
Abstract
Aims:
Cells have evolved a highly sophisticated web of cytoprotective systems to neutralize unwanted oxidative stress, but are challenged by unique modern day stresses such as cigarette smoking and ingestion of a high-fat diet (HFD). Age-related disease, such as age-related macular degeneration (AMD), the most common cause of blindness among the elderly in Western societies, develops in part, when oxidative stress overwhelms cytoprotective systems to injure tissue. Since most studies focus on the protection by a single protective system, the aim of this study was to investigate the impact of more than one cytoprotective system against oxidative stress.
Results:
Wingless (Wnt) and nuclear factor-erythroid 2-related factor 2 (Nrf2), two fundamental signaling systems that are vital to cell survival, decline after mice are exposed to chronic cigarette smoke and HFD, two established AMD risk factors, in a bidirectional feedback loop through phosphorylated glycogen synthase kinase 3 beta. Decreased Wnt and Nrf2 signaling leads to retinal pigment epithelial dysfunction and apoptosis, and a phenotype that is strikingly similar to geographic atrophy (GA), an advanced form of AMD with no effective treatment.
Innovation:
This study is the first to show that chronic oxidative stress from common modern day environmental exposures reduces two fundamental and vital cytoprotective networks in a bidirectional feedback loop, and their decline leads to advanced disease phenotype.
Conclusion:
Our data offer new insights into how combined modern oxidative stresses of cigarette smoking and HFD contribute to GA through an interactive decline in Wnt and Nrf2 signaling. Antioxid. Redox Signal. 29, 389–407.
Introduction
A
A prototypical, complex aging-related disease that is influenced by oxidative stress through cigarette smoke (CS) and HFD is age-related macular degeneration (AMD) (46, 61), the main cause for legal blindness in the industrialized world (8). By the mid-2000s, AMD developed into a major public health problem that has cost $30 billion annually in the United States (5), €597 million in Canada, €926 million in France, €1155 million in Germany, and €662 million in the United Kingdom (9). With the aging population, the number of people with advanced AMD in the United States alone will nearly double to 3 million people by 2020 (15). While treatment is available for the advanced wet form of AMD, no treatment exists for the advanced dry form of AMD called geographic atrophy (GA).
When evaluating protection against oxidative stress, most studies have focused on a single cytoprotective system. Since cytoprotective systems are likely an interactive web, we evaluated two essential systems and find that both the wingless (Wnt) and nuclear factor-erythroid 2-related factor 2 (Nrf2) pathways decline in a bidirectional feedback loop after chronic oxidative stress from cigarette smoke and high-fat diet. Importantly, this mechanism led to a phenotype strikingly reminiscent to geographic atrophy (GA), an advanced form of age-related macular degeneration without treatment, in mice. Future studies should consider assessing more than one cytoprotective system against oxidative stress. Our results provide potential treatment targets and a robust model for testing new therapy for GA.
A central feature of AMD is retinal pigment epithelium (RPE) degeneration. The RPE is a highly specialized cell that is sandwiched between the photoreceptors, which detect light, and the choriocapillaris, the highly vascular system that provides oxygen and essential nutrients to the photoreceptors. Because of its many specialized functions that maintain photoreceptor health, the RPE plays a pivotal role in maintaining normal vision and, in particular, central visual acuity from the macula.
Due to the high ambient oxygen tensions required for its high metabolism that is necessary to maintain the health and function of the overlying photoreceptors, and the unique, constant exposure to photo-oxidative stress, the RPE is armed with a robust antioxidant system (1, 57), making it a valuable model to study the oxidative stress response. Mild to moderate RPE cell dysfunction leads to photoreceptor impairment and visual loss in early AMD. As RPE dysfunction worsens, AMD severity progresses. When advanced, islands of RPE cell death develop, this defines GA (33). Since disease progression is related to the extent of the transition zone of damaged RPE surrounding the GA, understanding how the RPE degenerates will hasten therapeutic breakthroughs for GA.
We investigated the underlying molecular mechanism of how cytoprotective pathways guard the RPE against oxidative injury after exposure to CS and HFD, and the extent that a disease phenotype develops when these pathways become impaired. We chose to study wingless (Wnt) signaling because it is one of the first molecular responses to cellular stress, dysregulated Wnt signaling is implicated in age-related diseases (43, 69), and a single-nucleotide polymorphism in the Wnt receptor, lipoprotein receptor-related protein 6 (Lrp6) is associated with AMD risk (26), yet no studies have linked Wnt with AMD pathobiology.
In our study, we found that chronic CS and HFD treatment decreased Wnt signaling, which decreased the principal antioxidant network that revolves around the nuclear factor-erythroid 2-related factor 2 (Nrf2) transcription factor (TF) (27), and resulted in a phenotype strikingly similar to GA. Our data suggest that the combined modern oxidative stresses of CS and HFD contribute to a cooperative decline in Wnt and Nrf2 signaling and a severe age-related disease phenotype.
Results
Wnt5 is the predominant Wnt ligand expressed by the RPE/choroid in apoB100 mice
Our experimental plan was to chronically expose mice to a combination of HFD and CS to study the impact of chronic oxidative stress on cytoprotective pathways. Since mice predominantly express apolipoprotein (apo) B48 rather than apoB100, we selected “apoB100” mice, which have a mutation in the apoB48 editing codon that reduces the conversion of apoB100 to apoB48, to simulate in humans, lipid transport by apoB100 lipoproteins following an HFD (14). These mice produce physiological apoB100 lipoprotein levels in both the RPE and liver, which mimic humans (4, 16).
Wnt signaling is initiated after a Wnt ligand binds to the Frizzled-Lrp6 receptor complex. Since there are 19 known Wnt ligands, we used a Wnt PCR array to first identify the relative expression of relevant Wnt ligands that could activate Wnt signaling in the RPE/choroid of apoB100 mice relative to B6;129 wild-type (WT) mice, and secondarily, to determine the extent that Wnt ligands or other Wnt pathway genes have altered expression with aging. The mRNAs of all 17 Wnt ligands on the PCR array were detected and were not differentially expressed in the RPE/choroid of 2-month-old apoB100 and WT mice (Supplementary Table S1; Supplementary Data are available online at
Wnt5a, which can activate both canonical and noncanonical Wnt signaling (47), was the most abundantly expressed Wnt ligand in the RPE/choroid of 2-month-old apoB100 mice, and was decreased in 11-month-old apoB100 mice compared with 11-month-old WT mice. By Western blot analysis, Wnt5 protein was decreased in the RPE/choroid of 2-month-old apoB100 mice compared with WT mice (p < 0.01; Supplementary Fig. S1), while the Wnt3 protein, with low expression on the array, was not detected using two different antibodies. In general, Wnt pathway gene expression in the RPE/choroid was similar between 2-month-old apoB100 and WT mice, although Lrp6 was among several Wnt genes with decreased expression in apoB100 mice (Supplementary Table S2). Likewise, Wnt pathway expression was unchanged in the RPE/choroid with aging in old apoB100 mice, except for a small group of genes, including Lrp6, that were decreased (Supplementary Table S2).
Wnt5 signaling is impaired in apoB100 mice exposed to chronic oxidative stress
We next assessed the abundance of Wnt5 in the RPE/choroid of apoB100 mice given chronic oxidative stress. In apoB100 mice treated with CS + HFD for 6 months, Wnt5 protein in the RPE/choroid was decreased relative to apoB100 mice raised in air on a chow diet (p < 0.05; Fig. 1A and Supplementary Fig. S2). Lrp6 is phosphorylated after Wnt ligand binds to the Frizzled-Lrp receptor complex. The p-Lrp6/Lrp6 ratio was decreased in the RPE/choroid of apoB100 mice given CS + HFD, compared with mice treated with a normal diet and air (p < 0.05; Fig. 1B and Supplementary Fig. S3). p-glycogen synthase kinase 3 beta (Gsk3β) (Tyr216) regulates the stability of β-catenin. The RPE/choroid of apoB100 mice given CS + HFD had increased p-Gsk3β/Gsk3β (p < 0.05; Fig. 1C and Supplementary Fig. S4) and decreased β-catenin, compared with mice treated with a normal diet and air (p < 0.01; Fig. 1D and Supplementary Fig. S5).
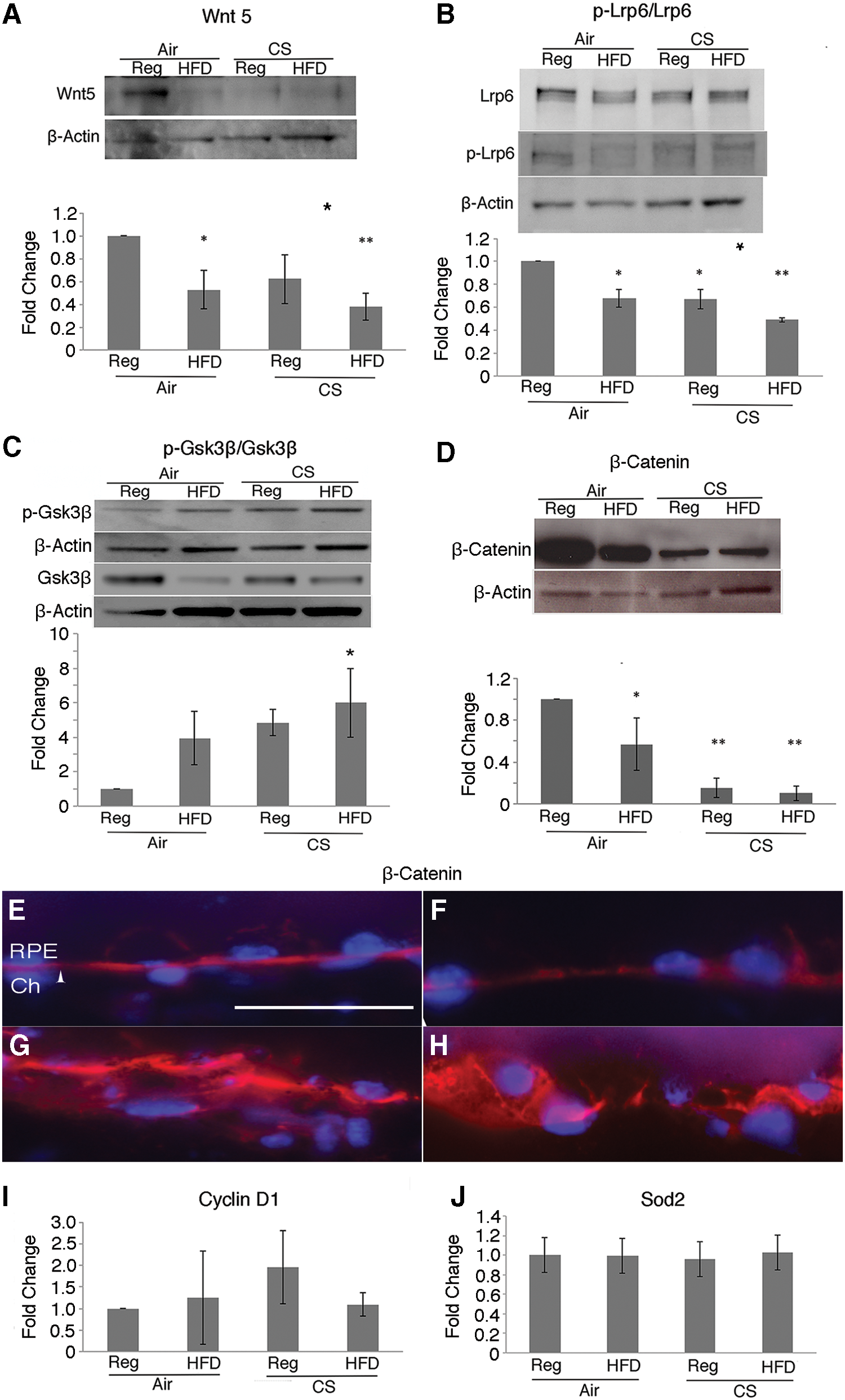
The distribution of β-catenin in the RPE changed with chronic oxidative stress. β-Catenin immunolabeling was distinct at the lateral and basal borders of the RPE from apoB100 mice raised in air on either a regular or HFD, but became diffusely and basally distributed in the RPE of apoB100 mice exposed to CS with or without HFD (Fig. 1E–H).
With Wnt activation, β-catenin interacts with lymphoid enhancer-binding factor 1/T cell factor (Lef1/Tcf) TFs to induce the expression of genes such as cyclinD1. Notably, cyclinD1 mRNA was not induced in the RPE/choroid after CS + HFD relative to mice treated with a normal chow diet and air (Fig. 1I). With oxidative stress, Wnt signaling can shift from Lef1/Tcf to forkhead box O (Foxo) TF cofactor mediated expression of antioxidant genes, such as superoxide dismutase 2 (Sod2) (2, 45). Similarly, Sod2 mRNA was not induced in the RPE/choroid of apoB100 mice given CS + HFD (Fig. 1J). Thus, all of the Wnt components tested were impaired by CS/HFD treatment while 50% (p-Lrp6, β-catenin) were impaired by CS/regular diet (Reg) treatment. In addition, the decrease for Wnt5 and p-Lrp6 was greater after chronic CS/HFD than CS-only exposure. Collectively, while CS impairs Wnt signaling, these results suggest that the combined stress of CS and HFD impairs Wnt signaling to a greater extent than CS treatment alone.
Nrf2 signaling is impaired in a bidirectional feedback loop with oxidative stress
p-Gsk3β (tyr216) can phosphorylate Nrf2 to promote Nrf2 degradation by a kelch-like ECH-associated protein 1 (Keap1)-independent pathway (55). Given the increased p-GSK3β in apoB100 mice treated with CS + HFD, we sought to determine Nrf2 abundance in our animal model. Nrf2 protein (p < 0.01) was decreased in the RPE/choroid of apoB100 mice treated with CS + HFD compared with mice given a normal diet and air (Fig. 2A and Supplementary Fig. S6). This decrease was associated with a blunted response of the Nrf2-mediated antioxidant genes, heme oxygenase 1 (Ho-1), glutamate-cysteine ligase modifier subunit (Gclm), and NAD(P)H quinone dehydrogenase 1 (Nqo-1), and decreased Ho-1 protein (Fig. 2A) in the RPE/choroid of mice treated with CS + HFD relative to mice on a normal diet and air (p < 0.01; Fig. 2B).
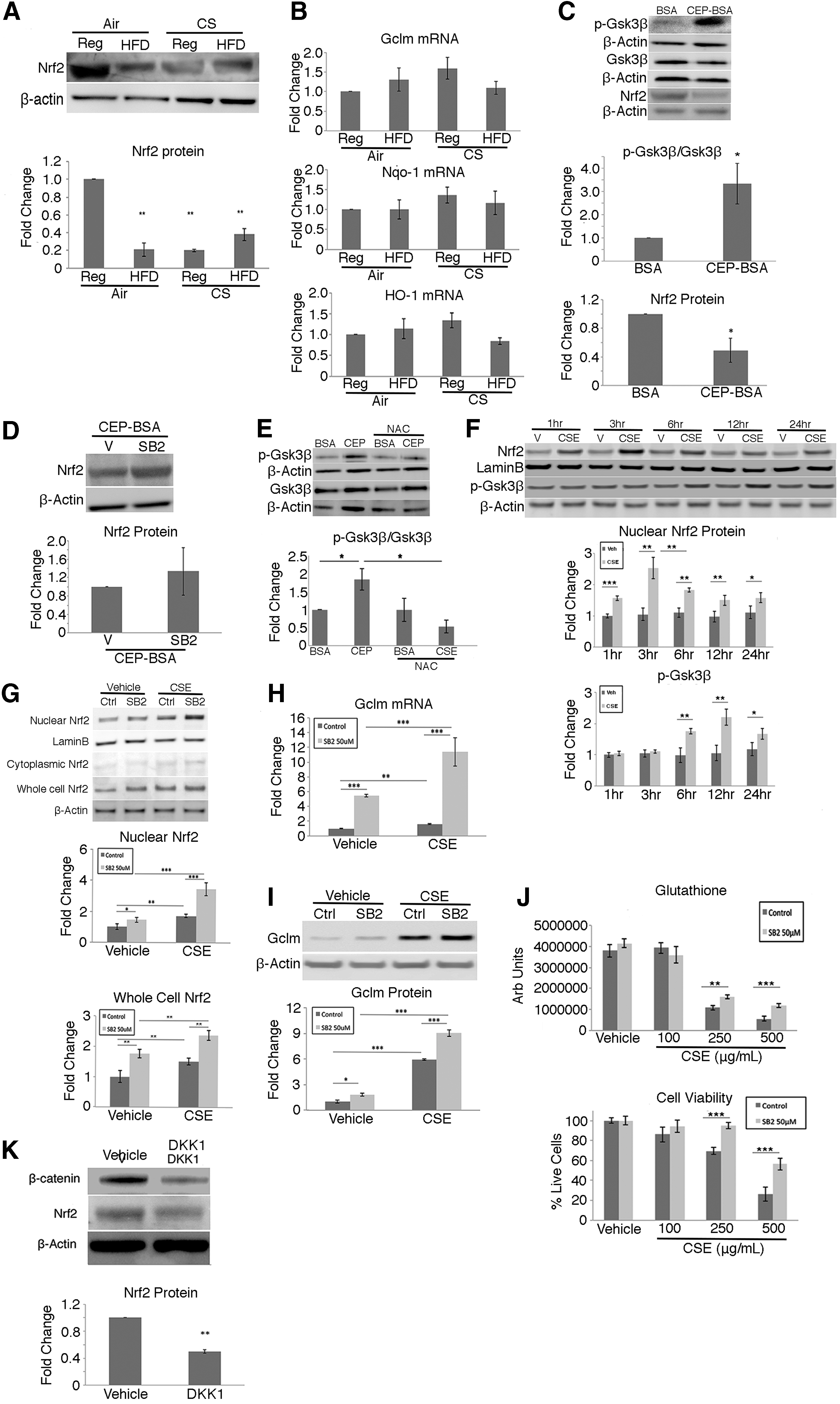
To demonstrate that p-Gsk3β influences Nrf2 signaling, apoB100 mice were given intravitreal (Ivt) cigarette smoke extract (CSE) or carboxyethylpyrrole (CEP-bovine serum albumin [BSA]), a specific oxidation product of docosahexaenoic acid that accumulates in the outer retina, RPE, and Bruch's membrane in AMD, to stress the RPE and induce Nrf2 (23). As expected, 6 h after Ivt CSE or CEP-BSA, Nrf2 protein from whole-cell lysates or nuclear extracts of RPE/choroid was increased compared to vehicle-injected mice (Supplementary Figs. S7 and S8). By 24 h, Nrf2 (p < 0.05) was decreased, while p-Gsk3β/Gsk3β (p < 0.05) was increased relative to BSA controls (Fig. 2C) and when 50 μM SB216763, a Gsk3β inhibitor, was Ivt injected with CEP-BSA, Nrf2 protein was restored to control levels that received Ivt CEP-BSA and vehicle (Fig. 2D). To demonstrate that oxidative stress mediates Gsk3β phosphorylation, after mice were injected with Ivt CEP-BSA in the presence of the antioxidant N-acetyl cysteine, the p-Gsk3β/Gsk3β was reduced (p < 0.05; Fig. 2E).
Furthermore, we were able to demonstrate similar events in an in vitro model. When human ARPE-19 cells were treated with CSE 100 μg/mL, nuclear Nrf2 protein was increased relative to vehicle-treated cells, in a time-dependent manner, peaking after 3 h, and the subsequent decline in Nrf2 (p < 0.01) coincided with induction of p-Gsk3β after 6 h (Fig. 2F). After ARPE-19 cells were treated with CSE 100 μg/mL and SB216763, Nrf2 was increased in both nuclear and whole-cell lysates, compared to controls, and this increase was magnified by cotreatment with CSE and Gsk3β inhibition (Fig. 2G). In addition, the mRNA (Fig. 2H) and protein (Fig. 2I and Supplementary Fig. S9) levels of Gclm, the rate-limiting enzyme in glutathione synthesis, were induced compared to control treated cells when Gsk3β was inhibited with SB216763, with or without CSE. Gsk3β inhibition with SB216763, as expected, increased total glutathione and protected against oxidant-induced cell death after CSE treatment (Fig. 2J).
Finally, when DKK1, a Wnt signaling inhibitor that is upstream of Gsk3β (21) was Ivt injected in one eye, and vehicle into the contralateral eye of apoB100 mice, β-catenin and Nrf2 were decreased in the RPE/choroid relative to controls (p < 0.01; Fig. 2K), suggesting that Wnt signaling can influence Nrf2 signaling.
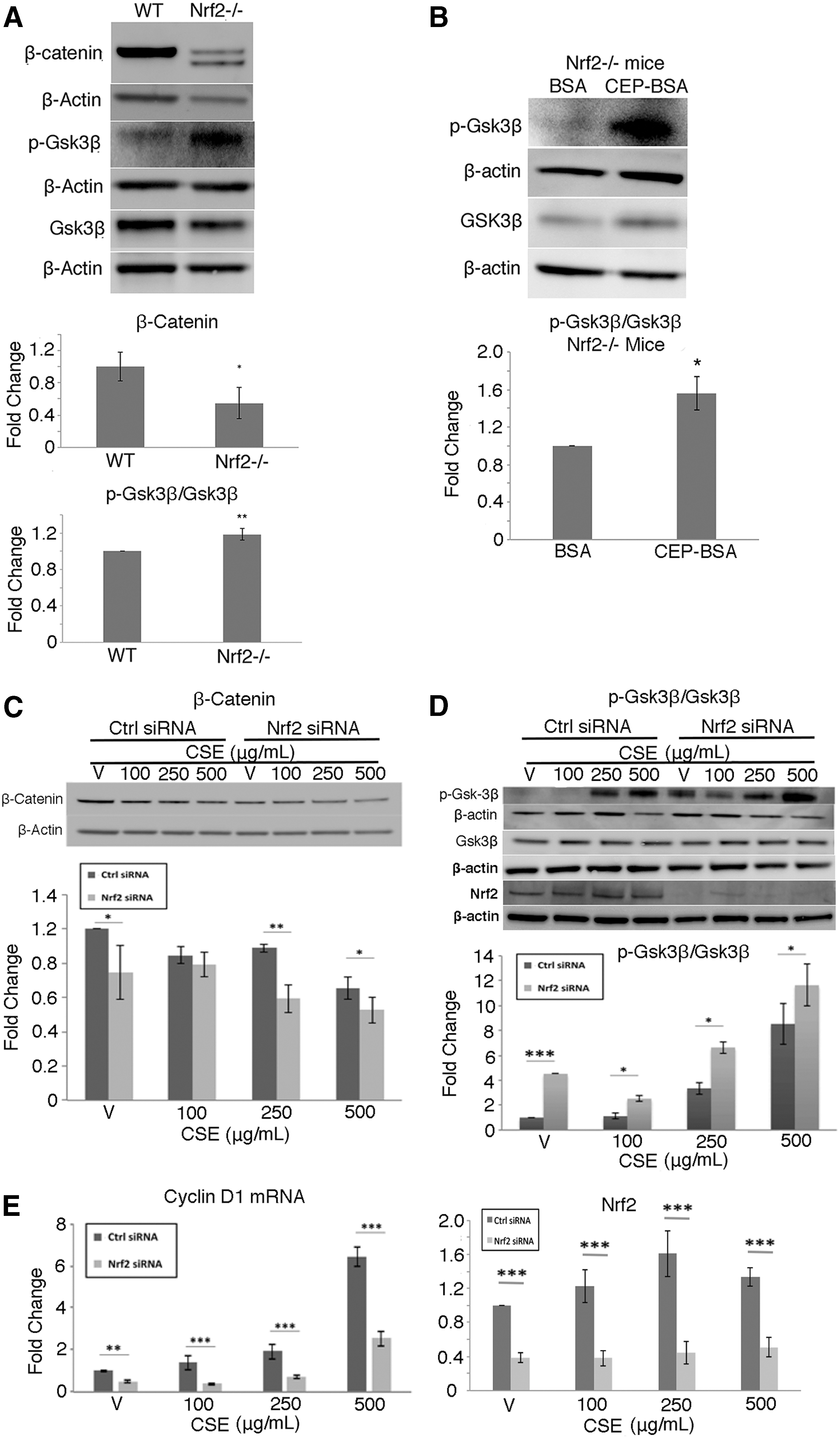
We next wanted to determine whether Nrf2 could influence Wnt signaling. In the RPE/choroid of Nrf2−/− mice compared with C57BL/6J WT mice, β-catenin protein was decreased and p-Gsk3β/Gsk3β was increased (p < 0.05; Fig. 3A). p-Gsk3β/Gsk3β was further induced in Nrf2−/− mice given Ivt CEP-BSA compared to BSA controls (p < 0.01; Fig. 3B). Likewise, in ARPE-19 cells given 0–500 μg/mL CSE, β-catenin was decreased (Fig. 3C), p-Gsk-3β/Gsk-3β was increased (Fig. 3D), and cyclin D1 mRNA was decreased (Fig. 3E) in a dose-dependent manner compared to vehicle controls, an effect that was magnified by Nrf2 knockdown. Collectively, these data identify a bidirectional feedback loop between Nrf2 and Wnt where impaired Nrf2 can decrease Wnt signaling, and decreased Wnt signaling can decrease Nrf2.
The RPE is dysfunctional and dies by apoptosis with posterior GA in apoB100 mice exposed to chronic oxidative stress
Rpe65, an integral visual cycle protein produced by the RPE that is suppressed by chronic stress, and a valuable indicator of RPE cell terminal differentiation (73), was decreased in the RPE/choroid of apoB100 mice treated with CS + HFD for 6 months relative to mice raised on a chow diet and air (Fig. 4A and Supplementary Fig. S10).

To demonstrate that RPE differentiation is impaired after reducing Nrf2 and Wnt, mice were treated with Ivt DKK1. Rpe65 and Lrat, another visual cycle component, were both decreased compared to controls (Fig. 4B and Supplementary Fig. S11). RPE apoptosis is a reported mechanism for RPE cell death in AMD (12). Using poly-ADP ribose polymerase (Parp) cleavage as a marker of apoptosis (71), we found that the 25 kDa cleaved product was increased in the RPE/choroid of apoB100 mice exposed to CS and HFD compared with mice treated with a regular diet and air (Fig. 4C and Supplementary Fig. S12). These results suggest that chronic CS and HFD exposure induces RPE dysfunction and cell death.
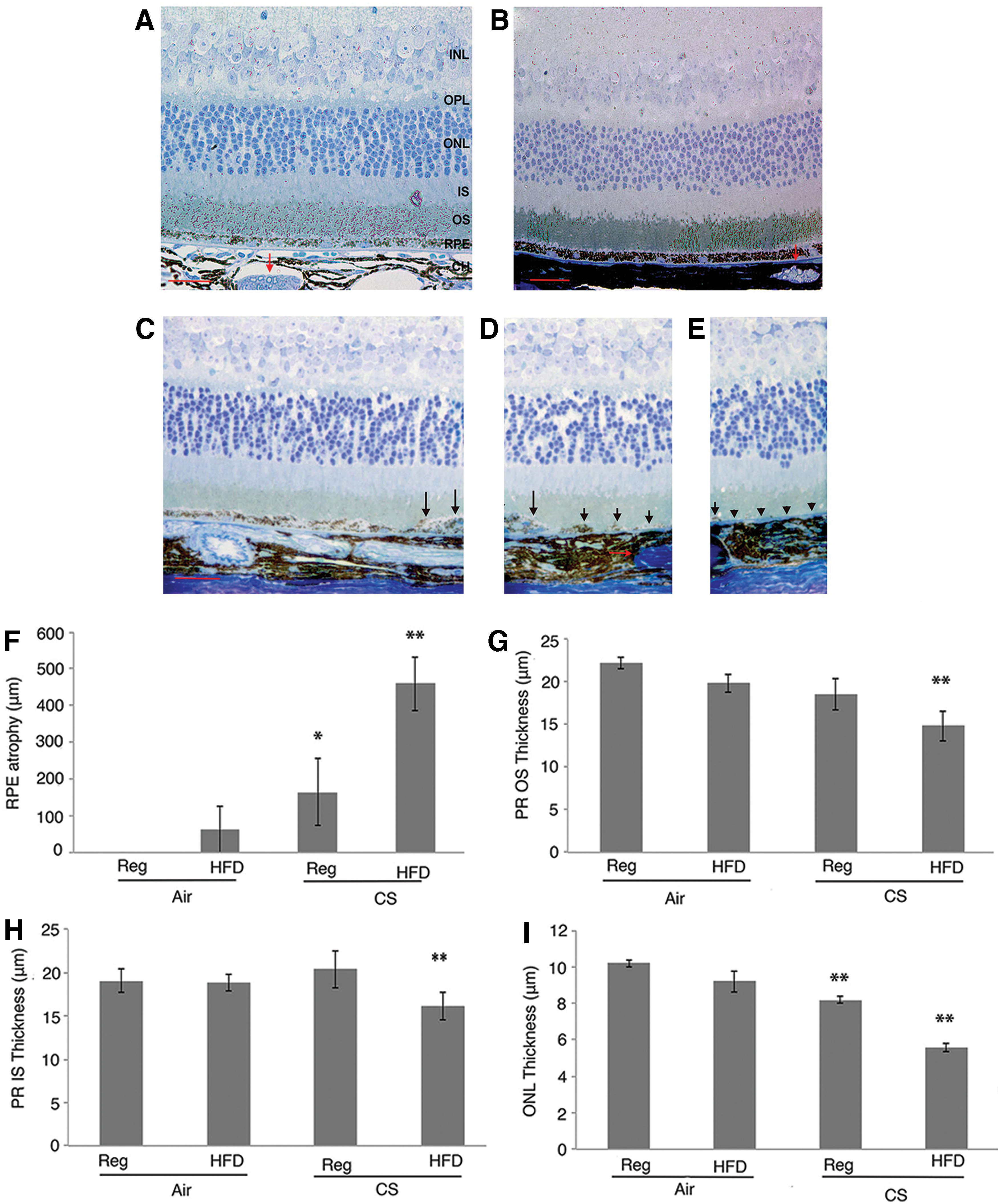
With molecular evidence of impaired Nrf2 and Wnt signaling, RPE dedifferentiation, and cell death, we next assessed the phenotype of apoB100 mice exposed to chronic CS and HFD. Mice raised in air and a normal or HFD did not show overt RPE morphologic changes. In contrast, areas of striking posterior RPE loss with an adjacent transition zone of severely dysmorphic RPE were identified from the optic nerve dorsally in 60% mice exposed to CS alone (n = 10), and 100% of mice exposed to HFD and CS (n = 10; Fig. 5A–E). The length of RPE loss from the optic nerve dorsally was significantly greater (p < 0.01; Fig. 5F), the outer nuclear layer was thinner (Fig. 5G), and the photoreceptor inner and outer segments (Fig. 5H, I) were shortened in mice treated with HFD and CS compared with mice treated with a normal diet and air.
Ultrastructural changes correlated with the morphological alterations. The RPE of apoB100 mice raised in air on a chow diet appeared normal with a normal array of melanin pigment, mitochondria, and intact apical microvilli and basolateral infoldings (Fig. 6A, B). Photoreceptor outer segments were interdigitated with RPE apical microvilli. Bruch's membrane was unthickened, and the choriocapillaris appeared normal. apoB100 mice raised in air on HFD also had normal ultrastructure (Fig. 6C, D). Occasionally, photoreceptor outer segments were truncated and Bruch's membrane had thin, homogeneous basal laminar deposits, which is typical of aging but not AMD (10).
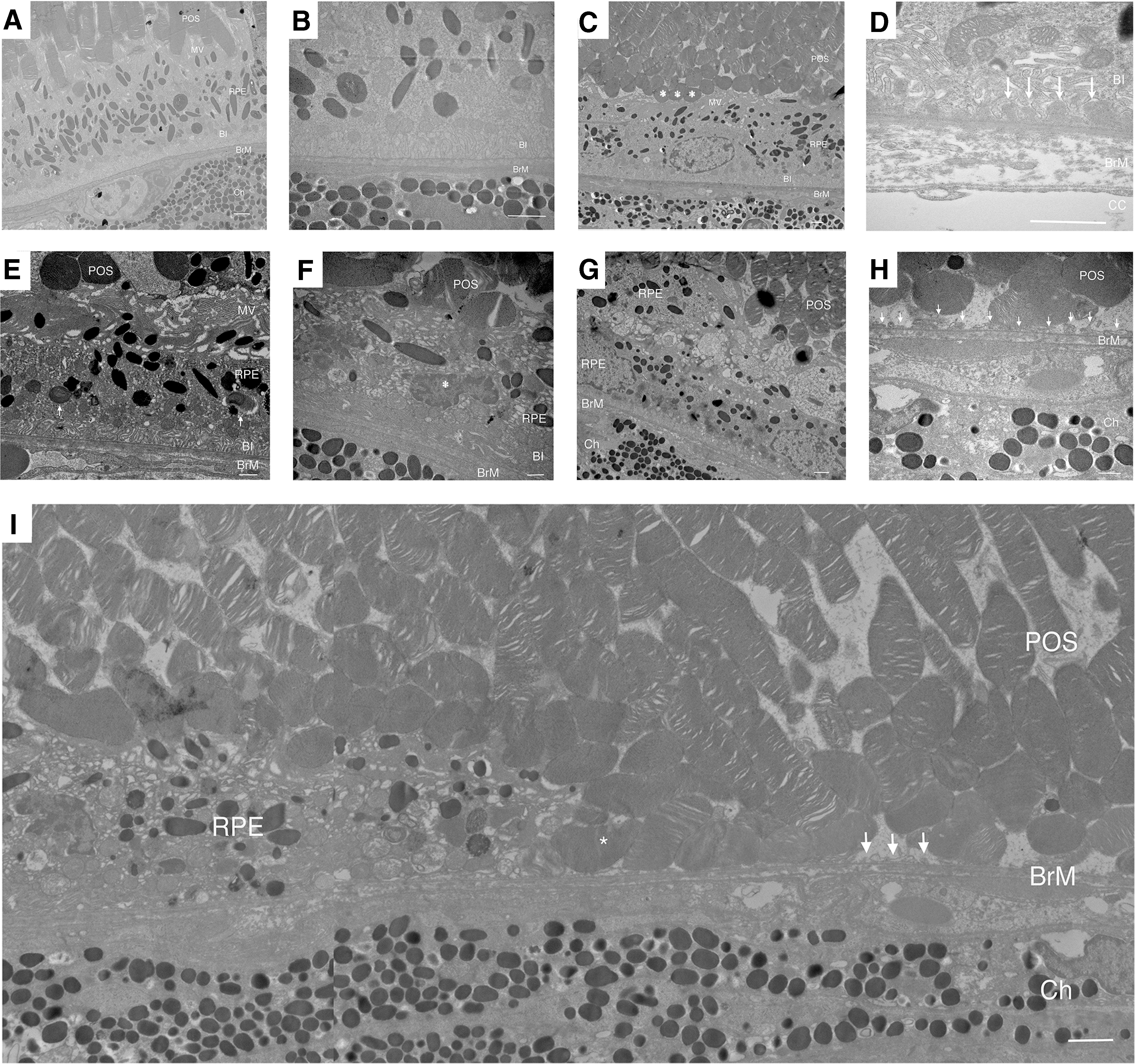
In apoB100 mice given HFD and CS for 6 months, the RPE was progressively deranged in the transition zone with closer proximity to the atrophy in 100% of mice examined (n = 10). At the distal edge, the RPE had a flattened cell shape and altered apical microvilli and cytoplasmic vacuoles, but preserved basal infoldings (Fig. 6E, F). In some cells, the nuclei were condensed with dense heterochromatin, mitochondria were swollen with dilated matrices, and the cytoplasm contained multiple membranous vacuoles, which are all suggestive of apoptosis (11, 19). In addition, the RPE basal infoldings were enlarged and truncated, seen in degenerated RPE in AMD (22).
Within the transition zone, the RPE was multilayered with flattened cell shape (Fig. 6G). Within the atrophy, only RPE cell remnants remained, and degenerated photoreceptor outer segments sit on Bruch's membrane (Fig. 6H, I). Similar features were seen in apoB100 mice given a normal diet and exposed to CS for 6 months (Supplementary Fig. S13).
β-Catenin and Gsk3β immunolabeling are altered in AMD
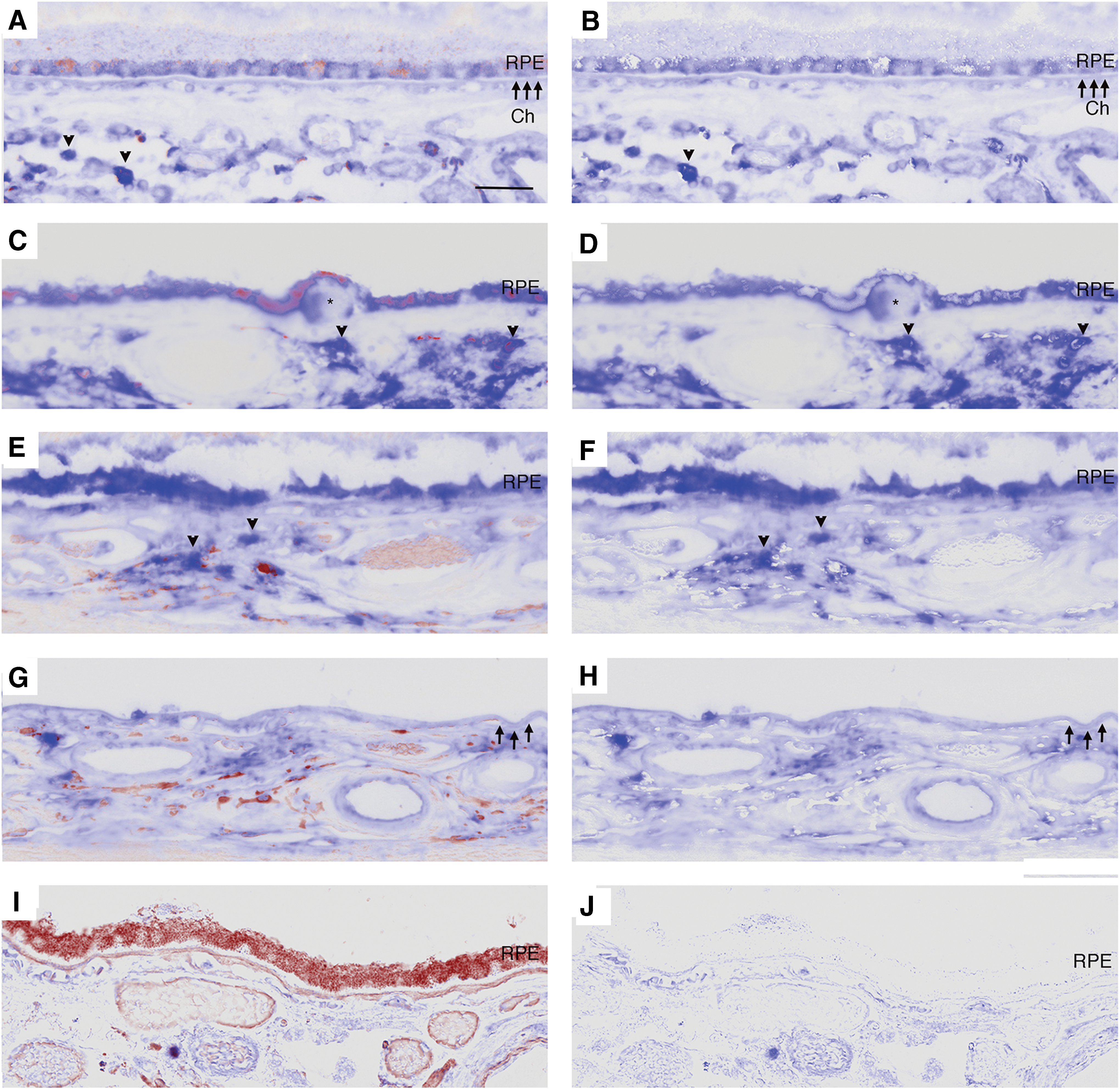
To provide relevance to human AMD, the distribution of β-catenin and Gsk3β in AMD samples was assessed by immunohistochemistry using validated antibodies (29, 38, 49, 50, 68, 70). In the RPE of control eyes, β-catenin staining appeared as a regular band at the basolateral aspect of normal appearing RPE cells in the macula and periphery (Fig. 7A, B). In early AMD (Fig. 7C–H), β-catenin labeling in morphologically preserved RPE adjacent to drusen maintained a regular band at the basolateral aspect of the cell while in dysmorphic, rounded RPE, as in apoB100 mice given CS with or without HFD, was thickened and globular in the lateral cell wall, and was prominent basally or in the sub-RPE space. β-Catenin was also found in basal deposits and drusen. In GA (Fig. 7I–N), the RPE without atrophy had a similar pattern of basolateral labeling, while dysmorphic or migrating RPE cells in the transition zone and atrophic areas had minimal to no β-catenin labeling (Table 1).

Immunolabeling was designated as “yes” when present or “no” when absent. When applicable, the immunolabeling pattern was designated as “globular” or “diffuse.”
AMD, age-related macular degeneration; CFH, Congestive Heart Failure; CVA, Cerebral Vascular accident; D–E, death to enucleation time in hours; MI, Myocardial infarction; N/A, Not available; NNV, Non-Neovascular; RPE, retinal pigment epithelium.
Compared to strongly labeled choroidal cells, Gsk3β immunolabeling was lightly stained and diffusely distributed in the cytoplasm of morphologically normal RPE. In dysmorphic RPE, whether adjacent to or overlying drusen, Gsk3β labeling was strong, consistent, and diffuse in the cytoplasm compared to choroidal cells in the same section. Prominent immunolabeling was observed in the RPE of early AMD, and was similar in dysmorphic and migrating RPE in the transition zone of GA (Fig. 8A–H). Strong Gsk3β staining was also observed in drusen and basal laminar deposits, as summarized in Table 2.
Immunolabeling was designated as “yes” when present or “no” when absent.
Discussion
Herein, we describe a new molecular mechanism of two critical cytoprotective pathways, Wnt and Nrf2, which are impaired in a bidirectional feedback loop in the RPE after apoB100 mice were exposed to two environmental AMD risk factors (Fig. 9). Importantly, the impairment of two, rather than a single cytoprotective pathway, contributed to a phenotype that vividly mimics human GA, including consistent RPE atrophy with overlying photoreceptor loss in the posterior fundus, and a surrounding transition zone of progressive RPE degeneration and heterogeneity, which is a hallmark change seen in human AMD (24). Importantly, in the transition zone, some dysmorphic RPE were multilayered, a key finding that to the best of knowledge, has not been reported in other mouse models, yet is a unique and critical feature in GA (58). Our immunohistochemical studies of aberrant β-catenin and GSK3β labeling in human AMD samples and our prior work showing decreased Nrf2 in dysmorphic RPE of AMD samples (67) suggest that a similar mechanism is involved in human AMD.

Currently, there is no treatment for GA due to a limited understanding of the critical events that lead to RPE atrophy and the absence of a model that mimics essential phenotypic features. The identification of two impaired, interacting cytoprotective pathways provides new insights into the mechanism of RPE degeneration. Since GA progression is related to the extent of damaged RPE in the transition zone (33), this model will be helpful in delineating how and why GA develops and enlarges, and will be valuable for testing new targets intended to either slow or reverse RPE degeneration.
Cell signaling pathways are organized as intertwined communication networks, which are finely tuned by regulators, and are a critical feature of physiological signaling designed to protect any given cell against endogenous or exogenous stressors. Both Wnt and Nrf2 responded to acute stressors in our experiments. Chronic stress can impair these protective systems. Indeed, chronic CS exposure was a driving factor in suppressing Nrf2 and Wnt signaling. The added burden of chronic HFD to CS exposure also impaired Nrf2 signaling and resulted in a more global suppression of Wnt signaling than CS alone. With chronic CS and HFD, 100% of the Wnt pathway components tested (Wnt5, p-LRP6, Gsk3β, and β-catenin) were decreased, while 50% of Wnt components (p-Lrp6, β-catenin) were impaired with CS only. In addition, the decrease in Wnt5 and p-Lrp6 was greater after chronic CS/HFD than CS-only treatment. The degree of Nrf2 and Wnt suppression correlated with the GA phenotype. With Nrf2 impairment and the more complete suppression of Wnt signaling by the combined stresses of CS and HFD, 100% of mice developed GA, while 60% of mice treated with CS only developed GA. These data support our theory that impairing two rather than a single cytoprotective pathway is more likely to induce GA.
Wnt and Nrf2 impairment contributed to each other's decline in a bidirectional feedback loop. L'Episcopo et al. found that impaired Nrf2 could deregulate Wnt signaling (37), while Rada et al. found that Wnt activated Nrf2 signaling by preventing Nrf2 phosphorylation through Gsk3β and Swi4-Swi6 cell-cycle box/β-transducin repeat containing protein (SCB/β-TrCP) mediated proteasomal degradation (55, 56). In our studies, Nrf2−/− mice had reduced β-catenin in the RPE compared with WT mice, an effect that was magnified by oxidative stress. In human ARPE-19 cells, Nrf2 knockdown also reduced β-catenin, increased p-Gsk3β, and reduced the expression of cyclin D, a downstream target of WNT signaling, to collectively suggest that Nrf2 influences WNT signaling. By inhibiting Wnt with DKK1, which is upstream of Gsk3β, Nrf2 was reduced, and inhibiting Gsk3β with SB216763 restored Nrf2 in apoB100 mice. Likewise, inhibiting Gsk3β with SB216763 in ARPE-19 cells restored Nrf2 levels and expression of Gclm, a known Nrf2 responsive antioxidant after an oxidative stress challenge. These results indicate that Wnt regulates Nrf2 signaling, in part, by modulating GSK3b activity, and Nrf2 regulates Wnt signaling.
Under basal conditions, Nrf2 is bound to Keap1 and is presented to the Cullin-3/Rbx1 complex for proteasomal degradation (27). With acute oxidative stress, several cysteines interact with electrophilic compounds to induce a conformational change in Keap1, releasing Nrf2 for translocation to the nucleus and the transcription of antioxidant genes. However, chronic oxidative stress, with persistently elevated basal electrophile levels, lacks an acute electrophile rise so that Nrf2 is not released from Keap1 (27). Under these conditions, Gsk3β can be activated to phosphorylate Nrf2, leading to β-TrCP/Cullin1/Rbx1 E3 ligase-mediated ubiquitinated proteasomal degradation (55, 56), acutely raising the electrophile level to release Nrf2 from Keap1. With excessive, constitutive Gsk3β activity, Nrf2 could remain decreased at levels that will prevent a cytoprotective response. This scenario is plausible with decreased Wnt signaling and chronic Gsk3β activation as in our study.
Several factors may contribute to impaired Wnt signaling in our model. First, the RPE of apoB100 mice, relative to WT mice that do not produce apoB100, had reduced expression of key Wnt molecules, notably Wnt5a and Lrp6. While inconsequential under basal conditions, their relatively suppressed expression might reduce Wnt signaling under duress. While Wnt5a was identified as the most abundantly expressed Wnt ligand, other Wnt ligands could be relevant, including Wnt5b, since the antibody used for this study, which is the only mouse Wnt5 antibody commercially available, recognizes both Wnt5a and 5b.
Second, oxidative stress can decrease Wnt transcription to blunt the Wnt signaling response (65, 74). Third, HFD itself can impair Wnt signaling (53). Fourth, the very low-density lipoprotein receptor (Vldlr) can form a heterodimer with Lrp6 to reduce Wnt signaling (41). Since either HFD or oxidative stress can induce Vldlr expression (36), it is possible that our conditions induced Vldlr, which suppressed Wnt signaling. Finally, just as impaired Wnt signaling contributed to Nrf2 decline, the decreased Nrf2 response likely contributed to decreased Wnt signaling.
Competent Wnt and Nrf2 signalings are essential for cell survival under oxidative stress because they regulate different cytoprotective networks, yet they are interconnected. β-Catenin classically binds to Tcf to induce the transcription of cell survival and differentiation genes such as cyclin D1, but oxidative stress induces a shift in cofactor activity to Foxo TF-mediated expression of antioxidant genes such as Sod2 (2, 45). While this switch is undoubtedly a cytoprotective response, in osteoblasts, the switch comes at the expense of Tcf-mediated transcription, reducing osteoblast differentiation that culminates in osteoporosis (2, 45). In our studies, neither cyclinD1 nor Sod2 was induced by chronic oxidative stress, which suggests that both Tcf and Foxo mediated gene expression, which could contribute to RPE dedifferentiation and reduce the antioxidant response, respectively, and are impaired. Furthermore, since Sod2 can also be regulated by Nrf2 (60), impaired Nrf2 signaling could also be a contributing factor. Under these conditions, the antioxidant response from two systems was inadequate to prevent RPE degeneration.
Our laboratory and other investigators have investigated the role of dysregulated complement, inflammasome activation, endoplasmic reticulum (ER) stress, and impaired autophagy in AMD (7, 13, 25, 30, 34, 35, 48, 59, 72). At present, it is unclear at what point during disease pathophysiology that these derangements contribute to AMD. Both Nrf2 and Wnt can influence complement (51) and inflammasome activation (66), ER stress (64), and autophagy (20, 52). The interaction of these systems with Wnt and Nrf2 signaling, and their impact on disease development, has not been explored to any significant extent. Given the influence of Wnt and Nrf2 on these systems, future investigation is warranted.
Research has uncovered impaired Wnt or Nrf2 signaling in a number of prominent complex diseases such as Alzheimer's (28, 31, 42), Parkinson's (3, 18, 40, 54), and Huntington's disease (62, 63), atherosclerosis and coronary artery disease (44), diabetes mellitus (32, 45), alcoholic liver disease (39), and age-related osteoporosis (2, 45). These studies have focused on one signaling system or the other. With the cross talk between Wnt and Nrf2 signaling in the RPE that resulted in impaired signaling of both cytoprotective pathways and mechanistically involved with an AMD phenotype, future research should consider the interaction and synergistic impact of impairment of both essential systems in these complex diseases.
Materials and Methods
Mice and animal care
All experiments were conducted according to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and the IRB at Johns Hopkins Medical Institutions approved the research. An equal number of male and female apoB100 mice, in a B6;129S background, and Nrf2−/− mice in a C57BL/6J background (27), without the RD8 mutation, were used for experiments. Mice were fed standard rodent chow or HFD consisting of 60% fat, 20% protein, and 20% carbohydrate (Research Diets, Inc., New Brunswick, NJ). Mice were given water ad libitum and kept in a 12-h light–12-h dark cycle. For some experiments, mice received an Ivt injection in one eye and an equal volume (1 μL) of vehicle control in the contralateral eye using a microinjection pump (Harvard Apparatus, Holliston, MA).
Exposure to cigarette smoke
At 8 weeks of age, mice were kept in a filtered air environment or placed in a smoking chamber for 2.5 h/day, 5 days/week, for 6 months, using our published protocol (17).
Tissue preparation
After mice were sacrificed, eyes were enucleated and the retina and RPE/choroid were dissected to extract protein or RNA or the eyes were fixed in 2% paraformaldehyde and cryopreserved for immunohistochemistry or 2.5% glutaraldehyde/1% paraformaldehyde in 0.08 M cacodylate buffer for electron microscopy.
Cell culture
Human ARPE-19 cells were grown in Dulbecco's modified eagle medium (DMEM)/F12 with 15 mM HEPES buffer, 10% fetal bovine serum, and 2 mM
RNA extraction
Total RNA was extracted using the RNeasy Mini Plus-kit (Qiagen, Inc., Gaithersburg, MD). RNA quality was confirmed using the Agilent Bioanalyzer (Agilent Technologies, Palo Alto, CA).
Quantitative real-time RT-PCR
Reverse transcription was performed using random hexamers and MultiScribe reverse transcriptase (Applied Biosystems). Quantitative real-time RT-PCR analyses were performed using Assay-on-Demand primer and probe sets (Applied Biosystems) with a StepOnePlus TaqMan system (Applied Biosystems). β-Actin was used for normalization.
Subcellular protein fractionation
Cell pellets or small tissue pieces were washed with cold phosphate-buffered saline, and lysed or homogenized in the cold CER I buffer, provided by the NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Thermo Fisher Scientific, Waltham, MA), with an additional Sigma FAST Protease Inhibitor added. Cytoplasmic and nuclear protein fractions were prepared sequentially, following the manufacturer's protocol, and quantified using the BioRad DC Protein Assay Kit.
Western blot analysis
Western blot analysis was performed as described (67). Membranes were incubated with the intended primary antibody (β-catenin, cleaved Parp, Lrp 6, GAPDH, p-Gsk3β [tyr216], Nrf2, Wnt3) (Abcam, Inc., San Francisco, CA) and p-Lrp6 (Ser1490), Gsk3β, Rpe65, Wnt5 (Cell Signaling Technology, Danvers, MA), Ho-1, Lrat (Santa Cruz Biotechnology, Santa Cruz, CA), Gclm (Abcam, Inc.). Signal was detected with a chemiluminescence detection system (Thermo Scientific, Waltham, MA). Blots were imaged with an ImageQuant LAS4000 scanner (GE Healthcare, Inc., Piscataway, NJ), and band intensity is reported as arbitrary densitometric units using ImageJ software. β-Actin or panactin was used for signal normalization.
Light and transmission electron microscopy
The posterior segments of eyes were postfixed with 1% osmium tetroxide, dehydrated, and embedded in Poly/Bed 812 resin (Polysciences, Inc., Warrington, PA). Semithin sections were stained with Toluidine blue. To quantify areas of GA, semithin sections were imaged with a BX50 light microscope (Olympus Optical Co., Tokyo, Japan) and images were acquired with a digital camera (DP22; Olympus Optical Co.). Temporally, the caliper was used to measure the length of RPE atrophy from the optic nerve, or the thickness of the inner and outer photoreceptor segment layers and outer nuclear layer within 1000 μm from optic nerve. As a comparative measure of outer nuclear layer thickness, the number of nuclei in each column was counted.
Ultrathin sections were stained with uranyl acetate and lead citrate, and examined with a JEM-100 CX electron microscope (JEOL, Tokyo, Japan) in the Wilmer Core Facility.
Immunohistochemistry
Mouse cryosections (10 μm) were blocked with 5% goat serum, incubated overnight with rabbit monoclonal anti-β-catenin (Abcam), and then with Alexa Fluor 594 goat anti-rabbit (Life Technologies) for 1 h, as previously described (6). Appropriate rabbit immunoglobulin G (IgG; Santa Cruz Biotech) was used as isotype control. Sections were imaged using a Zeiss LSM 710 confocal microscope.
Autopsy eyes (n = 31) were obtained from the Wilmer Eye Institute Pathology Division after approval from the Human Subjects Committee at Johns Hopkins University. “Unaffected” eyes (n = 12) had no AMD history or microscopic evidence of AMD. Early AMD donors had an AMD history and macular drusen (n = 16). Small drusen had a diameter of ≤4 RPE cells (<63 μm) and large drusen as >8 RPE cells (∼125 μm). GA samples (n = 3) had a known history and microscopic evidence of RPE atrophy.
Eyes were fixed in 4% formaldehyde, paraffin embedded, and sectioned at 4 μm thickness. Antigens were retrieved with the Target Retrieval System (Dako, Inc., Carpinteria, CA). Sections were incubated with goat blocking serum and then with validated antibodies, rabbit anti-human β-catenin monoclonal antibody (38, 50, 68, 70) (1:250 dilution; Abcam, Inc.) and anti-Gsk3β (29, 49) or isotype rabbit monoclonal IgG (1:250 dilution; Epitomics, Inc.; Burlingame, CA), overnight at 4°C. Sections were incubated with biotinylated anti-rabbit IgG and then with the ABC-AP reagent (Vector Labs). Slides were developed with blue substrate working solution (Vector Labs) supplemented with levamisole. Sections were imaged with a light microscope equipped with the Cri-Nuance system (Caliper Life Sciences, Inc., Hopkinton, MA) to subtract out the melanin pigment.
Statistical analysis
Statistical analysis was carried out using the Student's two-tailed unpaired t test, or for multiple comparisons, the Kruskal–Wallis test using GraphPad software (GraphPad Software, Inc., San Diego, CA). For in vitro experiments, each condition was tested in triplicate and each experiment was repeated at least three times. Blots are selected as the representative of the experiment, and graphs summarize the mean ± standard error of the mean of at least three independent experiments. For in vivo experiments, 3–6 mice were used for each condition, and 10 mice for transmission electron microscopy.
Footnotes
Acknowledgments
We thank Stephen Chan, Sonny Dike, Natalia Vergara, Christian Gutierrez, and Gillian Shaw for their technical expertise and guidance, and Akrit Sodhi, MD, PhD, and Debasish Sinha, PhD, for carefully critiquing the article. We thank Victor Perez, MD, for providing CEP-BSA. Supported by the NIH EY019904 (J.T.H.), EY14005 (J.T.H.), EY027691 (J.T.H.), EY 01765 (Wilmer Imaging Core grant), K12 EY108198 (K.B.E.), RPB Senior Scientist Award (J.T.H.), unrestricted grant from RBP, and gifts from the Merlau family and Aleda Wright.
Author Disclosure Statement
No competing financial interests exist.
Abbreviation Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.