the Gs-coupled signaling pathway, leading to increased intracellular cAMP (107, 113, 192, 193). To date, such high-affinity receptors have not been identified, although a 47 kDa membrane protein was found to bind to a 14,15-EET analog with a high affinity (37). In addition, a high-affinity receptor for 11,12-EET was also suggested by the following studies. In isolated rat small renal arteries, a nanomolar concentration of 11(R),12(S)-EET, but not 11(S),12(R)-EET or 14,15-EET, increased the activity of K+ channels in cell-attached patches obtained from VSMCs (218). However, this stimulatory effect was absent in excised inside-out or outside-out patches (218). A similar observation was made in cultured astrocytes where exogenous 11,12-EET (100 nM) only increased the NPo of both the 71 pS and 161 pS KCa channels in cell-attached but not in excised inside-out patches (196). Subsequently, it was demonstrated in isolated patches from VSMCs of small bovine coronary arteries that the stimulatory effect of a nanomolar concentration of 11,12-EET on the large conductance KCa channel requires the presence of Gsα, suggesting that 11,12-EET may interact with a Gs-coupled membrane receptor (107).
For HETEs isomers, GPR31 was identified as a high-affinity receptor for 12(S)-HETE (79). Similarly, a high-affinity binding site for 12-HETE was found in the human epidermal cell line (78), but it is unknown whether the receptor observed in this study is GPR31. The existence of a high-affinity 15-HETE receptor was demonstrated by the high-affinity specific binding of (3H)15-HETE in cultured PT-18 mast/basophil cells (142, 185). A high-affinity receptor for 20-HETE has also been suggested by functional readouts induced by nanomolar concentrations. For example, in rat middle cerebral arteries, 20-HETE induced a dose-dependent reduction in vessel diameter in the nanomolar range. Interestingly, this constrictive effect was blocked by 1 μM 15-HETE (69). This acute, dose-dependent constriction induced by 20-HETE is consistent with activation of a Gq-coupled receptor. The case for a putative 20-HETE receptor is also strengthened by the discovery of several 20-HETE antagonists such as 5(S)-HETE, 15(S)-HETE, 19(S)-HETE, and 6(Z),15(Z)-2-HEDE based on structure-activity studies (9).
Low-affinity receptors
We have recently identified several low-affinity receptors that were stimulated by 1 μM 14,15-EET (113). One of these low-affinity receptors, PTGER2, has been shown to mediate the effect of 14,15-EET in isolated rat mesenteric arteries in the micromolar range (197); 14,15-EET and other EETs regioisomers were also suggested to be agonists for a peroxisome proliferator-activated receptor (PPARγ) with micromolar efficacies based on the competitive bindings of EETs regioisomers toward the recombinant protein of the PPARγ ligand-binding domain (114). In addition, 14,15-EET has been suggested as an antagonist for thromboxane-prostanoid receptor (TP) based on the observation that 14,15-EET-inhibited TP agonist (U46619) induced vasoconstriction but not that induced by other non-TP agonists such as phenylephrine, endothelin-1 (19); 14,15-EET exhibited competitive binding toward TP in cultured CHO cells expressing TP with a Ki of 3.3 μM, which is nearly six times lower than the Ki toward PTGER2 determined in the same assay (19).
A low-affinity receptor for 19-HETE was identified as the prostacyclin receptor (179). Interestingly, in HEK cells overexpressing TP, 1 μM 12-HETE inhibited U46619-induced increase in intracellular Ca2+ and displaced specific binding toward SQ29548, thus acting as an antagonist for TP (162); 15-HETE was implicated as a TP agonist based on the observation that 15-HETE-induced constriction was prevented by SQ29548 in rabbit aortic rings (176). Furthermore, 12(S)-HETE and 15(S)-HETE have been implicated as PPARγ receptor agonists based on the observation that they inhibited ischemia-induced inflammation by activating PPARγ receptor, and their effects were reversed by the PPARγ antagonist GW9662 (167). In addition, 20-HETE has been suggested as an agonist for TP based on the observation that it induced contraction of isolated rat aortic rings or rat kidney arteries, both of which were abolished by pretreatment with SQ29548 (13, 56). However, this observation was in contrast to results from a study in dog renal arteries where the constriction induced by 20-HETE was not blocked by pretreatment with SQ29548 (116).
The apparent antagonistic nature of 15-HETE toward 20-HETE raises an interesting question as to whether 20-HETE exhibits a similar antagonistic function toward the putative 15-HETE receptor (185), and whether different HETEs regioisomers serve as agonists and antagonists for other putative regioisomer-specific receptors. In Table 1, we summarize the regioisomers of HETEs and EETs that are either associated with known receptors or their interaction with the corresponding putative receptors has been implicated by biochemical and pharmacological studies.
High affinity refers to effective concentrations <1 μM. Low affinity refers to effective concentrations ≤1 μM.
? indicates the existence of a receptor, although identity has not been confirmed.
Prostaglandin receptors: PTGER2, PTGER4, PTGDR, PTGFR, and PTGER3IV.
EETs, epoxyeicosatrienoic acids; HETE, hydroxyeicosatetraenoic acids; PPARγ, peroxisome proliferator-activated receptor γ; PTGIR, prostacyclin receptor; TP, thromboxane-prostanoid receptor.
Mechanisms of putative HETEs and EETs receptor activation
It is reasonable to expect that a certain level of specificity for the action of regioisomers of HETEs and EETs might be achieved based on the factors mentioned earlier, such as semi-selective CYP450 enzyme-catalyzed production of eicosanoids, differential incorporation of regioisomers into phospholipids, conversion of HETEs and EETs into secondary products, and cell-dependent receptor distribution. However, the next level determining the selectivity of HETEs and EETs is likely dictated by the modes of ligand-receptor interactions and the activation or inhibition of signaling pathways resulting from such interactions. Accumulating evidence indicates that TP interacts with multiple HETEs and EETs regioisomers to modulate vasoreactivity (13, 19, 56, 176) whereas PPARγ interacts with multiple HETEs and EETs regioisomers to modulate inflammation (129, 167).
Data in the literature point to three potential modes of ligand-receptor interactions (Fig. 4). These modes of action are not necessarily exclusive of each other. Mode A represents a simple ligand binding-induced activation of putative receptors for HETEs and EETs, respectively. Data in the literature indicate that HETEs stimulate an increase in intracellular Ca2+ whereas EETs stimulate an increase in intracellular cAMP (70, 107, 113, 192, 193, 206, 207). These simple mechanisms, while reasonable, may not provide the most efficient control to balance the opposing effects of HETEs and EETs as they must rely on the upstream events to control the local concentrations of these molecules and receptor distribution to achieve a fine balance between the opposing actions of these two classes of molecules.

Additional control, however, could be exerted by the actions of antagonists or partial agonists. For example, 15-HETE blocks 20-HETE-induced vasoconstriction in rat middle cerebral arteries (69) whereas 5-, 15-, and 19-HETE are demonstrated to block 20-HETE-induced vasoconstriction in rat renal arterioles (9).
Mode B depicts a dual-ligand interaction mechanism where HETEs and EETs compete for a single binding site on a single receptor. This scheme is proposed based on the observation that 20-HETE and 15-HETE are TP agonists (13, 56, 176) whereas 14,15-EET is an antagonist for TP (19), although it has not been shown experimentally whether 14,15-EET can actually block the agonist effect of 15- or 20-HETE on TP. Nevertheless, this type of shared binding site may pose a limitation on the binding affinity as the ligand coordination site has to be able to accommodate the binding of ligands that not only are similar, to some extent, but also differ structurally (Fig. 1), thus limiting the efficacy of the agonists or antagonists. It is not surprising then that the interactions between HETEs and EETs with TP tend to be of a low affinity (13, 19, 56, 176).
Mode C is proposed based on the general observation that HETEs and EETs tend to exert opposing effects in various tissues and conditions. It is curious to see whether they can bind to the same receptor but at separate binding sites to activate different signaling pathways. An example for this model is the orphan GPCR, GPR17, which was found to be responsive to two distinct unrelated ligands (43). GPR17 belongs to the rhodopsin family of GPCRs within the purin receptor cluster, along with purinergic receptors (P2Y) and cysteinyl leukotriene (cysLT) receptor (43, 65). The natural ligands for P2Y receptors are extracellular nucleotides (1, 27), whereas the natural ligand for cysLT receptor is cysLT (23). GPR17 was activated by both nucleotide and cysLT, leading to a decrease in cAMP and a transient increase in intracellular Ca2+ (43). The advantages of this mode of action are fast dynamic control by the relative concentrations of EETs versus HETEs, and having separate binding sites tailored for different ligands to achieve high-affinity binding for both ligands, thus enhancing the overall efficacy of the receptor.
HETEs, EETs, and ROS in Stroke
Ischemic stroke
Ischemic stroke is characterized by blood clots inside cerebral blood vessels that block blood flow to the brain. The sudden loss of blood supply deprives brain cells from essential oxygen and nutrients and triggers a cascade of cellular events that, if protracted, result in cell death. The most notable event is the generation of ROS (63, 130, 134), which mostly occurs during reperfusion, presumably due to the rapid increase in prooxidant mechanisms and the limited antioxidative capacity of cells (63, 111).
Although great progress has been made in understanding the roles of EETs and HETEs and their interplay with ROS in ischemia, the detailed mechanisms are still not fully characterized. Ischemic injury increases the activity of PLA2 (11), which hydrolyzes membrane phospholipids to liberate AA (17). An increase in AA promotes the subsequent production of HETEs and EETs along with other eicosanoids and the generation of ROS (82, 146). The activation of PLA2 after ischemia is, in part, attributed to increased glutamate release from neuronal cells (136), which has also been shown to stimulate AA release in mouse hippocampal slices and cultured mouse brain astrocytes (150, 164).
HETEs in ischemic stroke
The important role that HETEs play in ischemia is supported by their increased production after ischemia (12, 138, 167, 174). The specific regioisomer produced depends on the location, severity of injury, and method of induction of injury, among other factors. For example, in human plasma of patients who suffered from ischemic stroke and those with Parkinson's disease, the levels of 5(S)-, 12(S)-, 15(S)-, and 20-HETEs were increased, suggesting a role for both LOX and P450 hydroxylase metabolites (105). In another study, a significant positive correlation between lesion size and plasma 20-HETE was observed in patients suffering from ischemic stroke (190).
In a mouse bilateral occlusion model of transient global ischemia, 12/15-LOX upregulation correlated with injury, and 12/15-LOX pharmacological inhibition and gene deletion were protective against focal and global cerebral ischemia (182, 205). Similarly, in the mouse middle cerebral artery occlusion (MCAO) model, inhibition of 20-HETE synthesis reduced infarct volume and blocked the decrease in cerebral blood flow (CBF) during reperfusion (119).
In decapitation-induced global ischemia of the rat brain, levels of 5-HETE and 12-HETE increase along with increases in thromboxane B2 (TXB2) and ROS, suggesting activation of LOX and COX pathways (60). In the isolated perfused rat brain, levels of 5- and 15-HETE increased during brief ischemia, but levels normalized within 30 min of reperfusion, suggesting transient activation of the LOX pathway (174). In contrast, PGF2 alpha, PGE2, PGD2, 6-keto-PGF1α, and TXB2 did not change during ischemia, but increased during reperfusion, suggesting activation of the COX pathway by reoxygenation (174).
In the rat, 72 h after focal cerebral ischemia induced by MCAO, when brain edema was at its maximum, levels of 5-, 9-, 11-, and 15-HETEs increased, suggesting a role for LOX metabolites in brain edema (12, 181). In the anesthetized ischemic rat brain, the levels of 12(S)- and 15(S)-HETE increased along with increased expression of 12/15-LOX, predominantly in neurons (167). Interestingly, exogenous 12(S)- and 15(S)-HETE in this study were shown to be protective and anti-inflammatory, in part via PPARγ activation (167); 12/15-LOX total brain protein level increased significantly along with the level of 12-HETE after ischemia-reperfusion in rats (83).
This apparent contradiction in the effects of LOX metabolites may be related to the differences in mechanisms employed by endogenous and exogenous HETEs. For example, exogenous 15-HETE acts as an antagonist for 20-HETE in rat middle cerebral arteries to block 20-HETE-induced vasoconstriction (69) but it induces vasoconstriction in isolated rabbit aortic rings, and this vasoconstrictive effect was blocked by the TP antagonist, suggesting an interaction with TP (176). High concentrations of HETEs applied exogenously are more likely to induce their effect via both low- and high-affinity receptors whereas the low concentrations of HETEs produced endogenously might only interact with high-affinity receptors.
Genetic factors also contribute to the level of HETEs in ischemic stroke. For example, variations of the gene encoding h-CYP2C8, h-CYP4A11, and EPHX2 are significantly higher in patients who developed neurologic deterioration after acute stroke, which also coincided with an increased plasma 20-HETE concentration and a decreased level of EETs (204). Several mutations in the genes encoding h-CYP4F2 and h-CYP4A11, which catalyzed the production of 20-HETE, increased the risk of ischemic stroke, cerebral infarction in patients, especially in men (47, 62, 66).
In hypertensive stroke-prone rats, ischemic stroke-induced elevation of 20-HETE is associated with an increase in the expression of r-CYP4A protein, r-CYP4A1, and r-CYP4A8 mRNA (53). Gene interaction between the loci of h-CYP4A11 and h-CYP4F2 increases the level of plasma 20-HETE and the risk for ischemic stroke (110). In addition, variations of the gene ALOX5AP are associated with patients with ischemic stroke (49, 50, 88, 95, 115). This gene encodes the nuclear membrane-associated 5-LOX–activating protein required for the activation of 5-LOX and the production of 5-HETE (133).
The development of specific 20-HETE synthesis inhibitors, including N-hydroxy-N′-(4-butyl-2-methylphenyl)-formamidine (HET0016) (123) and N-(3-chloro-4-morpholin-4-yl) phenyl-N′-hydroxyimido formamide (TS-011) (122), played a major role in advancing our understanding of the role of 20-HETE in ischemia. HET0016 and TS-011 were originally shown to be more selective toward 20-HETE over 11,12-EET in isolated rat kidney microsomes (122, 123). The selectivity to other HETE regioisomers was later demonstrated in studies where HET0016 was shown to block the formation of 20-HETE but not 12-HETE in brain cortical tissue (138). TS-011 selectively blocked the formation of 20-HETE in cerebral arteries, with no effect on the production of 12-HETE, 11,12-EET, or 14,15-EET (171). Furthermore, in rats challenged with transient MCAO, pre-treatment with TS-011 significantly reduced the level of 20-HETE but not the levels of 5-, 12-, and 15-HETE (145), demonstrating its selectivity toward 20-HETE production over other regioisomers. HET0016 and TS-011 also selectively inhibited the formation of 20-HETE in rat renal microsomes and inhibited the synthesis of 20-HETE by recombinant h-CYP4A11, h-CYP4F2, or h-CYP4F3 (122).
Despite the fact that multiple HETE regioisomers have been reported to be altered by ischemia (12, 60, 137, 163, 167, 174), 20-HETE has been recognized as a major player in ischemic injury (47, 53, 132, 145, 146, 172). Recently, it was suggested to be an independent predictor of neurological deterioration in acute minor ischemic stroke (203). Earlier studies indicated that inhibition of 20-HETE production by HET0016 and TS-011 was effective in reducing ischemic brain damage, increasing CBF (53, 122, 145, 171) and improving neurological and functional outcomes (172). It was initially believed that increased 20-HETE production after ischemia may cause brain damage by reducing blood flow due to its vasoconstrictive properties (119, 138). However, a later study showed that inhibition of 20-HETE by HET0016 did not alter the diameter of pial arteries (33). Several other studies also failed to observe a difference in CBF between vehicle-treated and 20-HETE inhibitor-treated animals; 20-HETE inhibition, however, did reduce infarct size and neuronal damage, perhaps through modulating other signaling pathways (145, 200). For example, it was recently shown that in neurons the expression of m-CYP4A/4F-derived enzymes was significantly increased after oxygen-glucose deprivation (OGD) and inhibition of 20-HETE synthesis or antagonizing 20-HETE increased cell survival (212).
In addition to synthesis inhibitors, the identification of 20-HETE antagonists such as 5-HETE, 15-HETE, 19-HETE, and 6(Z),15(Z)-2-HEDE, also known as 20-hydroxyeicosa-6(Z),15(Z)-dienoic acid (WIT002) (9, 207), provided important tools for investigating the physiological function of 20-HETE. However, in the absence of a known putative receptor for 20-HETE, data using presumed 20-HETE antagonists should be interpreted with care. As discussed later, an antagonist for 14,15-EET (14,15-EEZE) that was initially identified by using similar structure-activity studies (67) was later found to have other activity besides being an antagonist for 14,15-EET (85, 113). In Figure 5, we compiled the known structures of 20-HETE along with its synthesis inhibitors and antagonists.

EETs in ischemic stroke
EETs are abundantly produced in the brain (8). Cells producing EETs in the brain include astrocytes, ECs, and neurons (8, 32, 68, 91, 92, 94, 128). EETs are rapidly hydrated to their corresponding vicinal diols (DHETs) by sEH (59, 121, 183, 211). Therefore, the reported cellular/plasma concentrations of EETs are generally low (6, 76, 98), and data on stroke-induced changes in EETs concentration are scarce. In one group of patients in Australia who suffered from acute ischemic stroke (<96 h), the plasma concentration of EETs was elevated to about 240 pM, which was more than double the value seen in control patients. The levels of 20-HETE and ROS were also increased in these patients, and plasma 20-HETE and EETs were attenuated 30 days after the stroke in a subset of patients (190).
Several studies demonstrated that EETs are protective in ischemic brain injury. In particular, the beneficial role of 14,15-EET has been elucidated by using genetic and pharmacological manipulations of CYP450 enzymes and sEH, which utilize 14,15-EET as their preferred substrate over other EETs regioisomers (121, 211). The first suggestion that EETs are protective in stroke came from preconditioning studies where protection by brief ischemia against subsequent prolonged ischemia in the rat (7) was accompanied by an increased expression of r-CYP2C11 epoxygenase (6, 8), suggesting that upregulation of EETs synthesis may serve as an endogenous protective mechanism in stroke (7).
We had previously identified astrocytes as the cellular source of expression of r-CYP2C11 in the rat brain (8). The mechanism of upregulation of CYP2C11 in astrocytes may be linked to either hypoxia-inducible factor 1α (HIF-1α) or glutamate. For example, we have shown that hypoxic preconditioning induces r-CYP2C11 mRNA expression in primary cultured astrocytes via HIF-1α (112). We have also shown that expression of r-CYP2C11 and EETs synthesis in primary cultured astrocytes are stimulated by glutamate (6), a major excitatory neurotransmitter that is significantly elevated in the ischemic brain. In cultured hippocampal astrocytes, brief hypoxia also increased the expression of r-CYP2C11 and the production of EETs (196).
In addition to astrocytic CYP450 isoforms, the human endothelial h-CYP2J2 and h-CYP2C8 isoforms also protect against stroke-related brain injury (96, 108). Interestingly, the protective effects of m-CYP2J2 and m-CYP2C8 are gender dependent, as demonstrated in a genetically engineered mouse model (Tie2-CYP2J2) where human h-CYP2J2 is specifically expressed in ECs (96, 108). Male Tie2-CYP2J2 mice sustain a greater reduction in infarct size, increased CBF, and reduced neuronal death and ROS production (96, 108). Overexpression of endothelial h-CYP2C8 isoform reduced infarct size, but by a different mechanism than that used by h-CYP2J2 where h-CYP2J2 exerts its protective effect by enhancing blood flow whereas h-CYP2C8 exerts its protective effect by suppressing inflammation (96).
Overall, elevating the level of 14,15-EET by inhibiting sEH, genetically deleting the gene encoding sEH, or exogenously administering 14,15-EET all protect against cerebral ischemic injury. In contrast to EETs synthetic enzymes, which are expressed in cells responding to changes in the surrounding environment (astrocytes and ECs), sEH seems to be expressed in target cells, such as neurons. For example, in the mouse brain, sEH is predominantly expressed in neuronal cell bodies and processes, and, to a lesser extent, in cerebral blood vessels (214). Pharmacological inhibition of sEH reduced infarct size after MCAO, and the protective effect was lost when EETs synthesis was also inhibited (214), suggesting that protection by sEH inactivation is specifically linked to EETs.
Similarly, deletion of the gene encoding sEH protected against stroke-induced brain damage. In the sEH knockout (sEHKO) mice, infarct size and neurological deficit were significantly smaller after MCAO compared with WT control mice (215), which was also associated with higher CBF in sEHKO mice. The total plasma concentration of sEH metabolite 14,15-DHET was significantly lower in sEHKO mice relative to controls, indicating a reduction in the conversion of 14,15-EET by sEH (215). In a co-culture of astrocytes and neurons, inhibition of sEH increased neuronal survival after OGD (216). Mutations in the sEH gene, designated ephx2, also affect its activity and alter the rate of neuronal survival, as seen in rat primary cortical neuronal culture transduced with human EPHX2 mutant proteins. Transduction of neuronal cells with these mutant proteins resulted in a variable rate of cell death induced by OGD, which was, in part, related to how the enzyme's hydrolase activity is affected, whereas pharmacological inhibition of sEH prevented the increase in cell death induced by transduction with WT sEH, indicating a protective role for EETs (100).
The mechanisms of protection by EETs in ischemic stroke are linked to changes in CBF, vasodilation, neuroprotection, promotion of angiogenesis, suppression of platelet aggregation, oxidative stress, and post-ischemic inflammation (90). Less clear are the molecular entities or pathways targeted by 14,15-EET to protect against ischemic stroke. As mentioned earlier, a specific receptor has been proposed to mediate the actions of 14,15-EET, but whether this putative receptor is involved here is less clear. This is especially true since the genetic and pharmacological methods mentioned earlier are all likely to increase the level of EETs above its normal physiological level. In cultured astrocytes, the concentration of EETs secreted into culture media was reported in the micromolar range (216).
Exogenous EETs treatment at concentrations between 250 nM and 10 μM before ischemic injury was found to be protective against ischemic brain injury (108, 215). In this range, not only the putative high-affinity receptor but also low-affinity receptors could potentially participate, which maybe important therapeutically (100, 112, 141, 175, 178). One such receptor is PTGER2 which is activated by 14,15-EET (113, 197), and activation of PTGER2 in cultured rat hippocampal slices by PGE2 and its selective agonist butaprost was neuroprotective against toxicity induced by glutamate receptor agonist NMDA. PTGER2 agonists were also protective against neuronal cell death induced by OGD (120). In addition, genetic deletion of the PTGER2 increased infarct volume after MCAO/reperfusion-induced transient cerebral ischemia in mice (120).
As with 20-HETE, for example, a presumed antagonist for 14,15-EET (14,15-EEZE) identified based on similar structure-activity studies (67) was found to exhibit agonist and partial agonist activities in the bovine coronary artery (26, 67) and mouse mesenteric arteries (85), which may be related to activation of PGE2 receptors. In support of the latter idea, we have recently shown that 14,15-EEZE behaved as an agonist or a partial agonist for PTGER2 using an exogenous expression system. In Xenopus oocytes coexpressing PTGER2 and CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) chloride channel, which served as a cAMP sensor because channel activation is dependent on the elevation of intracellular cAMP, exposure to 1 μM 14,15-EET or 14,15-EEZE resulted in a robust increase in chloride current, demonstrating the activation of PTGER2 by both compounds, especially given that these compounds had no effect on the conductance in the absence of PTGER2 expressed (113).
Interplay between EETs, HETEs, and ROS in ischemic stroke
Because of their reactive chemical nature, ROS generation results in damage to multiple cell components, including DNA alteration, lipid peroxidation (14, 86, 191), and protein modifications (64). DNA damage introduces mutations and alters normal protein synthesis. Modification of proteins by ROS generates diverse chemical species at the protein side chains, leading to a variety of functional modifications (64). Most notably, an ROS sensor has been identified as an ROS-activated, nonselective calcium permeable cation channel (transient receptor potential M2 [TRPM2]) (84). TRPM2 (also known as LTRPC2) is broadly expressed in neuronal cells, myocytes, β cells, and immune cells (195). It is activated by an H2O2-induced increase in NAD+ that binds directly to TRPM2 at the intracellular domain, which opens the channel and allows extracellular Ca2+ to rush into the cell in a voltage-independent manner, and Ca2+ overload resulting from TRPM2 activation leads to cell death (84). Not surprisingly, TRPM2 is found to be detrimental in transient cerebral ischemia (5, 202).
20-HETE production contributes to ROS generation. In brain slices obtained from the rat neocortex or hippocampus, increases in 20-HETE correlated with increases in ROS production (82, 146). In isolated rat brain slices, 1 μM 20-HETE induced a rapid increase in superoxide that reached a plateau within 3 min (82), and ROS production was reduced by pretreatment with 20-HETE synthesis inhibitor (82, 146). On the other hand, exogenously administered 14,15-EET opposes ROS production in primary cultures of cortical neurons. Pre-exposure to 20 nM 14,15-EET prevented the generation of ROS after OGD (189). This is, in part, linked to 14,15-EET-induced phosphorylation of Akt (p-Akt) (141), which participates in multiple cellular functions, including cell survival (2, 151).
The role of Akt in mediating the protective effect of EETs in ischemia was further confirmed by a recent study using the co-culture of primary astrocytes and neurons (216). That study made several interesting observations. First, it was observed that inhibition of sEH increased secretion of vascular endothelial growth factor (VEGF) by astrocytes into the culture media after OGD, and this increase was blocked by 14,15-EEZE. Second, exogenous 14,15-EET also increased VEGF release after OGD. Third, the addition of astrocyte-conditioned culture medium collected after OGD to neurons increased their survival. Fourth, the stimulation of VEGF receptor-2 in neurons by astrocyte- and OGD-conditioned medium increased p-Akt, suggesting that 14,15-EET secreted from astrocytes after OGD is involved in neuronal protection and it exerts its action via p-Akt (216).
One potential common mechanism linking HETEs and EETs with ROS is the TP, which is regulated by the two classes of P450 eicosanoids in opposite directions (13, 19, 56, 176). Activation of TP stimulates ROS production in isolated rabbit renal afferent arterioles and the mouse chronic kidney disease model (156, 188). Furthermore, a thromboxane A2 receptor antagonist, ONO-3708, has been shown to increase the survival rate of dogs after complete global brain ischemia, suggesting a detrimental role of TP activation in ischemia (170). Together, these observations suggest a potential role for TP in mediating the protective effect of 14,15-EET and the detrimental effect of 20-HETE in ischemia. These results do not necessarily exclude the potential involvement of other putative receptors for EETs and HETEs. Although 12-HETE has been shown to be an antagonist for TP (162), the inhibitory effect of 12-HETE on ROS production in ischemia has not been reported.
Hemorrhagic stroke
Hemorrhagic stroke is characterized by damage to brain cells due to blood vessel rupture and subsequent bleeding. The most common cause is aneurysm rupture, which leads to subarachnoid hemorrhage (SAH), and its most serious complication is cerebral vasospasm and the associated delayed cerebral ischemia (DCI) (137, 163). As with ischemic stroke, although both HETEs and EETs are implicated in hemorrhagic stroke, the specific regioisomer involved depends on the specific disease condition. In human SAH patients, 5-HETE was increased in cerebrospinal fluid (CSF), which coincided with the occurrence of cerebral vasospasm, suggesting a role for lipid peroxidation in the pathogenesis of chronic vasospasm after SAH (155, 168). In another study with two patients who suffered from SAH and developed cerebral vasospasm, levels of 12- and 20-HETE in CSF were significantly higher than those seen in matched controls who suffered SAH but did not develop vasospasm (137). Similarly, in another study, the level of 20-HETE increased immediately after SAH and by day 10 decreased to a level comparable to that seen in patients who did not develop DCI (163). After aneurysmal SAH, an increased CSF 20-HETE level was associated with unfavorable outcomes, including DCI and clinical neurologic deterioration (52).
20-HETE is also a key player in experimental SAH models. In a rat SAH model, the concentration of CSF 20-HETE increased more than 10-fold after SAH induced by injecting arterial blood into the cisterna magna, and inhibition of 20-HETE formation reduced the initial fall in CBF (99). Interestingly, along with an increase in the level of 20-HETE, SAH also increased the level of 5-hydroxytryptamine-1B (5-HT) in CSF (30). Furthermore, an intracisternal injection of 5-HT increased the level of CSF 20-HETE level and reduced CBF, whereas inhibition of 20-HETE synthesis prevented 5-HT-induced fall in CBF, suggesting an interaction between 20-HETE and 5-HT receptor after SAH (30).
In addition, HET0016 blocked the production of 20-HETE by r-CYP4A11, r-CYP4F2, and r-CYP4F3 in a concentration-dependent manner and administration of HET0016 after SAH induction returned CBF to the control level within 30 min whereas CBF of vehicle-treated rats remained at 30% below the control over the 2-h course of the experiment (99). The level of 20-HETE also increased in delayed vasospasm after SAH in a dog model where dual injections of blood into the cisterna magna on days 1 and 4 consistently induced delayed vasospasm (80). A decrease in the basilar artery diameter 3 days after the second injection of blood is accompanied by an increase in 20-HETE (80). Furthermore, acute inhibition of 20-HETE formation by TS-011 increased the diameter of the basilar artery, and chronic administration of TS-011 attenuated the development of delayed vasospasm, suggesting an important role of 20-HETE in SAH and subsequent injuries (80).
Among the regioisomers of EETs, only 14,15-EET has been implicated in SAH. In SAH patients, the level of 14,15-EET in CSF increased at 10 days after SAH, regardless of whether they develop DCI (163). Genetic variability of h-CYP2C8, h-CYP2J2, and h-CYP2C9 among patients with aneurysmal SAH was associated with different clinical outcomes depending on the level of EETs in CSF (51). In addition, genetic polymorphisms of sEH have also been linked to neurological outcome and survival after aneurysmal SAH (118).
ROS plays a critical role in SAH (199). ROS concentration increased in the primary cortical neuron SAH model after exposure to fresh blood obtained from veins of mice, which coincided with neuronal cell death (106). In a rat intracerebral hemorrhage model, a 10-min infusion with heparinized blood into the right caudate nucleus in cerebrum induced a significant increase in ROS production (77).
HETEs, EETs, and ROS in TBI
TBI refers to brain injury induced by external mechanical forces, which is characterized by primary mechanical damage as well as by secondary injury resulting from the initial injury (61, 144). The secondary injury is believed to largely result from glutamate-induced excitotoxicity, ischemia, and oxidative damage (187).
It has been demonstrated that the levels of 5- and 12-HETE dramatically increased in patients after TBI (61). Similarly, in a rat TBI model, EETs and HETEs were released within 24 h after TBI. In this study, the level of EETs was nearly fourfold higher than that of HETEs (74). However, in Ephx2-KO mice, only 8,9-EET level was increased due to TBI and these mice showed improved motor coordination (166). Furthermore, TBI induces an increase in ROS in both human and animal models (18, 39, 81, 101, 117, 173, 187) and the severity of injury correlated with the degree of oxidative stress (173). In animal TBI models, the generation of ROS leads to oxidation of the delayed rectifier potassium channel (KCNB1) and TRPM2, causing neuronal damage and apoptosis in the hippocampus, in addition to the induction of lipid peroxidation (208, 209).
Although ischemia is one of the major secondary injuries resulting from TBI (187), the roles of 20-HETE and 14,15-EET in TBI-induced secondary injury are not well described and the interplay between HETEs, EETs, and ROS is not well characterized in TBI.
HETES, EETS, and ROS in Neurodegenerative Diseases
Alzheimer's disease
AD is a chronic age-related neurodegenerative disorder that is associated with progressive cognitive impairment and memory loss (4). It is characterized by an overproduction and excessive accumulation of amyloid beta (Aβ), a 40–42 amino-acid-long peptide, and formation of neurofibrillary tangles resulting from hyper-phosphorylation of tau, a microtubule stabilizing protein (4, 22).
Although recent work suggest that tau is a better marker for tracking dementia status and a better predictor for cognitive performance than Aβ (22), extensive literature suggests that Aβ not only induces accumulation of ROS but also causes extensive cellular damage and modification of multiple targets, including lipid peroxidation, protein oxidation, and DNA and reactive nitrogen species (RNS) oxidation (28). Unfortunately, it was recently reported that an IgG1 monoclonal anti-Aβ antibody, known as solanezumab, did not provide a significant improvement in cognitive decline in patients (147). Failure of this drug only heightens the need to further understand the molecular and cellular mechanisms related to AD and to develop alternative therapies to treat AD.
ROS is an important player in AD. As a major intracellular organelle for oxygen-dependent ATP production, mitochondria stand as a major source for ROS. Not surprisingly, mitochondrial dysfunction in both neurons and astrocytes (3) plays an important role in AD (87). In co-culture of hippocampal neurons and astrocytes, mitochondrial membrane potential in astrocytes was altered by Aβ but not in neurons (3). Aβ-induced decrease in mitochondrial membrane potential causes an increase in the generation of ROS in astrocytes due to the activation of NADPH oxidase (3), which catalyzes one-electron reduction of oxygen to generate superoxide (O2 −) (15).
Considering that astrocytes also produce EETs, which are neuroprotective (8), the potential effect of EETs in AD was recently explored. In microsomes isolated from the rat cerebral cortex, Aβ reduced the concentrations of 14,15-EET and 11,12-EET in a region- and cell-specific manner (153, 154). Incubation with soluble Aβ led to a decrease in 14,15-EET production in the cerebrum, but not in the cerebellum, and Aβ had no effect on the production of 11,12-EET in these regions (153). A more detailed analysis showed that Aβ treatment decreased 14,15-EET production in astrocytes and neurons, but the production of 11,12-EET was affected by Aβ only in astrocytes (153). In the cortex and hippocampus, on the other hand, the production of 11,12-EET, but not 14,15-EET, was reduced by Aβ treatment (153).
Furthermore, blocking endogenous EETs production by an epoxygenase inhibitor aggravated the effect of Aβ on astrocyte mitochondrial dysfunction whereas pretreatment of primary hippocampal astrocyte culture with exogenous 11,12-EET or 14,15-EET for 24 h prevented Aβ-induced mitochondrial dysfunction (154). Although the molecular mechanisms underlining the protective effects of 11,12-EET and 14,15-EET are unknown, these results suggest that 11,12-EET, 14,15-EET, and other EETs may have therapeutic potential against Aβ−induced mitochondria dysfunction.
Other regioisomers have also been implicated in AD, with no consistent patterns of change in different studies. In AD patients, the levels of 12-HETE and 15-HETE were elevated (139, 201). In an AD mouse model, although the level of 12-HETE was elevated, the level of 15-HETE was found to be lower in 10 month-old AD mice than in the controls (169). In primary cultures of rat cortical neurons, 12-HETE was believed to be involved in inducing cell death because exposure to exogenous 12-HETE but not 5-HETE increased apoptosis (104). In contrast, in cultured human neuro-2A neuroblastoma cells expressing Aβ precursor, 5-HETE increases Aβ by activating cAMP-response element binding protein (CREB) (42). Factors such as species, cell types, and experimental procedures could potentially explain the differences observed among different studies. Stimulation of CREB by 5-HETE in the Aβ−expressing cell line is of particular interest because it suggests that Gs-coupled signaling might mediate the effect of 5-HETE.
Overall, the molecular and cellular mechanisms underlining the effects of EETs and HETEs in AD are not well understood. It is of interest to note that TP receptor is expressed in the brain (21) and activation of TP by isoprostanes, a marker for in vivo oxidative stress, increased the levels of amyloid precursor protein (APP) and Aβ production in a mouse AD model (161). Because both EETs and HETEs regioisomers can interact with TP, their effects to modulate Aβ production could potentially be mediated via their actions on TP.
Vascular cognitive impairment
VCI is the second most common cause of dementia after AD. It is defined as cognitive impairment due to vascular injury, with or without clinical stroke (75). VCI is a microvascular disease that is caused, in part, by microscopic bleeding or poor microvascular blood flow in the brain (177). The most severe form of VCI is vascular dementia (VD) (75). The mechanisms underlying VD are poorly understood, with no specific treatments currently available to prevent or treat VD and associated VCI. Because of the interdependence of cells within the neurovascular unit, damage to vascular cells will inevitably affect the health and function of neuronal cells (89).
Both HETEs and EETs have been implicated in VCI, supported by their dominant role in regulation of the microvasculation. Human autopsy brains with VCI had an increased ratio of DHETs to EETs, suggesting upregulation of sEH (126). The upregulation was localized by immunohistochemistry to the microvascular endothelium. In a separate study in mice, we found that high-fat diet-induced type 2 diabetes leads to impaired spatial memory (assessed by the Morris water maze) and upregulates cerebrovascular sEH expression in WT mice, suggesting that diabetes, a known risk factor for VCI, may, in part, be responsible for endothelial upregulation of sEH observed in the postmortem human VCI brain (220). sEH upregulation could potentially contribute to neuronal injury by causing a state of chronic hypoperfusion. In the mouse, chronic hypoferfusion can be induced by unilateral common carotid artery occlusion, which, we have shown, impairs cognition (221). In addition to decreases in EETs, 5-HETE, 12-HETE, 18-HETE, and 20-HETE were increased in VCI patients (126). VCI is also associated with increased free radicals (16). Several natural compounds such as Gastrodin, Betaine, and Puerarin have been shown to neutralize ROS and reduce the cognitive dysfunction in experimental VCI (109, 127, 213).
In Figure 6, we summarize potential interactions between ROS, HETEs, and EETs, along with enzymes that determine the production and conversion of HETEs and EETs in brain diseases and injuries discussed in this review. We also summarized the patterns of changes in the levels of HETEs and EETs in brain diseases/injury and protection in Table 2.
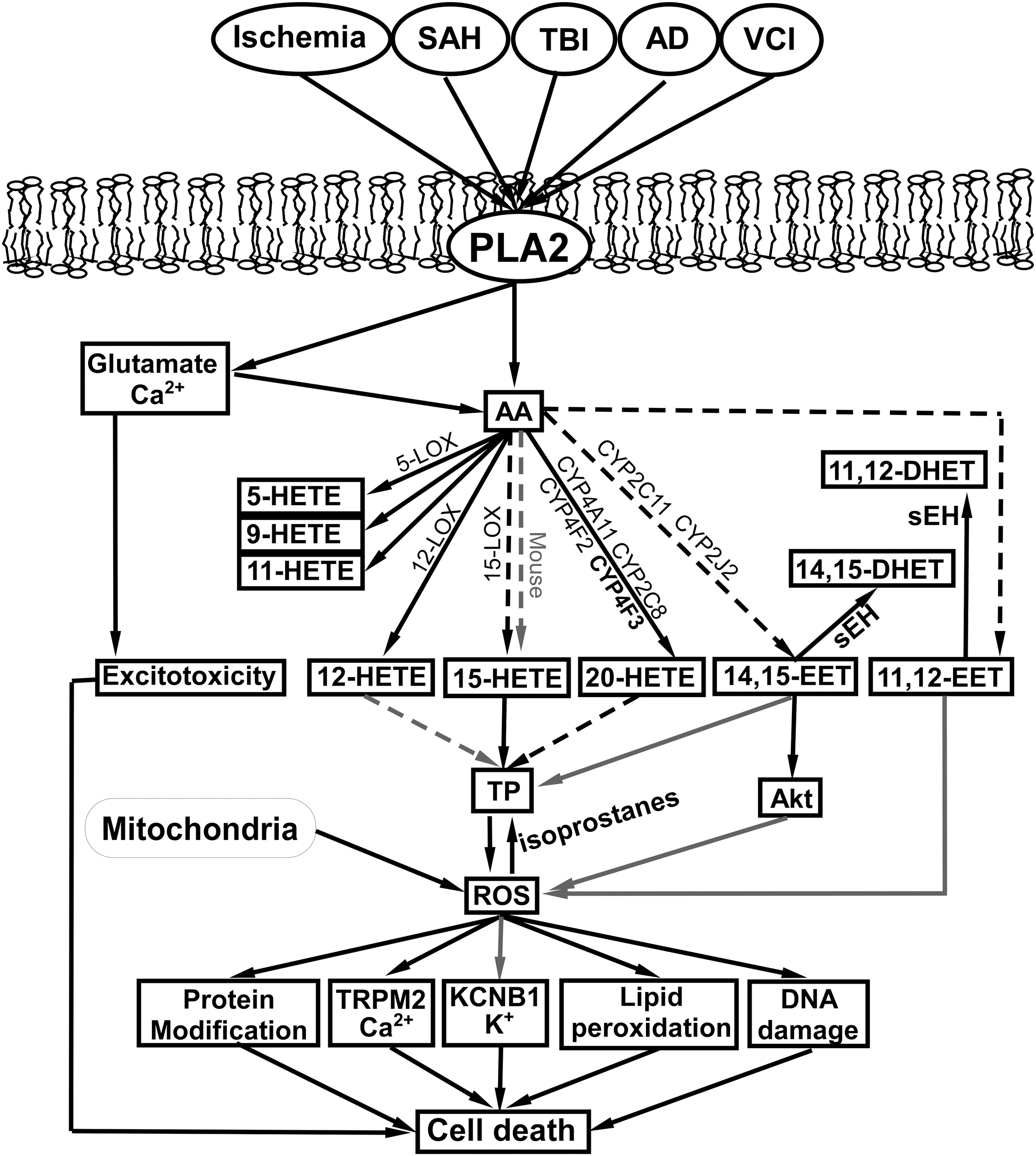
When reported results did not specify regioisomers, an increase in HETEs or EETs is assumed to include all regioisomers listed in the table. Superscripts next to the references identify the animal species used in the cited studies, where c = cat, d = dog, m = mouse, and r = rat.
AD, Alzheimer's disease; IS, ischemic stroke; LOX, lipoxygenases; OGD, oxygen-glucose deprivation; SAH, subarachnoid hemorrhage; TBI, traumatic brain injury; TIS, transient ischemic stroke; VCI, vascular cognitive impairment.
Conclusions and Future Directions
This review is aimed at highlighting the dual role of P450 enzymes and their metabolites in brain diseases and at providing an update on the molecular and cellular mechanisms underlying the effects of EETs and HETEs in various brain pathological conditions. In general, the two classes of P450 eicosanoids, EETs and HETEs, exert opposite effects on neuronal survival, and two potential convergence points for their antagonistic actions are ROS generation and TP activation (13, 19, 56, 176). Even though the interactions between EETs and HETEs with TP are of a low affinity (13, 19, 56), eicosanoid levels under pathological conditions and pharmacological interventions reach sufficiently high tissue concentrations to engage TP. The identification and role of any potential high-affinity receptors for EETs and HETEs remain elusive and limit our understanding of the roles of EETs and HETEs and their interplay with ROS in various disease states.
Footnotes
Acknowledgments
This work was supported by KCVI and NIH grants NS44313, NS070837, and AG043857 (Alkayed), and AHA GRNT31380022 (Alkayed).