
Abstract
Aims:
Mitochondrial ferritin (protein [FtMt]) is preferentially expressed in cell types of high metabolic activity and oxygen consumption, which is consistent with its role of sequestering iron and preventing oxygen-derived redox damage. As of yet, the mechanisms of FtMt regulation and the protection FtMt affords remain largely unknown.
Results:
Here, we report that hypoxia-inducible factor 1α (HIF-1α) can upregulate FtMt expression. We verify one functional hypoxia-response element (HRE) in the positive regulatory region and two HREs possessing HIF-1α binding activity in the minimal promoter region of the human FTMT gene. We also demonstrate that FtMt can alleviate hypoxia-induced brain cell death by sequestering uncommitted iron, whose levels increase with hypoxia in these cells.
Innovation:
In the absence of FtMt, this catalytic metal excess catalyzes the production of cytotoxic reactive oxygen species.
Conclusion:
Thus, the cell ability to increase expression of FtMt during hypoxia may be a skill to avoid tissue damage derived from oxygen limitation.
Introduction
M
Hypoxia is a paradigm of responses involving the whole organism. The function and regulation of mitochondrial ferritin (FtMt [protein]) have remained elusive. Our study describes the first functional regulatory axis with FtMt as the effector activating the hypoxia transcriptional program in oxygen deficiency. FtMt is a novel hypoxia-inducible factor 1α (HIF-1α) target gene. This finding that the protein eliminates the high oxidative stress resulting from hypoxia highlights the causes underlying FtMt's peculiar expression pattern. Furthermore, our results point to FtMt as a potential therapeutic target in situations of hypoxic challenge such as ischemic stroke.
Numerous reports have documented FtMt's involvement in several pathological processes. FtMt accumulates in high amounts in erythroblasts of subjects with impaired heme synthesis (26) and in ring sideroblasts from sideroblastic anemia patients (9). Our recent study reveals that FtMt can protect the murine myocardium from acute exhaustive exercise injury (59). Moreover, FtMt had a profound protective role in neurodegenerative diseases (61). Increased FtMt levels were detected in the human substantia nigra (SN) from restless legs syndrome (RLS) patients (49), in cardiomyocytes of Friedreich ataxia (FRDA) patients (24), and in the cerebral cortex of Alzheimer's disease (AD) patients (56). FtMt can also abrogate oxidative damage and rescue respiratory function in FRDA (6, 66) or attenuate the neurotoxicity induced by 6-hydroxydopamine (6-OHDA) (45), MPTP (63), and β-amyloid (Aβ) (60) both in Parkinson's disease and AD.
In addition, FTMT mutations were identified in patients suffering movement disorders (8), and in one case, age-related macular degeneration (50), suggesting that FtMt deficiency may decrease protection from iron-dependent oxidative stress in mitochondria. Increasing FtMt has been proposed as a therapeutic strategy to inhibit tumor growth by decreasing cytosolic iron levels (34, 46). All of these findings are indicative of vital roles for FtMt in antioxidant response and iron metabolism. The presence of free redox-active iron (Fe2+) in a cell can contribute to the formation of free radicals that increase cellular oxidative stress via the Fenton reaction, which entails Fe2+-catalyzed production of highly toxic hydroxyl radicals (52).
FtMt can sequester “uncommitted” iron in its spherical shells in a form that is not accessible for Fenton chemistry, thus it can protect mitochondria from oxidative damage. Therefore, it is not surprising that FtMt is preferentially expressed in cells with vigorous metabolic activity and oxygen consumption, which is associated with comparatively elevated production of reactive oxygen species (ROS). This is congruent with FtMt's role of mitigating redox damage from the combination of catalytic iron and oxygen (41, 48). However, overexpression of FtMt caused cytosolic iron deprivation (11) and sensitized cells to oxidative stress (31, 35, 42). Increased FtMt also may lead to pathogenic cytosolic iron deprivation in RLS (49).
In contrast to the relatively abundant functional studies, little attention has been devoted to FtMt regulation. The absence of functional iron responsive element sequences in FtMt messenger RNA (mRNA) is consistent with its iron-independent expression. Recently, published data shed light on the human FTMT promoter study in which the analyzed putative promoter region comprises the minimal promoter as well as a positive and a negative transcriptional factors target regions and provides new evidence as to the transcriptional and epigenetic regulatory mechanisms of this gene (17). However, the detailed mechanisms governing FTMT regulation are yet to be clarified. The preferential expression of the gene in cells with high oxygen consumption suggests an intimate relationship between FtMt and oxygen availability.
Appropriate responses to low ambient oxygen are largely mediated through transcriptional activation of genes by hypoxia-inducible factor (HIF) proteins. HIFs are heterodimers containing a HIF-α and a HIF-β subunit. The HIF-β subunit is a constitutively expressed partner for multiple basic-helix/loop/helix per-Arnt-sim (bHLH-PAS) transcription factors (TFs). The HIF-α subunit, however, is regulated in response to changes in cellular oxygen availability (54). HIF-1α, one subunit of the HIF-1 heterodimer, has been the most extensively studied. In hypoxic environments, or hypoxia-mimicking conditions in normoxia, HIF-1α is stabilized and translocated to the nucleus, binding, with HIF-1β, to hypoxia-response elements (HREs) of genes, thus activating the hypoxia transcriptional program (13, 18, 21, 47, 55).
In this study, we found that HIF-1α can activate the expression of FtMt in vitro and in vivo. Furthermore, depletion of HIF-1α by RNA interference (RNAi) in cells and conditional knockout (KO) of HIF-1α in mice abolished the upregulation of FtMt in response to hypoxic treatment. We, therefore, hypothesized that FtMt is a gene regulated by hypoxia. To explore the regulatory mechanisms, we examined the human FTMT promoter region and tested six potential HREs present in the 5′ untranslated region (UTR) upstream of ATG codon in silico. In addition, we show a stimulation of oxidative stress and cell death after hypoxia treatment. Altogether, here we show that HIF-1α is involved in the regulation of FtMt in hypoxia and FtMt plays an important protective role in hypoxia by reducing redox damage.
Results
FtMt levels are elevated under hypoxia in vivo and in vitro
To explore the roles that FtMt may play during hypoxia, six adult mice were kept under continuous hypobaric hypoxia (HH) for 18 h in a hypobaric chamber with a simulated altitude of 8000 m (oxygen content is around 8.5%, pO2 is about 7.5 kPa). We studied the cortex and hippocampus regions in the brains of mice due to the particular vulnerability of these regions to hypoxic stress (19, 67).
The level of mitochondrial ferritin (mouse and rat gene) [Ftmt] mRNA and FtMt protein in the cortices and hippocampi (Hippo) of mice treated with or without hypoxia was then measured by quantitative real-time polymerase chain reaction (qRT-PCR) and immunoblotting, respectively (Fig. 1A, B). Because of the high identity between FtMt and Ft-H, we validated the specificity of the FtMt antibody in immunoblots from rat testis lysate (Supplementary Fig. S1; Supplementary Data are available online at

In vitro, we observed the same phenotype in the C6 glioma cell line when maintained in 1% O2 for up to 12 h or with different concentrations of CoCl2, as CoCl2 is often used as a hypoxia-mimetic agent to simulate hypoxia under normal oxygen conditions (1); the level of Ftmt mRNA, as determined by reverse transcription-PCR, was increased in the treated cells compared with that in the controls (Fig. 1C, D), whereas the amount of FtMt protein was consistently changed along with the mRNA levels (Fig. 1E, F).
Increased FtMt expression under hypoxia is accompanied by changes in HIF-1α
HIF-1α has been largely studied for its role in cell survival under conditions of oxygen deprivation (hypoxia), wherein it helps to restore oxygen homeostasis by inducing glycolysis, erythropoiesis, and angiogenesis (20, 43, 58). HIF-1α can accumulate under hypoxia or treatment with a hypoxia-mimetic agent such as CoCl2. As expected, we found HIF-1α protein was elevated in C6 cells after treatment with hypoxia (1% O2) or with CoCl2 (Fig. 2A, B).
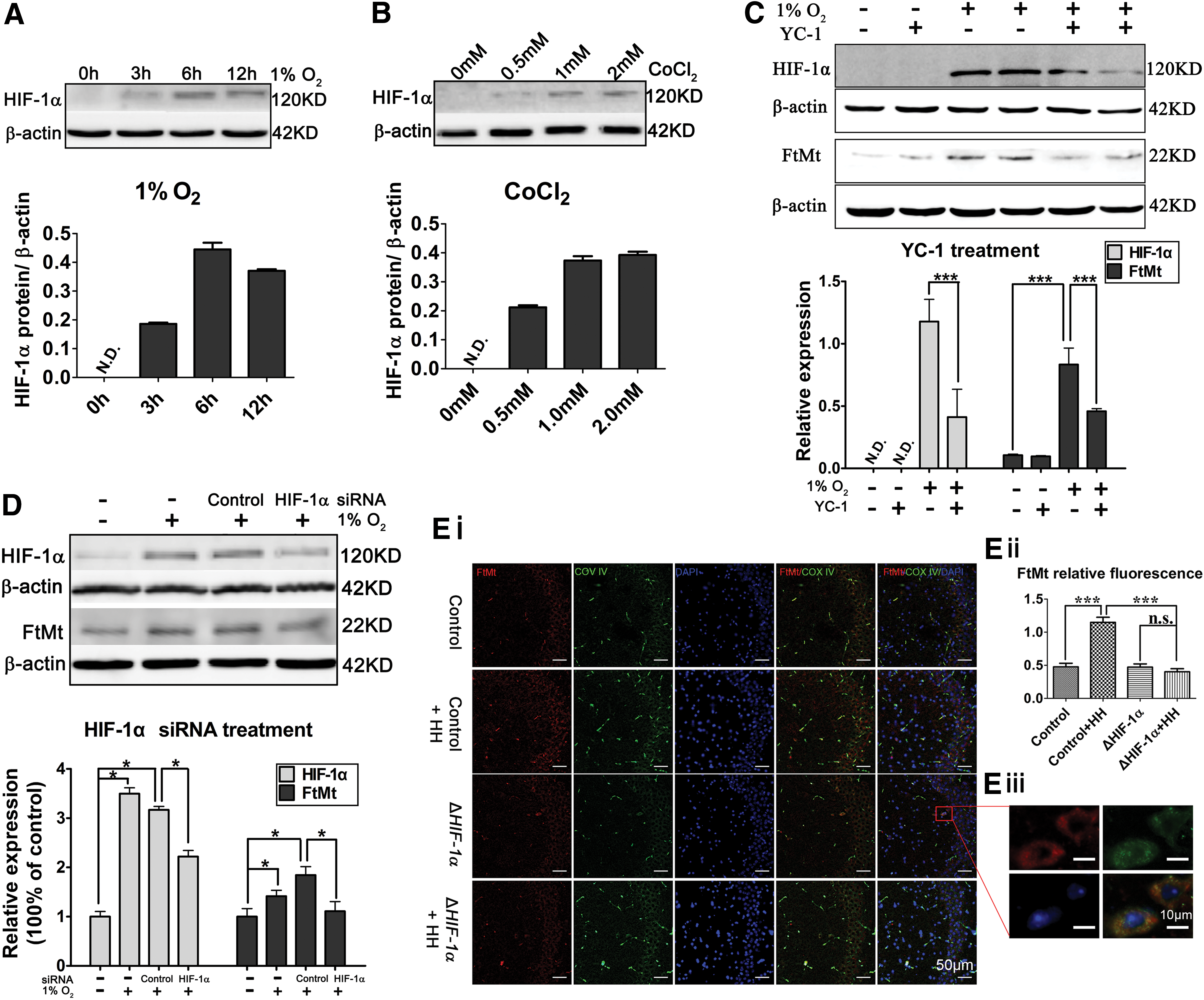
Previous reports have demonstrated that YC-1 [3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole] inhibits HIF-1α (3, 28). When C6 cells were treated with the YC-1 (30 μM) plus 1% O2 for 12 h, both FtMt and HIF-1α protein levels were decreased (by nearly 45% and 65%, respectively) compared with those in the untreated cells (Fig. 2C). Furthermore, when we depleted the HIF-1α protein level by RNAi, we also found an ∼40% reduction in FtMt protein amounts (Fig. 2D), which were consistent with the effects of HIF-1α inhibition with YC-1.
Conditional KO of HIF-1α in mouse neurons blocks hypoxia-induced elevation of FtMt in the hippocampus
HIF-1α was specifically inactivated in neurons by crossing C57BL/6 HIF-1αflox/flox mice (control) with Thy1-Cre C57BL/6 transgenic mice (ΔHIF-1α). The mice were then placed in a hypoxic chamber with 18 h continuous HH. We first checked the localization of FtMt in the hippocampi in the control groups (control, control+HH), using double immunostaining with antibodies specific for cellular markers in brain (green): NeuN for neurons, glial fibrillary acidic protein (GFAP) for astrocytes, and Iba1 for microglia, along with FtMt (red). These experiments revealed that FtMt is expressed mainly in the hippocampal neurons, suggesting a cell-specific regulation of FtMt (Supplementary Fig. S2).
In addition, immunostaining showed a higher FtMt expression (red) in the control+HH group (∼2.4-fold vs. the control group, Fig. 2Ei) than in the controls under normoxia. Importantly, we found no significant differences in FtMt expression in the ΔHIF-1α mice in the differing oxygen levels (Fig. 2Ei, Eii). Moreover, double immunostaining with the mitochondrial marker (COX IV, green) and FtMt showed that our FtMt antibody specifically recognizes a protein in mitochondria (Fig. 2Eiii).
HIF-1α activates FtMt expression via HRE sites in the FTMT promoter region
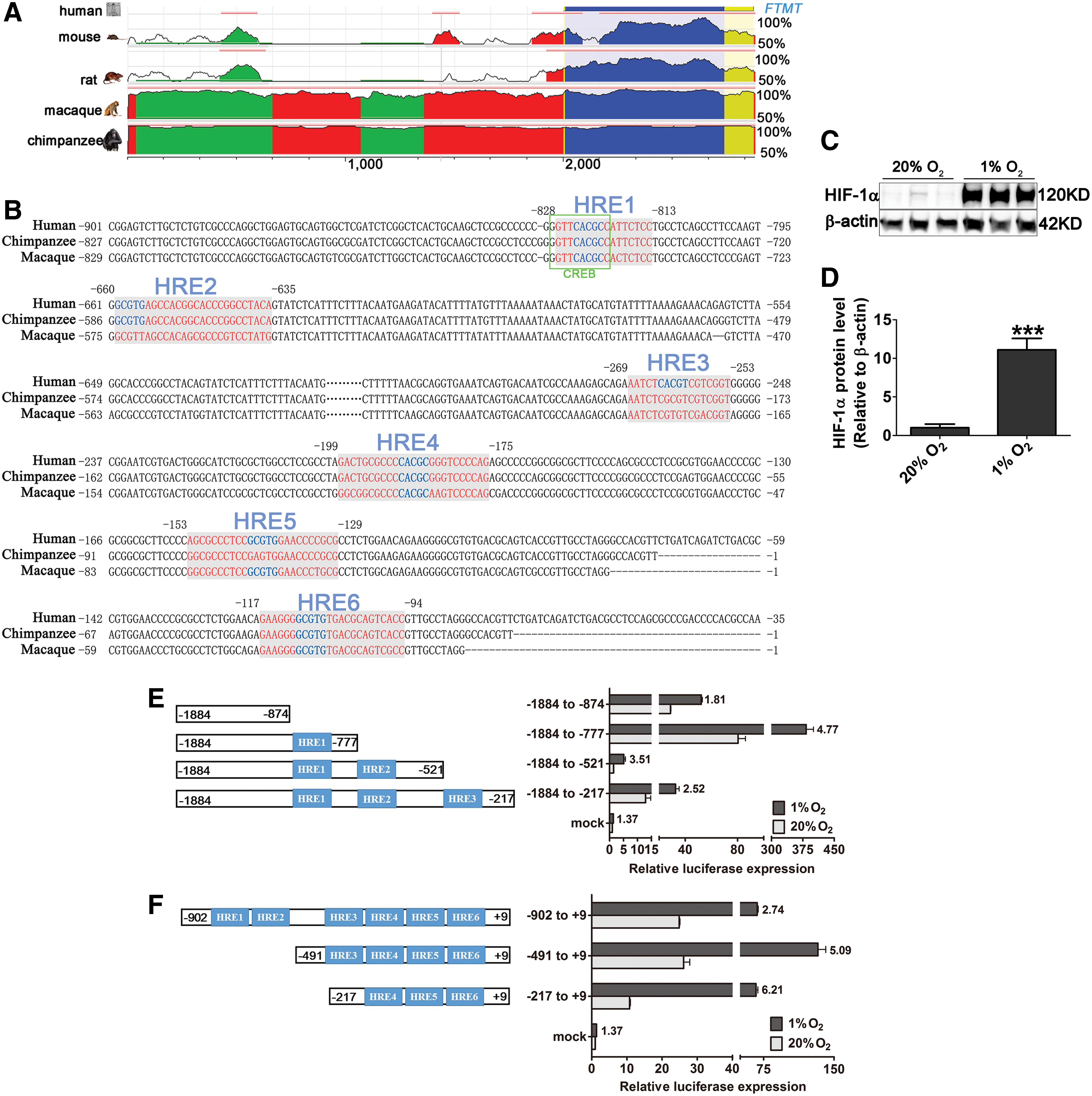
To investigate whether HREs may be present in the FTMT promoter region, we analyzed the evolutionarily conserved region upstream of the transcriptional starting site (TSS) in silico. In this region, the sequence similarity between human and rat or mouse was ∼70%, and >90% among the primates (Fig. 3A). We found six potential HREs upstream of the TSS in human FTMT (Fig. 3B). The matrix similarity from different databases is provided in Table 1.
Matrix Similarity of Predicted Hypoxia-Response Elements
HREs, hypoxia-response elements.
From the alignment, we can see the same location of HREs among the primates. Hence, we took the human FTMT promoter as a template and used a series of 3′ and 5′ deletions within the promoter to test the activities of each possible HRE using dual-luciferase reporter (DLR) assays in HeLa cells, whose responsiveness to the hypoxia treatment was followed by HIF-1α protein detection in immunoblot (Fig. 3C, D). We chose this cellular system because it was easier to transfect with varied reporter plasmids than SH-SY5Y or C6 cells.
In the 3′ deletion assay, the −1884/–777 group, which contained only the predicted HRE1 sequence, exhibited a nearly fivefold increase in luciferase activity after a 1% O2 treatment, with respect to the control group (20% O2). The −1884/−521 group, comprising predicted HRE1 and HRE2 sites, exhibited a 3.5-fold increase in hypoxia (Fig. 3E). In the 5′ deletion luciferase assays, the −491/+9 and −217/+9 groups, which include the predicted HRE4-HRE6 sequence, displayed the highest activities after hypoxia treatment (∼5-6-fold) (Fig. 3F). All the data suggest that the binding activities of HIF-1α to the HREs are located in the putative FTMT promoter region from −1884 to −521 and from −491 to +9.
To further investigate the HIF-1α binding abilities among the predicted HREs, we next performed electrophoretic mobility shift assays (EMSAs) to investigate the three HREs that are located in the minimal promoter region, which is from −491 to −1 of the FTMT promoter region (17) (HRE4, HRE5, and HRE6) and chromatin immunoprecipitation (ChIP) assays to study functional roles of HRE1, HRE2, and HRE3.
Consistent with our findings in C6 cells (Figs. 1E and 2A), we detected elevated levels of FtMt and HIF-1α proteins also in the human glioma cell line, U251, when exposed to 1% O2 for 12 h (Fig. 4A, B). From the EMSAs on U251 cell line, we found only the probes containing HRE5, and HRE6 showed binding activities, with higher levels of binding in extracts from cells treated with 1% O2, whereas the probe containing HRE4 gave negative results even with extracts from cells exposed to the hypoxic environment (Fig. 4C).

In addition, we analyzed the functions of HREs located in the putative promoter region from −1884 to −217, which includes three potential HREs (HRE1, −2, and −3) in K562 erythroleukemia cells, an easy cellular model for chromatin preparation. As with the other two cell lines, elevated protein levels of HIF-1α and FtMt were observed when K562 cells were exposed to 1% O2 for 14 h (Fig. 4D, E). By ChIP assay, we found another functional HRE (named HRE1 here) that can bind to HIF-1α in cells treated with 1% O2. In contrast, HRE2 and HRE3 did not exhibit HIF-1α binding activity (Fig. 4F). Taken together, our results show that HIF-1α protein, in response to hypoxia, transcriptionally enhances human FTMT expression through HREs located in the gene's promoter region (Fig. 4G).
Hypoxia-induced brain cell death is attenuated by overexpression of FtMt
It is well known that hypoxia can induce cell damage; however, little has been described about the relationship between FtMt and hypoxia-induced impairment in the brain. We used Ftmt −/− mice to study the effects of hypoxia on cell injury in the hippocampus and cortex of mice. After continuous HH, we confirmed an increase in the levels of HIF-1α in mice hippocampi (Fig. 5A, B). In the mouse hippocampus, hypoxia also induced marked cell death, which was exacerbated by the absence of FtMt (approximately two-fold increase, Fig. 5C, D). In the mouse cortex, FtMt KO can also worsen hypoxia-induced cell death (Supplementary Fig. S3).

We further investigated the protective effects of FtMt in a fly model. To achieve overexpression of FtMt in the Drosophila nervous system, we crossed the transgenic (Fer3HCH, expressing the orthologous human FTMT) lines with the elav-Gal4 (nerve system expressing) driver line. As a control, we crossed a strain lacking the transgenes of the same genetic background. After 5% O2 treatment, we observed a significantly higher death rate in the control group than in the FtMt overexpressing animals (Fig. 5E).
To validate these data in a human cell model, we exposed a neuroblastoma cell line, SH-SY5Y, stably overexpressing FtMt [FtMt-SY5Y (45)] to hypoxia. In addition, we further detected the endogenous expression of human FtMt using our mouse anti human monoclonal antibody (24), K562 Mt6 was used as a positive control (Fig. 6A). In a cell viability assay, control cells (pcDNA3.1-SY5Y) showed a significant, ∼50%, decrease after a 1% O2 treatment, whereas the FtMt-SY5Y cells showed only a slightly reduced cell viability (Fig. 6B). We confirmed this result using the Annexin V/PI staining method to detect apoptotic cells (Fig. 6C, D). These results demonstrate that FtMt has an indispensable role in preventing cell death in conditions of low oxygen.

FtMt inhibits Caspase 3-dependent apoptosis and blocks iron-dependent oxidative stress
To further investigate the apoptotic pathway induced by hypoxia, Ftmt −/− mice were placed into the hypoxic chamber for 18 h HH, after which apoptosis-related proteins were assayed. The ratio of Bcl-2/Bax (B-cell lymphoma-2/Bcl-2-associated X protein) dropped dramatically in the wild-type (WT) HH group compared with the WT control group. However, the Bcl-2/Bax ratio decreased even more in the Ftmt −/− HH group (KO HH). We found the same trend in the cleavage of Caspase 3, indicating that a lack of Ftmt may lead to severe neuronal apoptosis in the mouse brain (Fig. 7A–C) (see also Supplementary Fig. S4).

In in vitro experiments, hypoxia-exposed pcDNA3.1-SY5Y cells presented signs of apoptosis both through a decreased ratio of Bcl-2/Bax and increased cleavage of Caspase 3 (Fig. 7D–F), whereas in the hypoxic FtMt-SY5Y, the ratio of Bcl-2/Bax was increased compared with the hypoxic pcDNA3.1-SY5Y cells. In addition, the hypoxic FtMt overexpressing cells exhibited lower Caspase 3 activation than hypoxic controls (Fig. 7D–F).
Using the calcein-AM method (see Methods section), we detected a remarkably high level of cellular chelatable iron after hypoxia treatment (Fig. 7G). As expected, overexpression of FtMt reduced this chelatable iron level, thus maintaining the cytosolic free iron pool within a “safe” range. The role of hypoxia in increasing intracellular ROS levels is well established (5, 39). As expected, the level of ROS, as reflected by the 2,7-dichlorofluorescein diacetate (DCFH-DA) fluorescence level, was increased in pcDNA3.1-SY5Y cells under hypoxia. Again, FtMt exerted protective effects in these experiments, with the FtMt-SY5Y cells exhibiting lower DCFH-DA fluorescence levels under hypoxia (Fig. 7H).
To test whether FtMt has a protective role in mitochondria, we estimated mitochondrial membrane potential (MMP) by assaying the mitochondrial retention of rhodamine 123 in living cells, as previously described (45). In pcDNA3.1-SY5Y cells treated for 24 h with 1% O2, MMP was significantly decreased compared with that in untreated cells; this trend was weakened in FtMt-SY5Y cells (Fig. 7I). Moreover, the MMP was preserved in hypoxia-treated FtMt-SY5Y cells, providing additional evidence of FtMt's protective role in mitochondria.
Discussion
The preferential expression of FtMt in cell types with vigorous metabolic activity and high oxygen consumption inspired us to ask whether FTMT may be a HIF-1α target gene. For the first time, we show that FtMt expression was increased by both hypoxia and in chemically mimicked (CoCl2) hypoxia. In the nervous system, this increase in FtMt is accompanied by the activation of HIF-1α both in vitro and in vivo.
Recently published data characterized the human FTMT promoter, revealing the existence of one positive and one negative regulatory region with a minimal promoter region located in the 500 bp upstream of the TSS (17). In the same study, CREB, a binding site of which we have shown is located near the predicted HRE1 sequence in our study (the green frame in Fig. 3B), was identified as a TF that binds to the positive regulatory region in the human FTMT promoter (17). Another study also showed that CREB acts as one of the TFs involved in hypoxia transcriptional progress (13). Here we add new information on the regulation of FTMT with the identification and functional characterization of HREs in the FTMT promoter.
By sequence alignment, we found very high similarities among human, macaque, and chimpanzee in the DNA sequence 2000 bp upstream of the TSS (>90%). Among the six HREs identified and analyzed, HRE1, −4, and −6 were fully conserved among the three species. In the 3′-deletion DLR assays, as opposed to the −1884/−874 deletion, which contains none of the putative HREs, the −1884/−777 construct containing only HRE1 had the highest luciferase activity when the HeLa cells were exposed to 1% O2. Importantly, the −1884/−521 deletion exhibited the lowest luciferase activities in the normoxia condition, which is in support of the existence of a negative regulatory region in the promoter about 500 bp upstream of the TSS as previously reported (17). Nonetheless, increased expression in this construct can be still activated by incubation at 1% O2.
ChIP assays probing for the HRE1, HRE2, and HRE3 sequences showed that, in this more distal promoter region from −1884 to −217 upstream of the TSS, only HRE1, located in the positive regulatory region, possesses HIF-1α binding activity. Collectively, our data prove that the expression of FtMt is hypoxia inducible and, in a hypoxic environment, stabilized HIF-1α can bind to the HREs located in the positive regulatory and minimal promoter regions of human FTMT (Fig. 4G). FtMt protein levels changed together with the mRNA regulation. Our research provides a fresh view of the human FTMT promoter and highlights the mechanisms of FTMT regulation under hypoxia, which is congruent with an importance of FtMt in cell types of high oxygen consumption.
A recent study examining FtMt expression in hypoxia showed that the FtMt protein decreases after 48 h hypoxia in human ARPE-19 cells, without alterations in Ftmt mRNA levels (57). This may indicate that there are altered regulatory mechanisms in different cell types. In the same study, it was revealed that FtMt overexpression in ARPE-19 cells can stabilize HIF-1α protein under normoxia. Thus, an interesting regulatory loop may exist between FtMt and HIF-1α regulation.
We speculate that FtMt overexpression can cause iron deprivation in the cytosol and subsequently decrease intracellular iron availability, which is a critical cofactor in prolyl hydroxylase domain (PHD) enzymes. This inhibition of PHD maturation would thereby block HIF-1α degradation. Our hypothesis is supported by a publication in which lipopolysaccharide induces ferritin expression and lowers free available iron levels. This results in the deprivation of the essential PHD cofactor and HIF-1α stabilization in normal oxygen conditions (47). However, the detailed regulatory mechanisms still needed clarification in physiologically representative systems.
The induction of apoptosis by hypoxia has been revealed in numerous studies: hypoxic exposure of primary oligodendrocytes resulted in increased iron content and ROS levels (38); both apoptotic and necrotic cell death induced by intermittent hypoxia were observed in primary cultures of cerebellar granule cells (10). In addition, the energy decline accompanying hypoxia and deprivation of metabolic substrates was shown to predispose rat kidney proximal tubule cells to injury (40); loss of mitochondrial respiration and lethal cell injury in the ischemic myocardium has been detected during in vivo ischemia and hypoxic perfusion of hearts (33, 40, 64). In our study, we confirmed that hypoxia can increase brain cell death in vivo (in the mouse brain and in Drosophila) and in vitro (in SH-SY5Y cells).
A high metabolic rate is a common characteristic between tissues with high FtMt expression (41) (brain, heart, and kidney) and tissues sensitive to hypoxic injury. We, therefore, hypothesized that an increased expression of FtMt during hypoxia may be a response to avoid tissue damage derived from oxygen limitation. To prove our hypothesis, we used Ftmt −/− mice and FtMt-overexpressing SH-SY5Y cells and found that in Ftmt −/− mice, cell death caused by hypoxia was more severe while the overexpression of FtMt in SH-SY5Y cells significantly lessened the effects of hypoxia.
Furthermore, our findings in Drosophila overexpressing FtMt only in nervous system suggest that hypoxia-induced fly death seemed genotype dependent and gender-specific since (i) all the flies showed a higher death rate after hypoxia that was decreased by overexpression of Fer3HCH; (ii) both WT and Fer3HCH-overexpressing female flies exhibited higher survival ability than males in low oxygen conditions. The latter finding is consistent with a previous study that reported that Fer3HCH overexpression improves resistance to paraquat toxicity in female flies (32). Collectively, our data show that FtMt has a neuroprotective role in the context of hypoxia-induced brain cell death.
A sequence of events leads to apoptosis during prolonged or severe hypoxia. This cascade requires the release of anti- and proapototic proteins, such as the apoptotic effector Bax, Caspase 3, and Bcl-2 (16, 37). In our study, we observed the proapoptotic duo of a decreased ratio of Bcl-2/Bax and increased cleavage of Caspase 3 under hypoxia in both WT mice and in cells. This condition worsened with disruption of Ftmt in mice, and was abrogated with FtMt overexpression in SH-SY5Y cells.
Several reports demonstrate that ROS levels increase under hypoxic conditions, leading to programmed cell death (16, 22, 23, 36). We speculated that FtMt functions as an ROS scavenger during hypoxic injury in our cell model since FtMt can sequester iron from the cytosol in mitochondria (2). We observed elevated levels of ROS accompanied by elevated amounts of chelatable iron and decreased MMP in pcDNA3.1-SY5Y cells after hypoxia. In addition, the elevated level of transferritin receptor 1 (TfR1) expression during hypoxia can also result in increasing iron uptake into cells, which could be one of the resources of the increased chelatable iron in our cell model (30). All of these were significantly rescued by the overexpression of FtMt.
In light of this finding, we tentatively put forward a working model in which hypoxia increases uncommitted iron that, left unchecked, can stimulate ROS-derived mitochondrial dysfunction, leading to apoptosis. In this model, FtMt has a primary role of iron detoxification in cell types of high oxygen consumption, where iron sequestration is needed to decrease the catalytic iron-derived ROS toxicity under oxygen limitation.
This model may partially explain the extremely low level of FtMt in most solid tumor cell lines. In contrast to non-neoplastic cells in which severe hypoxia or anoxia initiates a cascade of events that leads to apoptosis that may be prevented by FtMt function, in solid tumors, the tissue has acclimated to hypoxia, having selected cells that are resistant to hypoxia and not as dependent on mitochondrial oxidative phosphorylation. Under these circumstances, FtMt may not be needed to mitigate iron-catalyzed ROS because the hypoxic resistance occurs through other mechanisms. This idea is supported by previous studies in which FtMt overexpression stunted in vivo tumor growth (34, 46).
In summary, our current study demonstrates that the expression of FtMt is regulated by HIF-1α in hypoxia; FTMT is a novel HIF-1α target that plays a neuroprotective role in the oxidative damage-induced cell death caused by hypoxia through reducing the ROS caused by elevated uncommitted iron levels in cells. Our findings not only offer an explanation for why FtMt levels are sustained in tachyaerobic organs but also may provide a therapeutic approach to diseases characterized by oxygen deprivation, as ischemic stroke.
Methods
In silico human FTMT promoter experiments
The FTMT 5′ evolutionarily conserved region alignments between human, mouse, rat, rhesus macaque, and chimpanzee were performed using the ECR Browser according to the instructions in the official website and Clustal Omega (29). The transcription binding sites and prediction of the TFs were analyzed by MatInspector (Genomatix Software GmbH) (7), PROMO (14), and by TFBIND (51).
Materials
Dulbecco's modified Eagle's medium (DMEM), modified Eagle's medium (MEM), and fetal calf serum were purchased from Gibco BRL (Grand Island, NY). The In Situ Cell Death Detection Kit, Fluorescein was purchased from Roche (Mannheim, Germany). Annexin V/PI, rhodamine 123, calcein-AM, anti-β-actin antibody, and DCFH-DA were purchased from Sigma (St. Louis, MO). MTT, TRIzol reagent, Lipofectamine 2000, and rat FtMt small interfering RNA (siRNA) were purchased from Invitrogen. Anti-Bcl-2 and anti-Bax antibodies were purchased from Santa Cruz Biotechnology. The anti-HIF-1α antibody and anti-TIM44 antibody were obtained from Novus Biologicals (Littleton, CO). Antibodies to Caspase 3 and Cleaved-Caspase 3 were purchased from Cell Signaling Technology. The anti-FtMt antibody was homemade as described in our previous publication (41).
Animals and HH treatment
Adult male C57BL/6J and Ftmt−/− mice were housed in stainless steel rust-free cages at 22–24°C with 45–55% relative humidity. All animals were provided free access to food and water. All experiments were approved by the institutional Animal Care and Use Committee (Hebei Normal University, Shijiazhuang, China). The animals were placed in a hypobaric chamber (Guizhou Fenglei) in our laboratory. The chamber can imitate HH; the simulated altitude (8000 m) was selected based on trials and previous reports (27, 44, 68) to create hypoxic conditions in the chamber at a velocity of ∼20 m/s, the O2 content in the 8000 m altitude is around 8.5% (O2 content was 20% in the control group), and the hypoxia exposure time was 18 h. For Drosophila maintenance, the procedures were as described previously (46), the O2 content for Drosophila was 5% for 18 h.
Cell culture and hypoxic treatment
The C6 and HeLa cell lines were maintained in DMEM; U251 cells were cultured in MEM; the human erythroleukemic K562 cells were cultured in RPMI (Lonza, Basel, Switzerland), all the cells were supplemented with 10% fetal calf serum and 100 U/mL penicillin/streptomycin at 37°C and 5% CO2. FtMt-overexpression (FtMt-SY5Y) cells and pcDNA3.1-SY5Y cells were obtained and cultured as previously described (45). The hypoxia treatment for all the cell lines was 1% O2, 5% CO2, and 94% N2. For CoCl2 treatments, CoCl2 was dissolved in sterile H2O and the cells were treated for the indicated time periods.
Immunoblotting, reverse transcription-PCR analysis, and qRT-PCR analysis
For detecting normal protein expression, immunoblotting was performed as previously described (46). For HIF-1α protein detection, PE-lysis buffer (6.65 M urea, 10% glycerol, 1% sodium dodecyl sulfate, Tris [tris(hydroxymethyl)aminomethane] HCl, pH 6.8, 5 mM DTT) was used to generate cell lysates according to methods described previously (47). Total RNA was extracted from cultured cells or tissues using TRIzol reagent according to the manufacturer's instructions. One microgram total RNA was reverse transcribed for reverse transcription-PCR (45) or qRT-PCR with an ABI 7900HT Fast Real Time PCR System (Applied Biosystems) using SYBR® Green Real-Time PCR Master Mix (Applied Biosystems). The primer sequences used are listed in Table 2. Data were analyzed using the ΔΔCT method. The normalized ratio of target mRNA to the internal control mRNA β-actin (Actb) was set to 1. The relative band intensities of the proteins are expressed as the ratio of each to β-actin.
Primers Used for Reverse Transcription-Polymerase Chain Reaction (Rat) and Quantitative Real-Time Polymerase Chain Reaction (Mouse)
ChIP, chromatin immunoprecipitation; Ftmt, mitochondrial ferritin gene for mouse and rat.
Detection of apoptosis in cultured cells and in mouse brain
Under anesthesia with 0.4% Nembutal, the mice were sacrificed, the brains were then removed, postfixed for 72–96 h, and then stored overnight in 30% sucrose. Serial coronal sections were cut at a thickness of 15 μm on a freezing microtome (Leica CM1950) and mounted onto a slide covered with APES (Beijing ZhongShan Biotechnology, Beijing, China). The presence of apoptosis in the mouse cortex and hippocampus regions after HH treatment was assessed by the terminal deoxynucleotidyl transferase-mediated Cyanine 3 (Cy 3)-dUDP nick-end labeling method using the One Step TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling assay) Apoptosis Assay Kit (Beyotime Biotechnology, Shanghai, China) following the manufacturer's protocol. Apoptosis in SH-SY5Y cells was measured by flow cytometry using Annexin V/PI staining as previously described (45).
Double immunofluorescence
The brain slices were washed three times with 0.01 M phosphate-buffered saline (PBS). Antigen retrieval was performed in a microwave oven for 10 min in 10 mM citrate buffer (pH 6.0). After blocking for 1 h with normal goat serum prepared in 0.01 M PBS, the slices were incubated overnight at 4°C with the mouse anti-NeuN monoclonal antibody (1:100; ab104224; Abcam, Abcam Trading [Shanghai] Company Ltd.), mouse anti-GFAP monoclonal antibody (1:500; Millipore Corporation, Temecula, CA), mouse anti-Iba1 monoclonal antibody (1:200; Millipore Corporation), and mouse anti-COX IV monoclonal antibody (1:200; ab14744, Abcam, Abcam Trading [Shanghai] Company Ltd), and FtMt antibody (1:200).
The slides were then washed three times for 5 min with 0.01 M PBS. The following secondary antibodies were used in 50 min incubations at 37°C: DyLight 549, goat antirabbit IgG (1:200; Abbkine Scientific Co., Ltd., Wuhan, China), and DyLight 488, goat antimouse IgG (1:200; Abbkine Scientific Co., Ltd.). Finally, after washing and mounting, the sections were analyzed with an OLYMPUS FV3000 confocal laser scanning microscope.
Transfection and DLR assay
The plasmids for 3′ deletion (−1884/−874, −1884/−777, −1884/−521, and −1884/−217) and 5′ deletion (−902/+9, −491/+9, and −217/+9) of the FTMT promoter were as previously described (17). HeLa cells were seeded in 48-well plates, grown to 90% confluence before transfection, and maintained in ambient air (20% O2). A DLR system (Promega and Abnova) was used. Cells were transiently transfected with pGL2-Basic (mock) or reporter constructs (0.4 μg) accompanied by Renilla luciferase (Rluc) control reporter vectors pRL-TK (20 ng). Transient transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. After 1% O2 treatment, cells were harvested and the luciferase activity was quantitated using a Victor 3 1420 Multilabel Counter (Perkin Elmer). The firefly luciferase activity was normalized to the control, Renilla luciferase activity.
EMSA and ChIP
Nuclear extracts from U251 cells were used for EMSA. The biotin labeled and unlabeled double-stranded oligonucleotide probes containing HIF-1α binding sites (hypoxia response element, HRE) were synthesized by Invitrogen. Nuclear extracts, biotin-labeled probe, 10 × binding buffer (100 mM Tris, 500 mM KCl, and 10 mM DTT, pH 7.5), 1% NP-40, 50% glycerol, 100 mM MgCl2, and Poly(dI·dC) were combined in 20 μL, and incubated for 20 min at room temperature. The EMSA was performed following the protocol of the LightShift Chemiluminescent EMSA kit (Thermo Fisher).
The ChIP experiments were performed according to a previously reported method (17). The HIF-1α antibody (NB100–134) from Novus Biologicals was used. Specific primers used to detect the amplified human FTMT promoter region are provided in Table 2.
Measurement of intracellular ROS, MMP, and chelatable iron
The level of ROS in pcDNA3.1-SY5Y and FtMt-SY5Y cells was quantified by assessing the fluorescence of DCFH-DA according to the methods of Bass et al. (4). Changes in MMP were analyzed by calculating the retention of rhodamine 123 as previously described (45). Chelatable iron levels in pcDNA3.1-SY5Y and FtMt-SY5Y cells were measured according to methods in the literature (45). For these studies, the cells were treated with 1% O2 for 24 h.
Statistical analysis
Results are expressed as means ± standard deviation. If not indicated otherwise, n represents biological samples obtained from N independent experiments or mice. Data were analyzed using Prism v5.0 (GraphPad Software). In general, for normally distributed data, two-tailed unpaired Student's t-test or analysis of variance was used. A p-value <0.05 was considered statistically significant.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant Nos. 31520103908, 31471035, 31300898, and 31271473) and by scholarships from the China Scholarship Council (CSC) awarded to Q.W. The authors wish to thank Alex Sheftel (High Impact Editing) for language editing and critical reading.
Author Disclosure Statement
The authors declare that they have no conflict of interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.