
Abstract
Significance:
Iron-dependent lipid peroxidation is a complex oxidative process where phospholipid hydroperoxides (PLOOH) are produced in membranes and finally transformed into a series of decomposition products, some of which are endowed with biological activity. It is specifically prevented by glutathione peroxidase 4 (GPx4), the selenoenzyme that reduces PLOOH by glutathione (GSH). PLOOH is both a product and the major initiator of peroxidative chain reactions, as well as an activator of lipoxygenases. α-Tocopherol both specifically breaks peroxidative chain propagation and inhibits lipoxygenases. Thus, GPx4, GSH, and α-tocopherol are integrated in a concerted anti-peroxidant mechanism.
Recent Advances:
Ferroptosis has been recently identified as a cell death subroutine that is specifically activated by missing GPx4 activity and inhibited by iron chelation or α-tocopherol supplementation. Ferroptosis induction may underlie spontaneous human diseases, such as major neurodegeneration and neuroinflammation, causing an excessive cell death. The basic mechanism of ferroptosis, therefore, fits the features of activation of lipid peroxidation.
Critical Issues:
Still lacking are convincing proofs that lipoxygenases are involved in ferroptosis. Also, unknown are the molecules eventually killing cells and the mechanisms underlying the drop of the cellular anti-peroxidant capacity.
Future Directions:
Molecular events and mechanisms of ferroptosis to be unraveled and validated on animal models are GPx4 inactivation, role of GSH concentration, increased iron availability, and membrane structure and composition. This is expected to drive drug discovery that is aimed at halting cell death in degenerative diseases or boosting it in cancer cells. Antioxid. Redox Signal. 29, 61–74.
Introduction
T
At the present level of knowledge, ferroptosis is seen as the final outcome of an imbalance between the rate of the oxidative reactions of lipid peroxidation and the rate of the metabolic pathways supporting the nucleophilic tone of the cell. Consistently, GPx4 activity emerges as a major player in neurodegeneration (17). Since cell death and inflammation are linked by a bi-univocal relationship (83), ferroptosis is a critical player in diseases that are collectively defined as neuro-inflammatory. Paradigmatic examples are Alzheimer (46, 48), Parkinson (8, 45, 49), and Huntington disease (22, 26).
Here, we will summarize the key features of lipid peroxidation describing the progressive achievements of knowledge that paved the road from rancidity of fat to the identification of GPx4 as a pharmacologically relevant target. Still open issues, requiring specifically addressed experimental work will be finally outlined.
Lipid Autoxidation and Peroxidation
Painters and carpenters know from centuries that vegetable oils, containing polyunsaturated fatty acids, harden when exposed to air and are warmed up, producing a film that is the ancestor of the modern polymeric resins. It is also known from centuries that foods, protected from faster microbiological degradation, for example, by salt, undergo a slow degradation process, leading to rancidity. The oxidative nature of these events was first pointed out by De Saussure in 1804 by manometric and gravimetric measurements showing that linseed oil binds oxygen (28). He also discussed the possibility that the oxidative polymerization was the actual mechanism of film formation in vegetable oils hardening. The term “peroxidation” was introduced later to focus on the notion that peroxides—lipid hydroperoxides indeed—are produced. Moreover, in the case of lipid peroxidation of biological membranes, structural constraints contribute to the control of the rate of the basic chemical reactions of autoxidation.
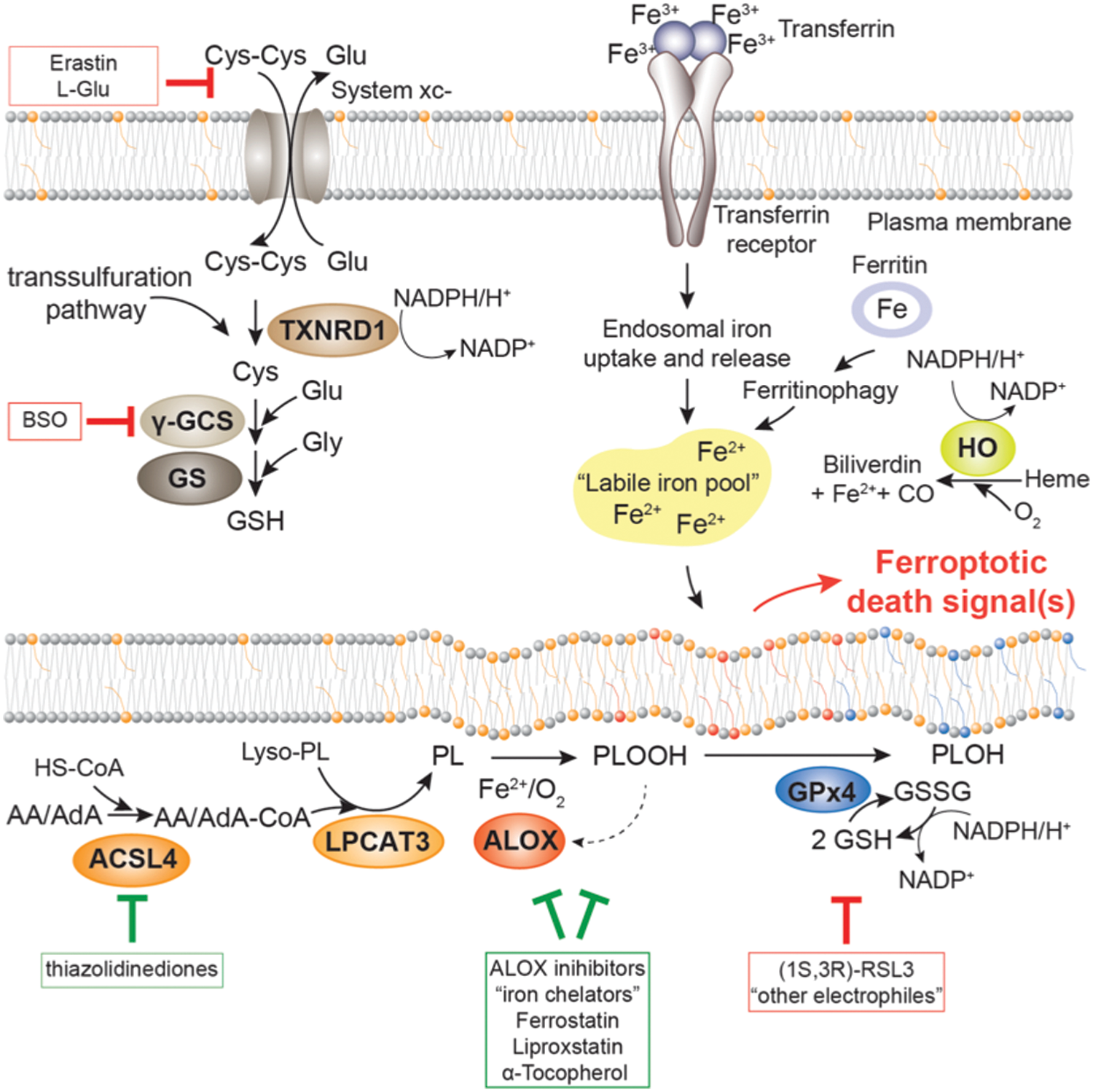
Iron-dependent lipid peroxidation encompasses free radical reactions that can be schematically summarized as: (i) initiation, which includes hydrogen atom abstraction from a phospholipid containing polyunsaturated fatty acid (PL) and oxygen addition to the phospholipid carbon centered radical (PL•); (ii) chain propagation yielding a phospholipid hydroperoxide (PLOOH) and a new PL• (PL’•); and (iii) arrest, by radical-radical interaction (Fig. 2). Notably, the O–O bond of PLOOH, the major stable (nonradical) product of the reaction, is, indeed, relatively unstable, driving new initiation reactions by decomposition and producing a plethora of small molecules, most frequently of an electrophilic nature (126).

Free radical reactions of lipid peroxidation are, in general, very fast, with a rate constant being in the range of 108 M −1 s−1 (2, 77, 126), with the exception of the rate-limiting chain propagation reactions. The H abstraction, indeed, from a methylene carbon of a polyunsaturated fatty acid by a lipid hydroperoxyl radical, forming a new carbon centered radical that propagates the peroxidative chain reaction and a hydroperoxide, occurs with a rate constant in the range of 103 M −1 s−1 (13) (Fig. 2). This relatively low rate constant permits the diffusion of phospholipid hydroperoxyl radical (PLOO•) within the membrane and toward the surface (the floating peroxyl radical theory) (7). Here, the fast interaction (∼106 M −1 s−1) takes place with the redox center of tocopherol (13), located at the water boundary. Thus, tocopherol is the most specific “chain-breaking antioxidant” (14), a concept that, while emphasizing the role in inhibiting chain propagation, limits the preventive antioxidant activity to conditions when lipid peroxidation is initiated by hydroperoxyl radicals. A typical example of the latter is the popular thermal decomposition of azo-initiators (126), which, on the other hand, has no suitable counterpart in biology. Even though recent computational quanto-mechanical analysis argues against the floating peroxyl radical theory (38), it seems reasonable assuming that in lipid peroxidation of membranes there must be a relevant trafficking of lipophilic and hydrophilic species from the core to the surface of membranes, where critical pro- and anti-peroxidant events take place.
The discovery of a peroxidation inhibiting protein (P.I.P.) was inspired by an observation coming from the laboratory of P. McCay, showing that, in vitro, in the presence of cytosol, GSH completely prevents iron-dependent microsomal lipid peroxidation (43). Neither the “classical” glutathione peroxidase (now known as GPx1, E.C.1.11.1.9) nor glutathione transferases, superoxide dismutases (SOD) or catalase, reproduce the effect of cytosol and GSH. To address the catalytic activity on PLOOH, purified P.I.P. was renamed as “Phospholipid Hydroperoxide Glutathione Peroxidase” (PHGPx, E.C. 1.11.1.12) (71, 110), and then systematically filed as the GPx4 gene product (44). In the in vitro studies mentioned earlier, the rather simple nature of the mixture of the pro- and anti-peroxidant components—adenine nucleotide, NADPH, GSH, membranes and cytosol, and, in some conditions, α-tocopherol—was already pointing out per se an aspect today emerging as a crucial feature of a specific form of programmed cell death (23, 29). Seemingly, lipid peroxidation is sparked by the inactivation of the anti-peroxidant mechanism based on the reduction of PLOO• and PLOOH, rather than by a sudden increase of an unknown oxidant.
Notably, to date, other enzymes have been shown to reduce esterified lipid hydroperoxide in vitro (namely, GPx3, Peroxiredoxin 6, and GPx7) (10, 34, 119) but apparently none of them could mimic the antioxidant effect of GPx4: reduction of PLOOH in membranes and inhibition of microsomal lipid peroxidation. This observation complies with the distinct phenotype obtained by individual gene silencing (80, 111, 112), where only that of GPx4 is indispensable (52, 125).
In biological membranes undergoing iron-dependent lipid peroxidation in vitro, the reduction of PLOOH into corresponding phospholipid alcohols (PLOH) by GPx4 and GSH (Fig. 3) integrates the antioxidant chain-breaking mechanism of α-tocopherol (Fig. 4). The latter, by scavenging the peroxidation-driving PLOO•, prevents chain propagation, producing a minute amount of PLOOH that can initiate, in the presence of ferrous iron, a new peroxidative chain reaction (109). The phospholipid alkoxyl radical (PLO•) produced starts a new chain reaction by attacking the methylene carbon of a polyunsaturated fatty acid, thus generating a PLOH and a new PL•. Remarkably, this reaction is so fast (2) that a competing reaction with a free radical scavenger, such as α-tocopherol, cannot produce any relevant antioxidant protection. It follows that the only way for preventing this initiation is the reduction of the precursor PLOOH (70). In summary, GPx4, GSH, and α-tocopherol are players of a concerted anti-peroxidant machinery where the synergy between one- and two-electron reactions—when PLOO• and PLOOH are reduced, respectively—accounts for the kinetic control of iron-dependent lipid peroxidation of membranes. Notably, an identical interplay between reactions quenching the propagation of hydroperoxyl radicals and “peroxidolytic reactions” (i.e., reduction of hydroperoxides) is the core element for the kinetic control of reaction rate in polymer chemistry (97).


It is worth mentioning that the synergistic anti-peroxidant mechanism, including GPx4 and tocopherol, also applies to a peroxidation that is produced by lipoxygenase (LOX) activity (66). In fact: (i) PLOOH (or a hydroperoxide, in general) activates LOX (92), and thus membranes exposed to GPx4 and GSH are resistant to 12/15 LOX-dependent lipid peroxidation (96); and (ii) tocopherol inhibits LOX activity, seemingly by keeping the catalytic iron reduced (20). The observation that GPx4 silencing activates ferroptosis, and that this cell death subroutine is rescued by α-tocopherol (18, 72, 98, 115), validates the concept of the synergy between the two antioxidant mechanisms supporting the hypothesis of a functional node in the process of ferroptosis encompassing GPx4, GSH, tocopherol, oxygen, iron, and, likely, LOX activity (Fig. 5).
Cell and Animal Studies on the Role of GPx4 in Cell Death
Almost 20 years after the discovery of GPx4, studies using different cell lines indicated its primary role for protection against oxidative damage (54, 116). These experiments, relying on GPx4 overexpression, provided the molecular explanation for the observation that selenium-supplemented cells, in selenium repletion studies, were more resistant than untreated cells to lipid hydroperoxide- and photooxidative-triggered cell death (42, 64). Successive studies finally pointed out to a specific role of GPx4 in preserving mitochondrial integrity (117).
Some sort of confusion about the potential role of different forms of GPx4 in cell death signaling came from overexpression studies suggesting that the mitochondrial form of GPx4 (mGPx4) rather than cytosolic GPx4 (cGPx4) was cytoprotective in response to cell stress challenges (3). mGPx4 distinguishes itself from cGPx4 by an N-terminal extension encoding a cognate mitochondrial targeting signal (4), which would confer localization to the mitochondrial matrix. mGPx4 overexpressing cells were also shown to be more resistant to 2-deoxyglucose-induced cell death by inhibiting cytochrome c release, nuclear fragmentation (78), adenine nucleotide translocator inactivation (53), and cardiolipin peroxidation (79), all of which are biochemical hallmarks of controlled cell death. Similar protective effects by mGPx4 were also observed in cells treated with pro-apoptotic cytotoxic compounds (78). However, in light of the specific expression patterns of the three different forms of GPx4 (86, 94), and follow-up studies using cells and mice with targeted loss of the entire GPx4 gene or specific mouse mutants, these early studies must be taken with care as mGPx4 (like nuclear GPx4) is only essential during a specific step in sperm maturation and does not contribute to the essential pro-survival function of the cytosolic form of GPx4 in cell and tissue protection (63, 93, 98).
The targeted loss of the GPx4 gene in mice eventually highlighted its outstanding importance for early development. Mice lacking GPx4 die, in fact, early in embryonic development, shortly after gastrulation (E7.5) (52, 125). Interestingly, the phenotype of the knockout of GPx4 resembles that of mice lacking the catalytic subunit of the enzyme catalyzing the first and rate-limiting step in GSH biosynthesis, γ-glutamyl-cysteine ligase (101). Moreover, knockout of the gene encoding the selenocysteine-specific tRNA (Trsp) also causes embryonic death at E6.5 (11), indicating that GPx4 is not only a limiting GSH-utilizing enzyme but also one or the most important selenoproteins.
Why nature selected selenium for some, although not all, peroxidases of vertebrates, evolving from a sulfur containing ancestor (106) has been recently unraveled. The reaction catalyzed by selenoperoxidases is faster (68), due to the better nucleophilic character of selenium in the oxidative part of the catalytic cycle and the better electrophilicity in the reductive part (90). This peculiar feature fits the result of the quanto-mechanical calculation of the energies of the transition states (81). Moreover, selenium, over sulfur, also offers the advantage of a better stability of the oxidized form of the enzyme. Here in fact, selenenic acid, when the reducing substrate is limited, reversibly forms a selenenylamide, whereas sulfur is irreversibly oxidized (81).
Due to the early embryonic lethal phenotype of homozygous GPx4 null mice, a series of early studies regarding its cell-protective functions have been performed with heterozygous cells and mice, as no other conditional knockout tools for GPx4 were available at that time (39, 88, 125). In essence, all these findings were in line with the previous overexpressing studies showing that cells and mice with only one functional GPx4 allele are more sensitive to a number of stress-inducing agents and conditions such as hydroperoxides, γ-irradiation, and paraquat. Notably, mice with only one functional GPx4 have decreased numbers of tumors and live somewhat longer than their wild-type counterparts (87). This suggests the hormetic effect of oxidants, where the loss of one GPx4 allele leads to an associated increase in the steady-state level of lipid oxidation products, thereby activating a still unknown protective mechanism. Notwithstanding, these early investigations firmly established an essential function of GPx4 in the protection against PLOOH and related cytotoxic agents, while leaving the issue of the final mechanisms of cell death unresolved.
GPx4 as a Master Regulator of a Novel form of Regulated Necrotic Cell Death
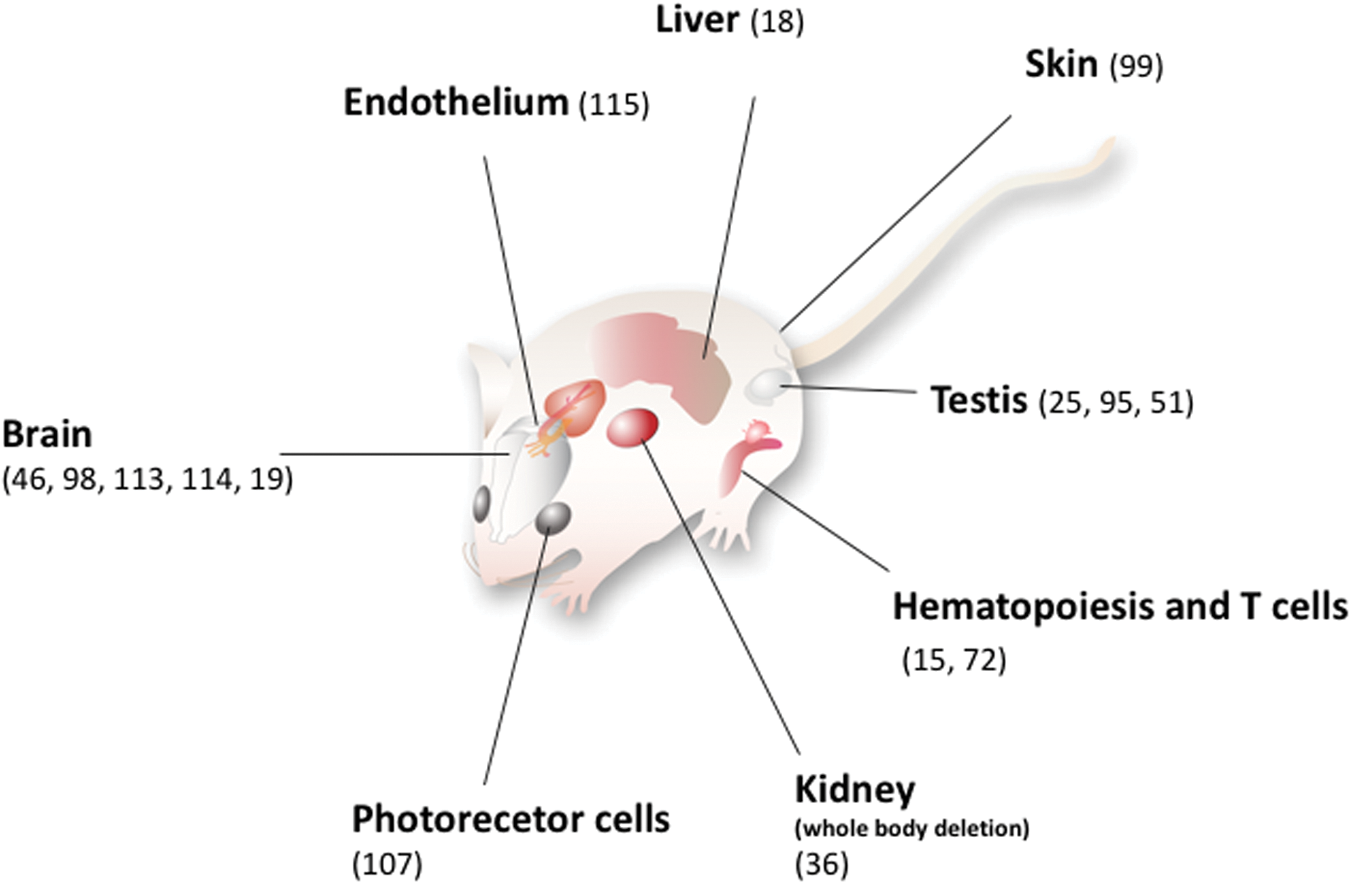
A major leap in the molecular understanding of how GPx4 controls cell death (Fig. 5) was made by the introduction of the first conditional knockout mouse model in 2008 (98). This model additionally served as an ideal toolbox to generate a cellular system derived from mouse embryonic fibroblasts with two loxP-flanked GPx4 alleles and stably expressing Tamoxifen-inducible Cre recombinase (now commonly referred to as “Pfa1 cells” (Fig. 6). Both conditional systems have been proved instrumental in unveiling the in vivo relevance of GPx4-dependent cell death and the underlying molecular mechanisms of this “yet-unrecognized cell-death pathway” (98). In this landmark study, not only could an essential neuroprotective role for GPx4 in hippocampal neurons and in the prevention of ataxia and seizures of young mice be unmasked, but also first insights could be achieved in terms of the mechanism of cell death triggered on GPx4 inactivation. Moreover, with the increasing availability of conditional knockout models for GPx4 (Fig. 1), these critical observations could then be expanded to parvalbumin-positive interneurons (114), photoreceptor cells (107), cerebellar Purkinje cells and granular cells (113), as well as motor neurons (19) and forebrain neurons (46). And in fact, using second-generation ferroptosis inhibitors, it could be already shown that ferrostatins are neuroprotective in an ex vivo model for Huntington's disease (103), although in vivo proof is still lacking that ferroptosis inhibition, indeed, confers neuroprotection. In this context, it is noteworthy that depletion of cystathionine γ-lyase (CSE), a key enzyme of the trans-sulfuration pathway as an alternate cysteine source, was unveiled to possibly mediate Huntington's disease pathophysiology (84).

A specific tissue damage due to GPx4 silencing was also observed in skin (99) and the hematopoietic system (15). Notably, the morphological and functional alterations observed in each tissue where GPx4 had been inactivated are practically overlapping those of spontaneous diseases. Thus, inactivation of the anti-peroxidant system based on GPx4 and the forthcoming ferroptotic death is seemingly the molecular mechanism of spontaneous diseases, against which therapeutic or preventive treatments could be specifically addressed.
The only caveat of the conditional knockout model is that it is impossible to specifically ablate just the cGPx4. Therefore, all three forms of GPx4 would always be concomitantly affected by the conditional knockout strategy of the entire GPx4 gene. To this end, a major effort was undertaken and two additional knockout mouse models, one specific for the nuclear form and one for the mitochondrial one, were established. These investigations finally demonstrated that the nuclear and mitochondrial forms only confer specific functions during spermatogenesis but are otherwise dispensable in somatic tissues and for embryo development (24, 25, 51, 93). Unequivocal genetic proof that it is the cytosolic form being essential for embryogenesis and adult somatic tissues was eventually provided by an elegant study by Liang et al., showing that transgenic expression of the cytosolic form in the GPx4−/− background allowed normal development and full viability of mice (63). Most interestingly, subcellular fractionations of cells and mitochondria from cGPx4-GPx4−/− transgenic liver and kidneys revealed that cytosolic GPx4 appears to be highly enriched in the intermembrane space of mitochondria (IMS) and absent in the mitochondrial matrix, suggesting a crucial role for the peroxidase in this particular subcellular location. Notably, an early study based on biochemical evidence of sub-cellular localization suggested that, in rat brain mitochondria, GPx4 is located in the inner mitochondrial membrane and contact sites (82). Together, these studies suggest that the GPx4 site of action is the outer face of the mitochondrial inner membrane. The mechanism of how “cytosolic” GPx4 enters the IMS, however, remains unknown, but it appears to be shared with other professional redox enzymes that are usually located in the cytosol, such as GPx1, SOD1, and thioredoxin reductase 1 (TXNRD1).
Remarkably, cell death triggered by the inducible deletion of GPx4 in Pfa1 cells as wells as ex vivo cultured cortical neurons from E15.5 embryos could be fully prevented by α-tocopherol (98), which was later demonstrated to be of utmost relevance for the in vivo situation. For instance, sufficient α-tocopherol content in the mouse chow or supra-nutritional α-tocopherol supplementation was demonstrated to compensate for the loss of GPx4 in endothelial cells (115), CD8+ T cells (72), and hepatocytes of newborn pups (18). Preliminary studies with the cellular Pfa1 system suggested the involvement of apoptosis-inducing factor (AIF; a misacronym for the involvement in “apoptosis”) and LOX-derived lipid hydroperoxides (in particularly that of 12/15-lipoxygenase) in the cell death mechanisms downstream of GPx4 deletion (98). However, these findings were largely based on inhibitor and cellular studies, with all the known caveats of possible unspecific side effects (e.g., radical trapping antioxidant activity).
In 2012, Stockwell's group introduced the term “ferroptosis,” a novel from of cell death induced by a class of compounds able to selectively kill tumor cells expressing an oncogenic RAS. Based on morphological, biochemical, and genetic traits, this new cell death modality emerged to be entirely different from apoptosis, necroptosis, autophagic cell death, and other forms of regulated necrotic cell death (29). The cell death induced by ferroptosis-inducing agents is characterized by an iron-dependent lipid peroxidation event detected as oxidation of C11-BODIPY (33), a probe reacting with free radicals produced during membrane peroxidation. Consistent with the involvement of lipid peroxidation, ferroptosis is inhibited by iron chelators and α-tocopherol. Ferrostatin-1 was also identified as a potent inhibitor of ferroptosis for which a free radical scavenging mechanism had been proposed (103).
As mentioned earlier, ferroptosis inhibitors were shown to be particularly effective in preventing cell death in organotypic brain slices exposed to a high concentration of glutamate. This is reminiscent of a previously described form of cell death, called oxytosis (104), which can be induced by millimolar concentrations of extracellular glutamate in neuronal and non-neuronal cells. A high extracellular concentration of glutamate blocks the cystine-glutamate antiporter, system xc −, thus leading to impaired cystine uptake and, consequently, impaired GSH biosynthesis.
In agreement with the mechanism described earlier, ferroptosis is also triggered in sensitive cells by the small molecule erastin (32), first described to interact with voltage-dependent anion channels (118), and then shown to inhibit system xc − (29). Two years later, in search of the target for (1S, 3R)-RSL3 (RSL3), a compound previously shown to induce ferroptosis without depleting the GSH pool, the same group identified GPx4 as the target that was efficiently inhibited in cells by this compound (121). RSL3-induced cancer cell death was feasible in a BJeLR xenograft cancer model and appeared to be highly effective in tumor cell lines of large B cell lymphoma and of renal cell carcinoma origins. However, whether GPx4 is really a valid target to efficiently eradicate tumor cells in vivo needs to be further explored, particularly in light of earlier data, which pointed to a dispensable role of GPx4 in cell survival and proliferation in a syngeneic tumor transplantation model of c-Myc/H-rasV12 -transformed GPx4-deficient fibroblasts (95). This dispensability of GPx4 is in line with earlier findings, where it was shown that GPx4 knockout cells are, indeed, able to proliferate under normal cell culture conditions when seeded at higher cell densities (98), or when grown as single cells in Matrigel (95). The evidence is consistent with the notion that oxygen tension and activation could become the rate-limiting determinant of oxidative reactions, eventually leading to cell death. A recent intriguing observation, in agreement with a major role of oxygen in cell death, is the apparent paradox of a protective effect of hypoxia in heart regeneration (76). Furthermore, the notion that ferroptosis is activated during neuronal reprogramming further contributes toward pointing out the relevance of lipid peroxidation in the control of efficiency of cellular reprogramming and proliferation (41).
In 2014, a first evidence that ferroptosis is relevant not only for cancer cell death but also in renal tubule cell death and in a mouse model of hepatic ischemia/reperfusion injury in vivo was provided (36). The tamoxifen-inducible deletion of GPx4 in adult mice was shown to cause acute renal failure and early death of mice, which could be delayed by liproxstatin-1, the first in vivo efficacious ferroptosis inhibitor. Liproxstatin-1 also proved to mitigate the damage inflicted by transient ischemia/reperfusion in the liver in wild-type mice while also firmly increasing neuronal reprogramming during fate conversion of astrocytes to functional neurons (40). Moreover, it was found that GPx4 inactivation-induced cell death presents as a major morphological alteration in the rupture of the outer mitochondrial membrane, which is accompanied by alterations in the oxi-lipidomic signature in cells and in knockout tissues; all the changes could be suppressed by ferrostatin-1, necrostatin-1 (an apparent off-target effect independently of its action toward receptor-interacting serine/threonine-protein kinase 1 [Ripk1]), and various lipoxygenase inhibitors (36). Thus, the observed morphological mitochondrial changes may well fit with the preferential localization of cGPx4 to the IMS and/or inner membrane outer surface (63, 82).
Most recently, another downstream ferroptosis player, acyl-CoA synthetase long-chain family member 4 (ACSL4), was unraveled and functionally characterized in the ferroptotic process (31, 58) (Fig. 5). Knockout of ACSL4 in Pfa1 cells or breast cancer cells as well as neuronal cells provided an unprecedented protection against ferroptosis induced by genetic GPx4 deletion or GPx4 inhibition (31). The same protective effect could be achieved when cells were treated with thiazolidinediones, an antidiabetic class of compounds that, as a side effect, also inhibit ACSL4 over other ACSL isoforms (60). The underlying mechanism of this robust anti-ferroptotic effect conferred by ACSL4 knockout or pharmacological inhibition was shown to rely on a strongly reduced incorporation of long ω-6 polyunsaturated fatty acids, such as arachidonic and adrenic acid, into a seemingly specific class of phospholipids (mainly phosphatidylethanolamine), thus dramatically lowering the susceptibility to lipid peroxidation events in membranes (31, 58). Hence, pharmacological targeting of ACSL4 in some degenerative disease contexts, such as neurodegeneration or ischemia/reperfusion scenarios, could provide beneficial effects. In addition, an alternative route for ferroptosis engagement was recently identified. The small molecule FIN56 leads to degradation of GPx4 through a not-fully understood mechanism that may involve activation of ACC1 (acetyl-CoA carboxylase), an enzyme involved in fatty acid synthesis (102).
In terms of the ferroptotic process per se, a number of reports published during the past few years have provided further upstream mechanistic details about the involvement of iron handling, p53-dependent regulation of SLC7A11 (the substrate-specific subunit of system xc −), and availability of cysteine via the trans-sulfuration pathway, which have been extensively illuminated in a recent comprehensive review article (16).
Major Open Issues and Future Directions
How is the “first” hydroperoxide formed?
The inhibition of iron-dependent lipid peroxidation by GPx4 and GSH points out the relevance of PLOOH but leaves open the question about the formation of the traces of PLOOH from which, in the absence of GPx4 activity, ferrous iron sparks lipid peroxidation.
Possible candidates for the formation of an initial PL• (Fig. 4), evolving into PLOO• and then PLOOH, are free radicals that are produced in living cells, such as the protonated superoxide anion (1) or the perferryl iron-oxygen complex (109) or the hydroxyl radical (126). The efficiency of these reactions in initiating lipid peroxidation, however, is extremely low in comparison to that of PLOOH-dependent initiation (105). No iron-dependent lipid peroxidation is, indeed, detectable when GSH and GPx4 are present, and traces of PLOOH formed are continuously reduced. Consistently, it seems reasonable to assume that traces of hydroxyl derivatives of membrane lipids (PLOH) are continuously produced and could be physiologically relevant, for instance, in controlling membrane lipid turnover.
Source of iron
Iron is indispensable for lipid peroxidation; however, although the name “ferroptosis” remarks this fact, very little is known about the form of cellular iron involved, besides the fact that chelation with desferoxamine prevents both lipid peroxidation and cell death (109, 122). The recent indication that the activity of the catalytic subunit of phosphorylase kinase (120) releases iron, although intriguing, does not contribute toward solving the issue. Typically, iron is released from ferritin by controlled proteasomal degradation (5), known as ferritinophagy, and participates in the heterogeneous “labile iron pool” (LIP) consisting of low-molecular-weight complexes from which iron enters into different metabolic pathways (100).
LIP can have a role in the activation of membrane peroxidation in two respects: by forming perferryl or hydroxyl species that are able to extract a hydrogen atom from a methylene carbon of a polyunsaturated fatty acid in PL and, much more efficiently, by participating in an organic Fenton reaction with a PLOOH (105, 109) (Fig. 4). In either case, oxygen concentration and the redox potential, type of coordination, and reducibility of the iron complex are the determinants of the actual pro-peroxidative effect. The relevance of the redox status of iron complexes is unquestionable in microsomal lipid peroxidation in vitro, where a continuous supply of electrons to the iron complex in indispensable. A pathophysiological evidence indirectly supporting the relevance of the redox status in vivo is the neurodegeneration associated to aceruloplasminemia, where lipid peroxidation is sparked by iron accumulating in the brain in the ferrous form due to absent ferroxidase activity of ceruloplasmin (73). Also the evidence that heme-oxygenase 1, which releases reduced iron, accelerates erastin-induced ferroptotic cell death (61) further confirms the relevance of iron concentration and sheds some light on a possible pro-ferroptotic effect of Nrf2 activation, which primes heme-oxygenase 1 expression.
Decrease of GSH
The decrease of GSH produced by erastin, which replenishes cells of cysteine for GSH biosynthesis (via cystine) (124), is the first mechanism of induction of ferroptosis identified in oncogenic RAS mutant cell lines. Consistently, GSH depletion by alkylation or inhibition of synthesis activates ferroptosis (65, 123). Thus, the question emerges about the nature of the physiological or pathological events leading to GSH depletion. One well-defined mechanism is related to the toxicity of glutamate competing for cystine uptake (59). However, other mechanisms have been described, such as the inhibition of the system xc − (56), GSH depletion by active efflux (35), or inhibition of CSE (84). In this respect, it is intriguing recalling that in the late phase of spermatogenesis GSH is almost completely depleted but this does not prime cell death but, instead, the moonlighting of GPx4, which becomes a cross-linked structural component of the mitochondrial capsule (108). Unraveling the analogies between two so different biological events will most likely shed some light on this unexpected similarity between cell death and sperm maturation.
Inhibition of GPx4
The clear-cut evidence that GPx4 silencing almost invariably causes ferroptosis stimulates the still unresolved question about a downregulation of the enzyme under specific physiological or pathological conditions, which might involve a post-translational modification and/or the activation of a proteolytic degradation. The inhibition of GPx4 in cells by the drug candidate RSL3 suggests the alkylation of the catalytic selenol as a suitable mechanism of irreversible inhibition of the peroxidase. Although such a mechanism frames the inhibition of GPx4 in the area of cancer pharmacology, it would be interesting knowing whether endogenously produced electrophiles could produce a similar effect.
Role of lipoxygenases
Despite the alleged role for one or more lipoxygenases in the cell death process downstream of GPx4 deletion/inactivation (36, 98), robust genetic proof that GPx4/lipoxygenase double knockout mutant are, in fact, protected against the lethality brought about by GPx4 deficiency is still lacking. Knockout of Alox15 (the gene encoding 12/15-lipoxygenase) in the GPx4−/− background, indeed, failed to rescue from early embryonic lethality (12), nor did it rescue acute renal failure and early death of adult tamoxifen-inducible GPx4 knockout mice (36). Hence, data showing an anti-ferroptotic effect by either deleting a distinctive lipoxygenase (i.e., 15-LOX) (98) or knocking down one or several LOX isoforms (120) are currently restricted to the cellular level and need validation in an in vivo relevant setting.
Although lipoxygenases are typically described as enzymes that are active on free fatty acids, in support of the activity on membrane lipids already observed for soybean LOX-2 (67) stands the recent evidence of the activity of 15-LOX-2 on membrane PL (9), the formation of eicosanoids of intact PL (21, 47, 74), and physiological function of the reticulocyte LOX (89). This enzyme is, indeed, competent by forming PLOOH for the destruction of mitochondria in the late phase of red blood cell maturation. Remarkably, it has been shown that this LOX active on phospholipid of sub-mitochondrial particles is fully inhibited by GPx4 (96). Moreover, in the presence of an appropriate GPx4 activity, the reticulocyte LOX produces stereo-specific PLOH. This suggests a possible specific function for these species, such as in membrane turnover and autophagy, as recently observed (75). In the area of neuroinflammation and neurodegeneration, the role of 5-LOX (57) and 12/15 LOX (85) has been convincingly demonstrated. Finally, the notion that LOX can be active on intact phospholipids raises the relevant question about the mechanism of the reaction. In fact, although it has been demonstrated that LOX can bind to bilayers (55), what is still unclear is how fatty acid chains esterified in PL could emerge from membrane and reach the catalytic center of the enzyme.
Role of α-tocopherol
The inhibition of ferroptosis by α-tocopherol (6, 98, 123) in the absence of GPx4 is still far from being clearly understood. In chemical terms, reduction of PLOO• by α-tocopherol cannot substitute for reduction of PLOOH. At least in vitro the two reactions are, instead, complementary in the concerted antioxidant mechanism (Fig. 4), where PLOOH, produced by one-electron transfer from PLOO•, are reduced by GPx4. An option, which can explain the data coming from animals or cells, is the inhibition of lipoxygenases (20). This effect, indeed, could give an account for the observed inhibition of ferroptosis. Moreover, this hypothesis would suggest the notion that ferroptosis can only take place after a consumption of α-tocopherol, which is expected to be oxidized while keeping reduced the catalytic iron of LOX. This could frame a new, interesting, role for the vitamin as controller that delays the programmed cell death execution. However, unfortunately, specifically addressed analytical evidence that could clarify this relevant issue is still missing.
Which are the species actually killing cells and which are the final events leading to cell death?
Granted the central role of PLOOH in the mechanism of ferroptosis, what is completely obscure is the series of final events. Are PLOOH the final agonists, possibly operating a redox transition on a target? Or are they the precursors of other species specifically committed for this function? Truncated cardiolipin is a possible reasonable candidate for this function, and, in fact, it had been reported to be involved in mitochondrial apoptosis (62), although this is, indeed, a form of cell death that is different from ferroptosis. Specific decomposition products of PLOOH containing ω-6 fatty acids may have a role, and, consistently, ACSL4, which enriches cellular membranes with ω-6 fatty acids, has been recently found as an essential component for ferroptosis execution (31). Remarkably, ω-6 fatty acid decomposition produces 4-hydroxynonenal (HNE) (27, 126) and the AKR1C1-3 (aldo-cheto reductase family 1 members C1-3) enzymes, which participate in the detoxification of alkenals (such as HNE), were found to be overexpressed in cells resistant to ferroptosis induced by erastin (30). However, although HNE has been shown to be competent for activation of a cell death pathway (27), the proof that it is the actual executor of ferroptosis is missing. At the present level of knowledge, we could just assume as likely the hypothesis that electrophiles produced by decomposition of PLOOH in membranes could participate in redox reactions of the death signaling pathway.
Conclusions
Iron-dependent lipid peroxidation, discovered several decades ago as an oxidative process where traces of iron and a source of reducing equivalents destroy microsomal membranes, has been rediscovered today as a specific form of cell death. The present set of evidences, indeed, revisits the biochemical events of lipid peroxidation in the frame of a controlled process of major biological relevance. The players of the pro- and anti-peroxidant mechanism are oxygen, membrane phospholipids, ferrous iron, GSH, GPx4, α-tocopherol, and, possibly, lipoxygenase(s). Available evidence indicates that it is the decrease of the efficiency of the anti-peroxidant mechanism that primes the formation of molecules—seemingly oxidized derivatives of membrane lipids—executing the death sentence. What we still do not know is the chemical nature of these species and the specific elements accounting for the variable sensitivity of different cells to ferroptosis, among which membrane composition and structural constraints are expected to play a relevant role. In addition, although the role of ferroptosis in cell death has been well documented in pathological conditions, it has been also suggested that the control by p53 of GSH level and thus of ferroptotic cell death participates in tissue plasticity (56), even though xCT (the gene product of SLC7A11) is dispensable for life in mice (91). In this light, what emerges today is that cell death by lipid peroxidation is not only confined to toxicology, but it is, indeed, a critical element of the balance between cell proliferation and death. This impacts life from embryogenesis to tissue homeostasis, likely affecting cellular reprogramming, as recently observed in the elegant study by Gascón et al., showing that inhibition of lipid peroxidation greatly enhances the efficiency of the conversion of astrocytes to functional neurons (41).
Footnotes
Acknowledgments
This study was supported by the Human Frontier Science Program, Grant RGP0013/2014 to F.U. and M.C. and Deutsche Forschungsgemeinschaft CO 291/2-3; CO 291/5-1 to M.C.