
Abstract
Significance:
This review evaluates the role of platelet-derived transforming growth factor (TGF)-β1 in oxidative stress-linked pathologic fibrosis, with an emphasis on the heart and kidney, by using ionizing radiation as a clinically relevant stimulus. Current radiation-induced organ fibrosis interventions focus on pan-neutralization of TGF-β or the use of anti-oxidants and anti-proliferative agents, with limited clinical efficacy.
Recent Advances:
Pathologic fibrosis represents excessive accumulation of collagen and other extracellular matrix (ECM) components after dysregulation of a balance between ECM synthesis and degradation. Targets based on endogenous carbon monoxide (CO) pathways and the use of redox modulators such as N-acetylcysteine present promising alternatives to current therapeutic regimens.
Critical Issues:
Ionizing radiation leads to direct DNA damage and generation of reactive oxygen species (ROS), with TGF-β1 activation via ROS, thrombin generation, platelet activation, and pro-inflammatory signaling promoting myofibroblast accumulation and ECM production. Feed-forward loops, as TGF-β1 promotes ROS, amplify these profibrotic signals, and persistent low-grade inflammation insures their perpetuation. We highlight differential roles for platelet- versus monocyte-derived TGF-β1, establishing links between canonical and noncanonical TGF-β1 signaling pathways in relationship to macrophage polarization and autophagy, and define points where pharmacologic agents can intervene.
Future Directions:
Additional studies are needed to understand mechanisms underlying the anti-fibrotic effects of current and proposed therapeutics, based on limiting platelet TGF-β1 activity, promotion of macrophage polarization, and facilitation of collagen autophagy. Models incorporating endogenous CO and selective TGF-β1 pathways that impact the initiation and progression of pathologic fibrosis, including nuclear factor erythroid 2-related factor (Nrf2) and redox, are of particular interest. Antioxid. Redox Signal. 27, 977–988.
Introduction
F
We here utilize ionizing radiation as a clinically relevant model for discussion of the pathophysiology of cardiac and renal fibrosis, with a focus on the regulation of TGF-β1-related pathways. We review recent promising interventions, including: redox-reactive agents such as N-acetylcysteine (NAC), which block TGF-β1 activation by inhibiting thiol-disulfide exchange (3); pirfenidone, an FDA-approved agent for IPF that inhibits TGF-β1 signaling (81, 82); tyrosine kinase inhibitors (51, 82); and carbon monoxide (CO). CO is generated endogenously in mammalian cells through heme catalysis by heme oxygenase (HO) (74, 89). HO and the stress inducible heme oxygenase (HO-1) play key physiologic roles in protecting against oxidative stress, but low-dose inhaled CO can fully substitute for the cytoprotective effects of endogenous HO-1 and CO through redox pathways (39, 100). The maintenance of an inhaled CO regimen is not practical, however, and chronic exposure to even very low-level inhaled CO can itself lead to myocardial injury (77). But a key regulator of the expression of genes coding for the majority of endogenous anti-oxidant and anti-inflammatory proteins is nuclear factor erythroid 2-related factor (Nrf2), and many of its functions are mimicked by HO-1 via induction of endogenous CO (56), suggesting novel therapeutics.
Pathologic Fibrosis Related to Radiation Therapy
The off-target effects of radiotherapy are of rising interest. Some 50% of cancer patients now receive radiation, alone or with chemo- or immune therapy (1), leading to a variety of radiation-induced fibrosis (RIF) syndromes (90). Indeed, fibrosis is the major dose-limiting consequence of radiotherapy that is used for many tumor targets (61). There are numerous potential pathways by which ionizing radiation could induce fibrosis leading to CVD and CKD (103). We concentrate on two prominent features of ionizing radiation. These concepts offer a testable model for novel interventions to diminish the contribution of persistent release and activation of TGF-β1 leading to organ fibrosis.
Points for Intervention in RIF
(1) Ionizing radiation initiates a “perfect storm” of profibrotic factors. First, there is acute generation of local and systemic ROS and reactive nitrogen species (29), and release of proinflammatory cytokines (12); these can induce tissue factor (TF) with thrombin generation, platelet activation, and release of LTGF-β1 (5). Subsequent activation of LTGF-β1 can occur by various mechanisms (3, 11, 26, 102, 107). We postulate that it is platelet-derived TGF-β1 that is primarily etiologic in radiation-associated cardiac and renal fibrosis and organ dysfunction. This suggests a need to focus on targeting platelet TGF-β1, rather than pan-inhibition of TGF-β. Indeed, ECM-preserving phenomena related to TGF-β1 and the myofibroblast, along with suppression of proinflammatory mediator synthesis, have been well described depending on the cellular source of TGF-β1 (11).
(2) Inflammatory cytokines such as interleukin (IL)-6, upregulated after ionizing radiation exposure, induce macrophage polarization toward proliferation of the M1 inflammatory subset; inhibition of autophagy-mediated collagen degradation via nitric oxide synthetase (NOS) and possibly other pathways may sustain these processes (67). Anti-inflammatory signals generated by TGF-β1 derived from M2c macrophages, leading to induction of CD4+CD25+Foxp3+ regulatory T cells (Treg) and initiation of collagen autophagy, may assist in the resolution of fibrosis (54, 92).
Radiation-Induced Cardiac Dysfunction: Clinical Impact and In Vivo Modeling
Cardiac and coronary artery exposures related to radiotherapy for a variety of malignancies can be very high with, for example, parts of the heart receiving >40 Gy in the course of treatment for Hodgkin's disease (98). But with the exception of pericarditis, occurring within weeks to months, most clinically apparent radiation-induced cardiac fibrosis occurs >10 years after exposure, regardless of dose (55). A radiation dose response was recently established by examining the incidence of heart failure after exposures of <15 versus 16–20, 21–25, and ≥26 Gy (98). This was accelerated in individuals treated with an additional oxidative stressor, anthracycline-based chemotherapy (98).
Recent advances in radiotherapy have limited total cardiac exposure after radiation for many malignancies. In breast cancer, exposures as low as 2.3 and 1.5 Gy for left- and right-sided breast lesions, respectively, and 7.6 and 1.6 Gy to the left anterior descending artery after treatment of left- and right-sided lesions, respectively, are now typical (61). But despite these reductions, nonbreast cancer-linked mortality among survivors of this malignancy has risen steadily, predominantly due to radiation-induced CVD, with a latency of 10–15 years (21). Regional cardiac perfusion defects have been observed in asymptomatic breast cancer patients within 6 months of radiotherapy, and these defects persist for years (95). There is also an increased CVD risk with very low cardiac radiation exposures, from <0.1 to 5 Gy, as demonstrated by the Japanese Atomic Bomb Survivor Life Span Study and the Canadian Nuclear Workers cohorts (55). The excess relative risk for ischemic heart disease per sievert (Sv) (where one Sv represents a 5.5% lifetime cancer risk based on a linear no-threshold model) ranged up to 12.1 (88).
In experimental models, TGF-β1 is critical to the induction of RIF. In one study, rats were irradiated with a single dose of 0, 15, 20, or 25 Gy to the heart (52). After doses ≤20 Gy, left ventricular (LV) TGF-β1 mRNA levels increased up to sixfold at days 1 and 12, then returned to control levels by 1 month. But at 25 Gy, there was a persistent elevation of TGF-β1 mRNA for >6 months, and mRNA for procollagen types I and III increased progressively beginning at month 6. Other investigators, using radiation exposures to 20 Gy, also documented microvascular endothelial cell damage with reduced myocardial capillary density and increased von Willebrand factor expression, accompanied by elevated levels of TGF-β1, angiotensin II, and aldosterone in the myocardium, and subsequent coronary artery disease (15). Damage to the microvascular network was progressive, suggesting a role for hypoxia in the fibrotic injury (95).
Very low-dose cardiac radiation also has a significant impact. ApoE−/− and wild-type (wt) mice were locally irradiated to the heart with 0.2 or 2 Gy and followed for 60 weeks (70). Both doses altered LV function, which was more severe at the 2 Gy dose, and induced an acute inflammatory infiltrate in scarring areas, with accumulation of M1 macrophages, and secretion of IL-6 and TGF-β1, in both types of mouse. IL-6 and TGF-β1 levels correlated with fibrosis scores. Increases in TGF-β1 occurred earlier in cardiomyocytes that were isolated from ApoE−/− rather than wild-type (wt) mice, and only the former suffered premature death (15). Smad7 expression, a part of the inhibitory pathway of TGF-β1 signaling (14, 92), was repressed in both mouse strains at 60 weeks postirradiation, suggesting suppression of this TGF-β1 inhibitory pathway during the late phase of cardiac pathology (70).
Radiation-Induced Nephropathy: Clinical Impact and In Vivo Modeling
Clinically, radiation nephropathy is a form of CKD beginning from 6 months to several years after renal irradiation or use of 90Y-tagged therapeutic radionuclides (22). All components of the kidney are affected, including glomeruli, tubular epithelial cells, the interstitium, and vasculature (23). In terms of high-dose therapeutic radiation, proteinuria appears within 6 weeks after a single 10 Gy dose of local irradiation, accompanied by glomerulosclerosis and tubulointerstitial fibrosis (23, 84). A decline in estimated glomerular filtration rate (eGFR) and development of hypertension follows within the next 6–9 weeks (23), with an incidence of 20% in one study utilizing 25 Gy directed to the kidney (60). Malignant hypertension occurs in some 30% of individuals after renal irradiation, as late as 11 years after the insult (60).
Experimental systems can model these injuries over much reduced time frames. In one study, unilaterally nephrectomized rats bearing a single hypertrophied kidney were subjected to single doses of ionizing radiation up to 20 Gy, and functional and histologic end points were assessed serially for 4–24 weeks (85). Time-dependent reduction in eGFR and increased blood urea nitrogen were paralleled by morphologic changes in the glomerular, tubular, and interstitial portions of the kidney, with dose-dependent changes observed in the tubulointerstitial compartments (85). These alterations appeared to be dependent on TGF-β1 activation. At 8 weeks postradiation, there was a marked elevation in tubular TGF-β staining; by weeks 16–24, there were substantial increases in interstitial TGF-β, with accumulation of collagen type III and fibronectin. These changes were accompanied by increased interstitial α-smooth muscle actin (SMA), suggesting the transformation of interstitial fibroblasts into myofibroblasts (60). In vitro irradiation of rat mesangial cells and tubule epithelial cells was similarly associated with increased fibronectin, collagen types I and III, and TGF-β deposition (84, 109, 110).
In agreement with studies of CVD after very low-dose exposure, Hiroshima-Nagasaki nuclear fall-out data suggest that a single fraction of total body irradiation of 1 Gy can initiate CKD with a latency period of years to decades (88).
RIF: Pathophysiologic Clues
TGF-β1 induction, canonical signaling, and ROS
Our understanding of the pathophysiology of pathologic fibrosis, in general, and its relationship to ROS continues to evolve. TGF-β1 signaling, oxidative stress, the antioxidant system, and chronic low-grade inflammation all appear to play important roles, a concept known as redox-fibrosis (43, 83). This is of particular interest to RIF, as the cardiac and renal radiation models reviewed earlier show evidence of prominent, persistent oxidative stress, including increased xanthine oxidase and adenosine deaminase activities, and show decrease in antioxidant enzymes such as glutathione peroxidase and superoxide dismutase (SOD) (9, 62). ROS and inflammation have been implicated independently in the development and progression of heart failure, given the fact that cardiomyocytes are abundant in phospholipids that are particularly sensitive to oxidative stress (60). Similarly, many acute and chronic renal injuries involve an imbalance among the molecular mechanisms that regulate oxidative stress, inflammation, and collagen autophagy (91). The major role of ionizing radiation, in both its anti-tumor effects and initiation of pathologic fibrosis, is the generation of ROS (105). Such oxidative stress is extremely efficient in the activation of latent TGF-β1 (29, 105).
Our synthesis of processes linking radiation injury to TGF-β1 release and activation, and development of organ fibrosis with consequent organ dysfunction, is outlined in Figures 1 and 2. In terms of homeostasis, collagen synthesis is usually controlled by two distinct TGF-β signaling pathways, Smad and TAK1/MKK3/p38 (Fig. 1) (14). The nuclear signaling adapter molecule TRAF6 (tumor necrosis factor receptor-associated factor-6) is critical to this process. It not only regulates the canonical Smad pathway, but is also required for signaling by many proinflammatory cytokines that are involved in the fibrosis cascade, particularly IL-6 and IL-8 (64, 67). TAK1/MKK3/p38 signaling is key to autophagy-dependent collagen degradation, helping to maintain a balance between collagen formation and its modeling. This has been established in vivo in murine models (41) as well as in vitro in cardiac myocytes and renal tubular cells (94). TRAF6 is involved via a negative feedback loop through MKK3 signaling and Beclin-1 upregulation (104) (Fig. 1).

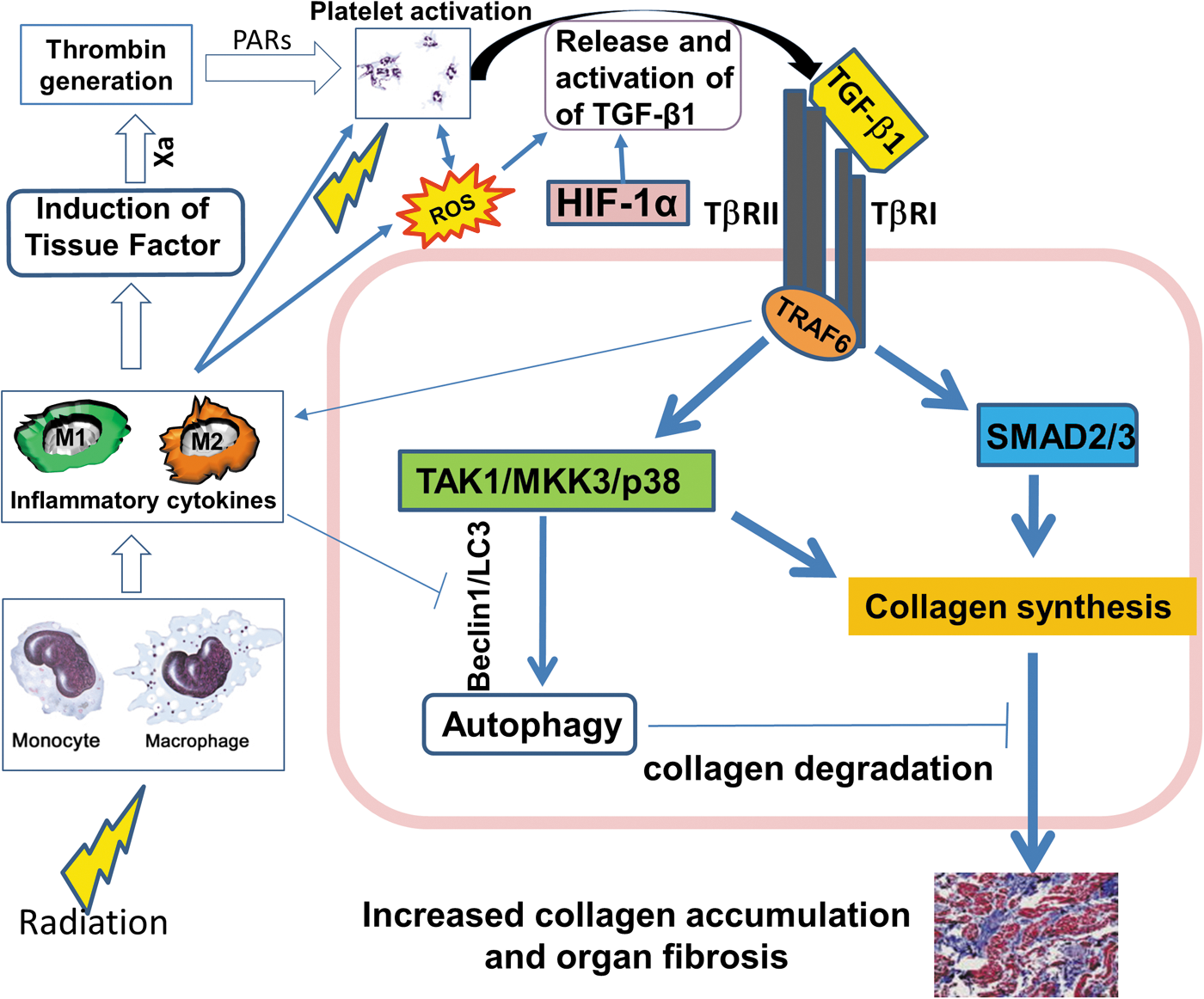
In terms of pathology, ionizing radiation leads to tissue injury, which augments release and activation of LTGF-β1, promoting pathologic fibrosis and organ dysfunction (Fig. 2). Immediate induction of ROS by radiation induces oxidative damage of DNA, protein, and lipid, along with a direct action on ECM, releasing TGF-β1 (105). TGF-β1 can be directly activated by local ROS (11), as integrin interactions enable traction forces among myofibroblasts (26, 102), and by changes in shear force due to endothelial damage and thrombus formation (3, 107). Increased levels of these integrins, particularly β6 and β8 and intracellular adhesion molecule I, are early consequences of ionizing radiation (95). Monocyte-derived TF-mediated thrombin generation activates platelets via protease-activated receptors (2, 13, 34), similarly leading to thrombin generation, platelet activation, and functional TGF-β1 formation (Fig. 2). TGF-β1 can also decrease synthesis of proteolytic enzymes that degrade matrix proteins and increase synthesis of protease inhibitors (103).
Single exposures of the heart or kidney to low-dose (0.2–2 Gy) or high-dose (>10 Gy) γ-radiation generates active TGF-β1 within an hour, concomitant with decreased LTGF-β1 immunostaining (7, 29). TGF-β1 plasma levels often correlate with the severity of radiation-induced injury in cancer patients (17, 111); however, regardless of circulating levels, irradiated organ vasculature shows increased immunostaining for TGF-β1 after radiation, sustained by 7 weeks postexposure, together with staining for phosphorylated Smad2,3 in the vascular smooth muscle (29).
In parallel with TGF-β1, IL-6 and IL-8 are elevated in vivo, systemically, locally, or both, after radiation exposure in clinical and preclinical models of radiation injury (12, 67, 111), including individuals exposed to low-dose radiation in Japanese atomic bomb cohorts (55), and in rodent models (110). These cytokines are also prominent inducers of platelet activation, with the potential for release and activation of platelet TGF-β1 (59), and their levels may be predictive for development of radiotherapy-induced fibrosis (10, 111). Radiation-induced microvascular endothelial cell injury also leads to hypoxia. This could sustain upregulation of collagen production via activated HIF-1α, either dependent on or independent of TGF-β1 signaling (105).
It is also important to note that regulation of TGF-β1 activation can occur without alterations in transcription or translation of involved proteins. This was documented by the persistent deregulation of TGF-β1 expression in myofibroblasts from irradiated tissue in vitro (63). In another study, there was no difference in intestinal TGF-β1 mRNA levels pre- or postradiation, despite elevated collagen synthesis and deposition in the bowel postradiation (105). We postulate that one pathway by which TGF-β1 signaling is enhanced by ionizing radiation exposure is through ROS-dependent activation of TRAF6, which, as outlined earlier, can facilitate signaling via TGF-β1 and other proinflammatory cytokines in the absence of alterations in the absolute levels of these factors.
Role of platelet activation and platelet-derived TGF-β1 in cardiac fibrosis
Platelets contain 40–100 times as much TGF-β1 as other cells, and they rapidly release it on activation (8). LTGF-β1 released from platelets becomes activated when subjected to flow-associated stress, particularly in the setting of turbulence, and thiol-disulfide exchange contributes to this process, providing potential mechanisms for physiologic control (3, 4). In terms of pathologic fibrosis, platelet TGF-β1 contributes to cardiac pathology in response to high shear states that are generated by surgically induced transverse aorta constriction (TAC) pressure overload in mice. This was documented by our group, showing the development of cardiac fibrosis and dysfunction in wt mice, but not in mice with targeted deletion of TGF-β1 in megakaryocytes and platelets (69). Four weeks after TAC surgery, the majority of wt mice had cardiac fibrosis, whereas mice deficient in TGF-β1 developed much less fibrosis in their hearts and were partially protected from cardiac dysfunction (69).
Since the TAC model requires surgery, and platelets can be activated by surgery itself, contributing to plasma TGF-β1 levels, we also used the hypercholesterolemic Ldlr−/−-ApoB100/100 mouse model (30). These animals develop aortic stenosis spontaneously when fed a high-fat diet (30). Aortic valve fibrosis directly correlated with increased systemic TGF-β1 levels and Smad2 signaling (101). Clinically, procoagulant TF is detectable in coronary artery thrombi from patients with ST wave elevation-associated myocardial infarctions, suggesting that TF expression leads to initiation of the coagulation cascade and generation of thrombin, which can further activate platelets during thrombus formation (76). Other fibrotic injuries characteristic of ionizing radiation, featuring persistent, low-level inflammation, also upregulate TF (34, 105).
TGF-β1, noncanonical signaling, and collagen autophagy
Downregulation of autophagy-mediated collagen degradation may further promote TGF-β1-related events in RIF. For example, treatment of cells with the autophagy inhibitors 3-methyladenine or chloroquine, or knockdown of Beclin-1, enhances tissue sensitivity to radiation (16). This is important to our model, as ionizing radiation generates both ROS and reactive nitrogen species, mainly as a result of the activation of NOS (80). Although ROS promote autophagy, generally in the damaged cell and by selective mitophagy, nitric oxide and nitrosative stress can inhibit autophagy via S-nitrosylation and subsequent blockade of transcription factors that regulate Beclin-1 (32).
Macrophage Polarization
Immune cells and platelets are present in the inflammatory milieu of organs postradiation, which is typical of most pathologic fibrosis models (105). Polarization of macrophage subsets in this environment regulates inflammation, and TGF-β1 contributes to this process. Several recent reviews of macrophage subsets, their phenotypic characterization and functional significance, have been published (41, 42, 57, 58, 66). There are four groups: • M1 cells express high levels of CD11b and F4-80/I-A,I-E. They are pro-inflammatory, induced by TNF-α, interferon (IFN)-γ, and IL-6, and produce high levels of these inflammatory cytokines. They appear early after an injury—within 7 days in the cases of fibrosis induced by infection (32)—and are prominent at the first time point examined, 20 weeks, post–low-dose cardiac radiation in ApoE−/− mice, together with an increase in total CD68+ macrophages (70). Of interest, although CD68+ cells were also increased in the irradiated wt mice, the M1 subtype was not specifically elevated, perhaps because ApoE is known to facilitate conversion from the pro-inflammatory M1 to anti-inflammatory M2 phenotypes (70). • At later time points after cardiac injury, M2 regulatory subsets predominate (41). They are F4-80/CD206+ and consist of three subtypes: M2a, b, and c, with functional differences between M2a and M2c cells that are of greatest significance in terms of their TGF-β production and regulation of inflammation. • M2a cells express F4-80/CD206. They are induced by IL-4, produce IL-10 and TGF-β1 at low levels, and do not induce anti-inflammatory Treg cells, as they lack the B7-H4-dependent ability to induce naïve CD4+ T cells into Treg (57). • M2c cells express B7-H4/CD163. They produce significant quantities of TGF-β1, and they potently induce Treg as an anti-inflammatory function (57).
These differences underlie the necessity to document the cellular sources of TGF-β1 at various points after ionizing radiation exposure, as well as to consider therapeutic interventions that do not pan-suppress TGF-β production and signaling, recognizing the anti-inflammatory and anti-fibrotic potential of M2c cells. Both pro-fibrotic and ECM-preserving phenomena related to TGF-β1 and the myofibroblast have been well described during the resolution phase of an inflammatory-based injury, indicating that TGF-β1 may play an important role in suppressing synthesis of proinflammatory mediators (11). This latter concept was emphasized in a recent study of the contribution of low levels of proinflammatory cytokines in promotion of myofibroblast persistence and cardiac fibrosis. It was concluded that, “Non-selective elimination of monocytes and macrophages leads to defective wound healing, whereas limiting monocyte influx into the heart is cytoprotective” (37). Another group continued this line of reasoning, noting that the cellular source of TGF-β1 “dictates its activity [so that] it remains unclear whether antagonism of the TGF-β1 signaling pathway will prove beneficial in humans” (103). Indeed, M2c cells appear to protect against renal fibrosis in murine models of oxidative stress (Adriamycin)-induced nephrosis and unilateral ureteral obstruction (UUO), via promotion of autophagy in the context of TGF-β1 secretion (57, 92). In addition, depending on cell type and context, TGF-β1 may activate autophagy via theTAK1/MKK3/p38 signaling axis (24).
Strategies to Mitigate the Pathologic Impact of TGF-β1 in RIF
In a recent review of RIF, a “bewildering range” of therapeutic targets was described, but clinically all were deemed to be “disappointing” (105). However, as noted in the Introduction, there are a few treatments currently FDA approved, or in advanced clinical trials, that specifically target at least portions of identified pathogenic mechanisms of fibrosis, regardless of the trigger (86), that should be applicable to RIF. NAC appears to have little efficacy in IPF (92, 96), possibly related to the fact that IPF is not predominantly a pro-inflammatory disorder (81) but, may have utility in RIF. At least two disease-modifying agents, FDA-approved pirfenidone, which is an ROS scavenger and decreases signaling pathways linked to IL-6, TNF-α, and TGF-β1 (27), and the tyrosine kinase inhibitor nintedanib, which blocks receptors for fibroblast growth factor, platelet-derived growth factor, and vascular endothelial growth factor, had consistent effects across IPF treatment subgroups (82). Pirfenidone and imatinib, another tyrosine kinase inhibitor, had some efficacy in renal fibrosis linked to CKD (51), and pirfenidone can reduce cardiac fibrosis in a variety of mouse models (87).
In RIF, we return to our pathophysiologic model to suggest a multi-factorial approach to inhibit the initiation and persistence of pathologic organ fibrosis. The redox system is an obvious initial therapeutic target (83). As noted earlier, TGF-β1 activation involves redox reactions, and thiol-reactive agents, including NAC, block TGF-β1 activation by inhibiting thiol-disulfide exchange (3, 4). CO similarly reacts with thiols, sulfides, and disulfides (39). These findings support the concept of blocking the redox system by using CO (Figs. 3 and 4) and NAC (Fig. 4).


Endogenous and exogenous CO: a model for a novel strategy combining anti-oxidant, anti-inflammatory, and pro-autophagy mechanisms
HO and HO-1 play key physiologic roles in protecting against oxidative stress. HO-1 and its constitutively expressed isozyme, heme oxygenase-2; catalyze the rate-limiting step in the conversion of heme to bilirubin IXα; ferrous iron, and CO (89). The bile pigments generated have antioxidant properties, although excess HO-1 production may be counterproductive due to a transient excess of reactive iron generated during heme metabolism, subsequent ferritin induction should compensate for this excess free iron (74, 89). Endogenous CO also induces SOD2 and mitochondrial H2O2, thereby activating Akt signaling and promoting a variety of anti-oxidant defenses (79).
Low-dose (100–250 ppm) exogenous CO can fully substitute for the cytoprotective effects of endogenous HO-1 both in vitro and in vivo (100). Inhaled CO suppresses TGF-β1-stimulated collagen expression in the kidney (48), inhibits bleomycin-induced pulmonary fibrosis (89), and protects against UUO-induced renal fibrosis (100), as reflected by decreased levels of fibronectin and type I collagen and decreased expression of both type I and II TGF-β receptors (100). In terms of radiation, exogenous CO liberated from CO-releasing molecules (CORM), which are based on transition metal carbonyls, can protect zebrafish embryos in vivo (20), and Chinese hamster ovary cells in vitro (36), from radiation-induced bystander effects. But there are no clinical studies of the impact of inhaled CO or CORM on RIF.
As outlined in Figure 4, exogenous CO appears to function via suppression of inflammation and fibrosis through its anti-apoptotic (47) and anti-proliferative (112) properties, as well as by promotion of collagen autophagy (48) and, via stimulation of cGMP, inhibition of platelet aggregation and promotion of vascular relaxation (89). CO-mediated autophagy is of particular interest with respect to TGF-β1. Depending on the signaling pathway involved, TGF-β1 can augment type I collagen synthesis or promote its degradation (Fig. 1). Using heterozygous Beclin-1 gene deletions in mice, and siRNA Beclin-1 knockdown experiments in vitro, it was shown that collagen degradation related to TGF-β1 is dependent on autophagy through TAK1/MKK3/p38 signaling and Beclin-1 induction (48). The anti-fibrotic effects of CO in the UUO kidney fibrosis model was MKK3 signaling dependent. Both wt and Mkk3−/− mice showed a UUO-induced increase in collagen deposition in kidneys, but CO failed to inhibit this deposition in the autophagy-deficient Mkk3−/− animals (100).
Potential toxicities of exogenous CO: back to the endogenous form, and its relationship to Nrf2
Myocardial injury is clearly associated with moderate-to-severe CO poisoning (38), and short-term exposure of rats to 1500 ppm CO—the LD50 for rodents is 3500 ppm—leads to significant electrocardiographic abnormalities (45). An early clinical study showed that inhalation of 1500 ppm CO 20 times per day for a week produced no cardiovascular effects (108), but more chronic exposures to even very low-dose inhaled CO can lead to significant myocardial damage in rodents and humans (77). In an attempt to mimic environmentally relevant CO levels that are consistent with urban pollution, rats were exposed for 4 weeks to 30 ppm CO, with five peaks of 100 ppm per 24 h period (6). Cardiac arrhythmias were noted along with LV fibrosis (6). In patients with existing CVD, continuous exposure to 50 ppm of CO for 8 days led to flattening or inversion of P waves (35). A multi-center clinical study (NCT01214187) for the treatment of IPF using CO inhalation, 250 ppm two times weekly, for 2 h per dose, for 12 weeks, is ongoing but closed to accrual. Results, and any side effects, have not yet been reported but in any event, long-term inhaled CO is not a practical clinical strategy in terms of adherence and possible cardiac toxicities. Currently available parenteral and oral CORMs have very short half-lives (48, 99) and relatively limited efficacy in vivo (48, 98, 99). Exogenous CO is highlighted here as a proof of concept to support further work on more practical interventions targeting those TGF-β1 signaling and autophagy pathways by which endogenous CO appears to act.
Future Therapeutic Strategies
ROS scavengers and suppression of ROS formation
NAC has been used as a scavenger of ROS to target mitochondrial oxidant stress, decreasing oxidant stress in the liver after acetaminophen poisoning (28). It is currently the only approved antidote for such an overdose, although other antioxidants, including Mito-Tempo, are under consideration (28). Acetaminophen hepatotoxicity is relevant to our general discussion of the role of macrophage polarization in fibrosis, as infiltrating M2 cells appear only past the peak of liver injury, suggesting that they are not responsible for the injury, but may be involved, most likely through M2c cell proliferation, in injury resolution (28, 40). NAC also has been shown to partially block shear-dependent platelet TGF-β1 activation in vitro by a mechanism involving thiol-disulfide exchange (3). Pharmacological doses of NAC are currently being evaluated in our murine aortic stenosis model, and our preliminary data indicate its efficacy in blocking platelet TGF-β1 activation (unpublished data; Fig. 4). One major impediment to the use of NAC or other agents with similar mechanisms of action based on the acetaminophen model is that patients who benefited most were those given NAC within 8 h after toxin ingestion (96). This may not be as great a concern in terms of radiation exposure, however, given the much more prolonged period for development of RIF.
SOD overexpression can decrease cardiac TGF-β1 staining, suppress cardiac fibrosis, and improve LV function in an aging mouse disease model (53), and SOD1−/− mice have increased generation of hydroxyl radicals and lipid peroxidation in an ischemia-reperfusion model (106). SOD1 mimetics (63, 72), pirfenidone (49, 87), and newer agents with potent anti-oxidative and anti-inflammatory properties such as fluorofenidone (19, 78, 93) have also been developed. Future research will be directed toward identifying specific target molecules and the mechanism by which these agents block organ fibrosis.
Nrf2 is a key regulator of the expression of genes coding for many, if not the majority of anti-oxidant and anti-inflammatory proteins (56). In endothelial cells, the Nrf2 pathway serves as a mechanosensitive transcriptional regulator of redox signaling (65; Fig. 3). The functions of Nrf2 are mimicked by one of its major dependent proteins, HO-1, and, as noted earlier, HO-1 acts through induction of endogenous CO (56). Nrf2−/− mice are highly susceptible to tissue fibrosis, and they have a reduced life span compared with wt mice after thoracic irradiation (97). In terms of alternatives to exogenous CO, S-adenosylmethionine (SAMe), the main endogenous methyl donor (75), is a nutraceutical in the United States but an EMA-approved drug in Europe. When given orally at 30 mg/kg/day, it can prevent and treat ethanol- and lipid-associated hepatic fibrosis in mice via Nrf2-mediated pathways (31).
Regulation of macrophage polarization and manipulation of Treg cells
An important criticism of studies attempting to relate macrophage phenotype to inflammation and inflammation resolution postradiation or other profibrotic injury is the fact that functional studies rely on in vitro assays of isolated subsets and do not reflect the fluidity of these subsets in vivo (41). However, the control of Treg by M2c versus M1 or M2a is clearly linked to differences in their surface receptors, and the therapeutic impact of Treg in organ fibrosis has been directly illustrated by using an aortic constriction-induced hypertension and cardiac fibrosis model in mice. Intravenous adoptive transfer of cultured, autologous Treg cells led to a decrease in LV fibrosis, suppressed inflammatory cell infiltration, decreased interstitial myofibroblasts, and decreased expression of TGF-β1 and its receptor by cardiac interstitial cells (44). Moreover, Treg has been shown to prevent organ fibrosis in models of acute liver (73), lung (71), kidney (50), and various cardiovascular disease models (44, 68), via mechanisms that are not yet established.
Autophagy pathways for resolution of inflammation and fibrosis
Apart from CO, other endogenous modulators can inhibit TGF-β1 signaling and increase autophagy flux. Preclinical data utilizing the secreted extracellular domain of the renal protein Klotho is of particular promise in this regard (92). The importance of Klotho in preventing cardiovascular fibrosis was recently documented in heterozygous klotho+/− mice, as they developed arterial stiffening and an increase in scleraxis, a transcription factor for collagen synthesis, compared with wt littermates (18). Klotho deficiency was associated with increased collagen and decreased elastin contents in the media of aortas, along with increases in TGF-β1 expression and myofibroblast differentiation (18). TGF-β1 has also been reported to decrease expression of Klotho protein by unclear mechanisms (25). This is a further argument for pursuit of novel treatment strategies in radiation fibrosis that are capable of influencing the many different, interacting pathways discussed earlier.
Future implications
Characterization of multiple mechanisms underlying pathological fibrosis should enable a more tailored approach to the management of patients with fibrotic diseases, regardless of the nature of the initial injury. Based on the in vitro and preclinical data reviewed here, highlighting the use of modulators of CO and redox pathways as a novel intervention in organ fibrosis, we postulate that new therapies should target specific molecules and/or signaling pathways related to platelet activation, with TGF-β1 release and activation, macrophage polarization in relationship to pro- and anti-inflammatory processes, and the importance of anti-oxidant and pro-autophagy mechanisms. It is also possible that measures of platelet-derived TGF-β1 in plasma or TGF-β1 signaling in tissue could serve as an early prognostic biomarker, or a surrogate marker for fibrosis development late postinjury.
Conclusion
A number of biological processes contribute to the pathogenesis of radiation-induced organ fibrosis. Simply describing fibrosis as a scar related to overproduction of collagen—a preprogrammed terminal event—fails to capture the complexity of the processes involved. Ionizing radiation injury, as many other physical, chemical, or biologic insults, may result in ROS generation and inflammation, with subsequent induction of TF-mediated thrombin generation, platelet activation, LTGF-β1 activation, and macrophage polarization. Feed-forward loops, as TGF-β1 promotes ROS formation, amplify these profibrotic signals in the promotion of myofibroblast accumulation and the production of ECM to cause fibrosis. We here emphasize the potential role for platelet-derived TGF-β1 in RIF. Further, in linking noncanonical TGF-β1 signaling pathways with macrophage polarization and autophagy, the concept of pan-TGF-β neutralization may be counter-productive. Finally, in vitro and preclinical work with carbon monoxide and redox pathways offers a novel approach to prevent, and treat, cardiac and renal fibrosis through intervention in multiple pathways that collectively lead to initiation and progression of pathologic fibrosis.
Footnotes
Acknowledgments
This work was supported by NIH grants R21 HL125044 (to J.L. and J.A.), R01 HL123605 (to J.A.), GM114731 and the Angelo Donghia Foundation (to J.L.).
Author Disclosure Statement
No competing financial interests exist.