
Abstract
Significance:
Mycothiol (MSH, AcCys-GlcN-Ins) is the main low-molecular weight (LMW) thiol of most Actinomycetes, including the human pathogen Mycobacterium tuberculosis that affects millions of people worldwide. Strains with decreased MSH content show increased susceptibilities to hydroperoxides and electrophilic compounds. In M. tuberculosis, MSH modulates the response to several antituberculosis drugs. Enzymatic routes involving MSH could provide clues for specific drug design.
Recent Advances:
Physicochemical data argue against a rapid, nonenzymatic reaction of MSH with oxidants, disulfides, or electrophiles. Moreover, exposure of the bacteria to high concentrations of two-electron oxidants resulted in protein mycothiolation. The recently described glutaredoxin-like protein mycoredoxin-1 (Mrx-1) provides a route for catalytic reduction of mycothiolated proteins, protecting critical cysteines from irreversible oxidation. The description of MSH/Mrx-1-dependent activities of peroxidases helped to explain the higher susceptibility to oxidants observed in Actinomycetes lacking MSH. Moreover, the first mycothiol-S-transferase, member of the DinB superfamily of proteins, was described. In Corynebacterium, both the MSH/Mrx-1 and the thioredoxin pathways reduce methionine sulfoxide reductase A. A novel tool for in vivo imaging of the MSH/mycothiol disulfide (MSSM) status allows following changes in the mycothiol redox state during macrophage infection and its relationship with antibiotic sensitivity.
Critical Issues:
Redundancy of MSH with other LMW thiols is starting to be unraveled and could help to rationalize the differences in the reported importance of MSH synthesis observed in vitro versus in animal infection models.
Future Directions:
Future work should be directed to establish the structural bases of the specificity of MSH-dependent enzymes, thus facilitating drug developments. Antioxid. Redox Signal. 28, 487–504.
Introduction
L
In most eukaryotes and Gram-negative bacteria, the tripeptide glutathione (GSH, γ-

Structure and Physicochemical Properties
MSH identification and structure resolution
MSH has been described in different Streptomyces species by high-performance liquid chromatography (HPLC) analysis of intracellular LMW thiols after their derivatization using monobromobimane, as a main peak eluting at t = 17 min using a reverse phase chromatographic method and was then named U17 (88). Previously, an unknown thiol designated as U25 (because of the elution of its monobromobimane derivative at 25 min using a different chromatographic method) was detected as a major thiol compound in the Actinomycetes Rhodococcus roseus and Streptomyces griseus (32). The fact that both U17 and U25 coelute when analyzed by the same chromatographic method suggested that they were the same compound (88). The production of MSH was later confirmed to be restricted to Actinomycetes, and among them, intracellular concentrations largely varied, being very abundant in mycobacteria, whereas it could not be detected in some of the actinobacterial species analyzed (81). In most Actinomycetes, GSH was undetectable. As exceptions, Rubrobacter radiotolerans and Rubrobacter xylanophilus produce GSH as main LMW thiol (58). In turn, Rhodococcus sp. AD45 and Nocardiopsis flava ATCC 29533 produce both high levels of MSH and significant amounts of GSH (58, 81, 147). Moreover, the MSH content changed during the different phases of bacterial growth, being in Streptomyces practically constant during the growth phase, while it increased greatly during the stationary phase (88). Comprehensive analysis of MSH isolated from different bacteria allowed to determine its components (N-acetyl
Acidity constant
Most of the biologically relevant reactions in which thiols participate are bimolecular nucleophilic substitutions (SN2), wherein the deprotonated forms of the thiols (thiolate anions) are the reactive species. Those include thiol–disulfide exchange reactions (18, 44): most one- as well as two-electron oxidations of thiols that yield thiyl radicals (RS•) and sulfenic acids (RSOH) (39, 105, 142), respectively, and thiol alkylation to produce thioethers (also referred as sulfides) (10, 43).
In those reactions wherein thiolate is the reactive species, the acidity constant of LMW thiols (KaSH) is an important determinant for the reactivity at physiological pH, because the apparent rate constant of the reaction at a given pH (k′) is determined by the pH-independent rate constant (k) (the reactivity of the thiolate) times the fraction of thiol as thiolate at that pH [Eq. (1)]:
Taking advantage of the thiolate anion absorption at 232 nm, the value of the pKaSH in MSH was recently determined as 8.76 ± 0.02 (Fig. 1) (129). This value is approximately 0.4–0.5 pH units higher than that of the first microscopic thiol pKa in cysteine * (8.38), probably because of the lack of a protonated amino group in MSH whose positive charge, in proximity to the sulfur, contributes to thiolate anion stabilization in aminothiols. It is slightly more acidic than the ammonium form of GSH, which holds two carboxylate groups (8.93). The pKa value of the thiol group in a modified form of MSH lacking the inositol aglycone is 8.91 ± 0.02, and this higher value was ascribed to the loss of intramolecular hydrogen bonds between inositol hydroxyls and the thiolate group when compared with nonmodified MSH (129). The pKaSH value of MSH indicates that 4.2% of its thiol group would be deprotonated at pH 7.4, in comparison with 2.9% of GSH and 11% of cysteine. It should be considered, however, that MSH concentrations in some Actinomycetes are very high, particularly in mycobacteria wherein it has been estimated to be in the range of 1–8 mM (86), indicating that the available MSH thiolate concentration at physiological pH would be ∼100 μM, being the most abundant LMW thiolate in these bacteria.
Thiolate nucleophilicity and pH-independent reactivity in LMW and some protein thiols show a Brønsted correlation with their conjugated acid pKa as indicated by Equation (2):
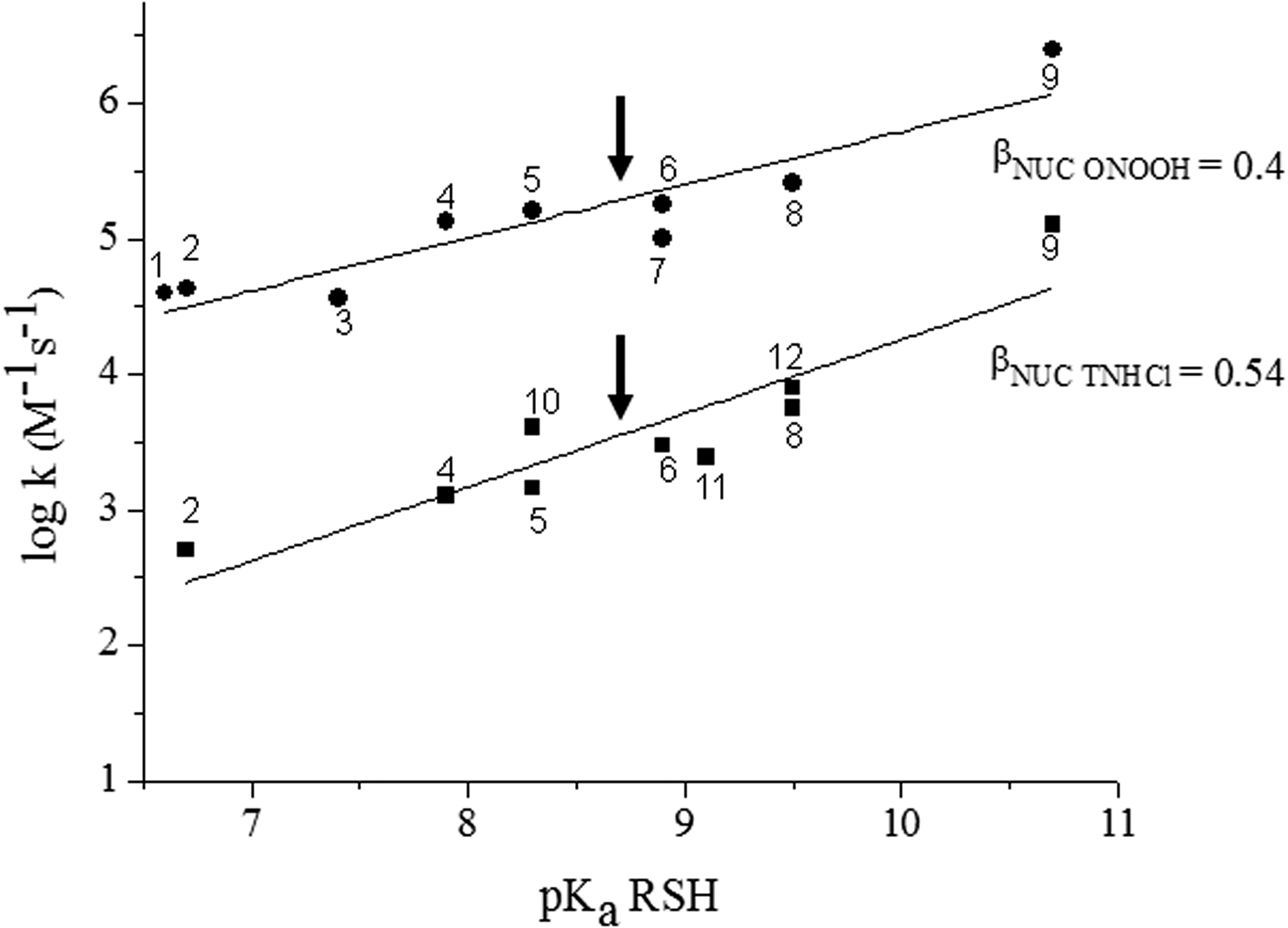
where βNuc is the nucleophilicity constant for the reaction of interest, thus suggesting that MSH nucleophilicity and pH-independent reactivity with two-electron oxidants [such as H2O2, βNuc = 0.27 (106); peroxynitrous acid, βNuc = 0.4 (143); taurine chloramine, (TNHCl) βNuc = 0.54 (37)], disulfides [βNuc values in the 0.2–0.6 range (18, 127)], and alkylating agents [βNuc values in the 0.2–0.52 range (106)] would be higher than those of cysteine and similar to those of GSH. Indeed, the rate constant of the reaction of MSH with H2O2 was reported as 0.60 ± 0.04 M −1s−1 at pH 7.4 and 25°C (103), which considering MSH thiol pKa translates into a pH-independent rate constant of 14.4 M −1s−1, in close agreement with the expected value (106). However, experimental data regarding the other chemical, nonenzymatic reactions of MSH are lacking so far. Figure 2 illustrates the expected reactivity of MSH with ONOOH and TNHCl [oxidants that can be formed by activated inflammatory cells during infection (5, 27)] calculated from MSH pKa value and reported Brønsted correlations.
Standard redox potential
The standard redox potential of MSH (E°′MSSM/MSH = −230 mV) (Fig. 1) was recently calculated relative to the standard redox potential of bacillithiol (BSH) [E°′BSSB/BSH = −221 mV (128)] from equilibrium mixtures of MSH/mycothiol disulfide (MSSM) and BSH/BSSB, measuring the concentration of the thiol/disulfide components at equilibrium by proton nuclear magnetic resonance (NMR) and using the Nernst equation (129). It is 10 mV higher than the standard redox potential of GSH (E°′GSSG/GSH = −240 mV) as shown in Figure 1 (124).
The thermodynamic parameter E°′MSSM/MSH is interesting as a means to predict the direction of electron flow from this redox pair to others, particularly when real concentrations of the different reacting species along with pH and temperature values are known, thus allowing real free energy calculations. However, it is the magnitude of the activation-free energy (and not free energy) that dictates the kinetics of reactions; because reactions involving MSH are mostly enzymatic processes, the kinetics of these reactions depends on the catalytic efficiency of the enzyme(s) involved (38).
Autoxidation
Copper-catalyzed MSH autoxidation was sevenfold slower than those of GSH or N-acetyl cysteine and ∼30-fold slower than cysteine autoxidation. The result was ascribed to a lower copper-binding ability of the thiol group of MSH because of its carbohydrate components that block the carboxyl group (83) (Fig. 1).
Biosynthetic Route
The synthesis of MSH consists of five steps revised in Jothivasan and Hamilton (59) and Newton et al. (84). In brief, 1L-myo-inositol-1-phosphate (Ins-P), which is produced from glucose 6-phosphate by inositol phosphate synthase (Ino1) (8), reacts with uridine diphosphate N-acetyl glucosamine (UDP-GlcNAc) to form 1-O-(2-acetamido-2-deoxi-α-
In M. tuberculosis and some other Actinomycetes, the genes encoding the enzymes responsible for MSH biosynthesis (except for the phosphatase MshA2) have been identified. The corresponding proteins were structurally and functionally characterized and have received considerable attention because they could constitute antimicrobial targets [for a comprehensive review on the structure and mechanisms of the MSH biosynthetic enzymes, see Fan et al. (33)]. Studies in Mycobacterium smegmatis indicated that the genes mshA and mshC are essential for MSH synthesis (90, 112). On the contrary, lack of functional mshB did not totally prevent MSH formation, implicating alternative routes of deacetylation (111), while lack of MshD activity caused decreased MSH levels and high production of novel thiols such as N-formyl-Cys-GlcN-Ins and N-succinyl-Cys-GlcN-Ins, derivatives of Cys-GlcN-Ins that could be reduced by mycothiol disulfide reductase (Mtr) or through thiol disulfide exchange reactions (16, 92).
In M. smegmatis, the genes involved in MSH biosynthesis are nonessential, although growth of mshA and mshC null-mutants was slightly slower than that of wild-type strains (161) and a reduced MSH content was associated with increased sensitivity to oxidative and acid stress, and to alkylating agents (110). Compensation mechanisms for the loss of MSH in this bacterium include the overproduction of ergothioneine (2-mercaptohistidine trimethylbetaine, ERG, † a less characterized LMW thiol synthesized by Actinomycetes and fungi) and increased expression of the organic hydroperoxide resistance protein [Ohr, a thiol-dependent peroxidase that reduces organic hydroperoxides using dihydrolipoamide as reducing substrate (3, 28, 29, 139)].
M. tuberculosis has no significant homologues for M. smegmatis Ohr (139). In the former bacterium, MSH synthesis was essential for growth in Erdman strains (122), in agreement with transposon site hybridization studies (123). MSH was also essential for growth of H37Rv or CDC 1551 strains in the absence of added catalase (151), but not for H37Rv strain in catalase-supplemented medium, indicating that MSH would not be essential in the presence of alternative routes of H2O2 (or peroxynitrite) detoxification as offered by catalase (42). Addition of oleic acid to the culture medium (as catalase, oleic acid is a component of the culture medium OACD frequently used for growth of these bacteria) had controversial results that could depend on the enzyme of MSH biosynthesis that was mutated (16, 42). In general, decreased MSH content is associated with higher susceptibility to oxidative and acidic stress (16, 17, 94). Furthermore, inhibitors of enzymes involved in MSH synthesis caused M. tuberculosis to die (48, 51, 85). To note, growth in the lungs of immunocompetent mice was only slightly defective in H37Rv mshA mutants lacking MSH, indicating the induction of compensatory mechanisms (151).
It was recently reported that levels of ERG substantially increased in M. tuberculosis, lacking functional mshA (118). Like MSH, ERG was able to protect M. tuberculosis from different oxidants and was important to maintain bioenergetic homeostasis. Although the role of ERG has only started to be unraveled, it has been reported to have antioxidant (2, 7, 126) and modulatory actions on immune host cell responses (108, 163) and seems to play overlapping but not equal roles as compared with MSH in the bacteria (118). To note, MSH/MSSM ratio was not affected in bacteria lacking ERG. ERG biosynthesis was recently reported to be critical for M. tuberculosis survival in both macrophages and mice (118). ERG reacts with different ROS (40, 50), and seems to be important for oxidant detoxification (in the form of exogenously added hydroperoxides as well as following exposure to redox cycling drugs), particularly in mycobacteria lacking MSH (118, 121). The role of ERG in protecting against antibiotics is controversial (118, 121), and if present, the mechanisms of such a protection are far from clear, probably involving a combination of ERG's antioxidant actions, immunomodulatory properties, and effects on the bacterial metabolism (108, 118, 163).
Understanding ERG function in mycobacteria and how it could overlap with MSH in some actions are not trivial. ERG exists in a tautomeric equilibrium between thiol and thione forms that favor the latter, thus sharing only some of other LMW thiol chemical reactions (50). With an E°′ of −60 mV, it is more resistant to autoxidation than MSH or GSH (23). In mycobacteria, concentration levels varied depending on the strain, culture medium, and growth phase. For example, Saini et al. (118) reported M. tuberculosis levels that ranged from 40 to >300 ng per 108 cells, which considering the mean reported cellular volume (∼0.3 fl) (162) translates into mM concentrations. ERG synthesis is regulated by the iron–sulfur redox regulator WhiB3, which is involved in maintaining bioenergetic homeostasis as well as in regulating gene expression in response to acidification, including the synthesis of Mtr (as indicated in the section “Reduction of MSH disulfide” 75, 118, 119). To date, no enzymatic pathway of ERG reduction has been identified. ERG seems to be actively secreted out of the cell (121). Thus, it would be interesting to know whether it can act as an extracellular antioxidant protecting M. tuberculosis from macrophage-derived reactive species that poorly permeate through membranes, such as carbonate radical (formed e.g., from peroxynitrite in the presence of CO2) (107) or taurine chloramine (derived from hypochlorite reaction with taurine) (47). Moreover, bearing in mind that ERG has immunomodulatory properties, its secretion might contribute to mycobacterial survival by modulating host immune mechanisms (108, 163).
Increased ERG synthesis may not be the only mechanism of survival of M. tuberculosis lacking MSH. Host GSH is likely available to the bacterium as an alternative LMW thiol, as was previously reported for Francisella tularensis (114). To note, a recent report indicated that decreased GSH in host macrophages correlated with increased M. tuberculosis MSH oxidation (21).
Regulation of MSH Biosynthesis
MSH synthesis is a regulated process. In Streptomyces coelicolor, a model antibiotic producing Actinomycete, several of the genes involved in MSH synthesis are regulated by sigma R (σR), an RNA polymerase sigma factor, and the antisigma factor RsrA. σR was first described in this bacterium as a sigma factor required for the induction of the thioredoxin reductase-thioredoxin operon by different oxidants (60, 98). RsrA is a redox-sensitive zinc-binding antisigma protein that in its reduced state specifically interacts with σR to form a 1:1 complex inhibiting its transcriptional activation activity (61, 69). Different one- and two-electron oxidants cause the oxidation of RsrA thiol groups and zinc release (9, 159), causing a conformational change that precludes interaction with σR, thus promoting transcription of target genes. Although sites of Zn binding and Cys residues involved in disulfide bond formation are controversial, it appears that the former involve Cys11, His37, Cys41, and Cys44, and disulfide formation between Cys11 and Cys44 results in zinc release (164). It should be noted that the response process of RsrA is probably more complex involving several steps (159). Mutants lacking functional σR have decreased MSH levels (97). Regulation of MSH synthesis by σR/RsrA occurs directly at the level of mshA gene, whereas indirect effects on mshB, C, and D gene expression have also been reported (87). In turn, MSH-deficient mutants exhibited increased expression of the σR/RsrA regulon (84, 100), because MSH, as well as thioredoxin/thioredoxin reductase, participates in RsrA reduction, promoting its binding to σR (87), therefore, providing a homeostatic feedback loop. More recently, many new genes of the σR regulon were identified in S. coelicolor.
Comparative genomics analysis revealed the existence of a core SigR regulon conserved across 42 selected Actinomycetes (63). Indeed, M. tuberculosis has a σH/RshA system similar to σR/RsrA of S. coelicolor (87). Moreover, long-term studies of the effects of σH on global transcription in M. tuberculosis revealed that several genes involved in sulfate acquisition and metabolism including cysteine synthesis were activated (74). In addition, other transcriptional factors are also involved in the regulation of MSH biosynthesis/reduction in this pathogen. For instance, the LacI-type regulator IpsA, conserved among mycobacteria and corynebacteria, activates Ino1 that encodes Ino1 required for the synthesis of Ins-P, precursor of MSH and of components of the cell wall (12).
Reduction of MSH Disulfide
MSSM is reduced by Mtr, a member of the class I flavoprotein disulfide reductases, a family of enzymes that also includes GSH reductase, among others (6). These are homodimeric proteins with a noncovalently but tightly bound flavin adenine dinucleotide (FAD) as well as one redox-active disulfide per subunit, which catalyze the reduction of their respective disulfide-bonded substrates. They share a characteristic histidine–glutamate ion pair essential for catalysis, and conserved regions for FAD (cofactor) and NADPH (substrate) binding (101), as well as similar catalytic mechanisms (102). Mtr catalyzes the reduction of MSSM in an NADPH-dependent manner, by a bisubstratic ping-pong kinetic mechanism. The reductive half-reaction involves the electron transfer from NADPH to the active site disulfide via FAD, which generates a two-electron reduced enzyme. In the oxidative half-reaction, the thiolate form of one of the redox active Cys in the reduced enzyme attacks MSSM to generate a mixed enzyme-substrate disulfide, releasing the first thiol product, MSH. The final step involves regeneration of the enzyme disulfide and release of the second MSH (102). Unlike other members of this family, wherein the oxidative half-reaction is usually rate limiting, rates for reduction and oxidation of Mtr are similar. However, it is not known whether this difference provides a catalytic advantage under physiological conditions (102).
In M. tuberculosis H37Rv strains, mtr was reported as essential according to Himar1-based transposon mutagenesis (123), whereas a Tn5370 transposon mutant in the mtr gene (also named as gorA) was viable (73).
Expression of Mtr of M. tuberculosis is regulated by the iron–sulfur containing intracellular redox sensor WhiB3, which is critical for pH-adaptive responses in the bacterium. Its mode of action has been recently described: modest increases in external acidity causes a significant reductive shift in EMSH of M. tuberculosis, which promotes WhiB3-dependent changes in gene expression (75).
To note, M. tuberculosis lacking MSH showed growth restriction in acidic medium and it was recently demonstrated that increased MSH levels protected Corynebacterium glutamicum (a nonpathogenic member of Actinomycetes and an industrial workhorse for amino acid and nucleotide production) from oxidative stress caused by low-pH conditions (16, 70). The overexpression of Mtr in C. glutamicum caused increased resistance to oxidants, bactericidal antibiotics, alkylating agents, and heavy metals compared with the wild-type strain. Mtr overexpression also leads to a major decrease in ROS levels and an increase in reduced MSH levels as well as in the activity of several antioxidant proteins (133). Moreover, Mtr has been shown to be involved in C. glutamicum resistance to arsenate, which is consistent with the role of MSH in the detoxification of this compound (see in section “Chemical Detoxification”) (95). In C. glutamicum, mtr was upregulated under cadmium stress (34) and in antisigma factor rshA deletion mutants (19).
In addition to Mtr, a novel mechanism of MSH recycling was described in M. smegmatis and M. tuberculosis. It involves a membrane-associated multiprotein complex (MRC) composed of iron-dependent superoxide dismutase A (SODA), an integral membrane protein (DoxX) and a predicted thiol oxidoreductase (SseA). Cells lacking a functional MRC showed a lower MSH/MSSM ratio and increased lipid peroxidation. These effects were reverted by sulfur supplementation (under the form of cysteine, hydrogen sulfide, or sulfate), probably involving increased MSH synthesis (78). The mechanism of MSH recycling by MRC is still unknown.
Protein S-Mycothiolation, a Reversible Post-Translational Modification
Upon oxidative challenge, protein S-glutathionylation (formation of a mixed disulfide between a protein cysteine residue and GSH) is a well-known redox-switch mechanism that protects protein cysteine thiols from irreversible oxidation and regulates protein activity (30). However, until very recently, there was no evidence of protein cysteine S-mycothiolation or its implication on the protein activity, nor on how protein S-mycothiolation is regulated. Protein S-mycothiolation (formation of a mixed disulfide between a protein cysteine residue and MSH) was first observed in C. glutamicum. Under sublethal oxidative conditions (15 min after 180 μM of sodium hypochlorite stress), 25 proteins were identified to be S-mycothiolated (25). The functions of these proteins are diverse, ranging from carbohydrate metabolism (maltodextrin phosphorylase, MalP) to the synthesis of inositol 3-phosphate (Ino1) and methionine (methionine synthase E, MetE) or methionine sulfoxide reduction (methionine sulfoxide reductase A, MsrA), as well as hydroperoxide detoxification (mycothiol peroxidase, Mpx, and thiol peroxidase, Tpx). The details of the S-mycothiolation mechanism were further investigated in some of these proteins, with special focus on MsrA, Mpx, and Tpx.
MsrA, the enzyme responsible for the reduction of the S-isomer of methionine sulfoxide, was studied using C. glutamicum and C. diphtheriae enzymes, which have 57% sequence identity and four cysteine residues located in similar positions. Although the study of Si et al. on C. glutamicum MsrA only detected S-mycothiolation of the nucleophilic Cys56 (132), Tossounian et al. showed that, upon H2O2 challenge, all four cysteines of C. diphtheriae MsrA are S-mycothiolated, and upon methionine sulfoxide reduction, the three catalytic cysteines (Cys52, Cys206, and Cys215) are S-mycothiolated (141). Furthermore, in the C52S mutant (Cys56 in C. glutamicum MsrA), the same methionine sulfoxide treatment did not lead to S-mycothiolation, indicating that there is an MSH redox relay, starting from Cys52 toward the other catalytic cysteines (141). It is very likely that the same mechanism happens in C. glutamicum MsrA, because of their high identity, but the S-mycothiolation on other cysteines was not investigated (132). So far, similar interactions between MSH and methionine sulfoxide reductase B (MsrB), the enzyme responsible for the reduction of the R-isomer of protein methionine sulfoxides that is expressed in most organisms, including Actinomycetes, have not been reported (31, 68, 136).
Mpx is a member of the Cys-containing GSH peroxidase family whose members form an intramolecular disulfide upon peroxide reduction, which is usually reduced by thioredoxin (138). The in vivo S-mycothiolation of its peroxidatic cysteine (Cys36) was in vitro confirmed in two parallel studies wherein H2O2 was used as oxidant, and both demonstrated that Cys36-Cys79 intramolecular disulfide formation competes with Cys36 S-mycothiolation (103, 131).
In the case of Tpx from C. glutamicum, which is an atypical two-Cys peroxiredoxin (Prx), in vivo S-mycothiolation was found on both the peroxidatic (Cys60) and the resolving (Cys94) cysteines (25). In addition, in vitro S-mycothiolation in the presence of H2O2 showed that the nonconserved Cys81 was also S-mycothiolated and that Tpx S-mycothiolation abrogates its thioredoxin peroxidase activity (25). The degree of C. glutamicum Tpx oxidation determined its oligomerization state (as indicated by native and nonreducing sodium dodecyl sulfate polyacrylamide gel electrophoresis [SDS-PAGE] techniques) as well as its activity. At the lowest tested H2O2 concentrations, C. glutamicum Tpx was monomeric and functioned as a thioredoxin peroxidase. However, at higher H2O2 concentrations, the enzyme dimerized, got S-mycothiolated, and had no thioredoxin peroxidase activity. When exposed to even higher levels of H2O2, the Tpx peroxidatic cysteine was overoxidized, leading to a tetramer with chaperone activity (130).
It is very likely that other proteins are S-mycothiolated under oxidative conditions, which were not yet detected in current S-mycothiolome studies (25). Such was the case for M. tuberculosis alkyl hydroperoxide reductase E (AhpE), a 1-Cys Prx in which MSH plays an important role during the reductive recycling mechanism (54). At pH 7.4 and 25°C, sulfenylated AhpE (AhpE-SOH) is S-mycothiolated with a rate constant of 237 M −1s−1, which is higher than its overoxidation rate constant (40 M −1s−1), indicating that MSH likely prevents AhpE overoxidation (AhpE-SO2H) (54, 103). Similar rate constants were obtained for the S-mycothiolation of C. glutamicum Mpx-SOH (620 M −1s−1) (103), and for S-glutathionylation of mammalian Prx2 (500 M −1s−1) (104). What all these proteins have in common is that S-mycothiolation generally occurs after the formation of a transient sulfenic acid, subsequent to the reduction of hypochlorite, peroxide, or methionine sulfoxide. Under in vitro reducing conditions, these protein cysteines are not S-mycothiolated. However, Mpx and Tpx were detected S-mycothiolated in vivo in the absence of external oxidant, so most likely the S-mycothiolation reflects the reactivity of these Tpxs to endogenously produced hydroperoxides (≥105 M −1s−1 reactivity with H2O2) (25, 57, 103). In addition, in the case of methionine sulfoxide reduction, the MSH is transferred between the catalytic cysteines via thiol disulfide exchange reactions (141).
Among the other proteins that have been found mycothiolated in vivo, MetE deserves special attention. The enzyme catalyzes the transfer of a methyl group from 5-methyltetrahydrofolate to homocysteine to form methionine, being vitamin B12 independent and essential according to Himar1-based transposon mutagenesis in H37Rv strains of M. tuberculosis (123). S-mycothiolation protects the enzyme from oxidants generated when C. glutamicum was exposed to acidic conditions (70).
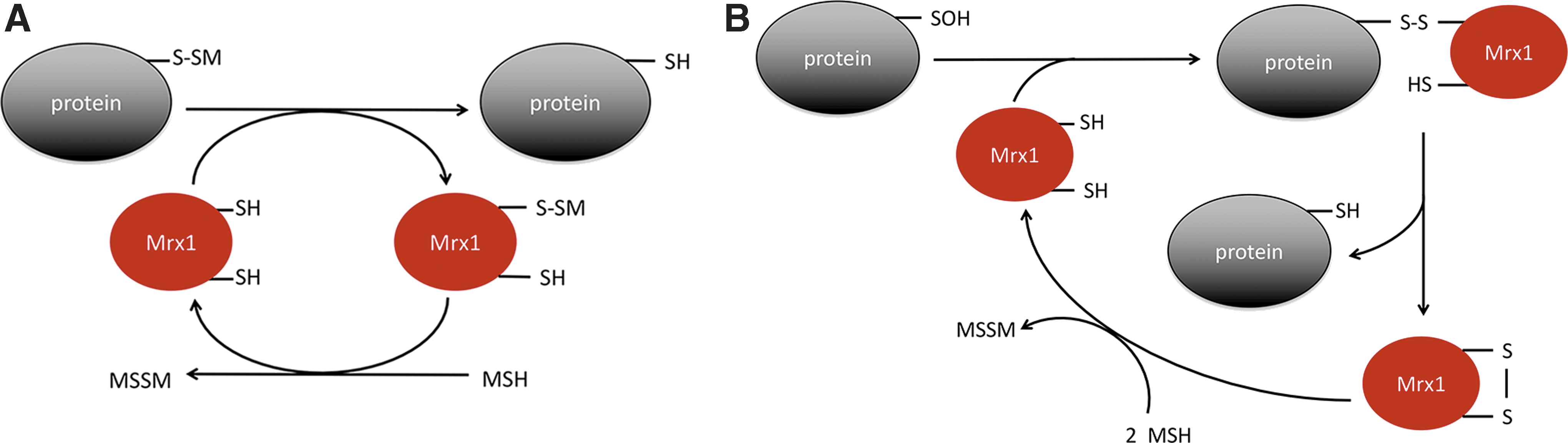
Protein S-mycothiolation is a reversible modification in which mycoredoxin-1 (Mrx-1), a glutaredoxin homologue, plays a central role, either using a monothiolic or a dithiolic mechanism (Fig. 3). Mrx-1 is an LMW thiol redoxin that is exclusively linked to the MSH electron transfer pathway, whose main function is to reduce protein–MSH mixed disulfides (148). Structures of reduced and oxidized Mrx-1 revealed a thioredoxin fold with a CGYC catalytic site motif (148). Mrx-1 activity was demonstrated with S-mycothiolated C. glutamicum Mpx and Tpx, C. glutamicum and C. diphtheriae MsrA and M. tuberculosis AhpE (25, 54, 103, 132, 141). Reduction of S-mycothiolated proteins operates via a monothiol mechanism, as mutation of the second cysteine of Mrx-1 does not affect its activity (54, 131, 148). Mrx-1 reduction of S-mycothiolated Mpx and MsrA is fast (104–105 M −1s−1), indicating reversibility of the modification (103, 131, 132). In the case of MsrA, there are different Mrx-1-mediated reduction mechanisms proposed: although Si et al. suggested that Mrx-1 reduces the S-mycothiolated Cys56 in C. glutamicum MsrA, Tossounian et al. showed that Mrx-1 is only able to reduce S-mycothiolated Cys206 (Cys204 in C. glutamicum MsrA) (132, 141). In the case of Mpx, not only Mrx-1 but also thioredoxin reduces S-mycothiolated Mpx. In this case, thioredoxin uses a dithiol mechanism in which thioredoxin attacks the sulfur of MSH in the mixed disulfide. A molecule of MSH is transferred to thioredoxin and, subsequently, reduced MSH is released after the formation of a disulfide on thioredoxin (103). This alternative reduction mechanism occurs, however, at a twofold slower rate (103). This finding opens the possibility that Trx reduces S-mycothiolated proteins under conditions in which the Mrx-1 pathway is inhibited, for instance, when reduced MSH is limiting. In agreement with this possibility, yeast S-glutathionylation levels are controlled by thioredoxins and not by glutaredoxins (46). In the case of M. tuberculosis AhpE, not only Mrx-1 reduces S-mycothiolated AhpE via a monothiol mechanism, but also sulfenylated AhpE can be reduced by Mrx-1 alone via a dithiol mechanism, and this with a rate constant of 1.6 × 103 M −1s−1 (54), which is 10-fold faster than the S-mycothiolation rate constant (Fig. 4). However, as the MSH concentration in the cell is in the millimolar range, the Mrx-1 cellular concentration (which is currently unknown) must be at least 50 μM to be a relevant competitor (54).
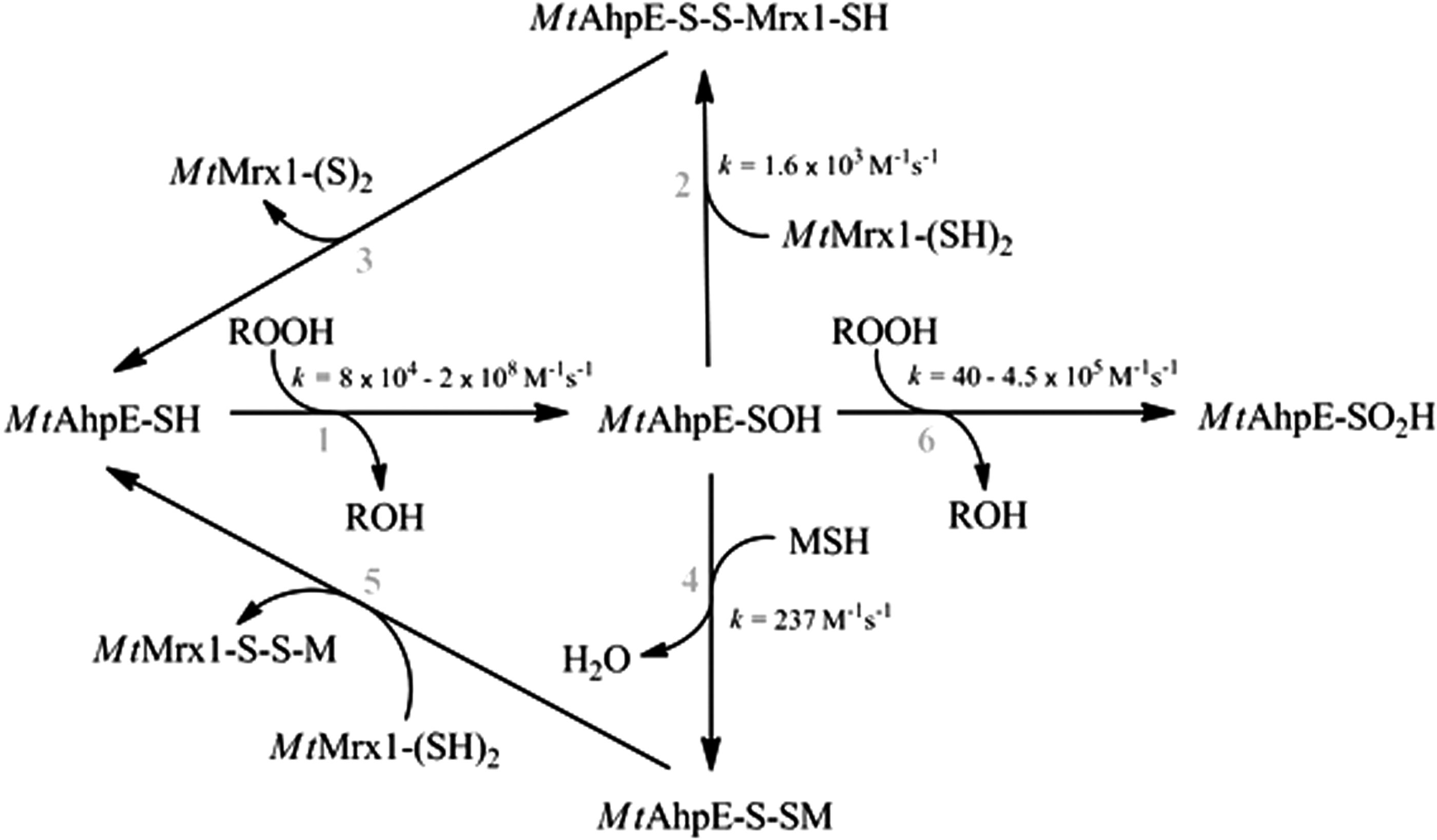
A recent study has proposed that MSH alone can reduce S-mycothiolated AhpE after 20 min incubation time (66). However, in the absence of Mrx-1, we observed no electron transfer between AhpE and the MSH/Mtr/NADPH pathway (54). We consider that this mechanism, although possible, is kinetically irrelevant. Indeed, the rate constant of Mpx demycothiolation by MSH alone was estimated to be 3.6 M −1s−1, which is 104-fold lower than for the same reaction in the presence of Mrx-1 (103). A model of the structure of S-mycothiolated AhpE was recently proposed (66).
Overall, these data provide a molecular link between MSH deficiency and the increased bacterial susceptibility to hydroperoxides (16, 112), and explain the fact that exposure of Mycobacterium bovis BCG in 0.9% saline solution to sublethal concentrations of H2O2 caused MSH oxidation (146), bearing in mind the slow uncatalyzed reactions already indicated. Thus, the protective effects of MSH against hydroperoxides are indirect, through MSH-dependent peroxidases. Indeed, the fact that M. smegmatis mutants devoid of MSH overexpressed Ohr had already suggested the existence of MSH-dependent peroxidases in the wild-type strains (139).
Among the different thiol-dependent peroxidases expressed in Actinomycetes, an MSH/Mrx-1-dependent catalytic activity was proven for M. tuberculosis AhpE and C. glutamicum Mpx, and considering the data already presented, it is also very likely in the case of Tpx (although studies under catalytic conditions are still lacking, and the enzyme can be efficiently reduced by thioredoxins B and C/thioredoxin reductase/NADPH). MSH plus Mtr/NADPH did not reduce the oxidized form of alkyl hydroperoxide reductase C (AhpC) from M. tuberculosis, for which two reducing systems, namely thioredoxin C/thioredoxin reductase/NADPH and AhpD/dihydrolipoamide dehydrogenase and dihydrolipoamide succinyl transferase/NADH, have been described (15, 57). In addition, although recent data indicate that M. tuberculosis PrxQ B is efficiently reduced by homologous thioredoxins B and C/thioredoxin reductase/NADPH (115), the role of MSH/Mrx-1 in the reduction of the PrxQ subfamily of Prxs was not investigated.
The interactions of MSH with catalase peroxidase (KatG), the main heme-dependent peroxidase expressed in mycobacteria that is responsible for the activation of the prodrug isoniazid (INH), remain to be investigated (52, 165). Indeed, several heme-dependent peroxidases can catalyze the one-electron oxidation of thiols to their corresponding thiyl radicals (67, 158). However, similar interactions of KatG compound I and/or II with MSH are presently unknown.
Thus, although recent findings have provided a grasp on how protein S-mycothiolation affects protein activity and cellular function, many studies are still required to identify novel targets of S-mycothiolation and to give a comprehensive picture of this post-translational modification.
S-Nitrosomycothiol
Several lines of evidence indicate that nitric oxide (•NO) and its derived reactive species are essential for host defense against M. tuberculosis (22, 79, 80). Among •NO-derived species, S-nitrosothiols have deserved special attention, because they could be responsible for some of the actions originally attributed to •NO as well as constitute a post-translational modification that can affect protein function (62). Indeed, phagocytosed M. tuberculosis by immunologically activated macrophages got S-nitrosated in different proteins (116). Furthermore, acidified nitrite and MSH lead to the formation of S-nitrosomycothiol (MSNO) in vitro, and a reductase with high specific activity toward MSNO was identified in mycobacteria and is conserved in some other Actinomycetes (84). The S-nitrosomycothiol reductase catalyzed the NADH-dependent reduction of MSNO to an unstable N-hydroxysulfenamide, which leads to the mycobacterial truncated hemoglobin N oxidation and nitrate formation (155). The enzyme also participated in formaldehyde detoxification pathways (24, 155). In M. smegmatis, MSH and MSNO are important for normal biofilm formation. Moreover, strains lacking mscR (which encodes the S-nitrosomycothiol reductase) and mshC are more susceptible to S-nitrosoglutathione and aldehydes when compared with wild-type strains (150).
Measurements of MSH Redox Potential Within Bacteria (EMSH)
MSH is present at millimolar concentrations in Actinomycetes, and this in a ratio of MSH and MSSM, buffering the cellular redox environment, which can be correlated to the redox potential (EMSH) via the Nernst equation [Eq. (4)]. This equation assumes an equilibrium, as the redox potential is a thermodynamic term that is directly correlated to the Gibbs-free energy. However, a living cell is not at equilibrium, but can rather be regarded as a steady-state condition. Nevertheless, scientists want to measure and label, to compare and understand, and preferably on a real-time scale. So genetically encoded fluorescent redox sensors were developed to correlate changes in the LMW thiol ratio to oxidative stress.
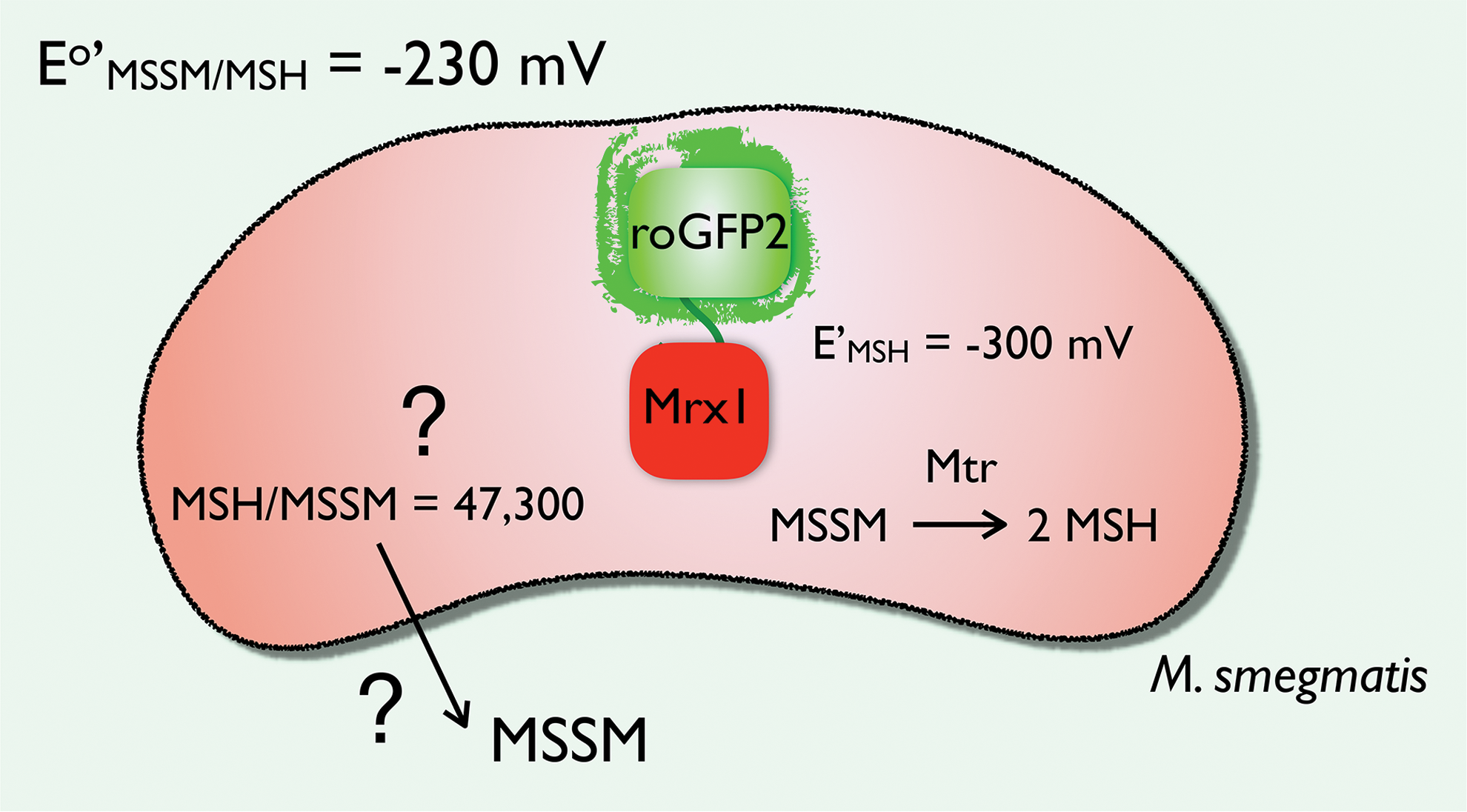
It was the group of Jakob Winther, who took the lead by coupling yeast glutaredoxin 1 via a short linker peptide to redox-sensitive yellow fluorescent protein (14), and his findings led to the development of several redox couple tailored probes during the past two decades. By combining several variants of redox-sensitive green fluorescent protein (roGFP) coupled to a family of glutaredoxins, the response of wild-type cells and their knockout variants upon oxidative challenge could be determined in a pH-independent and ratiometric manner. Here, the glutaredoxins are a kind of specific mediator between GSH and the fluorescent signal generated by roGFP to promote a fast response (49), as the response kinetics of the GSH/GSSG couple to roGFP are relatively slow and unselective (49). Analogous to the glutaredoxin-roGFP probe, a Mrx-1(Rv3198A)-roGFP2 probe was recently developed to measure the changes in MSH/MSSM ratio (13). This biosensor was expressed at submicromolar concentrations in M. smegmatis, suggesting that the introduced reducing power of the probe would not influence the in vivo situation. For wild-type M. smegmatis, reported EMSH values measured using the biosensor were (−300 ± 2 mV) (13) and (−296 ± 2 mV) (96), whereas it was more oxidized (−281 ± 3 mV) in strains resistant to isoniazid (96), and even more oxidized when the latter strains were treated with some compounds that caused oxidative stress in the bacteria such as ebselen (which was reported to inhibit thioredoxin reductase) (96).
The redox potential in the cell can be calculated using the Nernst equation [Eq. (4)]:
where R is the gas constant (8.314 J K−1 mol−1), F is the Faraday constant (9.65 × 104 coulombs mol−1), n is the number of electrons transferred (2), and T is the absolute temperature (310 K) at which the bacterium was grown for the original EMSH measurements. The change in EMSH is pH dependent: if the pH increases 1 unit at 37°C, this equates to −65.1 mV for a two-electron, two-proton thiol-disulfide redox process. With an Eo′ = −230 mV (129), a pH of 7.0 (109), an MSH concentration of 4 mM (92), and a ratio of MSH/MSSM of 500 (92), one would expect an EMSH of −239 mV. However, with the Mrx-1-roGFP biosensor, which has a midpoint potential of −280 mV, a value of approximately −300 mV was measured in M. smegmatis (13) (Fig. 5). As a consequence, small changes in MSSM concentration would have a huge effect on the redox potential. This interesting observation makes it clear that all pieces of the mechanism puzzle on how Actinomycetes maintain this low redox potential are yet not known. Again, a cell is not at equilibrium, and we do not know the rate at which the Mrx-1-roGFP biosensor reacts or competes with the reduction of MSSM by the NADPH-dependent flavoenzyme Mtr under nonlimiting NADPH conditions. In this way, the MSH/MSSM ratio can be 2 logs higher than what is currently measured, or alternatively it might also be a pH effect, if the pH were 0.9 pH units higher than pH 7.0. Further research is required to unravel this relatively hazy situation on the mechanisms used to keep the MSSM concentration low and as such the MSH redox potential in Actinomycetes reducing (Fig. 5). To note, M. smegmatis in stationary phase imports MSH from the culture medium by an active transport process. Uptake of MSSM also occurred but at slower rates (20).
In slow growing mycobacteria, EMSH values were very similar (−273 to −280 mV) for H37Rv, several multidrug-resistant M. tuberculosis, and M. bovis BCG strains, being more oxidized than those of M. smegmatis (13, 99). To note, the oxidizing shift in EMSH observed in isoniazid-resistant M. smegmatis strains was not observed in multidrug-resistant M. tuberculosis (which included resistance to isoniazid). The biosensor allowed to follow changes in EMSH in response to diverse oxidants and drugs, to report redox heterogeneity in M. tuberculosis populations during macrophage infection, and to detect oxidative changes caused by antibiotics with different mechanisms of action in M. tuberculosis during infection (13, 99).
Chemical Detoxification
MSH is crucial in the detoxification of alkylating agents and many antibiotics. First, an amidase activity able to cleave bimane derivatives of MSH to GlcN-Ins (retained in the cells) and the mercapturic acid N-acetylcysteine bimane (exported to the medium) was identified and purified from M. smegmatis (82). The functional characterization of the enzyme indicated that it was highly specific for S-conjugates of MSH, with a broader specificity toward the sulfur conjugate structure, and was named mycothiol S-conjugated amidase (Mca). M. smegmatis lacking a functional mca gene was more susceptible to alkylating agents, redox-cycling compounds, and the antibiotic streptomycin (113). Sequence homology analysis allowed the identification of the mca gene from M. tuberculosis (82). Inhibition studies indicated that Mca was a metalloenzyme (93), and protein analysis suggested that zinc was the native metal (137). Further studies showed that the metal dependency was based on the purification conditions: aerobically purified Mca contains stoichiometric amounts of Fe2+, whereas anaerobically purified Mca contains Zn2+. Thus, it has been proposed that Mca is mostly an Fe2+-dependent enzyme, whereas Zn2+-Mca arises during aerobic purification or in cells facing oxidative stress because of protein-bound Fe2+ oxidation and release (64). The impact of the nature of active site metal on enzymatic activity should deserve further investigation. Mca has weak deacetylase activity that partially maintains MSH synthesis in mshB mutants (161).
In eukaryotic cells, members of the GSH S-transferase (GST) superfamily catalyze the detoxification of electrophilic xenobiotics and the inactivation of several endogenous compounds. Many members of the subfamily have also been found in GSH-producing bacteria (4). The ability of M. smegmatis to rapidly conjugate monochlorobimane with MSH yielding a fluorescent product allowed the purification of the first mycothiol-S transferase (MST), product of the MSMEG_0887 gene, and the identification of Rv0443 as the gene encoding a 77% identical Mst in M. tuberculosis (91). MST is highly specific toward MSH, and showed <200-fold lower activity toward BSH or GSH. Analysis of MsMst sequence showed that it is a member of the DUF664 family belonging to the DinB superfamily, whose members are probably involved in thiol-dependent detoxification processes, and to the identification of different homologues in BSH- as well as GSH-producing organisms, unrelated to previously known BSH S-transferases (BSTs) or GSTs. Since MsMst acts on mitomycin C and BsBST acts on cerulenin, it has been suggested that these enzymes could play a role in antibiotic detoxification pathways (91). To note, the first member of the DinB superfamily to be identified was an MSH-dependent maleylpyruvate isomerase from C. glutamicum, which linked MSH biosynthesis and gentisate assimilation in this bacterium (35, 157).
Arsenate [As(V)] detoxification in bacteria occurs by intracellular arsenate reduction to arsenite [As(III)] that is subsequently extruded (1). Arsenate reduction is catalyzed by distinct arsenate reductases that use different reducing systems (154). C. glutamicum expresses both thioredoxin-dependent (Cg_ArsC1′) and MSH/Mrx-1-dependent (Cg_ArsC1 and Cg_ArsC2) arsenate reductases (95). The former is expressed constitutively, and once in contact with its substrate, it catalyzes the thioredoxin-dependent reduction of As(V) to As(III), which then induces the expression of the latter two (154). ArsC1 and ArsC2 catalyze the formation of an arsenic-MSH adduct, which is then reduced by Mrx-1, yielding a mixed disulfide between Mrx-1 and mycothiol (Mrx-1-S-SM) and As(III). A second moiety of MSH releases reduced Mrx-1 and MSSM, which, in turn, is reduced by Mtr at the expense of NADPH (95). In C. glutamicum, As(III) is transported out of the cell by two arsenite permeases of the Acr3 family (41). To note, C. glutamicum strains lacking mshA or mshC are extremely sensitive to As(V) (0.05 mM vs. 60 mM in the parental strain) and show hyperaccumulation of this metalloid, whereas strains lacking mshB or mshD have intermediate phenotypes (153).
Vitamin C was reported to kill drug-susceptible as well as multi- and extensively drug-resistant strains of M. tuberculosis. The mechanism of toxicity involved increased free iron content, increased production of ROS [particularly superoxide radical, H2O2 as well as hydroxyl radical formed through the Fenton reaction (56)] and DNA damage along with changes in cellular lipid composition. M. tuberculosis strains deficient in MSH biosynthesis showed extreme susceptibly to the prooxidant effect of vitamin C (152), consistent with the role of the thiol in hydroperoxide detoxification already indicated. Consistently, mycobacteria exposed to hydroquinone-based small compounds that generate superoxide radical (by direct reduction of oxygen and redox cycling) and hydrogen peroxide (derived from spontaneous or SOD-catalyzed superoxide radical dismutation) showed a significant oxidative shift in EMSH at doses that roughly correlated with those causing growth inhibition. To note, M. tuberculosis strains unable to synthesize MSH were extremely sensitive to these drugs (145).
Compounds that caused adenosine-5′-phosphosulfate reductase (APSR) ‡ inhibition have been recently reported to cause decreased intracellular levels of sulfite, cysteine, and MSH as well as an oxidative shift in EMSH and to show potent bactericidal activity against drug-susceptible and drug-resistant strains of M. tuberculosis (99).
Finally, 4-buty-4-hydroxyl-1-(4-hydroxyphenyl)-2-phenylpyrazolidine-3,5-dione (4-OH-OPB), an hydroxylation product of the nonsteroidal anti-inflammatory drug oxyphenbutazone (OPB), forms a covalent adduct with MSH and its precursor, N-acetyl cysteine, and was found to kill both replicating and nonreplicating mycobacteria (45).
MSH and Antibiotic Synthesis/Susceptibility
M. smegmatis devoid of MSH showed increased susceptibilities to several antibiotics, such as erythromycin, azithromycyn, rifamycin S, penicillin G, and vancomycin (112). In most cases, that is related to the antibiotic conjugation with MSH, now known to be potentially catalyzed by Mst (91), followed by conjugate hydrolysis by Mca and mercapturic acid elimination, as was first demonstrated for rifamycin S (137). Consistently, M. smegmatis lacking Mca failed to convert the adduct between MSH and rifamycin S into the corresponding mercapturic acid and showed increased susceptibility to streptomycin (113). Mca is also required for the synthesis of cyslabdan A, a mercapturic acid derivative of a diterpene compound that potentiates the β-lactam antibiotic imipenem, active against methicillin-resistant Staphylococus aureus (55). However, lower MSH content increased the resistance to isoniazid and to ethionamide in M. smegmatis, suggesting that MSH is involved in the activation of these prodrugs (151, 161). Treatment of M. tuberculosis-infected macrophages with several antibiotics of different modes of action induced a change in EMSH (measured using the Mrx-1-roGFP biosensor). To note, infection of macrophages induced heterogeneity in bacterial EMSH that correlated with differential susceptibility to antibiotics (13).
More recently, MSH and ERG were found to be used in Streptomyces lincolnensis for the synthesis of the sulfur-containing antibiotic lincomycin A [that has been used for the treatment of Gram-positive bacterial infections (72)], wherein MSH is the sulfur donor for a methylmercapto-group incorporation into lincomycyn, providing a novel route for the introduction of sulfur into biomolecules (166). A recent report indicated that in addition to MSH, ERG also participates in drug detoxification by mycobacteria, and that both thiols play overlapping but not completely redundant roles (118).
MSH Catabolism
MSH half-life was determined as 50 h in early stationary phase wild-type M. smegmatis and was reduced to 16 h in mshC-deficient mutants, wherein resynthesis from GlcN-Ins and cysteine is impaired (20). Catabolism of MSH is initiated by Mca that cleaves MSH to generate GlcN-Ins and N-acetyl cysteine (20, 82). The latter can be deacetylated to cysteine, thus providing a mechanism for generating the amino acid under specific metabolic circumstances, its intracellular accumulation is toxic because of its rapid autoxidation. GlcN-Ins and cysteine are then made available for MSH regeneration in the reaction catalyzed by MshC or are further metabolized. For instance, cysteine can be transformed into alanine by the cysteine desulfurase IscS from M. tuberculosis that is involved in iron–sulfur cluster assembly (117). It has been suggested that GlcN-Ins could also be used for producing cell envelope components derived from inositol (82).
Conclusions
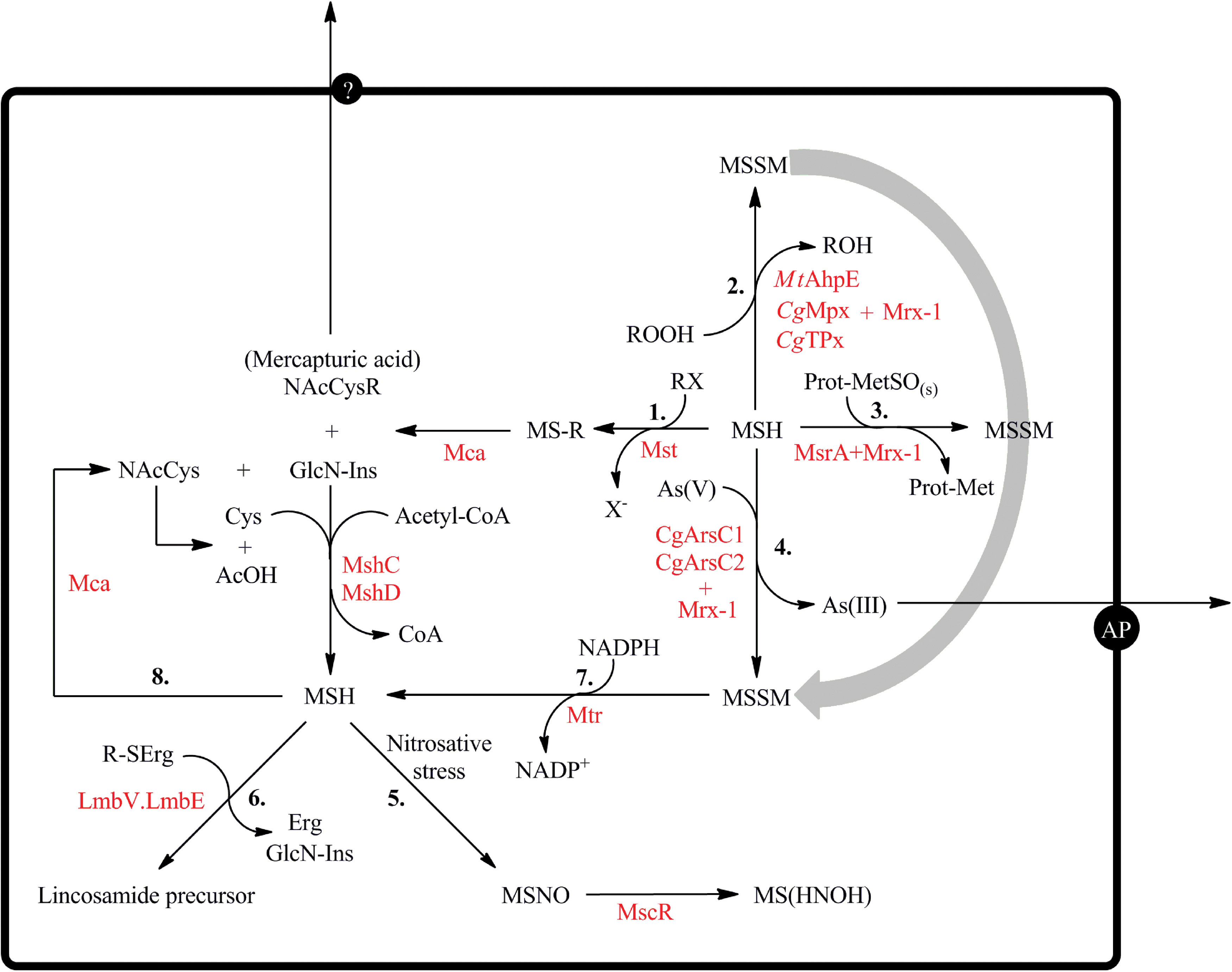
MSH, the main LMW thiol of Actinomycetes, plays most of the functions usually performed by GSH in eukaryotic cells and Gram-negative bacteria. Most chemical, nonenzymatic catalyzed reactions of thiols are slow, and they can only operate through enzymatic systems that have been described in some bacteria (Fig. 6). To date, many antioxidant systems have been shown to use MSH (plus Mrx-1) as reducing system, providing a molecular link between thiols, hydroperoxide detoxification, and protein repair pathways. Under conditions of nitrosative stress, MSH can be S-nitrosated, and enzymatic routes for MSNO biotransformation have also been described. Furthermore, MSH participates in chemical detoxification pathways such as of electrophilic compounds, As(V), formaldehyde, and many antibiotics. In contrast, a lower MSH content increases the resistance to isoniazid and to ethionamide in M. smegmatis, which suggested that MSH is involved in the activation of these prodrugs. These findings impose a selective approach when aiming for tackling the MSH-dependent metabolism for antibacterial purposes. Moreover, the ability of MSH to provide a methylmercapto group in the synthesis of lincomycin has been postulated as novel route for the incorporation of sulfur into biomolecules. MSH biosynthesis and MSSM reduction are regulated processes, and routes of regulation are starting to be unraveled in the different Actinomycetes. In vivo imaging indicated that the E′ values of the MSH/MSSM pair are in the −300 mV range in M. smegmatis, which suggests that the intracellular MSSM concentration is kept extremely low. Finally, Actinomycetes also synthesize other LMW thiols such as ERG, the function of which is less characterized. Further investigations are required to establish the extent to which other LMW thiols could take over some of the functions described for MSH.
Footnotes
Acknowledgments
A.M.R. was partially supported by a PhD scholarship from Universidad de la República-CAP (Montevideo, Uruguay), B.P. was supported by an IWT PhD fellowship from Flanders, Belgium, and M.A.T. was supported by an FWO PhD fellowship from Flanders, Belgium. This work was supported by grants from (i) Universidad de la República (CSIC Grupos 767) and Espacio Interdisciplinario, (ii) Agentschap voor Innovatie door Wetenschap en Technologie (IWT), and (iii) Vlaams Instituut voor Biotechnologie (VIB). Additional support was provided by the Programa de Desarrollo de Ciencias Básicas (PEDECIBA, Uruguay).