
Abstract
Significance:
Shortly after the discovery of the role of hydrogen sulfide (H2S) in many physiological and pathological processes, attempts were made to develop novel pharmaceuticals that may be of benefit for treatment or prevention of a wide range of disorders. The promise of H2S-based therapeutics is now being demonstrated in clinical trials.
Recent Advances:
H2S-releasing drugs, such as SG1002 for cardiovascular disorders, and ATB-346 for arthritis, have progressed into clinical trials and have shown considerable promise. Some older drugs, such as zofenopril, have now been recognized to produce at least some of the beneficial effects through release of H2S.
Critical Issues:
There remains a need to better understand the underlying mechanisms for some of the observed effects of H2S-releasing drugs in a clinical setting, such as the marked increase in analgesic potency that has been observed with ATB-346.
Future Directions:
The proof-of-concept clinical studies reviewed herein pave the way for examination, in a clinical setting, of several other potential applications of H2S-based drugs in a wide range of disorders, including diabetes, hypertension, and cancer chemoprevention. Antioxid. Redox Signal. 28, 1533–1540.
Introduction
T
Many early studies utilized “off the shelf” H2S donors [e.g., sodium hydrosulfide (NaHS) and sodium sulfide (Na2S)] and garlic-derived compounds (allyl disulfide) to investigate the potential for modulating disease processes in animal models. There was then a burst of activity in synthesizing novel H2S donors, often as hybrid molecules wherein an H2S moiety was covalently linked to an existing drug [e.g., to a nonsteroidal anti-inflammatory drug (NSAID)]. Such compounds were studied in a wide variety of preclinical models, with encouraging results in many cases (41, 45).
There are also a number of therapeutics on world markets that can generate H2S in vivo and where there is at least some evidence of H2S contributing to their therapeutic benefits. These drugs were not necessarily designed with an aim of delivering H2S, but there is, in at least some cases, good evidence to suggest that some of their beneficial clinical effects are attributable to H2S. For example, anethole trithione (brand names include Sialor and Sulfarlem) has been used for decades for treating xerostomia (dry mouth), including that associated with Sjögren's syndrome and rheumatoid arthritis. Gastric and duodenal bicarbonate secretion have been shown to be stimulated by H2S (5, 31, 42, 43), so there is a good possibility that the same would be true for salivary secretion. Indeed, several beneficial effects of H2S on periodontal health have been documented (22).
The anethole trithione moiety has been used by several groups in the design of H2S-releasing drugs for chemoprevention, reducing oxidative stress, and attenuating inflammation (12, 25, 30, 47). Oltipraz, a schistosomicide, is another member of the dithiolethione class. In rodent models, it has been shown to reduce the formation of various cancers (colon, bladder, blood, liver, kidney, pancreas, lung, and mammary) (25). There is evidence that the anticancer effects of oltipraz may be mediated through activation of Nfr2 (23, 50), a signaling pathway known to be activated by H2S (24).
This review is focused on drugs that have been developed in recent years specifically based on delivery of H2S for therapeutic benefit, and for which clinical data are available. The translation of a novel therapeutic from the preclinical through the first two stages of clinical development is the biggest hurdle in drug development. A small number of H2S-releasing drugs (some rationally designed as such) have now been studied in clinical trials, and important information has been generated that is, thus far, consistent with the promise of H2S-based drugs with reduced toxicity, enhanced efficacy, or both.
SG1002 (Sulfagenix, Inc.)
People suffering from congestive heart failure (CHF) have been shown to have a deficit of H2S in their blood, and there is an inverse correlation between plasma H2S levels and the severity of CHF (29, 34). Commercially available inorganic salts, such as Na2S and NaHS, can produce significant increases in plasma H2S levels in laboratory animals, but the increases are short lived and are unlikely to produce substantial benefits. Moreover, they have the potential to produce toxic effects (12). Studies in animals have clearly demonstrated that in addition to raising plasma H2S levels, administration of an H2S donor can increase plasma nitric oxide (NO) levels via stimulation of endothelial NO synthase (eNOS) (27, 28).
Sulfagenix is an Ohio-based company that is attempting to exploit these benefits of H2S and NO in the cardiovascular system. They have developed a prodrug (SG1002; sodium polysulthionate) that produces more sustained and consistent levels of H2S in plasma in the hope that it will be useful for treating conditions such as CHF. Preclinical studies demonstrated, using mice, that SG1002 attenuated cardiac dysfunction associated with a high-fat diet by reducing endoplasmic reticulum stress (36), protected against pressure overload-induced heart failure by upregulating eNOS (28). Beneficial effects of H2S of the ischemic myocardium were also demonstrated with another polysulfide, diallyl trisulfide (35). SG1002 is currently available as a prescription medical food under the trade name “Sulfzix.”
Sulfagenix has demonstrated that administration of SG1002 can restore to normal the plasma H2S and NO levels in CHF patients, thereby reducing the severity of, or preventing, heart failure (2). They have performed a phase 1 clinical trial in healthy volunteers and patients with CHF. The study compared the effects of various doses of SG1002 (200–800 mg twice-daily) versus placebo on plasma levels of H2S. The healthy volunteer studies examined safety and maximum tolerated dose of SG1002, as well as effects on plasma H2S and NO levels (2).
Doses of 200–800 mg twice daily for 7 days produced dose-dependent elevations of free H2S and sulfane sulfur in plasma, and significantly increased NO bioavailability (Fig. 1). Importantly, the levels of H2S in these subjects remained below cytotoxic concentrations (37).
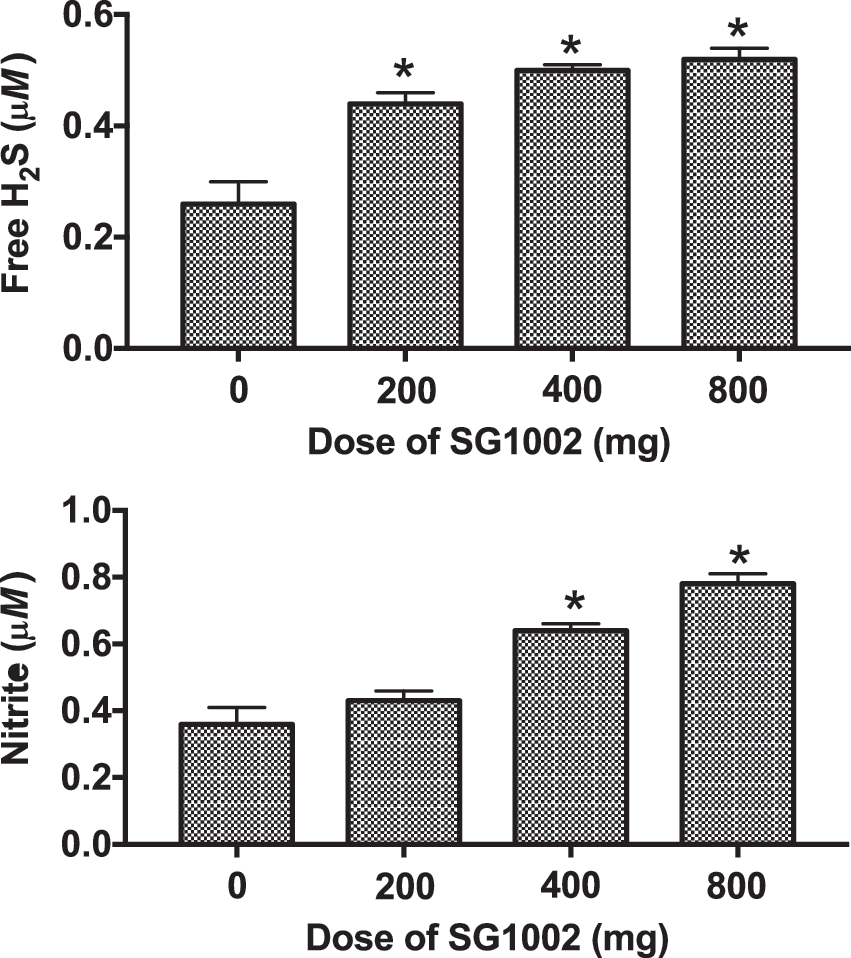
In heart failure patients, a similar dose-escalation study was performed, with 1 week of treatment with placebo followed by escalating doses of SG1002 for 1 week each (twice daily): 200, 400, and 800 mg (2). Larger increases in plasma H2S were observed in healthy volunteers than in the CHF patients, probably because of more rapid degradation of H2S as a consequence of the greater oxidative stress in the patients. SG1002 significantly increased plasma H2S at 400 and 800 mg, and produced sustained increases in plasma NO (2- to 2.4-fold). The increases in plasma H2S and NO in healthy volunteers and CHF patients were not accompanied by decreases in systemic blood pressure. The authors stated that based on preclinical studies (3), the increases in nitrite levels after SG1002 administration would be expected to have significant benefits in CHF patients.
The 800 mg BID dose was identified as the best “go-forward” dose for further phase 2 clinical studies. Those studies will examine whether or not SG1002 will significantly reduce free radical-mediated tissue damage.
Zofenopril (Menarini Group)
Zofenopril is an angiotensin-converting enzyme (ACE) inhibitor that entered the market a decade ago. It is a prodrug. Once absorbed, it undergoes complete and rapid hydrolysis to a sulfhydryl-containing active metabolite, zofenoprilat. It selectively inhibits cardiac ACE and exhibits potent antioxidant properties (16), attributed to the sulfhydryl moiety (13) and more specifically to the release of H2S (Fig. 2) (10).
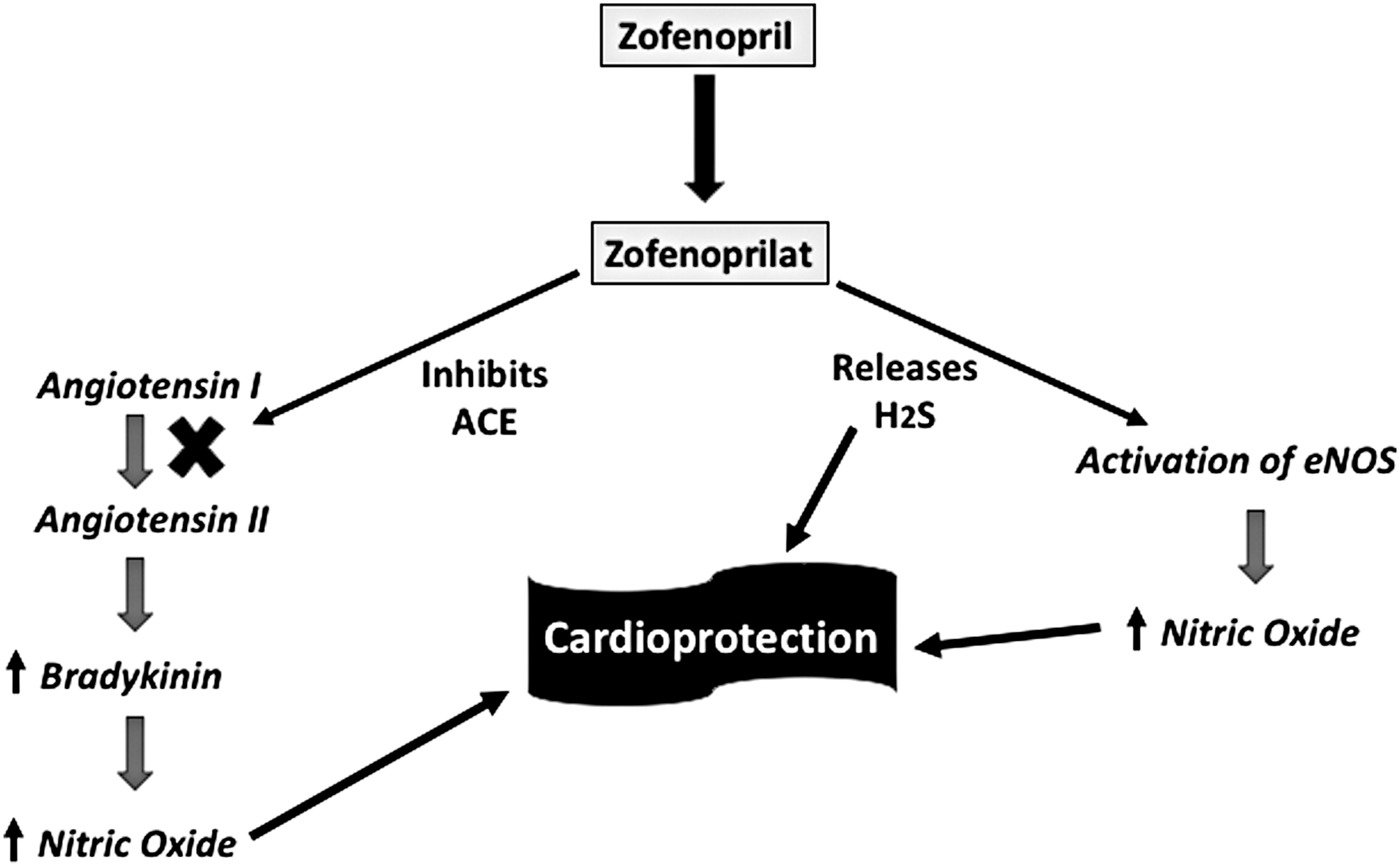
Antioxidant activities of zofenopril/zofenoprilat have been demonstrated in cardiac and vascular tissues, both in vitro and in vivo. In preclinical models, beneficial effects attributed to release of H2S have been shown to underlie prevention of endothelial dysfunction and ischemia, as well as reversal of nitrate tolerance, reversal of apoptosis, enhanced angiogenesis, and anti-inflammatory effects (9, 38, 39, 44). In both preclinical (8, 18) and clinical (11, 32) studies, zofenopril has been observed to exert cardioprotective and vasculoprotective effects that exceed those achieved by ACE inhibition, which are independent of the associated antihypertensive effects (6). For example, Bucci et al. (10) demonstrated that zofenopril significantly improved vascular function in a rat model of spontaneous hypertension, and the effect was shown to be a consequence of the H2S generated from this drug, and unrelated to suppression of ACE activity.
The potential contribution of H2S release from zofenopril in its beneficial cardiovascular effects was further examined by Bucci et al. (10). They studied spontaneously hypertensive mice and healthy controls. In in vitro studies, the vascular responses to acetylcholine in both the aorta and carotid artery were significantly impaired in the hypertensive rats. Exposure to zofenopril, but not enalapril, restored the responsiveness to normal. This group also demonstrated that the active metabolite of zofenapril (zofenoprilat) released H2S in a cell-free assay and concentration-dependently relaxed blood vessels. Moreover, the “R” stereoisomer of zofenoprilat, which has no inhibitory activity on ACE, produced beneficial effects on vascular function in the hypertensive rats and elevated tissue and plasma H2S levels (10).
Recent studies in pigs performed by Donnarumma et al. (17) clearly showed that zofenopril treatment resulted in significant elevations of both plasma and myocardial NO and plasma H2S (sulfane sulfur). The observed elevation of NO is consistent with previous studies, demonstrating that H2S donors can produce such an effect, and that the NO contributes additional beneficial effects to those provided by H2S. In studies in mice and pigs, zofenopril significantly reduced myocardial infarct size and cardiac troponin I levels after ischemia–reperfusion injury. Moreover, in the pig studies, zofenopril also preserved endocardial blood flow after ischemia (17).
ATB-346 (Antibe Therapeutics, Inc.)
NSAIDs are among the most commonly used medications, and their use is increasing with aging populations worldwide. Although effective in reducing pain and inflammation, NSAIDs carry a significant risk for gastrointestinal (GI) bleeding and ulceration, sometimes leading to death. Use of drugs that suppress gastric acid secretion has reduced NSAID-induced ulceration in the upper GI tract, but has little benefit, and may even worsen such damage in the remainder of the GI tract (48). With this in mind, and with extensive preclinical evidence for protective effects of H2S throughout the GI tract, Antibe Therapeutics has developed ATB-346, an H2S-releasing derivative of naproxen, one of the most commonly used NSAIDs. An extensive phase 1 trial of ATB-346 was completed in early 2015, and a phase 2 trial of this drug in osteoarthritis patients was completed in July of 2016.
Although an improvement of GI safety was the primary goal when ATB-346 was designed, the cardiovascular toxicity of NSAIDs has become much more appreciated since the introduction of selective cyclooxygenase (COX)-2 inhibitors (26). Thus, the selection of naproxen as the “base drug” in ATB-346 was made because it has been reported to be the most cardiovascular safe of the NSAIDs (26). NSAIDs can elevate blood pressure and this contributes significantly to cardiovascular adverse events. For this reason, particular attention has been paid to potential blood pressure effects of ATB-346 in clinical trials. Preclinical studies in dogs in which doses of up to 50 mg/kg of ATB-346 were administered, there were no significant changes in systolic or diastolic pressure, pulse or body temperature as compared with those in dogs treated with vehicle (unpublished).
In the phase 1 clinical trial of ATB-346, systolic and diastolic blood pressures were recorded before and several times (1, 3, 6, 12, and 24 h) after each dose of the drug. As shown in Figure 3, there were no significant changes in blood pressure as compared with placebo. Similarly, in the phase 2 clinical trial in osteoarthritis patients (described hereunder), no significant changes in systolic or diastolic pressure were observed.
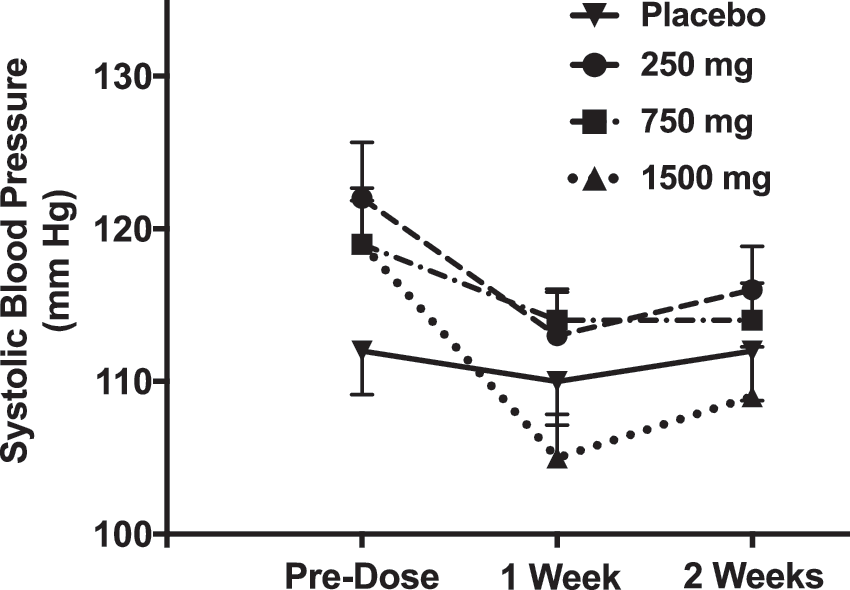
ATB-346 was safe and well tolerated in the single-ascending-dose portion of the phase 1 clinical trial. Doses from 25 to 2000 mg were assessed in groups of six subjects each, while two subjects received placebo. Rates of adverse effects did not differ significantly between the placebo- and ATB-346-treated subjects. Plasma naproxen levels after single administrations of ATB-346 increased in a dose-dependent manner. However, plasma levels of naproxen were consistently lower than what has been observed previously for equimolar doses of naproxen (14). For example, the peak plasma naproxen level after taking a 750 mg tablet of ATB-346 was 12 μg/mL, whereas plasma levels after taking an equimolar tablet of naproxen (500 mg) were ∼60 to 80 μg/mL (14). Also, the plasma half-life of naproxen derived from ATB-346 was significantly extended relative to what is typically seen with administration of naproxen.
The same general patterns emerged in the multiple-ascending-dose portion of the phase 1 clinical trial. As expected, plasma levels of naproxen increased with daily dosing of ATB-346 at 250 and 750 mg daily, reaching a plateau by day 7. Once again, the plasma naproxen levels were considerably lower than would typically be observed with naproxen administration (14), with an apparent increase in the plasma half-life of ATB-346-derived naproxen. In the final cohort of subjects, which was the first time that subjects received twice daily dosing with ATB-346 (750 mg BID), there were some dramatically different results from what had been observed up to that point. The twice daily dosing resulted in much greater increases in plasma naproxen levels. For example, a single dose of 1500 mg of ATB-346 resulted in average plasma naproxen levels 24 h later of 15 μg/mL.
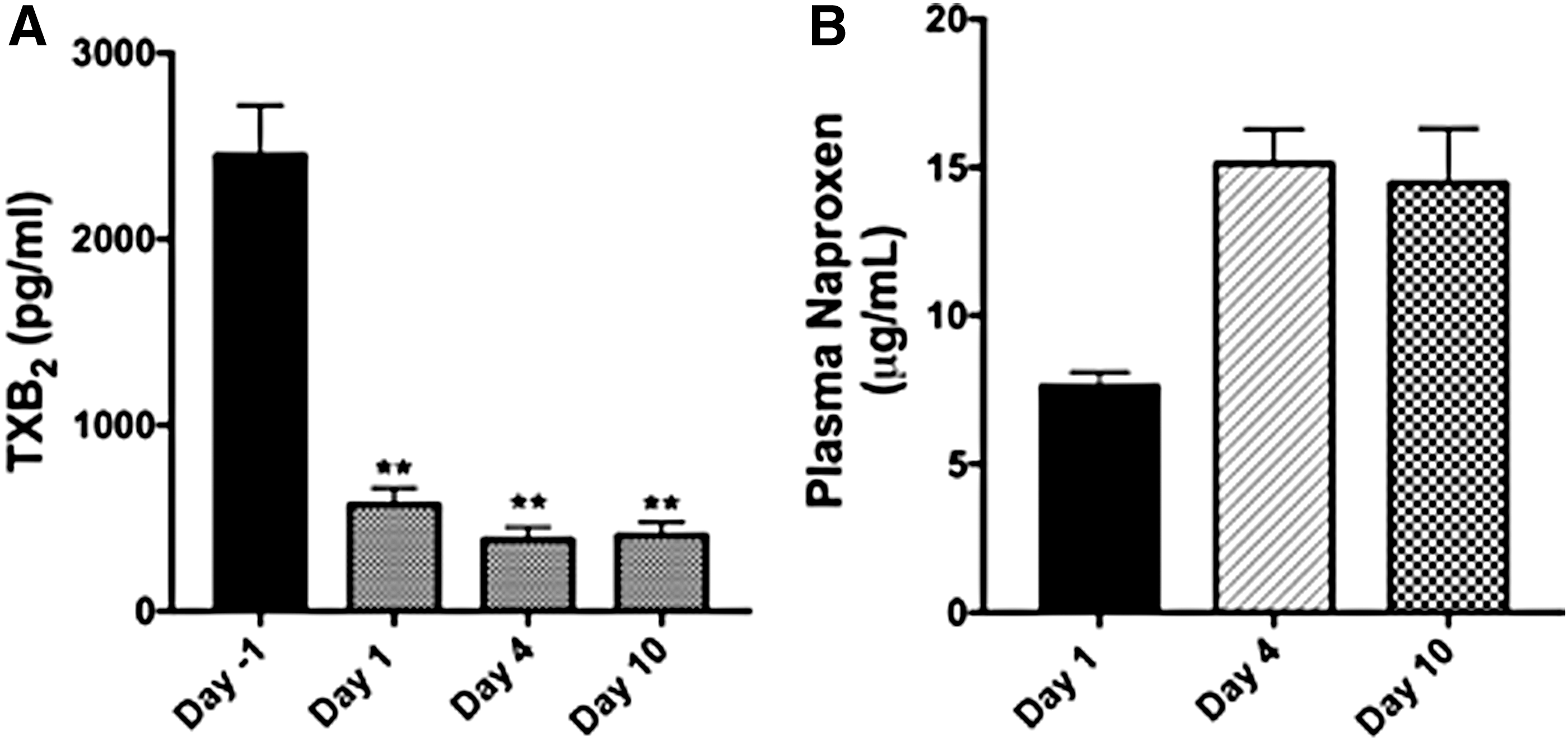
Surprisingly, administration of 750 mg of ATB-346 followed by a second administration of the same dose 12 h later resulted in plasma naproxen levels of 32 μg/mL. Figure 4A illustrates the marked accumulation of ATB-346-derived naproxen when the drug was administered twice daily. This accumulation was almost certainly a key factor in the occurrence of an adverse event in this cohort, which led to discontinuation of a subject's participation. On day 13 of the 14-day treatment period, this subject exhibited significantly raised liver enzymes (alanine transaminase and aspartate transaminase) and upper right quadrant abdominal pain. However, it is noteworthy that this subject was found to have a gallstone lodged in his bile duct (detected upon ultrasonic examination) that was deemed unrelated to the study medication. This subject also had a previous history of hepatitis, which he had denied during the clinical trial enrollment interview. There were two other cases of raised liver enzymes occurring within a week of completion of the study (750 mg twice daily dose of ATB-346). These events were characterized as classic liver responses to NSAID overexposure, and they were self-limiting, ultimately resolved without medical intervention. They are also consistent with the observed accumulation of the drug when administered at the highest dose twice daily (Fig. 4A).
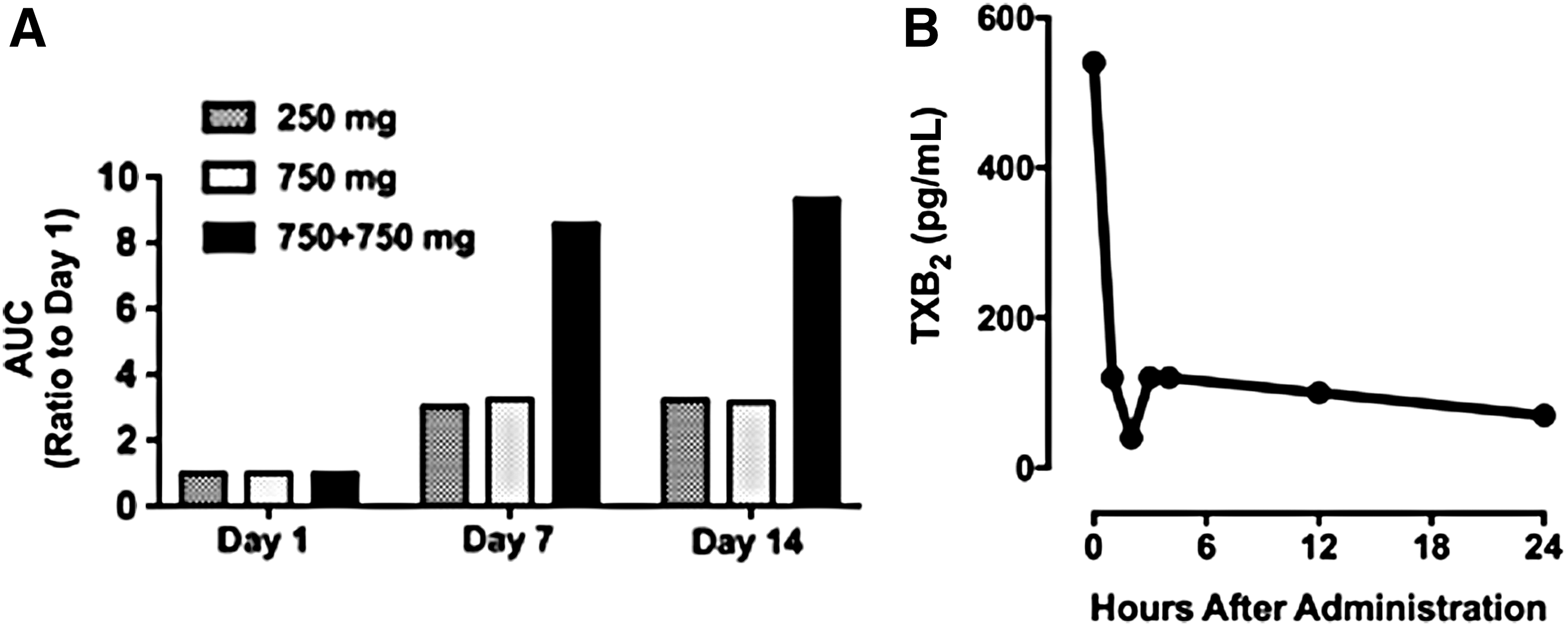
The results of the phase 1 trial demonstrated that naproxen derived from ATB-346 appeared in plasma at much lower concentrations than would be expected if naproxen itself had been administered, and persisted in plasma longer. Preclinical studies in various species had suggested that ATB-346 exerted anti-inflammatory and anticancer effects similar to, or greater than, equimolar doses of naproxen (15, 33, 46). The surprising pharmacokinetic findings in the phase 1 study prompted us to investigate the pharmacodynamics of ATB-346.
Blood samples taken during the trial were assayed for suppression of COX activity (prostaglandin E2 and thromboxane B2). Figure 4B illustrates a typical effect that was observed, which was surprising. A dose of ATB-346 of 250 mg once daily (the molar equivalent of only 166 mg of naproxen) produced a profound suppression of COX activity very quickly (within 1 h), and this effect was maintained for at least 24 h. Even at a dose as low as 75 mg, substantial inhibition of COX (>50%) was observed in the phase 1 trial.
These results strongly suggest, as had been observed in preclinical studies, that ATB-346 itself, and/or a rapidly formed metabolite, strongly inhibits COX activity for up to 24 h in vivo. To further test this hypothesis, a phase 2 clinical trial was performed in which a low dose of ATB-346 (250 mg once daily) was evaluated for its pain-relieving properties in patients with osteoarthritis of the knee. The Western Ontario-McMaster University Arthritis Index (WOMAC) pain score was the primary end point for this study. Subjects were taken off their usual medication for 5 days, and were eligible to participate in the trial if they exhibited an increase in the WOMAC score (0–20 scale) of at least 1 unit. One day after the initial score was recorded, the patients began taking ATB-346 each day for 10 days. The pain scores were recorded on days 1, 4, and 10. Blood samples for measurement of COX activity were drawn before starting the trial and on days 1, 4, and 10 of treatment.
This was an open-label trial, so there was no placebo or active comparator. However, there is extensive published experience with drugs such as celecoxib and naproxen in trials of this type, with this same end point. After 1 week of treatment twice daily, these drugs have consistently produced a mean decrease in the WOMAC pain scale of ∼4 units (7, 49). In the phase 2 trial of ATB-346, the mean decrease in the WOMAC pain scale after 4 days of treatment was 4.3 ± 1.0 (p < 0.01), whereas after 10 days of treatment, it was 7.6 ± 1.5 (p < 0.001) (Fig. 5). There were also significant decreases in other aspects of the WOMAC arthritis index: the scores for “stiffness” and “difficulty performing daily activities” were significantly reduced by ATB-346 on days 4 and 10 of treatment (Fig. 5).
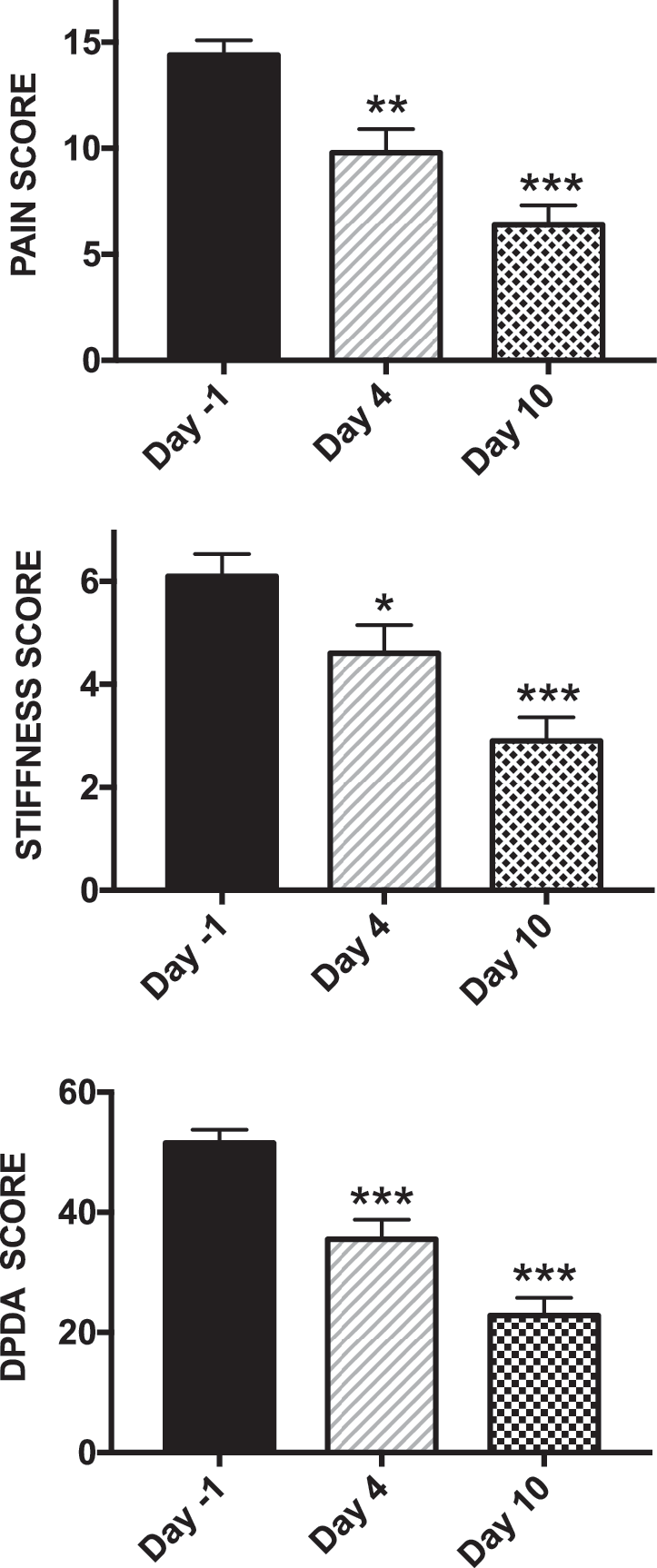
Consistent with the observations from the phase 1 trial, ATB-346 produced a profound inhibition of whole blood COX activity on the first day of treatment (Fig. 6). Plasma naproxen levels on day 1 of treatment averaged ∼8 μg/mL, and rose to ∼15 μg/mL on days 4 and 10 of treatment. Whole blood COX activity remained consistently and profoundly suppressed throughout the treatment period. This confirmed the phase 1 trial results that suggested that ATB-346 could suppress COX activity for 24 h at a dose much lower than had been anticipated from preclinical studies (4, 46). No elevations of liver enzymes or other adverse effects were observed in this phase 2 clinical trial.
Summary
In the years after the discoveries of roles of H2S in neuroprotection and vascular smooth muscle relaxation (1, 51), there was a burst of research activity aimed at developing novel H2S donors (12, 25, 30, 41, 45, 47). The potential therapeutic targets were broad, but many were focused on cardiovascular and GI targets, as well as anti-inflammatories, neuroprotectants, and chemopreventative agents. Concerns about H2S-related toxicity were no doubt an impediment to drug development in this field, but as more data emerged on the many physiological roles of H2S, these concerns have waned.
The endogenous pathways for H2S production have become well understood (1, 41, 45, 51), as have the contribution of the microbiome to “systemic” H2S levels (19, 20), and the important roles of H2S in driving mitochondrial respiration (40). Additional roles in promoting wound healing, reducing inflammation, protecting tissues from ischemia–reperfusion injury, and preventing cancers pointed to significant potential utility of H2S-releasing drugs. There is also potential for selective inhibitors of H2S synthesis as treatments for certain types of cancers (21). Although still in its infancy, there is now some solid evidence that H2S-based therapeutics have considerable promise for a number of indications. This review has focused on the few drugs for which clinical data are presently available. It seems very likely that the number of such drugs will increase substantially in the years to come.
Footnotes
Acknowledgments
This work was supported in part by a grant to Dr. Wallace from the Canadian Institutes of Health Research. The authors are grateful to Dr. Linda Vong and Dr. Philip Sherman for their assistance.