
Abstract
Significance:
Sphingolipids play critical roles in the membrane biology and intracellular signaling events that influence cellular behavior and function. Our review focuses on the cellular mechanisms and functional relevance of the cross talk between sphingolipids and redox signaling, which may be critically implicated in the pathogenesis of different renal diseases.
Recent Advances:
Reactive oxygen species (ROS) and sphingolipids can regulate cellular redox homeostasis through the regulation of NADPH oxidase, mitochondrial integrity, nitric oxide synthase (NOS), and antioxidant enzymes. Over the last two decades, there have been significant advancements in the field of sphingolipid research, and it was in 2010 for the first time that sphingolipid receptor modulator was exploited as a therapeutic in humans. The cross talk of sphingolipids with redox signaling pathways becomes an important mechanism in the development of many different diseases such as renal diseases.
Critical Issues:
The critical issues to be addressed in this review are how sphingolipids interact with the redox signaling pathway to regulate renal function and even result in chronic kidney diseases. Ceramide, sphingosine, and sphingosine-1-phosphate (S1P) as main signaling sphingolipids are discussed in more detail.
Future Directions:
Although sphingolipids and ROS may mediate or modulate cellular responses to physiological and pathological stimuli, more translational studies and mechanistic pursuit in a tissue- or cell-specific way are needed to enhance our understanding of this important topic and to develop effective therapeutic strategies to treat diseases associated with redox signaling and sphingolipid cross talk. Antioxid. Redox Signal. 28, 1008–1026.
Introduction
I
In particular, they are actively involved in various cellular stress responses and cell defending reactions such as oxidative stress and host response against bacterial or parasite invasion (165, 174). On a broad scale, sphingolipids are typically classified as CERs, sphingomyelins (SMs), or glycosphingolipids (GSL) and their metabolites. Among them, CERs are extensively studied, which have been shown to serve as a precursor for some other biologically active sphingolipids, including sphingosine (SPH), C1P, and S1P, and therefore, CER is often considered as a central molecule in metabolism and in the biological role of sphingolipids (Fig. 1) (117).
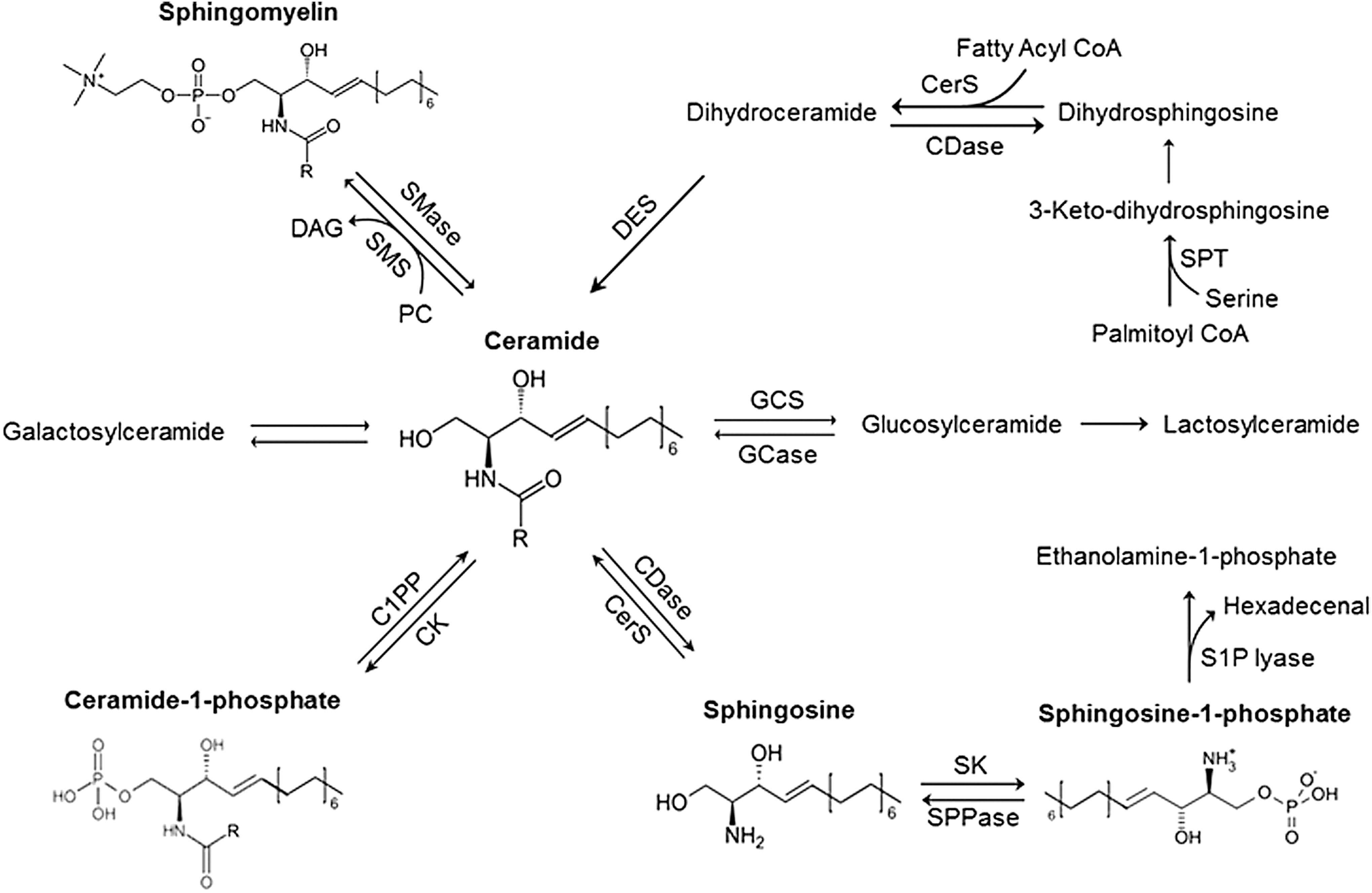
Several comprehensive reviews on sphingolipid biosynthesis and metabolism have been previously published (187). Here we will only briefly highlight the most common pathways for the production and metabolism of several important sphingolipids, which are related to the regulation of kidney function under physiological conditions and during some chronic kidney disease (CKD).
Sphingolipid Metabolism in the Kidney
In the kidney, sphingolipid metabolic pathway is also an important cellular process that represents high interconnections among various pathways, where CER plays a central role in metabolism (58). The process of sphingolipid metabolism is largely dependent on de novo synthesis in the endoplasmic reticulum (ER), CER transport from the ER to the Golgi, SM synthesis, conversion of SMs into CER, and the catabolism of CERs (46) (Fig. 2).
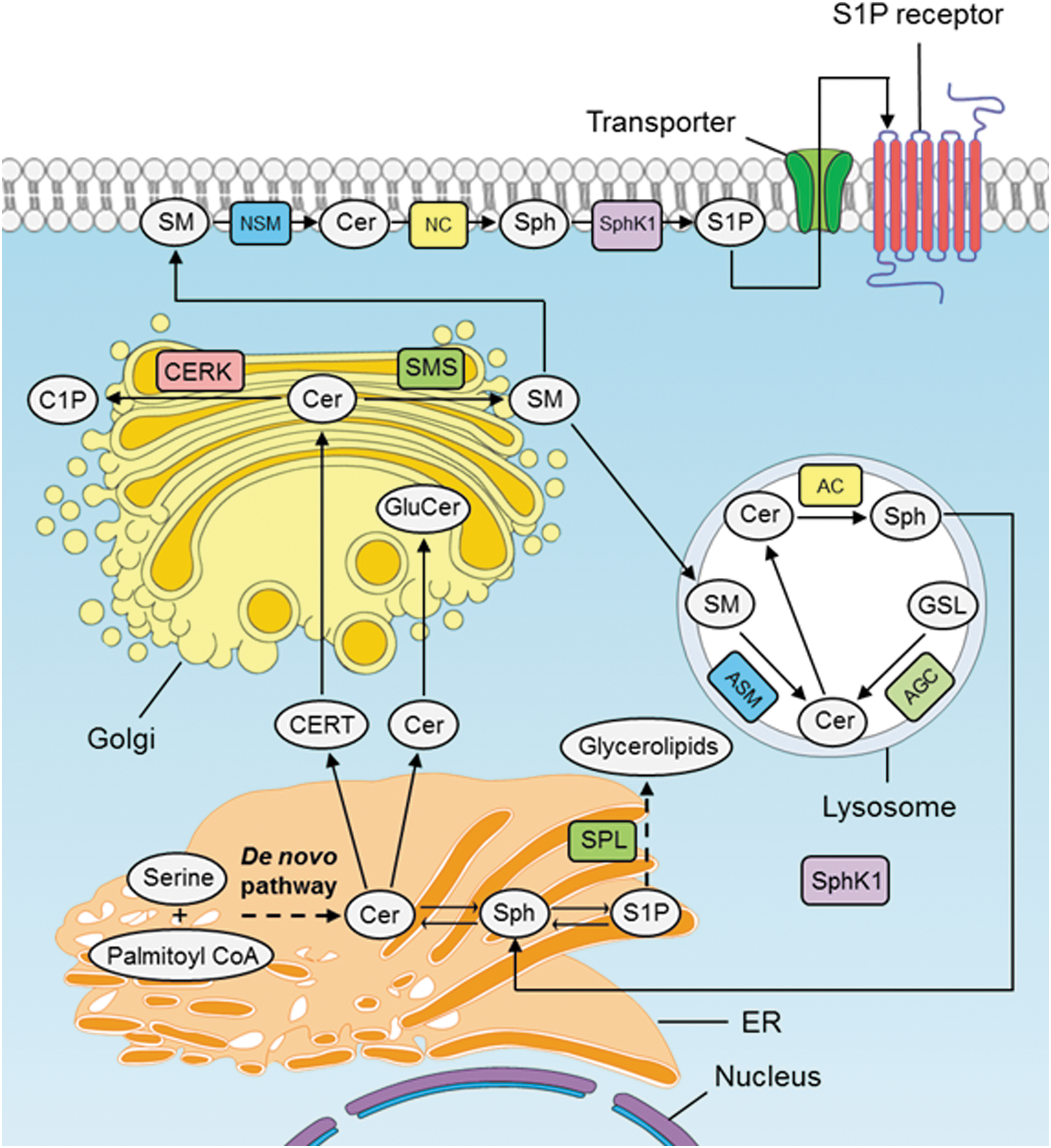
De novo synthesis in the ER
De novo biosynthesis of sphingolipids begins at the cytosolic leaflet of the ER where a set of four enzymes coordinately generate CERs of different acyl chain lengths from nonsphingolipid precursors (46). In brief, sphinganine (dihydrosphingosine) is acylated to dihydro-CER and further desaturated to form CER (145), which starts with the condensation of serine and palmitoyl CoA via serine palmitoyltransferase (56). Once CER is transported to the Golgi complex, various head groups can be added to produce more complicated forms of sphingolipids such as SM or GSL (124). Alteration of membrane dynamics or lipid signaling could be achieved by changing acyl chain length of CER and alpha hydroxylated fatty acids (46).
In glomerular podocytes, CER de novo synthesis was also demonstrated, which is linked to the pathogenesis of focal segmental glomerulosclerosis and other podocyte pathology. It is strongly suggested that the enzymes related to sphingolipid biosynthesis, in particular, CER synthesis or degradation, might play an important role in the SM to CER conversion in podocytes.
Enhanced de novo synthesis of CER and consequent production of its metabolites were also found in renal epithelial cells in response to a variety of stimuli such as angiotensin II type 2 (AT2) receptor activation (70), hyperglycemia (48), radiocontrast-induced renal tubular cell (RTC) injury (67), and cadmium (Cd2+) toxicity (29). This altered CER de novo synthesis is also an important pathogenic mechanism responsible for renal tubular dysfunction or injury.
CER transport from the ER to the Golgi
CER, which is generated in ER as a membrane-bound molecule, is less soluble in aqueous environment and needs to be transported to the Golgi complex for further modification into SMs or GSL. Therefore, cell utilizes two major mechanisms for its transportation, one is vesicular transport and another is a nonvesicular transport system through the CER transfer protein (126). CERT is a cytosolic protein transferring CER from the ER, where it is generated, to the Golgi apparatus where it serves as a substrate for production of complex sphingolipids such as SM and GSL (46). CERT usually transfers CER species with acyl chains less than C22, although it transfers C22 and C24:1 CER but with less efficiency (40%) (85). CER transported by CERT from ER to the Golgi is preferentially incorporated into SM over GSL (57).
SM synthesis
SM is the most abundant complex sphingolipid in mammals. In cell culture studies, mammalian and yeast cells fail to survive due to inability to produce SM either because of CERT mutation or defects in de novo sphingolipid synthesis (148). The precise single function that SM fulfills, which is absolutely necessary for cell survival, is not clear due to many known functions of SMs in membrane biology.
SM synthases catalyze the synthesis of SM. In most mammalian species, there are at least two members of the SM synthase family SM synthases 1 and 2 (62), and possibly a third family member known as SM synthase related (SMSr) (149). Both SMS 1 and 2 are present in the trans-Golgi, although SMS2 is also localized to the plasma membrane. Hence, SMS2 also maintains SM content directly at the plasma membrane. Transfer of a phosphocholine headgroup from phosphatidylcholine to CER yields diacylglycerol (49) and SM. Recent studies showed that SMS2 contributes to renal sphingolipid metabolism using SMS2-deficient mice under either high-fat or normal diet condition (144).
Conversion of SMs into CER
Sphingomyelinase (SMase) family catalyzes the breakdown of SM into CER and free phosphocholine through the hydrolysis of the phosphocholine headgroups. Based on their pH optimum, mammalian SMases were classified into three major categories: acid SMase, alkaline SMase, and the neutral SMases. Even though all the three forms of SMases catalyze a similar reaction, they are evolutionarily unrelated and have different subcellular distributions. Alkaline SMase is expressed exclusively in the intestine and liver where it plays an important role in the digestion of dietary SM (39). Both acid SMase and neutral SMase serve as the major regulators of SM catabolism in most tissues and are ubiquitously expressed.
Acid SMase (aSMase), encoded by the gene Smpd1, a lysosomal protein was the first to be characterized in mammalian cells, which metabolizes SM present on endosomal membranes. aSMase secreted into the extracellular space known as secretory SMase. It metabolizes the SM-containing lipoproteins that are abundant in the plasma and also in outer leaflet SM on the plasma membrane (133).
Within the past decade, among three different mammalian neutral SMase (nSMase) genes identified, Smpd2, Smpd3, and Smpd4, the first nSMase gene discovered was Smpd2. nSMase 2 is the best characterized nSMase to date. Under subconfluent conditions, it localizes to the Golgi, however, on reaching confluence, nSMase2 translocates to the plasma membrane (103). Overexpression of nSMase2 causes degradation of SM into CER with a preference for C24:0 and C24:1 species (103). nSMase3 is encoded by the Smpd4 gene, which is predominantly expressed in skeletal and cardiac muscle and is localized to the ER and possibly the Golgi (83).
There are many studies showing that SMases (23), including aSMase and nSMase (28, 59, 97, 186) and CER synthases (14, 158, 159), are related to CER production in RTCs in response to a variety of stimuli such as endotoxin, fluorinated anesthetic exposure hypoxia/reoxygenation, or oxidant injury. It has been also shown that during ischemia/reperfusion (I/R) injury, CER conversion from SM hydrolysis is increased leading to elevated CER levels in RTCs in vivo (186). On the contrary, there is increased activity of SMases or ceramidase (CDase) to catabolize SM or CER when the kidney is exposed to the anti-GBM antibody (186) or carbon tetrachloride intoxication (64).
Catabolism or degradation of CERs
All sphingolipids are catabolized to CER, SPH, and finally, S1P. The deacylation of CER species is catalyzed by the family of enzymes known as ceramidases, which have organelle-specific expression. Acid ceramidase (AC), encoded by Asah genes, is a lysosomal enzyme that deacylates CER species produced from the degradation of sphingolipids from plasma membrane. Although the levels of SPH and CER were normal, Asah2−/− mice showed a lack of metabolizing dietary CERs in the kidneys. It is still unknown, however, what roles CDase plays in the regulation of kidney function due to the lack of visible pathological evidences in the Asah2−/− mice (80).
In addition, there is evidence that CER conversion into complex sphingolipids such as globotriaosyl-CER (Gb3) and galabiosyl-CERs also occurs in the mouse kidney. In the α-Gal A KO (with Fabry disease) mouse model (84), Gb3 is highly accumulated in the renal cortex compared to the medulla. There are increased Gb3 isoforms and CER dihexosides (composed mostly of galabiosyl-CERs) in this Fabry disease model compared to wild-type mice. It has been suggested that CER distribution in the cortical region of the kidney with high concentrations of sphingolipids may reflect the CER's role in the biosynthesis or degradation of these complex sphingolipids (46).
Neutral ceramidase, the most active ceramidase at neutral pH, is associated with the plasma membrane. It regulates SPH and S1P production and release (153). The alkaline ceramidases contain three separate family members: alkaline ceramidases 1, 2, and 3 activated by calcium ion in vitro (101). After deacylation of CER into SPH by ceramidases, shingosine kinase 1 (SPHK1) and 2 localized in the cytosol or peripherally associated with specific membrane compartments catalyze the conversion of SPH to S1P. In the final step, S1P lyase present in the ER degrades S1P to produce hexadecenal and phosphoethanolamine. S1P can be degraded through the SM cycle, while SPH can be transported via recycling mechanism from membranes or lysosomes to ER, re-entering into the SM cycle to form CER by the salvage pathway, namely, reacylated by CER synthases (CerSs) (Fig. 2).
Every step in this SM cycle can be changed by different pathological stimuli that interfere with sphingolipid metabolism or enhance the production of various bioactive lipids that may serve as pathogenic factors or be the therapeutic targets for treatment or prevention of related diseases (120).
In addition, CER can also be phosphorylated to produce C1P. CER kinase (CERK) phosphorylates CER into C1P, which is mainly produced in the trans-Golgi, and potentially the plasma membrane. CERK belongs to the diacylglycerol (DAG) kinase family, which was originally identified based on its homology to SPH kinase. However, CERK only utilizes CER as a substrate and has no activity for DAG or sphingosine (146) and it has a preference with acyl chain lengths greater than 12 carbons long (170).
Cross Talk of Sphingolipids with Redox Signaling
There is increasing evidence that oxidants such as reactive oxygen species (ROS) and other free radicals may regulate or modulate sphingolipid metabolism (24) and they also serve as downstream messengers to mediate sphingolipid signaling (125). For example, the depletion of cellular reduced glutathione (GSH) by increased ROS and reactive nitrogen species (48) regulates enzymatic activities of SMases and CDases (63), while sphingolipids, including CER, sphingosine, and S1P, have the ability to change cellular redox homeostasis through regulation of NADPH oxidase (Nox) (193), mitochondrial integrity (44), nitric oxide synthase (NOS) (172), or antioxidant enzymes.
In the next several sections, we focus on several important signaling sphingolipids to summarize their interactions with redox signaling pathways. Since the redox signaling participates in a variety of cellular activities, including cell proliferation (19), differentiation (129), and apoptosis (60), imbalanced redox status in cells or tissue, namely, oxidative stress is frequently involved in various pathophysiological processes such as inflammation (10), senescence (31), I/R (52), and hypoxia (99) leading to different diseases such as hypertension (36), shock (47), atherosclerosis (116), diabetes mellitus (11), Alzheimer's disease (118), cancer (9), and even infectious diseases (71). Therefore, during discussion of the interactions of sphingolipids with redox molecules, we link their relevance to related kidney diseases.
CER and redox regulation
Among various sphingolipids, CER is one of the important family members of sphingolipids. It is now known that the cross talk between CER and redox signaling importantly contributes to the regulatory or signaling activity associated with CER.
CER activation of redox signaling
In many cell, animal, and human studies, CER was demonstrated to activate ROS-generating enzymes such as Nox, xanthine oxidase, uncoupled NOS, and enzymes in the mitochondrial respiratory chain, which will enhance ROS production in cells or tissues (34). In particular, CER has been reported to mediate the fusion of small membrane raft (MR) domains to form CER-enriched membrane platforms that cluster subunits of Nox, forming an active enzyme complex to produce O2 .−. CER-enriched membrane platform with Nox subunit assembling and enzyme activation is referred to as an MR redox signaling platform (MRRSP).
Although it has been reported that CER can interact with the mitochondrial electron transport chain to generate ROS (34), the MRRSP associated with CER has been intensively studied as a regulator or pathogenic mechanism in different cells because Nox is one of the most important redox signaling enzymes that contribute to many physiological and pathological processes (42).
The MRRSP is dependent on the activation of SMase to produce CER in cellular membrane, including plasma membrane and other membranes. CER production is stimulated or enhanced by various physiological or pathological stimuli. In such MRRSPs, redox molecules such as Nox subunits or cofactors can be assembled into a redox enzyme complex and thereby activate its enzyme activity resulting in production of O2 .− (73, 190).
It has been reported that the MRRSPs may serve as a signaling platform for many factors such as death receptor ligands namely CD95 ligand, TNF-α, or endostatin (72, 73, 190), ultraviolet irradiation (27), L-Hcy, and M1-receptor agonists (191). A general working model of MRRSP is presented in a previous review (92). In brief, aSMase synthesized from the ER will be transported through the Golgi apparatus into lysosomes proximal to the cell membrane. In response to various stimuli such as death receptor activation, lysosomes are mobilized to move and fuse to the cell membrane, where aSMase from lysosomes is activated to produce CER leading to MR clustering. During MR clustering, Nox subunits or related cofactors are aggregated and thereby assembled to form MRRSPs producing O2 .−, conducting transmembrane signals (Fig. 3).
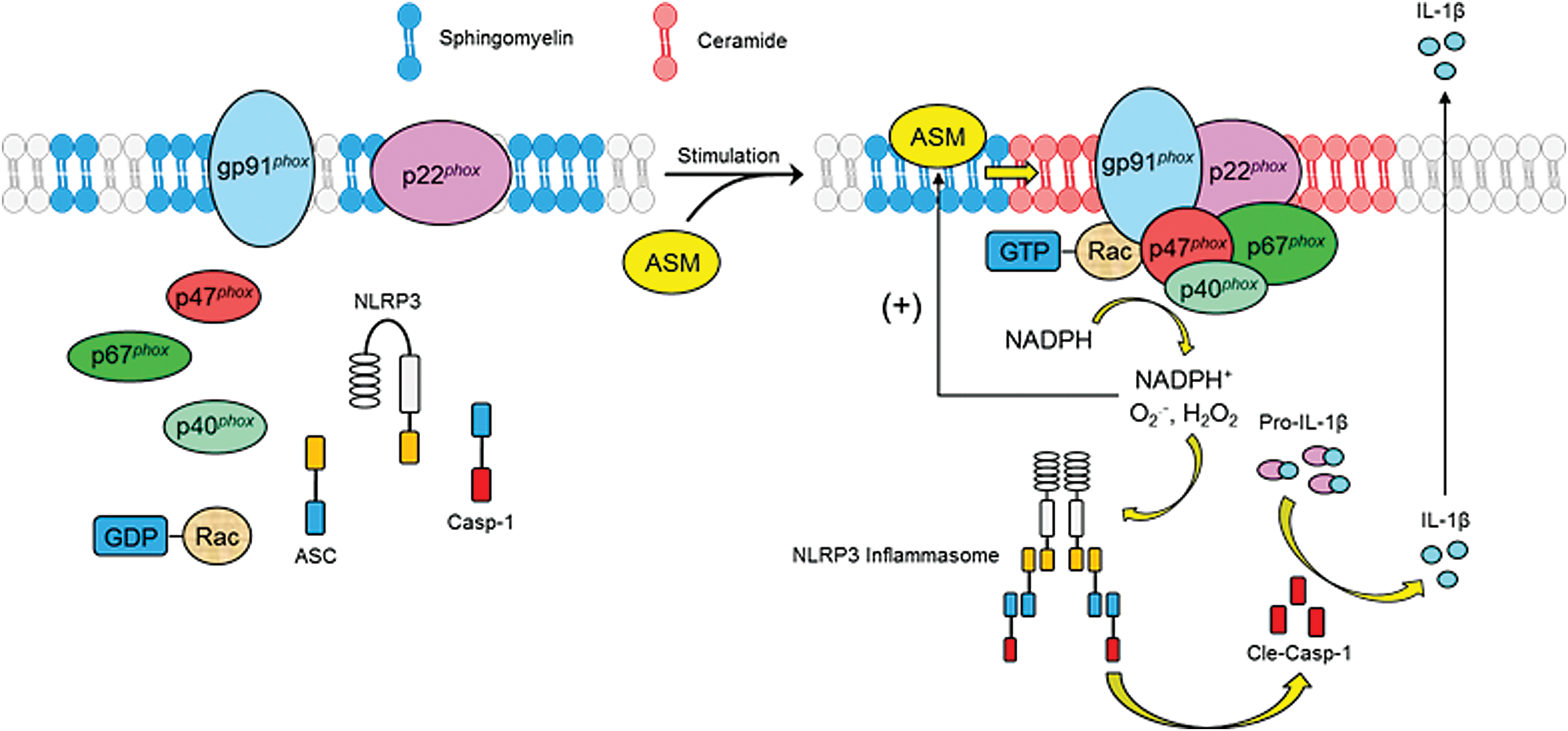
More recently, MRRSP-mediated ROS production has been shown to contribute to NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome activation, which serves as intracellular machinery to turn on the inflammatory response in many different cells, including renal podocytes and tubular cells (1). This redox activation of inflammasome, and thereby inflammatory response, is now considered as an underlying mechanism mediating the progression of many chronic degenerative diseases such as glomerular sclerosis and consequent end-stage renal disease (ESRD) (38, 88).
In addition, MRRSPs with Nox subunits are importantly involved in the functional regulation of renal proximal tubule (RPT) cells. In tubular cells, disruption of MR association with Nox subunits activates this enzyme to produce O2 .− (55), which is different from podocytes. It is clear that different cells may link MRs to conduct redox signaling in very different ways (182). For example, dopamine receptor activation associated with MRs inhibits ROS production in RPT cells (178) and receptor agonist such as fenoldopam was found to decrease Nox2 and Ras-related C3 botulinum toxin substrate 1 (Rac1) proteins in MRs. The receptor agonist action may lead to redistribution of Nox2, Nox4, and Rac1 from MRs to non-MR fractions thereby maintaining Nox in an inactive state (55).
Redox regulation of CER metabolism and raft clustering
CER mediates MRRSP formation, and CER-mediated signaling is influenced by ROS. It has been shown that MRRSP formation in endothelial cell (EC) membrane is reduced by superoxide dismutase (SOD) but enhanced by ROS production (189). H2O2 was demonstrated to change MR activity, which in turn activates PI3 kinase/Akt and extracellular signal-regulated kinases 1/2 (51, 175). O2 .− produced from xanthine/xanthine oxidase can activate CER-enriched membrane platform formation in ECs (122, 189). In addition, lipid peroxides are shown to be a potent promoter of CER-enriched membrane platforms (8). With respect to the mechanisms of redox regulation on CER-mediated signaling, it has been reported that ROS not only alter MR clustering or formation of macrodomains (98) but also directly change MR constituents such as caveolin-1, cholesterol, and related raft proteins (107).
ROS play an important role in CER-associated signaling by regulating the enzyme activity of CER production or sphingolipid metabolism such as SMase. Many studies have shown that the generation of ROS is implicated in the sphingolipid metabolism (26, 40). For example, there is evidence that production of ROS is required for activation of aSMase and the consequent CER generation and CD95 clustering (132) and that aSMase activation by ultraviolet-C light (3) can be blocked by an antioxidant, pyrrolidine dithiocarbamate (PDTC). Several molecular mechanisms have been assumed to mediate ROS regulation of CER production via SMase activity.
It has been reported that the redox regulation of SMase may be associated with the cPLA2-arachidonic acid pathway that is sensitive to redox molecules (100). It has been shown that aSMase activity needs zinc and related C-terminal cysteine modification. Zinc serves as a coordinator to interact with a water molecule that constitutes an optimal structure or microenvironment for aSMase activity (161). GSH regulation is considered as an important redox regulatory mechanism of various SMases (96, 104).
There is evidence that glutathione, its analogs, and individual fragments may inhibit the activity of various SMase isoforms (95, 96). The g-glutamyl-group of GSH was found to have an inhibitory effect on the neutral Mg2+−dependent SMase, and the ability of GSH to inhibit oxidative processes in the cell may exert its role to inhibit SMase activity (157). Moreover, intracellular GSH concentration is found important to SMase gene expression and thereby alters its activity (184). Feedforward amplifying mechanism represents a cross talk between ROS and CER signaling.
Another mechanism for degradation of sphingolipids may be direct oxidation of these lipids by ROS as do other cellular lipids such as glycerophospholipids. However, since the fatty acid domain of sphingolipids consists of mainly saturated residues, they are poor substrates for peroxidation in cells or tissues when interacting with ROS.
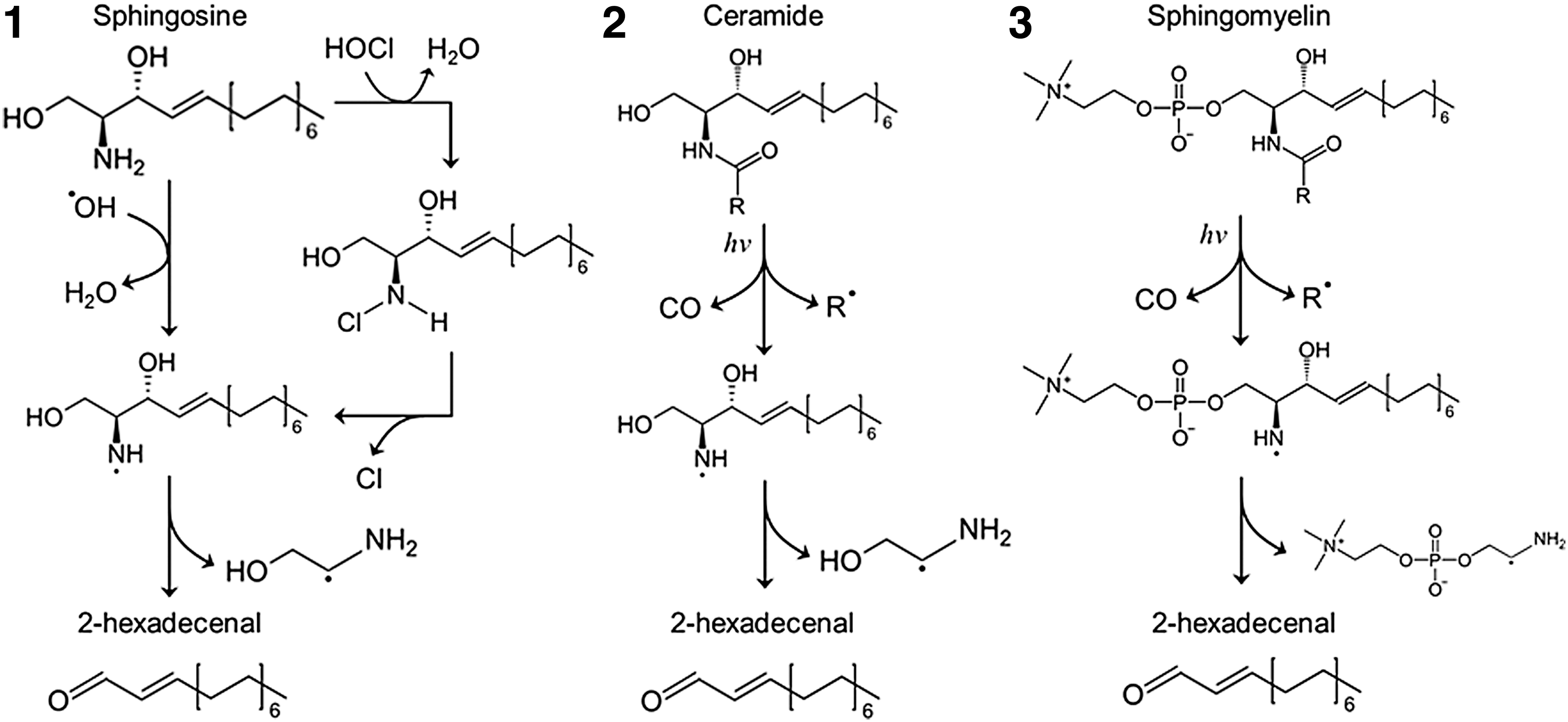
In recent studies, sphingolipids were found to interact with free radicals or ROS via fragmentation, but not oxidation. ROS-induced fragmentation is mainly associated with transformations of hydroxyl-containing glycerophospholipids and sphingolipids. It has been demonstrated that there are diverse types of sphingolipid fragmentation, depending on whether it is a radiolysis or photolysis of sphingolipids. Free radical fragmentation may lead to the formation of a 2-hexadecenal (Hex) as shown during sphingosylphosphorylcholine (SPC) radiolysis and photolysis of N-acetylated sphingolipids. There are reports that Hex formation from the degradation of S1P or other sphingolipids may undergo an enzymatic reaction, and therefore, these lipids will be irreversibly destructed. Enzyme may be S1P lyase or other lyases, which can catalyze to form Hex and aminoethanol phosphate (137).
Hex is found to alter many biological activities in different cells such as reorganization of the cell cytoskeleton, apoptosis, and production of adducts with DNA leading to mutagenic consequences (87, 160). It is now known that free radical destructive processes occur in various sphingolipids such CER, SPH, and SM under different conditions such as radiation or HOCl stimulation. The reactions of free radical fragmentation for several sphingolipids are summarized (Fig. 4).
SPH and redox regulation
Effects of SPH on redox regulation
During cellular oxidative stress, lipid peroxidation is an important biologically relevant event. In the sphingolipid pathway, SPH and sphinganine have a relatively low blood level and their effects have been overlooked, because they are easily converted to S1P by blood cells (177). However, there is increasing evidence that SPH within cells interacts with the redox signaling pathway to exert regulatory action in different cellular activities and organ functions.
In this regard, Wilson et al. in 1986 reported that SPH and sphingoid-based dihydrosphingosine reduce O2 .− production via inhibition of protein kinase C (PKC) in intact neutrophils (171). The positive charge of SPH was found to inhibit Nox activity (130). SPH can also suppress translocation of p47 phox , through which the assembling of the Nox complex is blocked. Moreover, SPH and its CER analogs (N-acetyl sphingosine and N-hexanoyl sphingosine) may not only reduce ROS production by inhibition of Nox but also by direct scavenging of some ROS (108).
Pharmacologically, safingol ([2S, 3S]-2-amino-1,3-octadecanediol) is a saturated analog of SPH (136), and it is currently under clinical trials to treat advanced solid tumors in combination with cisplatin (94). It has been demonstrated that ROS have a key role in mediating the cytotoxicity of safingol in cancer cells. At high concentrations ≥10 mM, however, safingol treatment may cause increased production of ROS leading to cell death by necrosis producing toxicity in normal cells or tissues. Another important action of SPH on redox signaling relates to its effect to induce extensive aggregation of peroxidized vesicles, facilitated by electrostatic interaction between the positively charged SPH and the cell membrane bilayer. During tissue oxidative stress, peroxidation-induced vesicle aggregation may serve as an important working model for lipoprotein aggregation (152).
Redox regulation of SPH production and action
As discussed above, GSL are degraded via one or more glycosidases, whereas an acidic CDase causes degradation of CER to produce SPH and a fatty acid. Desaturation of dihydro-CER also eventually generates SPH, where dihydro-CER Δ4-desaturase (DES) is a critical enzyme. It belongs to the desaturase family that converts dihydrosphingosine backbone within CER into SPH (20) In DES-mediated catalyzing reactions, molecular oxygen and hydroxyl species play a critical role (131).
In addition, ROS have been reported to induce acidic CDase degradation and thereby reduce SPH production. Action of ROS on acidic CDase may be due to cathepsin B activation. N-acetylcysteine (37) as a precursor of glutathione is an intracellular antioxidant, which protects cells from acidic CDase degradation in stimulation of a drug against metastatic melanoma, dacarbazine (DTIC). It has been suggested that increased production of ROS during DTIC treatment is critically responsible for the degradation of acidic CDase due to their effects to activate cathepsin B (15).
S1P and redox regulation
S1P is a widely studied bioactive sphingolipid that is derived from SPH—the backbone of most sphingolipids. So far it has been known that S1P is produced in a variety of mammalian cells or tissues such as platelets (179), red cells (66), ECs (163) and kidney tissues (195). S1P is also demonstrated to play various regulatory roles on different cell or organ functions (91, 150) through an S1P receptor family comprising five members termed as S1P1-5 (119). For more information about the role of S1P in renal and cardiovascular regulation and disease, readers are directed to another comprehensive review published recently (91). Here we mainly focus on the cross talk between S1P and redox signaling and associated functional relevance.
S1P action on redox signaling
Recent studies have reported that S1P can stimulate ROS production in different cells via Nox2 or Nox4 and thereby induce lipid peroxidation resulting in production of bioactive aldehydes. There is evidence that S1P increases Ca2+ sensitization in vascular smooth muscle cells (VSMCs) due to its effects on Rho A/Rho kinase to result in Nox activation and ROS production. Moreover, S1P was found to promote the egress of hematopoietic progenitor cells from the bone marrow via S1P receptor 1 (S1PR1) in ROS-dependent pathway (50). In the heart, cardiac fibrosis in SPHK1 transgenic mice may be mediated by enhanced local oxidative stress (151). Mechanistically, S1P was found to promote p47 phox translocation thereby leading to activation of Nox to produce O2 .−. S1P-stimulated p47 phox translocation is mediated by a PKCd/PYK2 pathway (25).
In the kidney, S1P is considered as a potent vasoconstrictor of the preglomerular microvasculature, which was enhanced in I/R. I/R-enhanced vasoconstriction is accompanied by an increase in S1P2 receptor protein expression in renal microvessels. When rats were administrated JTE-013, an S1P2 selective inhibitor, much less necrosis and cell damage induced by I/R in the proximal tubules were observed. By analysis of renal ROS levels, it was found that increases in ROS induced by renal I/R were almost completely blocked, suggesting that S1P2 receptors mediate I/R-induced ROS production and related renal I/R injury (13, 54, 90), relating to reduction of ROS levels.
Redox regulation of S1P production and actions
Accumulating evidence suggests that the S1P production or its biological actions are reciprocally regulated by redox molecules. It has been reported that under different conditions such as hyperglycemia (185), hypoxia (3), angiogenesis (22), and exogenous administration of H2O2 (30), an increase in ROS, more precisely increased H2O2, activates SPHK1 to produce S1P in different cells or tissues. In VSMCs, low concentration of H2O2 and oxidized low-density lipids (ox-LDLs) resulted in SPHK1 activation, through which S1P generation leads to SMC proliferation (30). Inhibition of SPHK1 activation and S1P generation induces an imbalance of the CER/S1P ratio, which produces growth arrest and cell death (141).
On the contrary, low concentrations of ox-LDL have been reported to induce angiogenesis, which is also associated with ROS production and related oxidative effects on the nSMase2/SK1/S1P signaling pathway. It was shown that pharmacological inhibition of nSMase2 and SPHK1 by sphingomab, an anti-S1P monoclonal antibody, remarkably attenuated the capillary tube formation in vitro and angiogenesis in vivo (21).
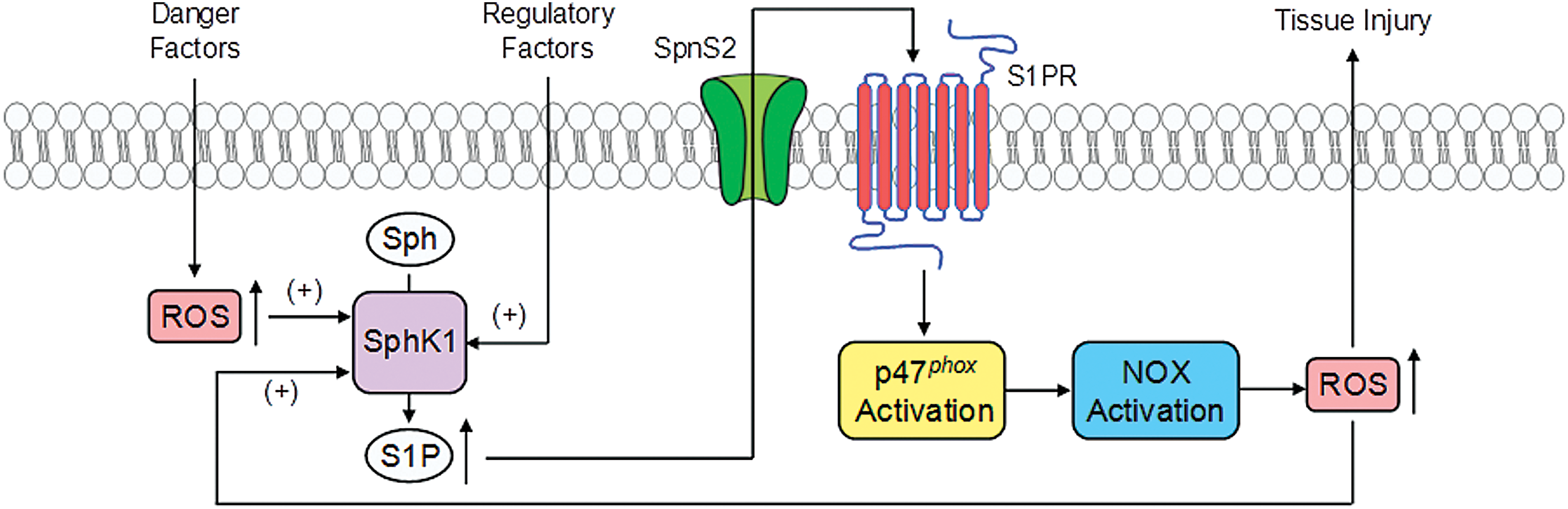
Based on current literature, the cross talk between S1P and redox signaling is summarized (Fig. 5). In response to various stimuli such as different danger factors, ROS can be generated from different resources such as MRRSP and mitochondrial respiratory chain, which in turn may activate SPHK1 to generate S1P to produce intracellular signaling action regulating the cellular activity or organ function. S1P may also be transported out of cells through its transporter such as the S1P transporter 2 (Spns2), where it binds to its receptor to exert regulatory role such as S1P1. The binding of S1P to S1P1 receptor may activate Nox4 to produce O2 .−, leading to redox signaling or regulation, even causing oxidative injuries in the kidney or other tissues.
Other sphingolipids and redox regulation
Although many other sphingolipids may also be more or less implicated in the regulation of cell or organ function, studies on their interactions with redox signaling are much less than three main signaling sphingolipids discussed above. It should be noted that the interaction between sphingolipids and ROS was first reported in type I Gaucher's disease, a lysosomal storage disease, where Nox activity is linked to the effects of sphingolipid accumulation in many cells (93). It is now known that in type I Gaucher's disease, many pathobiological changes are due to the accumulation of glucosyl-CER (glucocerebroside) and consequent suppression of Nox-mediated O2 .− generation. The latter in turn leads to monocyte dysfunction (93).
In addition, ganglioside GD3 accumulation causes lysosome storage disease, which may interfere with complex III of the electron transport chain in mitochondria, which may increase ROS production leading to mitochondrial permeability transition pore opening and cytochrome c release, thereby producing lysosome storage disturbance (45, 127).
Interactions Between Sphingolipids and Redox Signaling in Chronic Glomerular and Interstitial Injury or Diseases
Sphingolipids as bioactive lipids are involved in a number of kidney diseases, which are well summarized and discussed in recent review (135). Here we briefly highlight two major renal pathological processes, namely, glomerular and interstitial injury or lesions in the kidney, in which the cross talk between some specific sphingolipids and redox signaling may play important roles as a pathogenic mechanism.
Glomerular injuries and diseases
Interactions between sphingolipids and redox signaling have been reported to contribute to the development of glomerular injury or sclerosis developing into ESRD under different pathological conditions. We have demonstrated that during glomerular injury induced by hyperhomocysteinemia (hHcy), the interplay between CER and redox is critical, which may be one of the most important early events over the development of ESRD (180, 181, 183). Using fumonisin B1 and myriocin, inhibitors of the de novo synthesis pathway of CER in mesangial cells or in animals, it was found that Hcy-induced mesangial cell dysfunction and injury and ultimately glomerular injury and proteinuria were substantially attenuated (181, 183), suggesting that CER is a critical factor in hHcy-induced glomerular injury. Further studies have demonstrated that CER-mediated glomerular injury is associated with the enhancement of Nox activity and production of O2 .− in the kidney (183).
In podocytes and glomerular capillary ECs, the formation of MRRSPs activated by CER production was observed (182, 192). It is now clear that MRRSPs and related local oxidative stress due to Nox activation may mediate progressive glomerular injuries or glomerulosclerosis during hHcy and other diseases such as diabetes and hypertension (183).
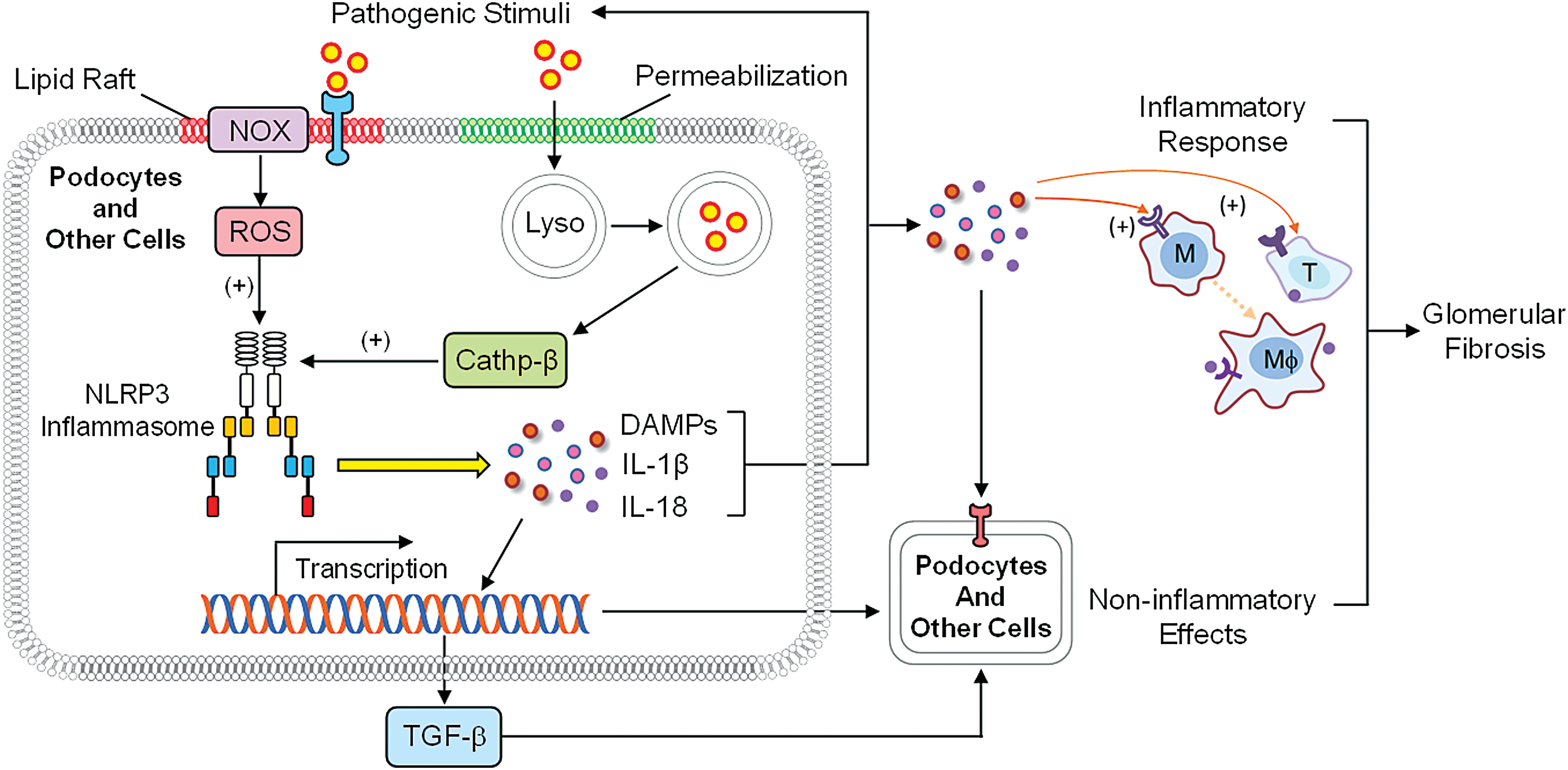
More recently, CER has been reported to be involved in the activation of inflammasome, in particular, the NLRP3 inflammasome. Inflammasome functions as an intracellular inflammatory machinery and its activation not only triggers the inflammatory responses but also produces various uncanonical effects such as cell membrane injury, cell death, metabolic disturbance, and transdifferentiation. This inflammation triggering action and the uncanonical effects of NLRP3 inflammasomes may work together to result in different tissue injuries and organ diseases (1, 173).
It has been reported that NLRP3 can sense CER signaling to directly activate NLRP3 inflammasomes (79, 162) and that CER can form MRRSPs to produce ROS resulting in NLRP3 inflammasome through thioredoxin dissociation with NLRP3, which leads to inflammasome formation and activation (194). In addition, lysosomal CER produced via aSMase in response to different stimuli may regulate cathepsin B release, which may induce NLRP3 inflammasome activation to trigger inflammatory response and tissue injury, including podocyte damage and other cell dysfunctions (32, 33), together leading to glomerular fibrosis and ultimately ESRD (Fig. 6).
Diabetic nephropathy (DN) (157) is the major cause of ESRD, which is characterized by hypertension, glomerular filtration, microalbuminuria, and proteinuria. One of the important mechanisms is abnormal sphingolipid metabolism and its regulatory role in diabetes mellitus and associated complications, including diabetic nephropathy. It is now evident that a dysfunctional sphingolipid metabolism generated specific sphingolipid species, which may cause diabetic-related complications (43).
The cross talk between sphingolipids and redox signaling was also found to be involved in the pathogenesis or progression of DN. Some in vitro and in vivo studies reported that the generation of ROS caused by CER further enhances oxidative stress, which may result in GSL production participating in DN. There is evidence that pyridoxamine, an ROS inhibitor, attenuated the accumulation of various sphingolipids in glomeruli of db/db diabetic mice and thereby prevented DN (53). This is consistent with several other studies showing that pyridoxamine is indeed effective in prevention or suppression of DN in animal models of diabetes or in diabetic patients (35).
Other studies reported that a cross talk between sphingolipids and ROS production in DN may include activation of Nox by sphingolipids such as CER or S1P and a decrease in the antioxidant defenses such as reduction of NO and SOD activity (41, 143). There is enhanced turnover of complex sphingolipids into bioactive metabolites, including CER, sphingosine, and S1P that can induce ROS production and vice versa during DN (48, 92). However, more studies are needed to investigate temperospatial roles of the interactions between sphingolipids and redox signaling in the pathogenesis of DN.
Sphingolipids have also been emerging as promising candidates, potential biomarkers, or mediators of obesity onset and progression, including their roles in associated complications such as atherosclerosis, hypertension, and kidney injuries (168). It has been reported that MRs and their clustering with Nox, namely, MRRSPs and other signaling molecules, are involved in the development of obesity and related complications such as CKD, type 2 diabetes mellitus, and cardiovascular diseases.
In several studies from our laboratory, we demonstrated that the excessive accumulation of sphingolipids such as CER and their metabolites leads to the development of obesity and associated kidney damages. There were significantly increased plasma total CER levels in high-fat diet-fed mice, blocked by aSMase inhibitor (amitriptyline) treatment or its gene deletion (18). We also found that in addition to the role of aSMase in obesity, CER producing enzyme is also locally involved in renal glomerular injury by stimulation of ROS production, suggesting that enhanced CER biosynthesis and its action to activate ROS production may play a prominent role in the development of obesity, metabolic syndrome, and associated kidney damages (18).
It has been demonstrated that glomerular injury associated with obesity is attributed to the MRRSPs in glomerular capillary endothelial cells (GECs). This is because visfatin, an adipokine, stimulates aSMase activity and thereby results in the aggregation of CER with Nox subunits, increasing O2 .− production. Large production of O2 .− leads to local oxidative stress increasing glomerular permeability by disruption of microtubule networks in GECs and podocyte dysfunction, ultimately causing local inflammatory response and glomerular injury till fibrosis and sclerosis (17). In addition, CER or its analogs were shown to inhibit complex III of mitochondrial electron transport, which in turn leads to increased ROS production. Inhibition of complex III by agents such as TNF, adriamycin, or tamoxifen is mediated by increased mitochondrial CER levels (147). These factors together enhanced glomerular endothelial and podocyte dysfunction leading to glomerular disease.
On the contrary, glomerular sphingolipid and its metabolites may also be enhanced by local oxidative stress. Increased ROS levels in local tissues were found to stimulate turnover of complex sphingolipids thereby generating bioactive sphingolipid metabolites, including CER, sphingosine, and S1P. It is clear that the cross talk between sphingolipids and redox signaling may critically contribute to local oxidative stress in glomeruli, which may be one of the important mechanisms responsible for the onset and progression of obesity-associated CKDs.
Tubulointerstitial lesions and fibrosis
It has been reported that tubulointerstitial injury might be one of the major causal events associated with progressive kidney dysfunction in many forms of CKD. Several general mechanisms are taken into consideration by which tubulointerstitial injury occurs and thereby leads to progressive CKD.
For example, Kriz and LeHir (81) proposed that chronic damage to the glomerulus causes foot process effacement and, eventually, loss of podocytes, which may result in adhesion of glomerular basement membrane to parietal epithelial cells, forming a bridge between the glomerular and parietal basement membranes. Such bridge may lead to the formation of adhesion or synechia between the glomerular tuft and Bowman's capsule at the affected site. At such adhesion site, the filtrate is extruded directly into the periglomerular interstitium, resulting in recruitment of interstitial fibroblasts that proliferate at the site of the transmutation in response to plasma proteins. Ultimately, interstitial inflammation and fibrosis occur around the glomerulus.
Another type of tubulointerstitial injury and fibrosis relates to obstructed flow of filtrate from glomerulus. Under different pathological conditions such as glomerulonephritis or diabetic nephropathy, inflammation in glomeruli leads to extracapillary cell proliferation or transformation of parietal cells, resulting in crescent pathological changes in glomeruli. The occurrence of Bowman's capsular pathology together with inflammatory cell infiltration may block the outflow of filtrate toward proximal tubules associated with obstruction of the glomerulotubular junction or initial segment of the proximal tubules. Ultimately, tubular atrophy and degeneration are developed and fibrosis is activated (81, 89).
In addition, increased delivery of proteins or other physiologically active molecules to the tubular lumen may also cause tubulointerstitial injury, which is associated with a decline in GFR and proteinuria and often called proteinuria tubulointerstitial injury (111). It has been suggested that the “severity” of proteinuria and its specific characteristics such as the nature of the molecules that are present in the filtrate are essential in the development of tubulointerstitial lesions.
There is evidence that ROS or local oxidative stress is involved in tubulointerstitial injury. In response to albuminuria, for example, the generation of ROS increases in some tubular segments, which may further activate or promote tubulointerstitial injury. It has been shown that during the albumin endocytosis process, the reabsorption of albumin in the proximal tubule cells produces concomitant activation of Nox and ROS generation through the activation of Rac1, a GTPase (169), which may be due to the action of both albumin and other albumin-bound molecules as a tubular toxin/activator (139). During kidney diseases, tubular lumen has an acidic environment and exposure to oleic acid-bound albumin enhances ROS production.
Another common redox-associated factor in triggering or promoting tubulointerstitial lesions is ox-LDL. Some reports have indicated that ox-LDL may activate inflammation in the renal interstitium in both T cell-dependent and T cell-independent mechanisms (166). Binding and uptake of ox-LDL to scavenger receptors, CXCL16, or CD36 in different cells such as macrophages and tubular cells also play a critical role in lipotoxicity in tubulointerstitial tissue leading to lesions (115, 128).
The deposition of interstitial matrix, particularly the presence of collagenous fibers along with infiltration of inflammatory cells, tubular cell loss, and fibroblast accumulation, is essential to the tubulointerstitial fibrosis (16). In this regard, damaged tubular epithelial cells trigger fibrogenesis via release of proinflammatory chemokines and cytokines that induce mononuclear inflammation and the formation of fibroblasts (12, 69). It is the release of these cytokines that locally provides critical signals, triggering and mediating interstitial injury and ultimately leading to fibrosis (134).
In addition, tubular mechanical stretch due to obstruction also induces TGF-β and NF-κB, which initiates the downstream release of plasminogen activator inhibitor-1 or other chemokines and growth factors (196). Inhibition of NF-κB, TNF-α, or CCR1 was found to successfully attenuate the tubulointerstitial injury (75, 76, 164). Also, several studies reported that the epithelial-to-mesenchymal transformation of tubular epithelial cells has an active role in the progression of tubulointerstitial fibrosis (188). It has also been revealed that on injury in the tubulointerstitial area, profibrotic factors such as transforming growth factor-β (TGF-β) 1 and connective tissue growth factor (CTGF) are upregulated, leading to renal tubulointerstitial fibrosis (121).
Yang et al. (176) reported that G2/M stage cell cycle arrest of proximal tubular cells after injury produced profibrogenic growth factors such as TGF-β1 and CTGF that are capable of stimulating fibroblast proliferation and collagen production. Furthermore, the study demonstrated that the onset of G2/M arrest activated c-jun NH2-terminal kinase (JNK), which is known to enhance TGF-β1 and CTGF gene transcription and thereby upregulate profibrotic gene expression in vitro and collagen production in vivo. It is clear that tubular cell cycle changes during pathogenic stimulation and related gene transcriptional regulation of different cytokines or injury factors are an important initiating mechanism of renal tubulointerstitial fibrosis.
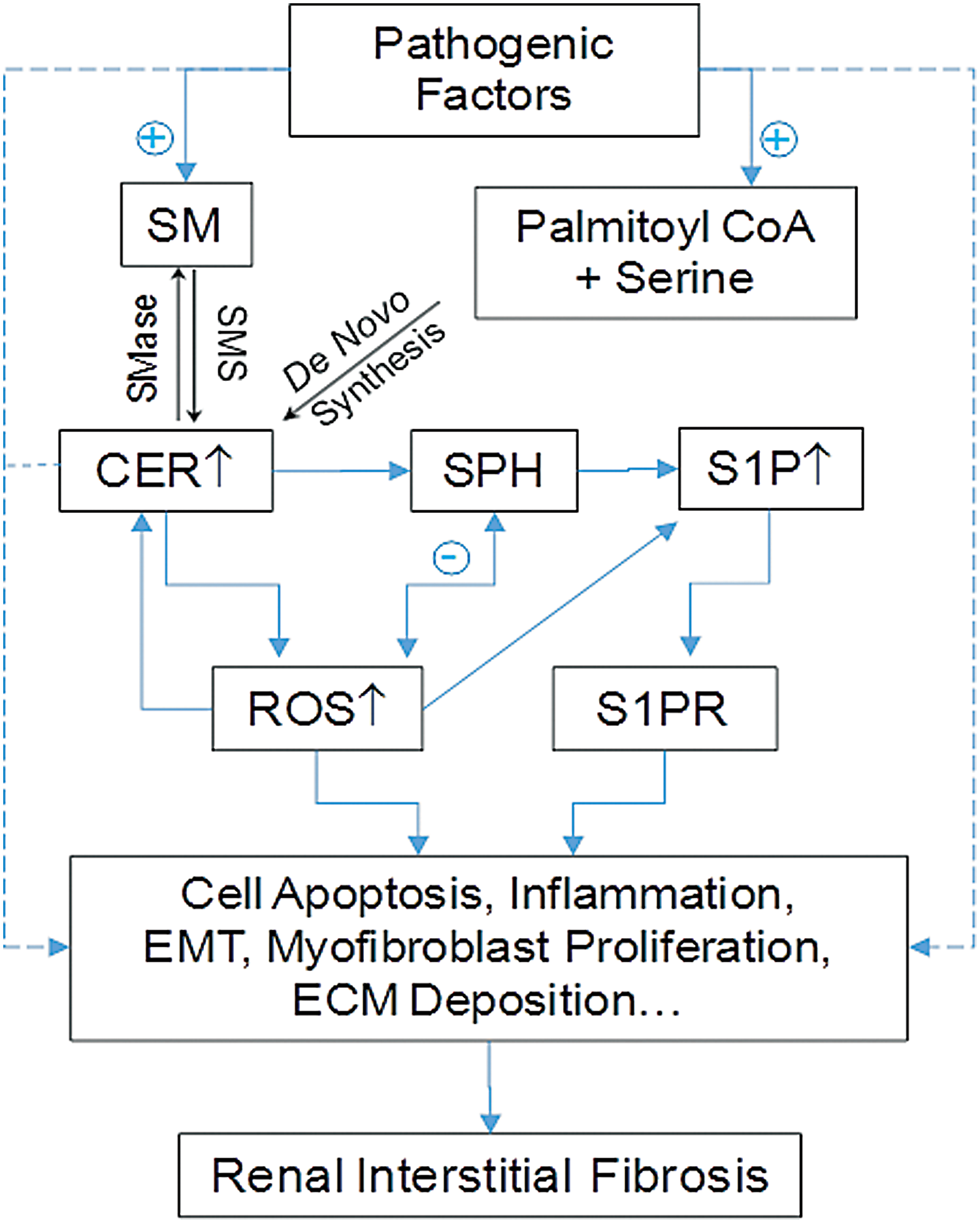
The important role of sphingolipids in tubulointerstitial injury or fibrosis has also been reported. As we discussed above, sphingolipids can interact with ROS in different ways and with different species in two families. It has been reported that hypoxia/reoxygenation or radiocontrast increased CER synthase activity, which may lead to tissue CER increase and consequent apoptotic change of the renal epithelial cells. In addition, aSMase inhibitor, amitriptyline, was shown to inhibit inflammation and myofibroblast formation, blocking renal fibrosis in an ureterally obstructed mouse model. The beneficial effect of amitriptyline is attributed to reduced CER production, which reduces tubular cell apoptosis, interstitial inflammation, and myofibroblast formation (2, 140).
There is a wealth of evidence that SPH kinase-1 (SK1) and its product, S1P, are implicated in the induction of CTGF and TGF-β, leading to accumulation of extracellular matrix proteins and ultimately renal interstitial fibrosis. Blockade of S1P production via inhibition of SK1 was found to reduce tubulointerstitial renal inflammation and fibrosis in DN (6, 112, 113). Using FTY720, an S1P receptor agonist and FDA-approved drug to treat multiple sclerosis, it has been demonstrated that S1P may reduce renal inflammation (106) and attenuate renal profibrotic events such as inhibition of myofibroblast activation and collagen deposition during different coronary heart diseases (112, 113, 138).
Especially in an albumin overloaded rat model of nephropathy, FTY720 was found to reduce proteinuria and decrease infiltration of inflammatory cells into tubulointerstitium with suppressed expression of monocyte chemotactic protein-1. Similar results were also found in angiotensin II (Ang II)-induced progressive kidney disease, suggesting the critical role of S1P in Ang II-induced renal interstitial injury (142).
In other experimental models of kidney interstitial inflammation such as unilateral ureteral obstruction, FTY720 was also shown to ameliorate interstitial inflammation, interstitial fibrosis, and tubular atrophy (138, 154, 156). These effects of FTY720 are mainly associated with their action on S1P receptors and related downstream signaling pathways. However, there are also some reports indicating that the intracellular action of S1P is to promote an antifibrotic effect, which is opposite to an extracellular profibrotic response of S1P (78). In Figure 7, we summarize the effects of these sphingolipids and ROS on the development of renal interstitial injury or fibrosis, where the interactions between ROS and S1P or other sphingolipids with each other may need further studies to dissect the causative or effective contribution of ROS or sphingolipids.
Therapeutic Perspective
More recently, it has become clear that the intracellular sphingolipid composition in podocytes and other cells of the kidney may contribute to the pathogenesis and progression of the disease. Their increase or decrease in different cell types is involved in the development or progression of CKD. The major strategies using these sphingolipids as therapeutic targets are activation or replacement of their producing enzymes or use of agonists of downstream products if specific diseases are due to the defect of these enzymes. If some diseases are related to the accumulation of sphingolipids, however, inhibitors of their producing enzymes, activators of their metabolizing enzymes, and the receptor antagonists of the enzyme product may be used.
In CKDs, some approaches have been used as therapeutic strategies based on the pathogenic role of sphingolipids. For example, Fabry disease is caused by systemic accumulation of globotriaosylceramide (Gb3) and related GSL mainly in the brain, heart, and kidney (110). Patients with Fabry disease have elevated levels of Gb3 in their plasma or urine (82) and highly increased levels of globotriaosylsphingosine (lyso-Gb3) (51). Gb3 accumulation in the kidney was observed mainly within lysosomal, ER, and nuclear markers of renal glomerular cells such as podocytes and mesangial cells (5), which lead to hypertrophic podocytes with foamy appearing vacuoles, inclusion bodies of glycolipids in podocytes (zebra bodies), and mesangial widening (4). Ultimately, accumulation of Gb3 and associated GSL results in podocyte injury such as albuminuria and foot process effacement (109, 155).
A successful treatment to reduce Gb3 accumulation even in human patients with Fabry disease is the enzyme replacement therapy (ERT), by which recombinant human α-GalA is used to hydrolyze CER trihexoside and reduce Gb3 accumulation. This ERT markedly attenuates renal complications and prevents renal failure in Fabry disease (123, 155). The ERT has been also used for treatment of Niemann-Pick disease-A and -B type, which are caused due to Smpd1 gene mutation, deficient aSMase activity, and consequent accumulation of SM in the affected tissues, including the kidney. Both patients with Niemann-Pick disease-A and -B and Smpd1 knockout mice revealed the presence of lipid-laden macrophages resembling foam cells in different cells, including renal cells (86).
It has been observed that in mice and patients, ERT with recombinant human aSMase significantly improved the pathological changes in the reticuloendothelial system and kidneys (105). Another promising therapeutic strategy to target sphingolipids is to use S1P receptor agonists, such as FTY720 (an unselective S1P receptor agonist) and SEW2871 (a selective S1P1 receptor agonists), to protect from renal ischemia/reperfusion injury (7, 77, 114) and CKDs (74). In DN, activation of Rho kinase by excessiveS1P/S1P2 receptor pathway in RTCs leads to renal fibrosis, including glomerular and interstitial fibrosis (65).
Lupus nephritis, an inflammation of the kidney caused by systemic lupus erythematosus, leads to increased circulating S1P levels (167). Recent studies have indicated that the progression of glomerular or tubulointerstitial injuries can be blocked or reversed by FTY720. S1P agonists may be one of the effective antifibrotic remedies for treatment of CKDs.
Concluding Remarks
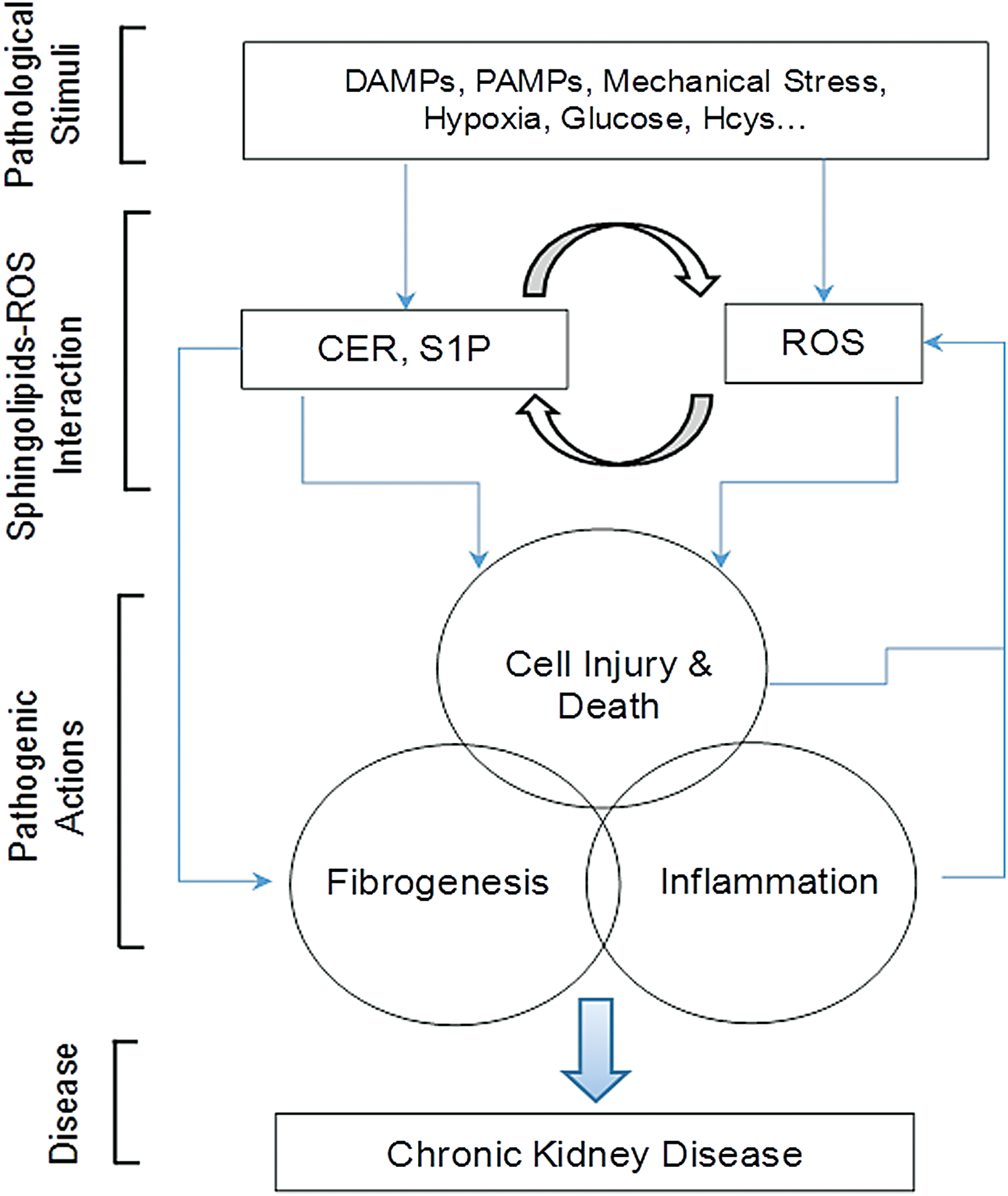
This review briefly presents current evidence about the metabolism of sphingolipids in the kidney and their interaction with redox signaling pathway, which may importantly contribute to the regulation of renal function and play important roles in the progress of CKDs. It has been indicated that sphingolipids may interact with various redox signaling molecules such as O2 .−, H2O2, ox-LDL and related signaling platforms such as MR clusters, NADPH oxidase-associated signalosomes, and inflammasomes, result in physiological and pathological changes in cellular activity and function. Moreover, changes in redox molecules may alter sphingolipid metabolism or actions, thereby influencing major biological processes such as inflammation, lipid metabolism, and insulin sensitivity in renal glomerular or tubular cells.
This cross talk between sphingolipid and redox signaling pathways may be activated by a number of pathological stimuli such as hHcys, hyperglycemia, hypercholesterolemia, hypoxia, radiocontrast, mechanical stress, cytokines, adipokines, pathogen-associated molecular patterns, or damage-associated molecular patterns, and therefore, it has been implicated in the development of a variety of renal diseases, in particular, chronic degenerative kidney disease due to their actions to induce cellular dysfunction and to activate profibrogenic process, which results in glomerular sclerosis and tubulointerstitial lesion, ultimately leading to ESRD (Fig. 8).
Although some new therapeutic strategies to target sphingolipids or ROS may be directed toward clinical use in the treatment of CKDs, approaches to targeting the cross talk or interactions between sphingolipids and redox signaling will be imperative, which may promote development of more effective therapies for prevention or treatment of ESRD.
Footnotes
Acknowledgment
Most of the works cited in this review from authors' laboratories were supported by the National Institute of Health grants (DK054927, HL057244, HL075316, and HL122937).