
Abstract
Significance:
Oxidative stress increases in the brain with aging and neurodegenerative diseases. Previous work emphasized irreversible oxidative damage in relation to cognitive impairment. This research has evolved to consider a continuum of alterations, from redox signaling to oxidative damage, which provides a basis for understanding the onset and progression of cognitive impairment. This review provides an update on research linking redox signaling to altered function of neural circuits involved in information processing and memory.
Recent Advances:
Starting in middle age, redox signaling triggers changes in nervous system physiology described as senescent physiology. Hippocampal senescent physiology involves decreased cell excitability, altered synaptic plasticity, and decreased synaptic transmission. Recent studies indicate N-methyl-
Critical Issues:
We review redox homeostasis mechanisms and consider the chemical character of reactive oxygen and nitrogen species and their role in regulating different transmitter systems. In this regard, senescent physiology may represent the co-opting of pathways normally responsible for feedback regulation of synaptic transmission. Furthermore, differences across transmitter systems may underlie differential vulnerability of brain regions and neuronal circuits to aging and disease.
Future Directions:
It will be important to identify the intrinsic mechanisms for the shift in oxidative/reductive processes. Intrinsic mechanism will depend on the transmitter system, oxidative stressors, and expression/activity of antioxidant enzymes. In addition, it will be important to identify how intrinsic processes interact with other aging factors, including changes in inflammatory or hormonal signals. Antioxid. Redox Signal. 28, 1724–1745.
From Oxidative Signaling to Oxidative Stress
R
The major metabolic forms of ROS include superoxide (
Similarly, nitric oxide (NO) is a free radical synthesized by nitric oxide synthase (NOS) and induces thiol S-nitrosylation of cysteine residues. Like the disulfide bonds induced by hydrogen peroxide, S-nitrosylation is rapid and reversible and acts as a signaling mechanism. However, under oxidative stress, the increased level of superoxide reacts with nitric oxide to produce the highly reactive RNS, peroxynitrite (ONOO
In contrast to reversible modification induced by hydrogen peroxide and nitric oxide, the highly reactive peroxynitrite, superoxide, and hydroxyl radical produce irreversible damage to DNA (e.g., 8-hydroxy-2′-deoxyguanosine), lipids (e.g., 4-hydroxy-2,3-nonenal), and proteins (e.g., carbonyls and 3-nitrotyrosination) (22, 35). In this case, the molecules must be removed and replaced to maintain functional integrity. Similar to other organs, oxidative damage increases in the brain with age (8, 44, 66, 112, 192, 256, 286). Age-related vulnerability to oxidative stress and the level of oxidative damage vary by brain region (1, 63, 189, 224, 256). Previous work suggests that regional differences in the accumulation of damaged molecules may contribute to age-related impairment of sensory-motor function (66, 162) and memory impairments (40, 81, 137, 194, 195). Interestingly, despite increased levels of lipid peroxidation and protein carbonylation, nitrotyrosine levels may not accumulate in the brain with age or may be increased only in selective brain regions (44, 78, 154). Finally, for neurodegenerative diseases, levels of oxidative damage are increased above age-matched controls (253, 267). In some cases, the oxidative damage may be a consequence of the disease; however, evidence indicates that the products of lipid peroxidation can cause cell death (267).
Redox Signaling
Mounting evidence indicates that an oxidized redox state or redox stress precedes accumulation of oxidative damage and the shift in redox state underlies physiological characteristics of neuronal senescence (90, 102, 153, 207). A mild, yet sustained, increase in ROS/RNS levels can induce an incessant increase in the activity of redox signaling cascades to influence cell function. As such, an oxidized redox state likely represents an early marker of Alzheimer's disease by contributing to the emergence of impaired synaptic function before frank histopathology (90). Indeed, the oxidized redox state may pave the way for increasing oxidative stress and the accumulation of oxidative damage, which precedes aggregation of molecules that normally characterize neurodegenerative diseases (218). The results suggest a continuum of changes due to increasing oxidative stress with advancing age and disease (Fig. 1). As the redox state becomes increasingly oxidized, functional disruption develops, and depending on genetic or environmental factors, the oxidized redox state will contribute to the accumulation of malformed or damaged molecules.

An understanding of the role of the redox environment in cellular processes depends on the tools for measurement and manipulation of redox state. In biological systems, redox state is commonly measured as the balance of redox couples GSH/glutathione disulfide (GSSG), nicotinamide adenine dinucleotide (NAD+)/(NADH), and NADP+/NADPH. In most cases, GSH/GSSG is employed as the measure of redox state due to greater abundance and because GSH acts as the reducing agent for many other molecules (103, 240). A decline in free GSH or rise in GSSG is observed in middle age for several vulnerable brain regions, including the cortex, hippocampus, thalamus, striatum, and cerebellum (75, 212, 223 –225, 303). In contrast, the level of GSH may remain constant in the brain of animals that maintain cognitive function over the course of aging (187). Consistent with a continuum of redox changes, the decline in GSH/GSSH may precede mitochondrial dysfunction (47, 173). Finally, GSH levels in astrocytes are higher than those observed in neurons such that regional differences in astrocyte number or function could influence vulnerability to oxidative stress (158).
In addition to measures of GSH, the role of redox state in cell function can be tested by examining the effects of reducing and oxidizing agents on physiology. Some specificity can be achieved in identifying intracellular or extracellular redox changes by using membrane-impermeable agents, particularly oxidized or reduced GSH (31, 57, 226, 234, 268). Membrane-permeable oxidants include 5,5-dithio-bis-2-nitrobenzoic acid (DTNB) and hydrogen peroxide. While hydrogen peroxide lacks specificity, application of these oxidizing agents to neurons from young animals can mimic the effects of aging (24, 31, 296). Similarly, the membrane-permeable reducing agent, dithiothreitol (DTT), has a rejuvenating effect on physiology in cells from aged animals (31, 32, 42, 108, 144).
Sources for ROS and RNS in the Brain
The vulnerability of different cell types, cellular compartments, and brain regions depends on the chemical character of the oxidative stressor, level of ROS production, and expression/activity of antioxidant enzymes. Superoxide is generated in mitochondria during oxidative phosphorylation, and the production of mitochondrial superoxide and hydrogen peroxide is inversely related to longevity (115, 258). Due to its high reactivity, this radical does not travel far, which may limit where damage occurs. The mitochondrial free radical theory of aging suggests that the production of superoxide by mitochondria results in impaired mitochondrial function (101, 115). Indeed, relative to other molecules, enhanced damage to mitochondrial DNA (181) and increased mitochondrial protein carbonyls (256, 294, 295) can be observed in models of aging and neurodegenerative diseases. However, others have not observed an age-related increase in mitochondrial damage, possibly due to differences in brain region vulnerability, rate of protein removal, or specificity of the cellular compartment (68, 104). As discussed in Antioxidant Enzymes section, increased expression of mitochondrial antioxidant enzymes decreases oxidative damage and may be protective against neurodegenerative diseases; however, decreasing mitochondrial superoxide does not appear to protect cognitive function during normal aging. Rather, treatments that influence hydrogen peroxide-mediated redox signaling may be more important for age-related cognitive decline (123, 152, 153).
As noted above, most of the mitochondrial-generated superoxide is converted to the less reactive hydrogen peroxide. Unlike superoxide, hydrogen peroxide can readily cross membranes to travel a greater distance. However, hydrogen peroxide interacts with iron or copper to form the highly reactive hydroxyl radical. Thus, regional difference in the level of reactive metals is another factor that may contribute to specificity of oxidative damage and brain aging (231, 298, 300). Interestingly, oligodendrocytes and microglia accumulate iron (21). Furthermore, relative to astrocytes, these glial cells exhibit reduced mitochondrial DNA repair capacity (119, 149), which could underlie the notable white matter damage associated with aging (77).
NADPH oxidase (NOX) provides another source for superoxide. NOX1 and NOX4 are expressed in neurons and are involved in synaptic plasticity as well as cerebral blood flow (136). NOX2 is primarily located in microglia and provides a source for superoxide associated with neuroinflammation (55, 107, 172, 236). NOX activity is increased during neuroinflammation and over the course of aging (73, 110, 146, 193), and increased NOX activity is thought to contribute to the progression of neurodegenerative diseases (54).
In the vascular system, nitration reactions can result from peroxide enzyme activity (283). In the nervous system, the formation of nitric oxide by NOS interacts with superoxide to form the highly reactive peroxynitrite, which underlies much of the neurotoxicity associated with increased production of nitric oxide (204). Normally, nitric oxide synthesis is tightly regulated such that neuronal NOS (nNOS) or endothelial NOS (eNOS) generates nitric oxide as a signaling molecule to influence synaptic function and cerebral blood flow. However, toxins and cytokines associated with inflammation initiate the generation of an inducible isoform (inducible nitric oxide synthase [iNOS]) in astrocytes and microglia (45, 46). Furthermore, increased expression of nNOS has been reported for neurodegenerative diseases (204). Thus, regional differences in neuroinflammation can shift cells from rapid and reversible redox signaling toward oxidative damage and may contribute to regional specificity in the onset or progression of brain aging and neurodegenerative diseases.
Antioxidant Enzymes
To act as a signaling molecule, ROS signals must be concluded in a timely manner. Antioxidant molecules and enzymes balance the biological activity of ROS to regulate ROS signaling and minimize toxicity. Thus, the ability to regulate the function of antioxidant defense mechanisms is a factor to consider in the onset and progression of cognitive decline associated with aging and diseases. The antioxidant enzymes include superoxide dismutase (SOD), which catalyzes superoxide into hydrogen peroxide, and catalase (CAT) or glutathione peroxidase (GPx), which convert hydrogen peroxide to water and oxygen. SODs are classified according to their metal cofactors and cellular localization. Cu/Zn-SOD (SOD1) is distributed throughout the cytoplasm, nucleus, and inner membrane space of mitochondria. Cu/Zn-SOD (SOD3) is located in the extracellular space, and Mn-SOD (SOD2) is restricted to the mitochondrial matrix. The literature concerning the brain expression or activity of antioxidant enzymes over the course of aging is mixed (249). For example, Cu/Zn-SOD activity is either decreased or unchanged and Mn-SOD activity is either increased or unchanged in the brain with aging.
Transgenic mice have been employed to address questions of how antioxidant enzymes contribute to oxidative damage, vulnerability to disease, and aging. Mice that exhibit knockout (SOD3) or knockdown (SOD1, SOD2) of SOD isoforms exhibit increased oxidative damage and increased vulnerability to age-related diseases, without necessarily affecting longevity or age-related cognitive decline (27, 150, 188, 211, 247, 284). The results suggest that a decline in SOD activity may render animals more vulnerable to age-related diseases; however, these studies leave open the question of the role of oxidative damage in cognitive decline associated with normal aging.
If superoxide and oxidative damage mediate age-related cognitive decline, then upregulation of SOD should reduce oxidative damage and be protective against aging. Upregulation of mitochondrial SOD2 reduces mitochondrial superoxide, but does not protect against cognitive decline associated with normal aging (123, 153). Rather, crossing mice that overexpress SOD2 with a transgenic mouse model of Alzheimer's disease indicates that SOD2 is protective against the onset of cognitive impairment associated with expression of the genes linked to Alzheimer's disease (74, 175). Again, the results are consistent with the idea that SOD activity is protective against diseases of aging and leaves open the question of the role of oxidative damage in mediating cognitive decline associated with normal aging.
In mice, overexpression of SOD1 or SOD3 impairs hippocampal-dependent memory and synaptic plasticity in adult mice (95, 156, 274). Because these were transgenic animals, it is possible that memory changes were due to developmental influences of SOD overexpression. While expression was increased in all tissues, examination of hippocampal physiology suggested a possible SOD effect specific to hippocampal synaptic plasticity. The role of ROS in regulating synaptic plasticity has recently been reviewed (174). Briefly, long-term potentiation (LTP) is a long-term increase in synaptic transmission in response to patterned synaptic activity. LTP represents one mechanism for learning and memory, and the redox-mediated disruption of LTP during aging is thought to contribute to cognitive impairment (87). At hippocampal CA1 synapses, the induction of LTP requires a transitory increase in superoxide and induction is impaired by a sustained increase in hydrogen peroxide. In the case of SOD3 overexpression, LTP was rescued by inhibition of SOD, but not by application of CAT, suggesting that the impairment was due to a loss of superoxide signaling rather than elevated hydrogen peroxide (274). Using the radial arm maze as a test of learning and memory, it was shown that adult SOD3-overexpressing mutant mice exhibit deficits that depend on motivational state (156). With advanced age, these mutant mice exhibited improved learning and memory on the radial arm maze (157). It is curious that improved behavior was not associated with a decrease in oxidative stress in the hippocampus and was dependent on motivational state.
In contrast to SOD3, impaired LTP associated with increased SOD1 expression is thought to result from excess hydrogen peroxide (134, 152). The specificity of SOD1 overexpression on memory and synaptic function was examined in a series of studies that employed a viral vector-mediated delivery system (152, 153). SOD1 expression was limited to region CA1 of the hippocampus of rats at specific ages (152, 153). Overexpression reduced age-related oxidative damage, indicating that increasing SOD1 activity can reverse the accumulation of damaged molecules, specifically when expressed in older animals (Fig. 2A, B). Importantly, SOD1 expression and the decrease in oxidative damage were associated with impaired cognitive function and impaired synaptic plasticity in middle-aged and aged animals (Fig. 2C–E). This would seem counterintuitive that reduced oxidative damage in older animals is associated with impaired cognition and synaptic plasticity. The answer to this conundrum was revealed by examination of the redox state. In addition to decreased oxidative damage, expression of SOD1 was associated with a decrease in GSH and an increase in expression of GPx1. Excess hydrogen peroxide can deplete the level of GSH and initiate a compensatory increase in GPx expression (197, 230). The idea that SOD1 was producing excess hydrogen peroxide was confirmed by overexpression of CAT, which rescued cognition and synaptic plasticity (Fig. 2C–E). The results emphasize the contribution of hydrogen peroxide and an oxidized redox state, rather than oxidative damage, as the mechanisms for age-related cognitive impairment.
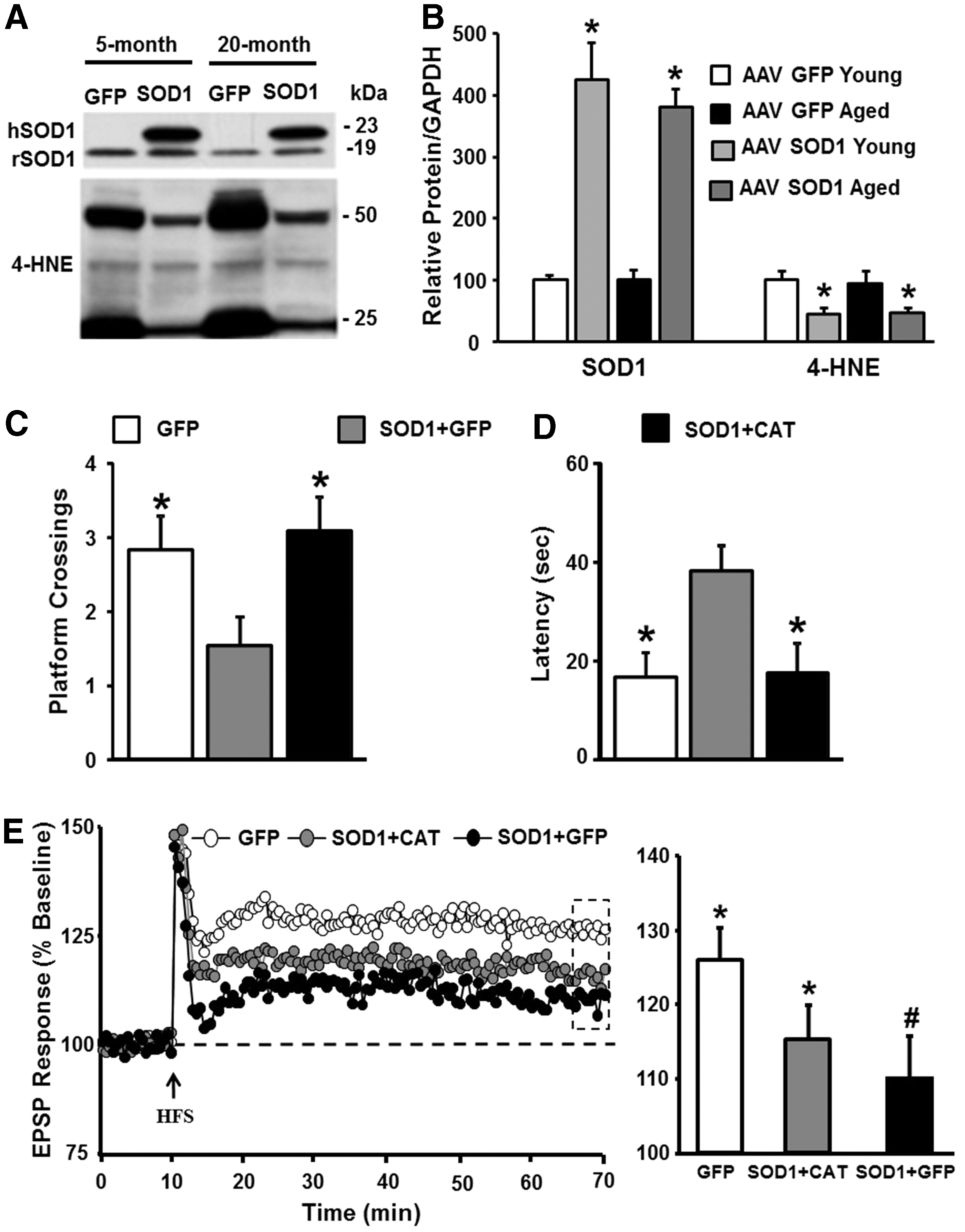
Similar to SOD, knockout and overexpression of GPx influence the susceptibility to nervous system oxidative damage, without necessarily influencing normal aging (155). Knockout of CAT is relatively benign, possibly due to compensation by GPx, although overexpression of CAT is neuroprotective against ischemia (11). Interestingly, transgenic mice, which express CAT, specifically in the mitochondria, exhibit reduced effects of aging on heart, muscle, and metabolism, as well as increased longevity (67, 151, 244, 277, 280) and enhanced hippocampal-dependent memory (203). However, the study of hippocampal memory was limited to adult mice (5–6 months). Enhanced memory was not associated with a shift in markers of oxidative damage, suggesting a possible redox signaling mechanism. Finally, viral-mediated expression of CAT in the hippocampus provides protection from impairments associated with overexpression of SOD1 alone, and upregulation of SOD1+CAT was protective against cognitive impairments in advanced age (153). The results point to disruption of hydrogen peroxide signaling and redox state as a critical factor for age-related cognitive decline.
Redox Stress and Senescent Physiology
Senescent physiology
Senescent physiology of the hippocampus, examined in vitro, has been well characterized as a decrease in cell excitability, altered synaptic plasticity, and decreased synaptic transmission (85 –87, 89, 91, 140, 200, 273). It is important to emphasize that senescent physiology of hippocampal circuits is observed starting in middle age in association with the onset of cognitive impairments (87, 98, 108, 144). Much of the work has focused on impaired LTP as a hypothesized mechanism for age-related deficits in learning and memory (14, 71, 82, 83, 87, 90, 91, 141, 249). As described below, there are several redox-sensitive mechanisms that underlie the decrease in cell excitability and impaired induction of LTP during aging.
Redox sensitivity of N-methyl-D-aspartate receptor function
N-methyl-D-aspartate (NMDA) receptor activity is critical for induction of LTP, and NMDA receptor hypofunction is observed with advancing age (Fig. 3A, B). The effect of oxidizing and reducing agents on NMDA receptor responses is age dependent such that oxidizing agent, xanthine/xanthine oxidase or hydrogen peroxide, induces a decrease in the NMDA receptor synaptic response in hippocampal slices from young animals (3, 4, 24, 31, 160, 272). In contrast, bath application of the reducing agent DTT enhances NMDA receptor synaptic responses specifically in older animals (31, 108, 144, 152) (Fig. 3C).

Several studies have linked a redox-mediated NMDA receptor hypofunction with age-associated cognitive impairment (108, 144, 152). First, the decline in NMDA receptor-mediated synaptic transmission in the hippocampus and prefrontal cortex (PFC) is observed in middle-aged rats that exhibit deficits on tasks dependent on these regions. In addition, the magnitude of DTT-induced growth of the NMDA receptor synaptic response at CA3-CA1 and PFC synapses correlates with impaired spatial memory and executive function (108, 144). The fact that the NMDA response can be enhanced by DTT suggests that hydrogen peroxide and an oxidized redox state mediate the NMDA receptor hypofunction. This was confirmed by viral expression of SOD1, which impaired cognitive function and promoted senescent physiology observed as a DTT-sensitive decrease in the NMDA receptor-mediated synaptic response (152). Finally, viral expression of CAT rescued the NMDA receptor hypofunction in aged animals and in young overexpressing SOD1, indicating that excess hydrogen peroxide mediates impaired NMDA receptor function in senescence (152, 153).
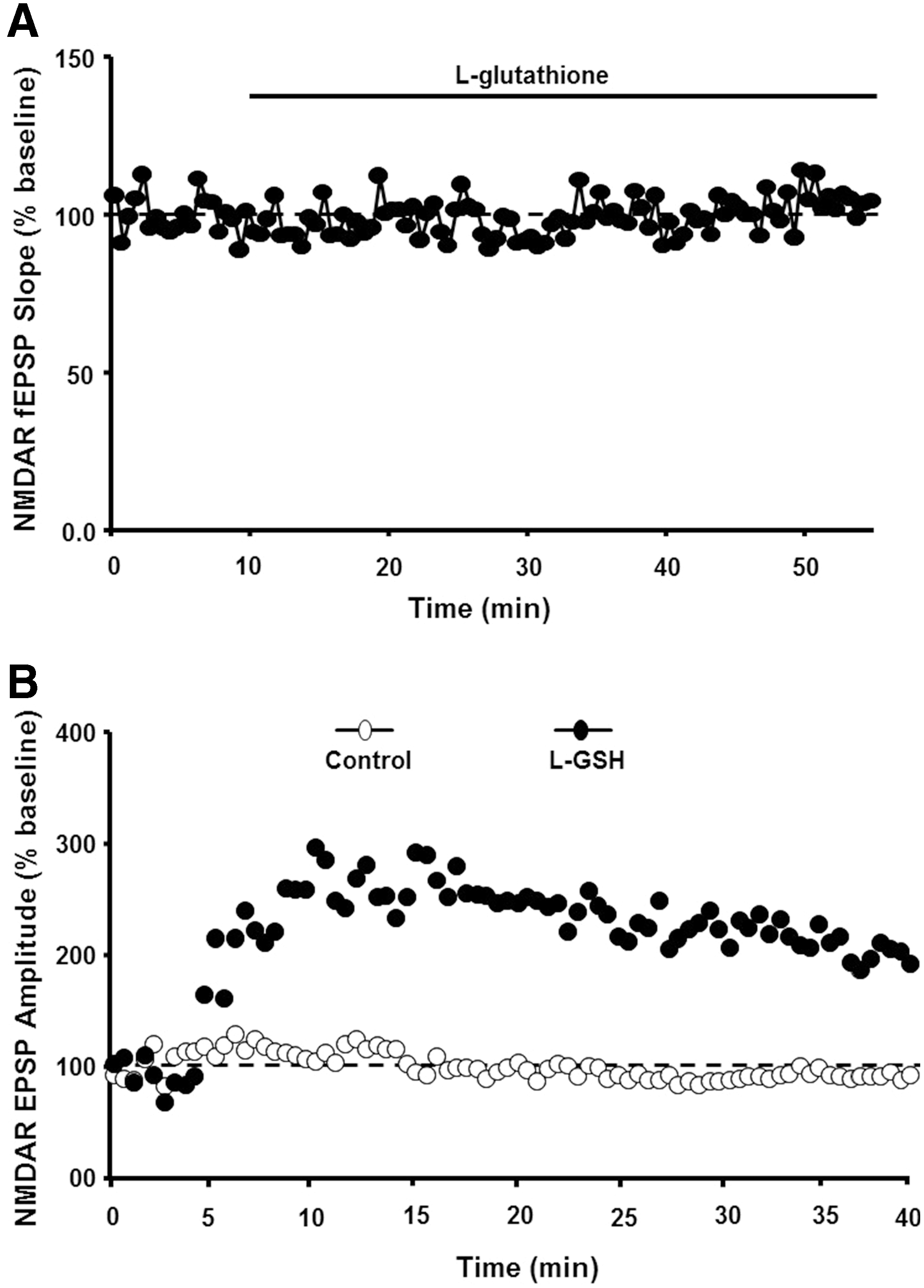
There are several redox mechanisms for regulation of NMDA receptor function. For example, NMDA receptors contain extracellular cysteine residues, which may interact under oxidizing condition to form disulfide bonds, decreasing receptor function through a shift in receptor confirmation (160). However, in the case of aging, an increase in NMDA receptor function is observed when reduced GSH is delivered intracellularly and is not observed following extracellular application of GSH (31) (Fig. 4). Furthermore, NMDA receptor hypofunction is not ameliorated by antioxidants vitamin C and the vitamin E analog, trolox, which would be expected to reduce membrane and extracellular oxidative stress (296). However, NMDA receptor hypofunction can be reversed by dietary supplementation with the GSH precursor, N-acetylcysteine (117, 227). These results support the idea that intracellular redox state, rather than oxidative damage of the membrane, mediates NMDA receptor hypofunction.
Using various phosphatase and kinase inhibitors, we observed that the DTT-mediated enhancement of the NMDA receptor response was blocked by specific inhibition of Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity (Fig. 5). Moreover, using cytosolic extracts, we observed that DTT increased CaMKII activity in an age-dependent manner (31) (Fig. 5C). Likely candidates for redox modulation of CaMKII activity include cysteines on CaMKII, calmodulin, and neurogranin. Recent reports indicate that prolonged exposure to nitric oxide inhibits CaMKII through S-nitrosylation of CaMKII (60, 259). Calmodulin is also sensitive to redox state (26, 257) and oxidation of calmodulin would limit CaMKII activation and subsequent CaMKII regulation of NMDA receptor function (31, 228). In addition, neurogranin binds calmodulin to localize it near the synapse, and this binding is reduced by redox-mediated formation of neurogranin disulfide bonds (124, 171, 252), which could limit the responsiveness of calmodulin signaling.

Under normal physiological conditions, NMDA receptor activity induces the production of nitric oxide, which can give feedback to reduce NMDA receptor activity through S-nitrosylation of cysteines on NMDA receptor subunits, NR1 and NR2A (160), and a prolonged increase in nitric oxide can reduce CaMKII activity (60, 259), which would decrease NMDA activity (31). However, with increased oxidative stress, production of nitric oxide through NMDA receptor activation can induce the formation of peroxynitrite and neurotoxicity associated with oxidative damage (159). In addition, as oxidative damage accumulates, NMDA receptor hypofunction may reverse. In cultured neurons, addition of the lipid peroxidation product, 4-hydroxynonenal (4-HNE), rapidly enhanced NMDA receptor responses (165). The increased Ca2+ from NMDA receptors may underlie the enhanced induction of LTP in the dentate gyrus following 4-HNE application to hippocampal slices (5, 6). If 4-HNE influences NMDA receptor function during aging, then it might be expected that effects of 4-HNE on LTP would be occluded in aged animals; however, this has not been investigated.
Redox sensitivity of intracellular Ca2+ stores
The other major characteristic of senescent neurons, a decrease in cell excitability, involves an increase in the magnitude of slow, Ca2+-dependent K+-mediated afterhyperpolarization (sAHP) (32, 98, 141, 142, 147, 186, 275). Several pieces of evidence indicate that the age-related increase in sAHP (Fig. 6A) results from a redox-mediated increase in Ca2+ release from intracellular Ca2+ stores (ICS) (32). Evidence for redox regulation of the sAHP amplitude comes from studies that observe age-dependent effects of reducing and oxidizing agents on sAHP. First, DTT application reduced the sAHP amplitude (∼50%), specifically in aged cells, such that sAHP was similar to that of young cells (Fig. 6A, B). Second, increasing oxidative stress through the application of xanthine/xanthine oxidase mimicked the effects of aging, increasing sAHP in young animals (Fig. 6C). The specificity and magnitude of DTT effects on sAHP suggest that redox stress mediates age differences in sAHP (32).

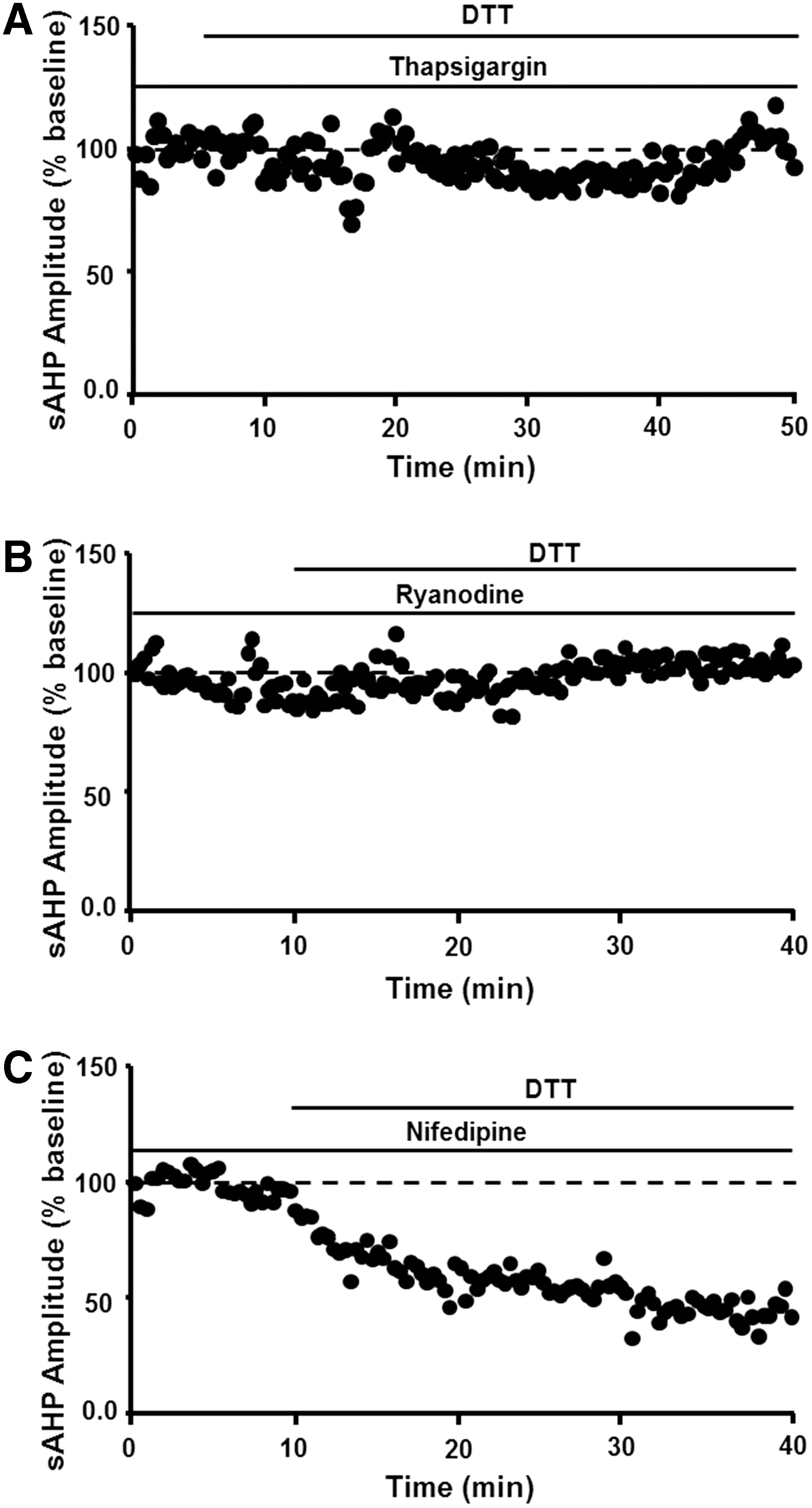
The redox mechanism involves ICS such that decreasing Ca2+ release from intracellular stores through depletion of intracellular stores by thapsigargin or blockade of release by ryanodine prevented DTT-mediated reduction of sAHP (Fig. 7A, B). In contrast, blockade of voltage-dependent L-channels reduces the magnitude of sAHP by ∼30% in young and aged rats (199, 217), and L-channel blockade did not occlude the effects of DTT (Fig. 7C). The results are consistent with the finding that cysteine residues within the ryanodine receptor are highly sensitive to redox conditions, indicating that an oxidized redox state underlies increased release of Ca2+ from ICS (209). Finally, addition of 4-HNE to hippocampal cell cultures increased the activity of voltage-dependent Ca2+ channels, suggesting that accumulation of oxidative damage during aging or during progression of neurodegenerative diseases could further alter Ca2+ regulation (164).
It might be expected that a decrease in cell excitability would lower cell discharge rates. However, for the CA1 region, the level of discharge activity of pyramidal cells recorded in behaving animals is not dramatically altered during aging (39, 178, 202). Rather, aged cognitively impaired animals exhibit a deficiency in the ability to modify firing or maintain changes in firing associated with learning and memory. It is likely that the loss of modifiability is due to redox-mediated impairment in synaptic plasticity. In the case of sAHP, greater hyperpolarization disrupts the integration of depolarizing postsynaptic potentials needed to activate NMDA receptors for induction of LTP, and treatments to reduce sAHP unmask LTP in aged animals (31, 143, 199). Thus, the oxidized redox state impairs synaptic plasticity either directly through NMDA receptor hypofunction or indirectly through hyperpolarization, which would limit NMDA receptor activation. Again, physiology may change as oxidative stress increases and oxidative damage accumulates. There is an indication that lipid peroxidation is associated with an overall decrease in discharge activity of hippocampal CA3 neurons recorded in awake rats (251). Regardless, results suggest that the emergence of cognitive deficits is associated with the rise in an oxidized redox state, which impairs synaptic plasticity, decreases synaptic throughput, reduces the modifiability of cell discharge activity, and the accumulation of oxidative damage may further compromise cognitive function.
Redox Regulation of Neurotransmitter Systems
Progression of functional alterations related to the level of oxidative stress is evident in several neurotransmitter systems. In some cases, the activity of the transmitter will influence redox state and, under physiological conditions, the shift in redox state can act as a rapid and reversible feedback mechanism on transmitter activity, influencing transmitter release and receptor function. Senescent physiology may represent the co-opting of redox signaling pathways, which are normally responsible for feedback regulation of synaptic transmission. In the case of the NMDA receptor, a mild increase in oxidative stress during aging may result in a constitutive or prolonged activation of these feedback mechanisms, resulting in senescent physiology (Fig. 8). Finally, the site of redox modification on vulnerable molecules may undergo oxidative damage, resulting in an irreversible inhibitory action on proteins involved in synaptic transmission, as well as promoting mitochondrial dysfunction and excitotoxicity associated with neurodegenerative diseases (Figs. 1 and 8). Below are a few examples that emphasize the role of the transmitter system in regulating ROS and the range of changes associated with increasing oxidative stress.

Glutamate
Glutamate is the main excitatory transmitter of the central nervous system and acts on metabotropic and ionotropic receptors, including the NMDA receptor. Both NMDA receptor activation (36) and blockade (19) are reported to induce oxidative stress through an increase in NOX activity. As detailed above, increased ROS results in NMDA receptor hypofunction and the production of nitric oxide can reduce NMDA receptor activity through S-nitrosylation of cysteines on NMDA receptor subunits. Similarly, S-nitrosylation of serine racemase inhibits formation of the NMDA receptor coagonist,
In addition to NMDA receptors, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors represent the other major class of ionic glutamate receptors. Under physiological conditions, protein S-nitrosylation influences the surface expression of AMPA receptor subunits, GluR1 and GluR2, at the synapse (125, 245, 260). N-ethylmaleimide-sensitive factor is also redox sensitive and involved in delivery of AMPA receptors to the membrane (176). Synaptic transmission mediated by AMPA receptors is decreased with age and its attenuation is associated with cognitive impairment (144). However, AMPA receptor function is not altered by DTT, suggesting that the decrease is not directly related to redox state (31). Furthermore, application of 4-HNE does not alter the AMPA receptor synaptic response, suggesting that oxidative damage of membrane lipids does not influence AMPA receptor-mediated synaptic transmission (165). Rather, the decrease in AMPA receptor-mediated synaptic transmission during aging is ancillary to NMDA receptor hypofunction and impaired synaptic plasticity. As such, the decrease in AMPA receptor synaptic transmission is associated with a shift in the activity of kinases/phosphatases that mediate NMDA receptor-dependent synaptic plasticity, indicating a decrease in the endogenous level of LTP of older animals (92, 198).
Under normal physiological conditions, nitric oxide is generated by NMDA receptor-mediated activation of nNOS (99). In turn, nitric oxide regulates glutamate metabolism, the release of glutamate at the synapse, and transport out of the synapse (35, 221). An increase in protein S-nitrosylation can prolong inhibition of glutamate release, decreasing synaptic strength (232). This inhibition of glutamate release may represent a protective feedback mechanism since glutamate transporters and the removal of glutamate from the synapse are also inhibited by oxidizing conditions (hydrogen peroxide or DTNB) (279). Importantly, inhibition of glutamate uptake by an oxidized redox state is readily reversible by DTT. In contrast, with high levels of oxidative/nitrosative stress, inhibition of uptake by peroxynitrite is not reversed by DTT. Thus, an oxidized redox state would inhibit glutamatergic transmission; however, a rise in reactive ROS/RNS during ischemia or neurodegenerative diseases would result in oxidative damage and excess or prolonged glutamate in the synapse and overactivation of glutamate receptors, contributing to excitotoxicity of neurons and glia (33, 56, 177).
Gamma-aminobutyric acid
Gamma-aminobutyric acid (GABA) is the main inhibitory transmitter in the central nervous system, and GABAergic synaptic transmission is modified by several redox-sensitive mechanisms. In the case of GABA receptor function, redox state may have opposing effects on GABAB and GABAA receptors. The response of GABAB receptors may be decreased under reducing conditions (49, 96), and GABAB-coupled K+ channels are altered by intracellular redox state (301). Like synaptic NMDA receptors, synaptic GABAA receptor function recorded in vitro can be weakened under oxidizing conditions (10, 205); however, in the hippocampus, tonic currents are increased by hydrogen peroxide (210). Nitric oxide is thought to inhibit tonic and phasic GABAergic responses in the hippocampus (100), and S-nitrosylation of the GABA receptor scaffolding protein, gephyrin, reduces the surface expression of GABAA (70).
There is no consensus concerning the influence of oxidative stress on GABA release, which may depend on the brain region examined (35). For example, mounting evidence suggests that redox changes of NMDA receptor synaptic input to inhibitory interneurons can decrease the release of GABA and induce the loss of inhibitory interneurons in the PFC. Indeed, NMDA receptor hypofunction and the loss of GABAergic neurons form the basis for recent models of schizophrenia (19, 161, 289). Electrophysiological recordings indicate that activity-dependent NMDA receptor channel blockers reduce the discharge activity of the fast-spiking interneurons, in turn releasing cortical pyramidal cells from inhibition (120, 216). Furthermore, NMDA receptor antagonists induce a persistent increase in ROS through NOX activation, and the increase in oxidative stress results in the loss of fast-spiking inhibitory neurons (19). Thus, a redox-mediated NMDA receptor hypofunction could shift the balance of excitatory/inhibitory synaptic activity in the PFC. The shift in balance of excitatory/inhibitory synaptic activity may contribute to increased activation of the PFC in older individuals (77) and increased expression of activity-related genes in the PFC of aged animals that are impaired on PFC-dependent tasks (127). It is not clear if a similar mechanism occurs in other brain regions; however, the ability of NMDA receptors to drive GABA release in the hippocampus is redox sensitive (129).
Finally, oxidative damage can decrease the synthesis of GABA. Normally, glutamine synthase in astrocytes converts glutamate to glutamine. The glutamine is then transported to neurons where it is used in the synthesis of glutamate and GABA. Aging and Alzheimer's disease are associated with carbonyl modification of glutamine synthase (50, 214). Moreover, regional vulnerability exists such that the decrease in glutamine synthase activity is particularly evident in the frontal cortex (256) and hippocampus (229).
Catecholamines
High concentrations of dopamine (DA) are associated with neurotoxicity via shifts in redox homeostasis (116, 182). In particular, catabolism of DA shifts the cellular redox state toward oxidative stress through the production of superoxide, hydrogen peroxide, quinones, and quinoprotein adducts (166, 183, 305). Monoamine oxidase catalyzes the oxidation of DA to form hydrogen peroxide and DA undergoes nonenzymatic oxidation to produce DA-o-quinone, which acts on cysteinyl proteins forming quinoprotein adducts. DA metabolites also interact with trace metals, such as Fe(III) and Cu(II), within cells to produce additional ROS and further exacerbate redox imbalance (261, 305). The level of ROS, accumulation of adducts, and DNA damage increased by DA transporter activity (76, 292). Once DA is transported into the cell, it can be oxidized by monoamine oxidase, inordinately influencing accumulation of adducts in dopaminergic cells (287). Thus, regions rich in DA may exhibit increased vulnerability to ROS and quinoprotein adducts. In aging, levels of monoamine oxidase and quinoprotein adducts increase in the brain with significant adduct expression in the substantia nigra pars compacta and, to a lesser extent, the PFC and caudate nucleus (9, 133, 196, 287). In addition, levels of iron and ferritin, which store iron, are both elevated in older rats and humans, with greater accumulation occurring in the substantia nigra and striatum compared with the thalamus, globus pallidus, cerebellum, and cortex (231, 298, 300).
Feedback through redox-sensitive mechanisms may help to limit oxidative damage associated with DA metabolism. For example, oxidative modification of the DA transporter can inhibit activity, limiting uptake, and subsequent metabolism (2, 23). Similar to the synthesis of GABA, DA synthesis is also decreased as oxidative stress increases. The decrease in synthesis is mediated by inhibition of tyrosine hydroxylase, the synthesizing enzyme, and rate-limiting step of DA production. Reversible inhibition can occur through disulfide bond formation on cysteines (34). As oxidative stress increases, formation of 3-nitrotyrosine by peroxynitrite may result in an irreversible inhibition of tyrosine hydroxylase (30).
In contrast to DA, low concentrations of norepinephrine can elicit neuroprotective effects for dopaminergic, cholinergic, hippocampal, and cortical neurons in vitro (61, 169, 170, 276, 278). The mechanism appears to involve decreased neuroinflammation, inhibition of microglial NOX, and inhibition of induction of iNOS (52, 131, 185). In addition, extracellular norepinephrine may exhibit antioxidant properties (276, 278). It should be noted that the above studies of neuroprotective effects of norepinephrine have mainly been performed in vitro. The ability of endogenous norepinephrine to protect neurons is also suggested by studies that report lesions of the locus coeruleus or inhibition of norepinephrine synthesis enhances inflammation and oxidative stress induced by stressors (118, 126, 130, 290).
Acetylcholine
Cholinergic integrity is crucial to cognitive processes that decline with age, including attention and working memory (58, 65, 179, 238). The basal forebrain is a rich source of cholinergic neurons, which may be preferentially lost in Alzheimer's disease. Indeed, this brain region may exhibit increased vulnerability to oxidative stress, with higher levels of peroxides and oxidized GSH (48). In agreement with this idea, cholinergic neurons are highly susceptible to cell death following treatment with oxidized lipids (38). The oxidized lipid, 4-HNE, disrupts coupling of muscarinic cholinergic receptors to phospholipase C-linked GTP-binding proteins (29). The effect likely involves redox signaling since the process is reversed by GSH. A similar decline was observed for activation of metabotropic glutamate receptors and Gαq/11 binds 4-HNE suggesting a common mechanism (29).
The loss of cholinergic input may provide two hits on vulnerable brain regions. In addition to loss of information provided by cholinergic input, cholinergic activity is neuroprotective. Under conditions of injury (i.e., ischemia and hemorrhage), cholinergic stimulation, specifically of α7 nicotinic acetylcholine receptors (nAChRs), elicits neuroprotective effects by inhibiting proapoptotic signaling cascades within neurons (64, 138) and activating the nuclear factor erythroid 2-related factor (Nrf2) pathway in microglia, which reduces ROS and tumor necrosis factor levels via increased heme oxygenase-1 expression (206). Nrf2 and its downstream products are crucial in maintaining proper redox homeostasis, and age-related changes in this pathway contribute to increased vulnerability due to heightened oxidative stress. For example, Nrf2 messenger RNA (mRNA) levels and transcriptional activity decline over the course of aging, resulting in decreased synthesis of GSH (266). Moreover, the decline in function renders neural circuits of middle-aged and aged animals more susceptible to environmental toxins that contribute to neurodegenerative diseases (145). Finally, aging may shift the expression of α7 nAChRs on astrocytes and inhibitory interneurons in the hippocampal CA1 region (94), and reduced expression of α7 nAChRs has been reported in the frontal cortex in elderly humans (282). The loss of cholinergic input, α7 nAChRs, and redox-mediated decrease in muscarinic function in the PFC would likely influence cognitive processes (e.g., attention and working memory) that depend on this region.
K+ channels
In addition to transmitter systems, an age-related change in redox state is likely to influence physiology by modifying voltage-dependent channels, either directly or through redox-sensitive signaling cascades. The function of several K+ channel classes is redox sensitivity, altering activity with a rise in hydrogen peroxide, or following application of DTT or GSH (233, 234, 250, 301). In particular, a shift in the activity of the Ca2+ and voltage-gated BK channel (237, 271, 302) or G protein-coupled inwardly rectifying potassium channels (GIRKs) and M-channels (97, 301) could influence cell excitability through an increase in the amplitude of the sAHP or increase the response of inhibitory transmitters. Finally, oxidation of the voltage-gated K+ channel subfamily B member 1 (KCNB1) will increase neuronal vulnerability to apoptosis (59, 291, 299), suggesting that this mechanism could interact with the increased oxidative stress associated with neurodegenerative diseases.
Interaction of Oxidative Stress with Other Aging Mechanisms
As discussed in Sources for ROS and RNS in the Brain section, the vulnerability of different cell types, cellular compartments, and brain regions depends on the chemical character of the oxidative stressor, level of ROS production, and expression/activity of antioxidant enzymes. In addition to oxidative stress, aging encompasses many other biochemical, physical, and functional changes. In addition to oxidative stress, mitochondrial dysfunction, Ca2+ dyshomeostasis, inflammation, and disruption of neuroendocrine regulation have been proposed as aging mechanisms and common risk factors for neurodegenerative diseases. It is important to recognize that these risk factors interact and that the vulnerability of different brain regions may be explained by specific risk factors. For example, as discussed in Redox Stress and Senescent Physiology section, redox regulation influences Ca2+ homeostasis through the release of Ca2+ from intracellular stores and modulation of NMDA receptor function. Several recent reviews discuss factors that determine regional vulnerability/susceptibility to Ca2+ dyshomeostasis (140, 288) and relate how an oxidized redox state influences Ca2+ homeostasis, which precedes the accumulation of oxidative damage during aging and neurodegenerative diseases (72, 90, 102, 209).
Inflammation
A persistent low-level increase in serum markers of inflammation is consistently observed during aging and is thought to contribute to age-related diseases (51, 121, 255), including Alzheimer's disease, and age-related memory deficits (25, 53, 62, 105, 168, 190, 219, 241). Proinflammatory cytokines, IL-1β, IL-6, and TNF-α, cross the blood–brain barrier to activate glia and increase ROS production (12, 13, 73, 109). Furthermore, the increase in ROS with age may contribute to this process as ROS activates inflammatory signaling through stress kinases (JNK, p38) and transcription of proinflammatory mediators through NF-kappaB (122, 220).
Markers of inflammation and oxidative stress vary across brain regions (16, 127, 241, 246, 254, 286). The mechanisms for differential susceptibility to inflammation are unknown, but may be due to the distribution of microglia or differential expression of receptors for cytokines that cross the blood–brain barrier (93, 106, 148, 243). The relationship between oxidative stress and markers of neuroinflammation in different brain regions and disruption of specific cognitive processes mediated by these brain regions is complex. Microglial activation may be beneficial or detrimental to neurons. Neuroinflammation arises as part of a process to initiate tissue repair and support neurons exposed to oxidative stress (264, 265). Regional differences in the expression of proteins or genes that signify microglial activation (e.g., major histocompatibility complex II antigens) are not necessarily linked to cognitive impairment (28, 127, 285). In contrast, senescent microglia release of proinflammatory cytokines and chemokines that induce oxidative stress (265) and local elevation of these signaling molecules are linked to impaired cognition (241). Thus, the activity of signaling molecules and level of oxidative stress should be assessed when considering the role of neuroinflammation in brain function.
Neuroendrocrine regulation of brain aging
Similar to systemic inflammation, aging is associated with an increase in plasma levels of glucocorticoids (167, 180). Normally, glucocorticoid receptor (GR) transcriptional activity mediates a reduction in inflammation and an associated reduction in oxidative stress (15). However, the release of high levels of glucocorticoids during behavioral stress can induce an increase in NOX activity, decrease the levels of the reduced form of GSH, and induce oxidative damage (41, 239, 248). Brain regions that are vulnerable to behavioral stress include the striatum, hippocampus, and PFC (262). Furthermore, under oxidizing conditions, GR activity is decreased due to redox regulation of ligand binding, decreased transport of the receptor into the nucleus, and redox regulation of DNA binding (201, 270).
Sex steroids decline with age, and sex steroids interact with brain aging mechanisms. In particular, estrogen has effects on physiology that are diametrically opposite to senescent physiology (84), including the ability to increase NMDA receptor function and decrease the amplitude of the sAHP (18, 142). While it is clear that estrogens can reduce oxidative damage, the mechanism is less clear. Estrogens may act to promote antioxidant capacity by regulating antioxidant enzyme activity or through effects on mitochondrial function (132, 135, 163, 184, 297). Finally, there are several estrogen receptors, and effects of estrogen treatments likely reflect age and regional differences in receptor expression (17, 88, 113, 139).
Finally, food restriction is the most reliable method for extending life span, which has led to an extensive body of research on gene mutations linked to insulin and insulin-like growth factor signaling. In general, reduced insulin receptor signaling is associated with increased life span. Normally, insulin induces an increase in hydrogen peroxide, which results in autophosphorylation of the insulin receptor (263). Under these relatively mild oxidative conditions, phosphatases are inhibited through cysteine modifications, while the insulin receptor tyrosine kinase is activated (72, 242). With an increase in oxidative stress, S-nitrosylation of the insulin-degrading enzyme will inhibit the breakdown of several peptides, including insulin and amyloid (7). Thus, similar to the effects of redox state on synaptic function, an oxidized redox state may be associated with a prolonged on state for insulin receptor signaling (37). In turn, disruption of insulin signaling will disrupt energy homeostasis and could contribute to altered synaptic transmission and neurodegeneration (69, 213, 304).
Summary
Redox signaling depends on the reversible modification of redox-sensitive proteins. During aging, a rise in hydrogen peroxide or nitric oxide results in constitutive or prolonged activation of redox signaling and the emergence of senescent physiology linked to impaired cognition. Due to the reversible nature of redox reactions, senescent physiology may be modifiable; however, unchecked, oxidative stress may progress, resulting in increasing levels of highly reactive forms of ROS and RNS and the accumulation of oxidative damage. In turn, oxidative damage may further alter physiological processes and contribute to neurodegeneration. This continuum of functional alterations, from redox signaling to oxidative damage, can be described for several transmitter systems. For each system, senescent physiology may represent the co-opting of transmitter signaling pathways that are normally responsible for feedback regulation of synaptic function. The vulnerability of transmitter systems, neural circuits, and brain regions depends on the chemical character of the oxidative stressor, level of ROS production, and expression/activity of antioxidant enzymes or redox buffering. In addition, the liability of specific systems or circuits is influenced by transmitters that induce ROS production during synaptic activity (e.g., NMDA receptors) or as part of transmitter breakdown (e.g., DA). Finally, vulnerability to oxidative stress will depend on the concentration of reactive metals, function of local astrocytes and microglia, and response to inflammatory or hormonal signals.
Footnotes
Acknowledgments
Financial support by National Institutes of Aging Grants, R01AG037984, R37AG036800, R01AG049711, and RO1AG052258, and the Evelyn F. McKnight Brain Research Foundation is highly appreciated.