
Abstract
Significance:
Major pathogenic enterobacteria and protozoan parasites from the phylum Euglenozoa, such as trypanosomatids, are endowed with glutathione (GSH)-spermidine (Sp) derivatives that play important roles in signaling and metal and thiol-redox homeostasis. For some Euglenozoa lineages, the GSH-Sp conjugates represent the main redox cosubstrates around which entire new redox systems have evolved. Several proteins underwent molecular adaptations to synthesize and utilize the new polyamine-based thiols.
Recent Advances:
The genomes of closely related organisms have recently been sequenced, which allows mining and analysis of gene sequences that belong to these peculiar redox systems. Similarly, the three-dimensional structures of several of these proteins have been solved, which allows for comparison with their counterparts in classical redox systems that rely on GSH/glutaredoxin and thioredoxin.
Critical Issues:
The evolutionary and structural aspects related to the emergence and use of GSH-Sp conjugates in Euglenozoa are reviewed focusing on unique structural specializations that proteins developed to use N 1,N 8-bisglutathionylspermidine (trypanothione) as redox cosubstrate. An updated overview on the biochemical and biological significance of the major enzymatic activities is also provided.
Future Directions:
A thiol-redox system strictly dependent on trypanothione is a feature unique to trypanosomatids. The physicochemical properties of the polyamine-GSH conjugates were a major driving force for structural adaptation of proteins that use these thiols as ligand and redox cofactor. In fact, the structural differences of indispensable components of this system can be exploited toward selective drug development. Future research should clarify whether additional cellular processes are regulated by the trypanothione system. Antioxid. Redox Signal. 28, 463–486.
Trypanosomatids and Their Polyamine-Based Thiol System
T
As an indication of the dramatic evolutionary adaptation to parasitism, comparison of the genomes of Bodo saltans [a free-living kinetoplastid bacterivore that inhabits fresh and marine water (85)] and parasitic trypanosomatids revealed that acquisition of parasitism entailed the loss of nearly 50% of protein-coding genes (121). Yet, all kinetoplastids share a significant number of common and unique molecular, structural and metabolic traits (121). A few of these features are briefly outlined.
These organisms lack individual transcriptional regulation of gene expression, and hence, protein-coding sequences are transcribed as long polycistronic units that are posttranscriptionally edited. In addition, messenger RNAs (mRNAs) for mitochondrial proteins must undergo posttranscriptional editing that includes uridine insertion or deletion. The guide RNAs, which drive the editing process, are encoded in thousands of small circular DNA molecules (minicircles), while larger DNA molecules (maxicircles) encode for a few mRNAs. Both DNA structures are concatenated forming the kinetoplast or mitochondrial DNA. Another peculiarity is the compartmentalization of most glycolytic enzymes within peroxisome-like organelles called glycosomes.
Regarding the redox system, the pioneer biochemical studies of Dr. Stoppani highlighted several peculiarities common to trypanosomatids, such as the lack of catalase and glutathione (GSH) peroxidase activities and a lower GSH content compared to mammals, which render these parasites deficient in coping with hydrogen peroxide (22, 23). Years later, the team lead by Dr. Cerami reported on the presence of a novel, low-molecular-weight, heat-stable cofactor containing reducible thiol group(s) in soluble extracts from different trypanosomatid species (56). The cofactor was isolated from the non pathogenic trypanosomatid Crithidia fasciculata (Cf) and identified as a spermidine (Sp) conjugate of GSH—N 1,N 8-bis(glutathionyl)-spermidine—for which the trivial name of trypanothione * was coined (55) (Fig. 1A).

Further studies demonstrated the ubiquity of trypanothione in trypanosomatids (143). The occurrence of trypanothione can be traced back to the root of Euglenozoa (113, 120), and its precursor, monoglutathionyl spermidine (Gsp), to the nonrelated γ-proteobacteria Escherichia coli (Ec) (49, 157).
Trypanothione utilization required the specialization of key redox proteins and led to the gradual loss of the major NADPH reductases glutathione reductase (GR) and thioredoxin reductase (TrxR) genes, which are absent in all known genomes of trypanosomatids [(17, 53, 84);
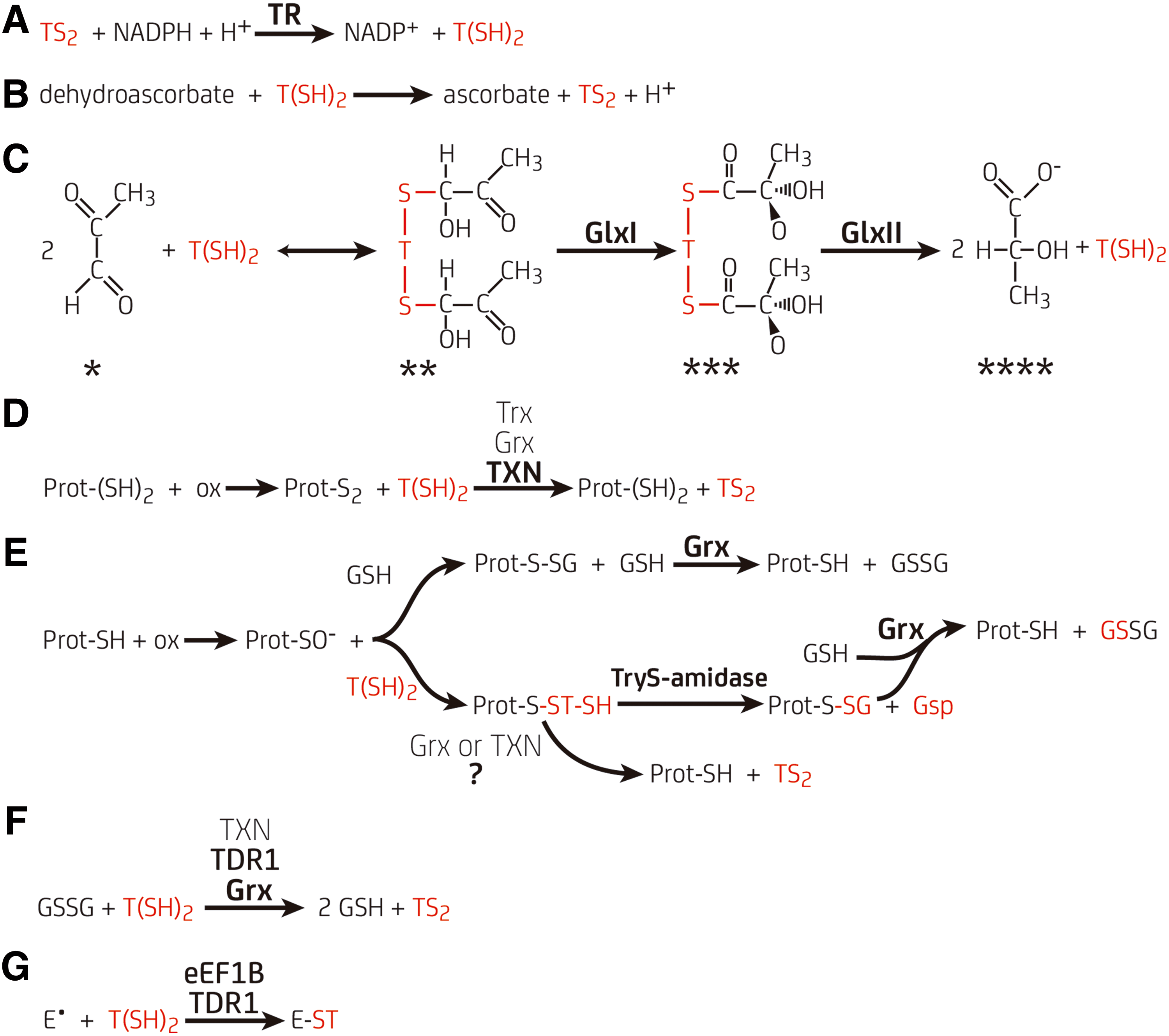
GR and TrxR provide reducing power to the GSH/glutaredoxin (Grx) and the thioredoxin (Trx) pathways that regulate different cellular functions in most living organisms (42). Instead, trypanosomatids rely on a minimalistic system composed of trypanothione synthetase (TryS), trypanothione reductase (TR), dihydrotrypanothione [T(SH)2], and the kinetoplastid-specific Trx-like tryparedoxin (TXN) (30, 39, 135, 142) (Fig. 1B), while the oxidoreductase activity of Grx and Trx, or even selenoproteins, appears dispensable, at least for African trypanosomes (20, 117, 141).
A comprehensive overview of the molecular adaptations that several proteins from the thiol-dependent redox system of trypanosomatids underwent to use trypanothione is discussed in the following sections in a biochemical, evolutionary, and biological context. For more general reviews on trypanothione metabolism, the reader can refer to Comini and Flohé (37), Irigoín et al. (82), Krauth-Siegel and Comini (93), Krauth-Siegel and Leroux (94), and Manta et al. (107).
Molecular and Evolutionary Aspects of Polyamine-Thiol Biosynthesis
The biosynthesis of T(SH)2 involves the conjugation of two GSH molecules to the free amine groups of Sp in sequential reactions catalyzed by monoglutathionyl spermidine synthetase (GspS) and/or TryS (Fig. 1B). GspS from E. coli (18, 100) and GspS or TryS from different kinetoplastids (16, 35, 36, 101, 123 –126, 148) have been characterized.
These enzymes are a fusion of an N-terminal peptidase domain (amidase module) presenting a papain-like fold and a C-terminal peptide/amide-forming catalytic domain (synthetase module). The amidase module has homology to the C-terminal domain present in the cell wall-associated cysteine peptidase domain (NlpC/p60) protein superfamily and the synthetase module resembles a circularly permutated ATP-grasp domain. Both paralog enzymes catalyze the ATP-dependent phosphorylation of the glycyl carboxylate of GSH and favor the nucleophilic attack of the free amine from Sp or Gsp (only TryS) on the activated carbon atom by positioning both substrates in close proximity. Gsp is the final product of the reaction catalyzed by GspS. In TryS, Gsp can bind in an orientation suitable for conjugation of its remaining free amine with the second preactivated GSH to produce T(SH)2 (36, 65).
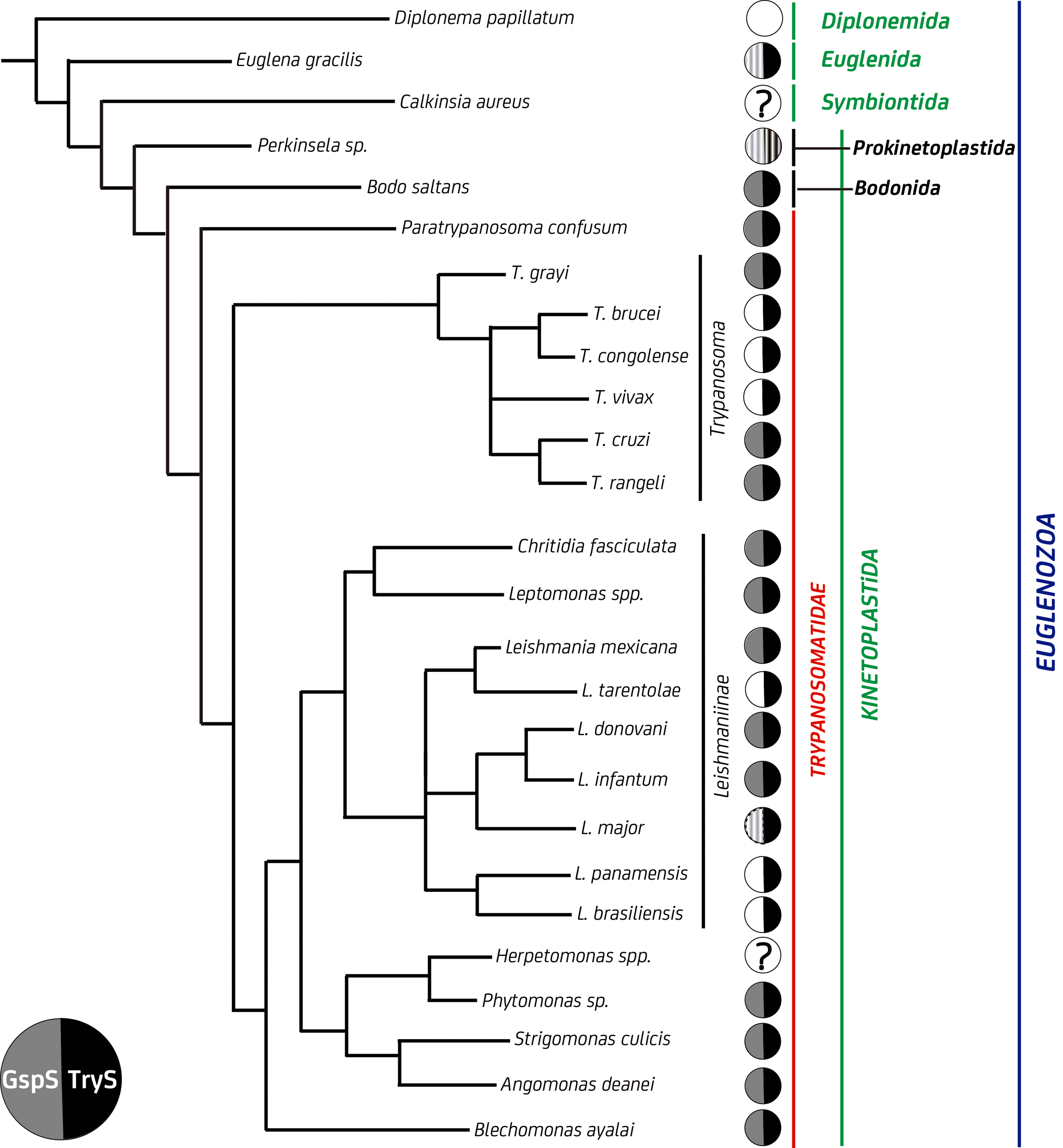
The presence of a GspS gene is not ubiquitous to all kinetoplatids (Fig. 2). Thus far, the expression of a functional GspS, which likely provides an independent source of Gsp for TryS (see substrate Michaelis–Menten constant [K M] values in Table 1), has been demonstrated only in C. fasciculata (36, 92). Strikingly, pathogenic trypanosomatids appear to lack an independent GspS activity because most of them harbor GspS pseudogenes or genes whose expression is fully silenced (149). This can partly be explained by the 5- to 195-fold lower K M for Sp of TryS from pathogenic species (16), which warrants the parasites a proper supply of Gsp for T(SH)2 synthesis.
CDNB, 1-choro-2,4-dinitrobenzene; Cf, Crithidia fasciculata; DTNB, dithionitrobenzoate; E 0′, standard redox potential; Ec, Escherichia coli; GR, glutathione reductase; GSH, glutathione; GSSG, glutathione disulfide; Gspox, monoglutathionyl spermidine disulfide; GspS, monoglutathionyl spermidine synthetase; Hs, Homo sapiens; k cat, catalytic constant; K M, Michaelis–Menten constant; LMWT, low-molecular-weight thiols; ns, the compound is not a substrate of the enzyme; Sp, spermidine; Tc, Trypanosoma cruzi; TryS, trypanothione synthetase; TS2, trypanothione disulfide; T(SH)2, dihydrotrypanothione.
TryS has been shown to be essential for the survival of trypanosomatids lacking [T. brucei; (38, 162, 171)] or having [Leishmania infantum; (149)] an additional GspS sequence. RNA interference (RNAi) silencing of TryS in African trypanosomes led to depletion of Gsp and T(SH)2, in vitro and in vivo cell death, and enhanced sensitivity toward oxidants, despite the GSH content being increased to low mM levels (7, 38, 171). This demonstrated the entire dependence of trypanosomatids on T(SH)2 and the lack of functional complementation by GSH.
Despite the current evidence supporting the nonessentiality of GspS, the protein has likely been a key player in the evolution of the redox metabolism of trypanosomatids. A recent study addressing GspS phylogeny reported the presence of GspS homologues only in Eubacteria (except for the group Chlamydiae), several kinetoplastids, and the related pseudofungi from the Oomycetes class (filamentous protists) (31). Our search for GspS and TryS sequences in the recently sequenced genomes from phylogenetically related organisms confirmed the lineage-specific distribution of these proteins (Fig. 2) and that the loss of the GspS gene occurred later and independently when trypanosomatids radiated into different species.
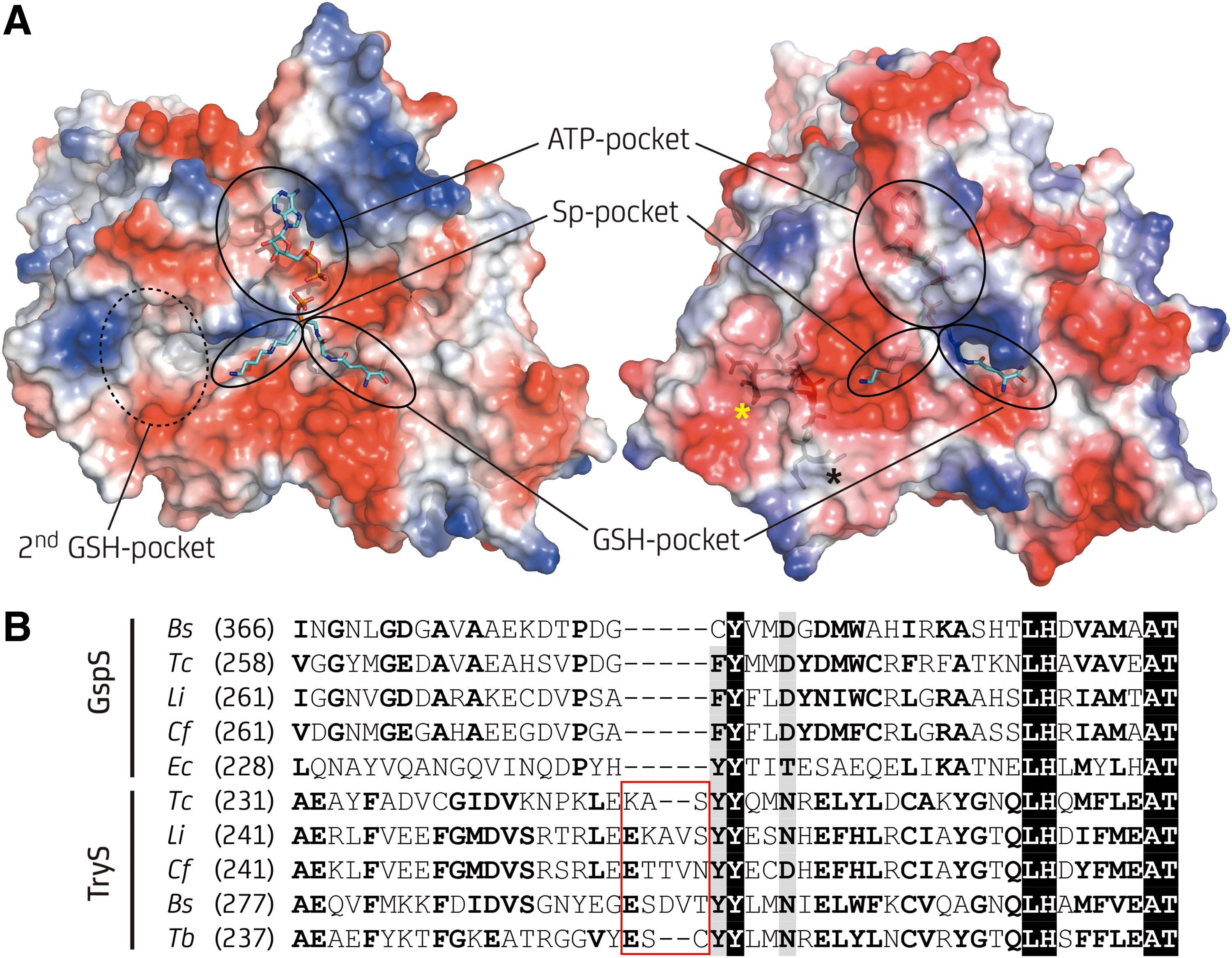
Kinetoplastid TryS presents higher identity with bacterial GspS (29% average amino acid identity) than with kinetoplastid GspS (24% average amino acid identity), mainly due to the presence of several large insertions in the kinetoplastid GspS that are absent in the bacterial homologues and in TryS. This suggests that a bacterial GspS and not a kinetoplastid one was the template from which TryS may have evolved by gene duplication followed by neospecialization.
In this regard, TryS presents a strictly conserved insertion within a loop [Gly250-Val262 for Leishmania major TryS: LmTryS; (65)] (Fig. 3A), which, according to molecular dynamic simulation, adopts a rigid conformation that allows access to a cavity that extends from the Sp binding pocket in a direction opposite to the catalytic site (where the first GSH-binding pocket is located) and suitable to host a second GSH molecule (91). In the second step of the biosynthetic reaction catalyzed by TryS, the GSH moiety of Gsp binds to this second GSH pocket, while the polyamine moiety occupies the Sp pocket with its free amine pointing toward the catalytic site. In fact, all TryS so far characterized display substrate inhibition by GSH (K i GSH = 0.036–1.7 mM depending on parasite species) due to occupancy of this second GSH pocket (16). The shorter loop of EcGspS [residues Gly242-Pro249; (129)] occludes and distorts this region abolishing Gsp binding. Consistently, all GspS from kinetoplastids lack the conserved five-residue insertion of TryS and inhibition by GSH has not been reported (124) (Fig. 3B).
As noted above, GspS and TryS harbor a cysteine, histidine-dependent amidohydrolase/peptidase (CHAP) domain that hydrolyzes Gsp and T(SH)2 to yield GSH and Sp (Fig. 1B). Given the absolute dependence of trypanosomatids on T(SH)2 and not on GSH, except as a trypanothione precursor, the amidase activity may contribute to polyamine homeostasis. In C. fasciculata, the growth phase-dependent redistribution of free and GSH-bound Sp was not associated with de novo synthesis of polyamines but ascribed to the amidase activity of GspS and TryS (143). A similar role in the regulation of the intracellular pool of polyamines can be envisaged for the TryS/GspS-amidase of T. cruzi and Leishmania spp., which are, respectively, fully or partially dependent on polyamine uptake from extracellular medium (5, 29, 73, 74). The biological role of the TryS-amidase activity in polyamine homeostasis appears less clear for African trypanosomes, which can synthesize Sp de novo (158) and have a low capacity to take up polyamines from the medium (170). On the one hand, metabolic studies on parasites with impaired Sp biosynthesis suggested that Gsp and T(SH)2 catabolism may contribute to maintain the Sp pool (170). On the other hand, the in vivo growth deficiency of a cell line devoid of TryS-amidase activity could not be restored on infected animals receiving Sp (171) at a dose that proved effective in protecting parasites against chemical inhibition of Sp biosynthesis (118). Thus, functions beyond maintenance of polyamine homeostasis can be envisaged for the amidase activity of TryS. In this regard, a recent study shows that TryS-amidase from African trypanosomes is involved in the hydrolysis of protein-bound thiol–polyamine complexes (164), as shown previously for EcGspS (33).
Physicochemical Properties of Trypanothione as Driver of Molecular Adaptations
From the chemical point of view, GSH and T(SH)2 are equally able to participate in thiol/disulfide exchange reactions. However, T(SH)2 outstands for its high efficiency in several redox assays with physiological oxidants and proteins. For instance, T(SH)2 reacts faster than GSH with hydrogen peroxide and, more remarkably, with peroxynitrite (Table 1) (6, 133, 163). T(SH)2 proved also to be two to three orders of magnitude more efficient than GSH in the reduction of dehydroascorbate (95) (Fig. 4) and ribonucleotide reductase (RnR) (48, 96). Although trypanosomal Trx and Grx lack conspicuous molecular adaptions to use T(SH)2 (see Trx of Trypanosomatids, an Orphan Redoxin section and Class I Grx, Redox Hubs Between GSH and Trypanothione Metabolism section), both proteins are more efficiently reduced by T(SH)2 than by GSH (30, 135, 142).
GSH and T(SH)2 have similar redox potentials (E 0′, Table 1) (57). Moreover, GSH is a better nucleophile than T(SH)2, as demonstrated by its higher pH-independent second-order rate constant against electrophiles (Table 1) (115). However, GSH and T(SH)2 differ by 1–1.5 units in the pKa of their cysteine(s) (Table 1), with the dithiol being half deprotonated at pH 7.4 due to the contribution of the adjacent ammonium group of the Sp bridge, which stabilizes the thiolate species (115, 132). Thus, under physiological conditions, a higher fraction of T(SH)2 will be deprotonated, which represents the nucleophilic species.
Another important difference is that T(SH)2 is a dithiol, whereas GSH is a monothiol, and formation of an intramolecular disulfide is, in general, kinetically and thermodynamically favored when compared to the intermolecular oxidation of two GSH molecules (70, 115). From the structural point of view, trypanothione has an intrinsic flexibility and lacks major conformational constraints. Hence, it may adopt conformations suitable for reaction with different disulfide or sulfenic substrates. Importantly, T(SH)2 and GSH differ in their net charges at physiological pH (+1 and −2, respectively) and in the absence of a free carboxylate glycine in the first. Thus, negative charges near substrate binding pockets may not be a major obstacle for reactivity of proteins with T(SH)2, while they could interfere with GSH binding.
Enzymatic Recycling of Trypanothione by TR and Substrate Selectivity
TR is an NADPH-dependent flavoprotein that has been genetically and chemically validated as drug target due to its indispensability for the survival of trypanosomatids (50, 97, 102). The protein shares >40% sequence identity with GR, including active site motifs and catalytic mechanism (99). Nonetheless, TR and GR present mutually exclusive substrate specificity for trypanothione disulfide (TS2) and glutathione disulfide (GSSG), respectively (152). TR displays a dimeric conformation with two symmetrical active sites. Each subunit in the dimer consists of three domains: the N-terminal FAD-binding domain, the NADPH-binding domain, and the C-terminal interface domain involved in dimer assembly (79, 99) (Fig. 5A).
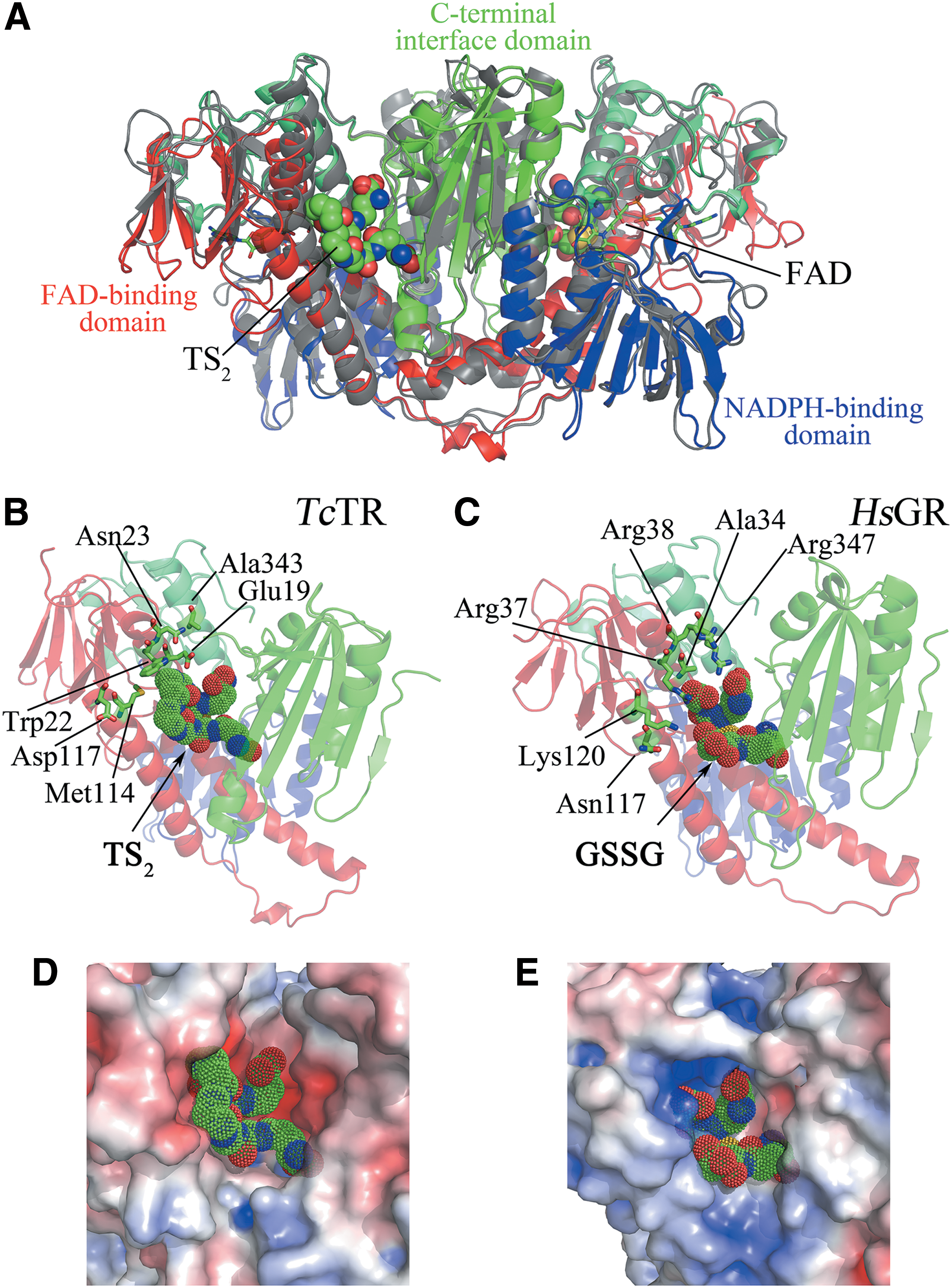
The substrate binding site of TR and Homo sapiens GR (HsGR) consists of a deep crevice with the redox active disulfides (Cys53-Cys58, numbering according to Trypanosoma cruzi [Tc]TR) and the active site base (His461) located at its bottom, with the flavin ring below them. One side of the crevice includes three helices from the FAD domain of one subunit, and the other side is formed by part of the interface domain of the other subunit. Most of the residues that are within 8 Å from the flavin ring system in TR are strictly conserved in HsGR, which explains the very similar binding conformation of the γ-Glu-Cys moiety of TS2 in TR (Fig. 5B) and of GSSG in GR (Fig. 5C) (19).
The residues conferring discrimination for substrate are located in the Sp-binding region of TR. The side chains of Trp22 (Arg37 in HsGR) and Met114 (Asn117 in HsGR) form a hydrophobic patch suitable to bind Sp and provide stacking interactions along the long axis of the tryptophan ring, while repelling the carboxylates of GSSG. In EcGR, these two residues are replaced by Asn21 and Val102, respectively, leading to a reduction in positive charge and a larger active site compared to HsGR. This explains why the bacterial enzyme is able to reduce TS2 (75, 147), while HsGR does not (24) (Table 1). In TcTR, the carboxylate of Glu19 (Ala34 in HsGR) is hydrogen bonded to the amide nitrogen of the Sp-GSH linkage in a conformation stabilized by Asn23, but does not interact directly with the positively charged amino group of the polyamine (no other negatively charged side chain is positioned to interact with the ionized N 4 secondary amino group of TS2).
Nevertheless, Glu19 together with Asp117 (Lys120 in HsGR) provides a negatively charged environment close to the Sp-binding site of TR. This contrasts with the cluster of positively charged residues in HsGR (Arg37, Arg38, and Arg347 substituted by Trp22, Asn23, and Ala343, respectively, in TcTR) that are located adjacent to the negatively charged glycine carboxylates of GSSG. As matter of fact, replacement of Ala34→Glu and Arg347→Trp in HsGR decreased 10,000-fold and increased 400-fold the catalytic efficiency for GSSG and TS2, respectively, compared to wild-type HsGR (24) (Table 1). Similar results were obtained for EcGR mutants resembling trypanosomal TR (75). Structural analysis revealed that the steric hindrance introduced with the Ala34→Glu substitution caused a large displacement of the side chain of Arg347 and enlarged the binding pocket of HsGR.
Comparative inhibition experiments performed with CfTR, TcTR, and HsGR further supported that electrostatic interactions are the major discriminating factor for TR and GR specificity, while hydrophobic interactions are less important but contribute to the binding affinity for the substrates (54). Interestingly, the charge complementarity for the substrates is not confined to the binding site but extends out and around the active site cleft, suggesting that long-range electrostatic interactions can contribute to enzyme specificity (10). TR has a ring of acidic residues outside the binding pocket, at the side that binds Sp (Fig. 5D), whereas GR has a patch of basic residues, able to interact with the negatively charged glycine carboxylates of GSSG (Fig. 5E). It has been proposed that these charges may attract the respective substrates and, probably, preorient them to tunnel into the active site (10).
Notably, on binding of the large disulfide substrates, no major rearrangements have been observed in the conformation of the enzymes, indicating that the active site has a well-defined rigid shape, with chemical properties varying in different positions of the cavity. Accordingly, the interaction of TR with its substrate has been described as mold-and-met fit (10) in which the very flexible (molten) substrate diffuses into the mold and is forced by noncovalent interactions into the correct orientation and specific conformation needed for the binding and subsequent catalysis. Nevertheless, cocrystallization of Trypanosoma brucei (Tb)TR (130) and Leishmania infantum (Li)TR (11) with small ligands has shown that, on noncovalent binding, the enzyme undergoes subtle changes in side chains allowing the formation of a new, small binding pocket in the otherwise large, solvent-exposed active site, which is occupied by an aryl moiety of the ligand. This small cavity is formed also by residues Trp22, Met114, and Glu19, which, as discussed previously in this section, are crucial for substrate selectivity (11, 130).
In both GR and TR, a hydrogen bond occurs between the Cɛ1-H of the active-site histidine and a carbonyl of the bound substrate. This interaction stabilizes the protonated and catalytic histidine. Thus, whereas the enzyme/ligand interactions position the substrate for catalysis, the substrate primes the active-site histidine for catalysis. This mechanism, known as electronic induced fit, may also contribute to specificity because each enzyme can be optimally aligned for reaction only when its cognate substrate is bound in a catalytically suitable orientation (19).
Conjugation of Trypanothione by Glyoxalases
Methylglyoxal is a cytotoxic by-product of several metabolic pathways, glycolysis being the most relevant. In organisms with GSH-based redox systems, methylglyoxal reacts spontaneously with GSH to form a GSH hemithioacetal that is isomerized by lactoylglutathione lyase or glyoxalase I (GlxI) to form S-D-lactoylglutathione, an intermediate product that is subsequently hydrolyzed by hydroxyacylglutathione hydrolase or glyoxalase II (GlxII) to produce GSH and
GlxII, glyoxalase II; LiGlxII, Leishmania infantum glyoxalase II; LmGlxI, Leishmania major glyoxalase I; nd, not determined; ns, the compound is not a substrate of the enzyme; TbGlxII, Trypanosoma brucei glyoxalase II.
Strikingly, the biological relevance of the Glx system for each trypanosomatid species does not keep pace with our current understanding of parasite metabolism. On one hand, bloodstream African trypanosomes, which have a high glycolytic rate and display null to negligible levels of NADPH/NADH-dependent methylglyoxal reductase activities, lack a GlxI gene and are able to survive if the GlxII gene is knocked out (169). On the other hand, GlxI proved to be essential for Leishmania donovani (32), a parasite with a comparatively low rate of glucose consumption. Nonetheless, the efficient hydrolase activity displayed by GlxII of T. brucei with different thioesters of trypanothione (169) opens the possibility for functions of this protein unrelated to methylglyoxal metabolism.
Binding of trypanothione by GlxI and GlxII
GlxI from both prokaryotes and eukaryotes (including leishmanial GlxI) are homodimeric enzymes with each monomer having two βαβββ domains and the active site composed by residues from both subunits (Fig. 6A). Sequence and structure comparison revealed that Leishmania major (Lm)GlxI and TcGlxI are more similar to bacterial GlxI than to their mammalian counterparts (72, 166), mainly because they lack an insertion between β3 and β4 that is typical of eukaryotic forms. Similar to bacterial GlxI, trypanosomal GlxI presented a marked specificity for nickel over zinc as metal cofactor. Strikingly, LmGlxI differs from other GlxI in containing a shorter loop between β6 and β7 and a relatively long C-terminal helix, absent in EcGlxI, and shorter and differently oriented in HsGlxI. In GSH-dependent GlxI, residues from the loop between β6 and β7 form a roof over the active site that influences substrate accessibility and contributes to anchoring (via their backbone nitrogens) the glycyl-carboxylate of GSH. In LmGlxI, the shorter β6–β7 loop results in a more open and accessible active site cleft that favors accommodation of larger substrates such as the Gsp- or T(SH)2-hemithioacetal (8) (Fig. 6B, C).
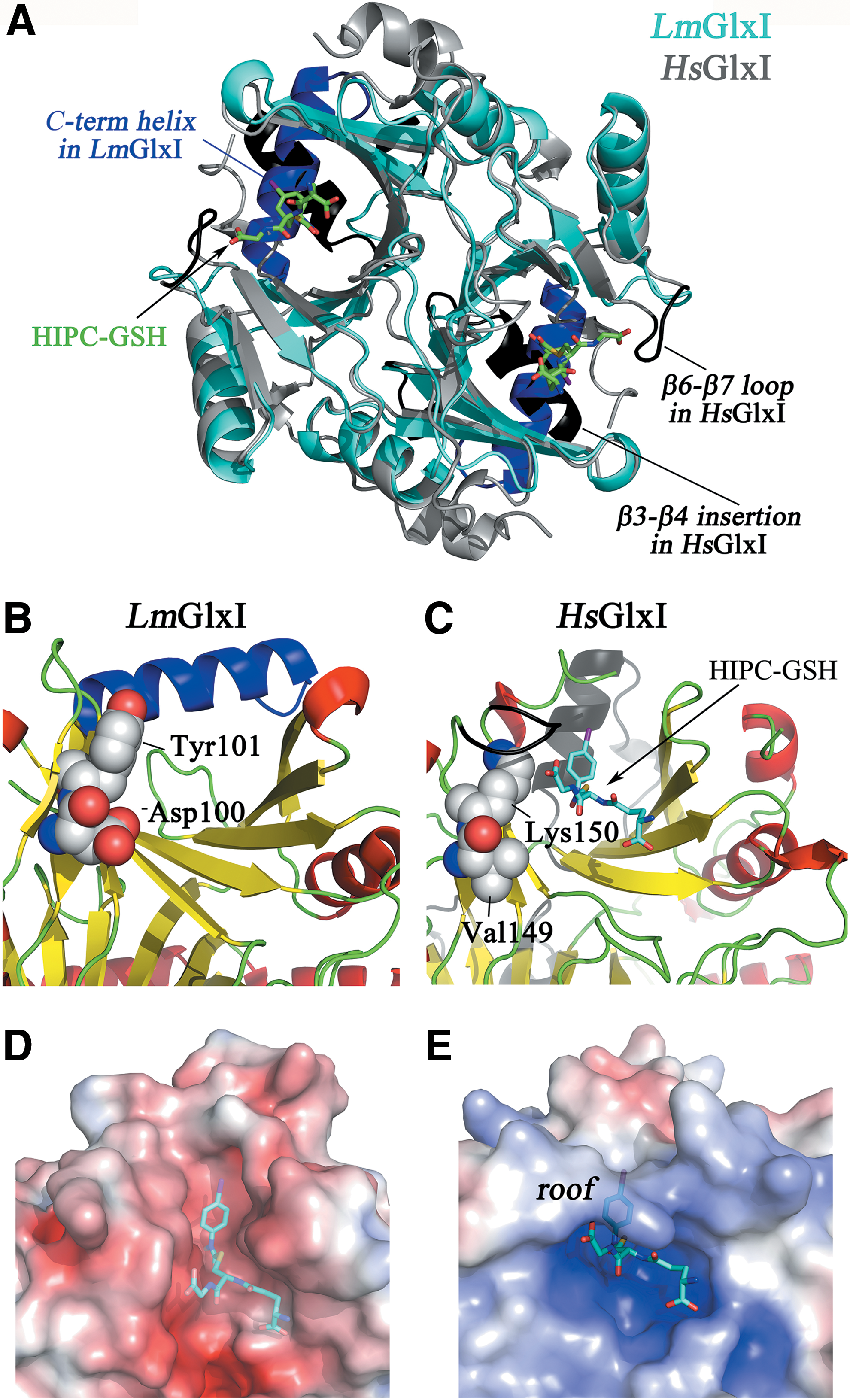
The structure of HsGlxI with bound S-(N-hydroxy-N-p-iodophenylcarbamoyl)glutathione, a transition-state analogue inhibitor, has been solved (26) and its superposition with LmGlxI allows to infer the position of the glycyl and γ-glutamate moieties of GSH, and the putative position of Sp (Fig. 6D, E) (8). Comparative analysis of the structural models shows that Val149 and Lys150 from HsGlxI are substituted in LmGlxI by Asp100 and Tyr101, both positioned at the C-terminal end of β6. The acidic side chain of Asp100 precludes interaction with a negatively charged group of the thiol substrate and probably explains the refractoriness of LmGlxI to bind and catalyze GSH hemithioacetal isomerization. Because HsGlx1 lacks a positive charged residue in the analogous position, it tolerates a neutral amide linkage in the substrate, explaining why the human enzyme is less discriminating against glycyl amides and esters (8). Moreover, Asp100 and Tyr101 are believed to favor the interaction with the positive/aliphatic polyamine linker, facilitating binding of Sp in a conformation where its N8 group points toward the methylglyoxal pocket (8). The fact that these amino acids are strictly conserved in the homologous proteins from trypanosomatids (72, 128) reinforces their role in the specificity of parasite GlxI toward hemithioacetal derivatives of Gsp or T(SH)2.
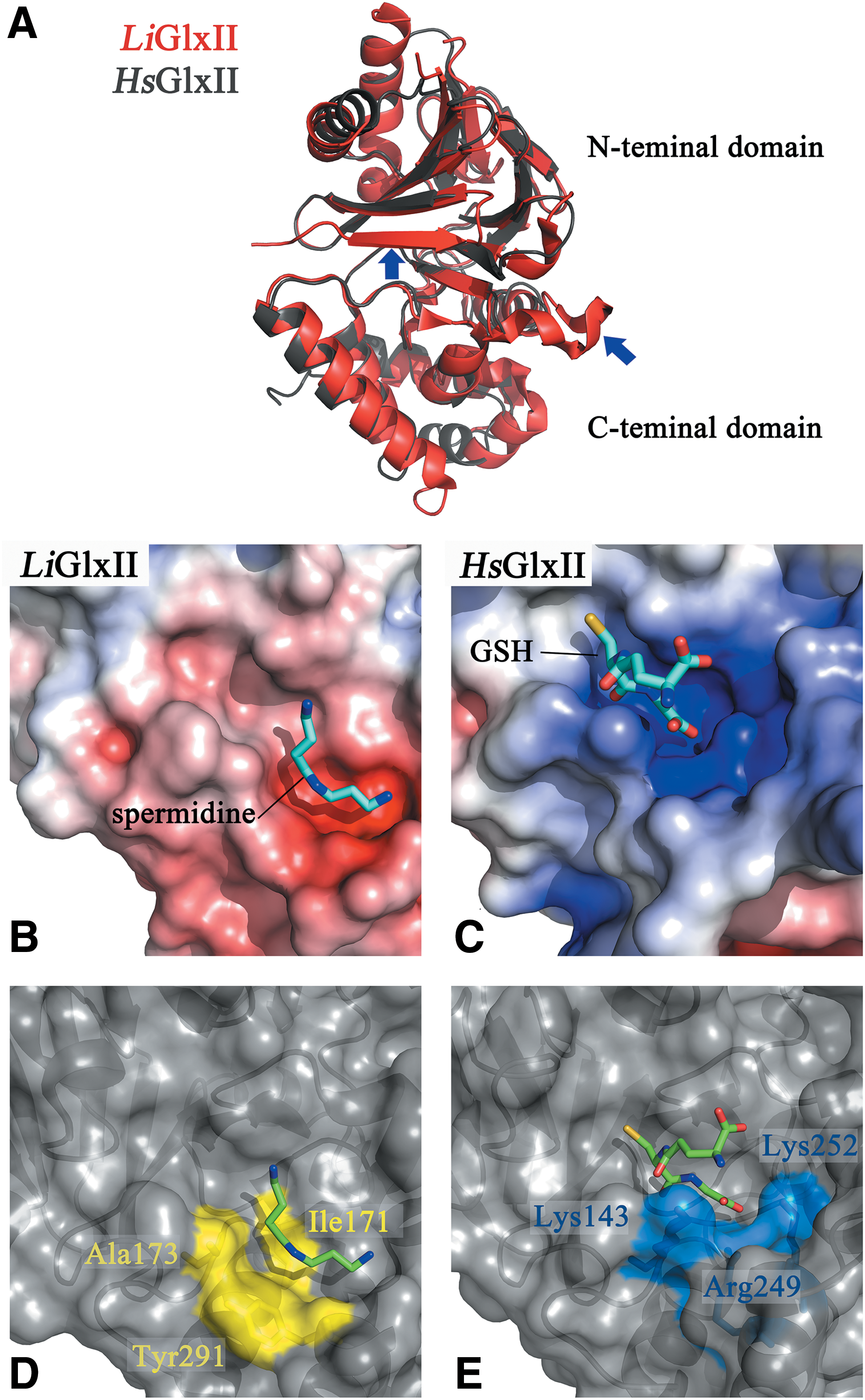
The overall structure of Leishmania infantum (Li)GlxII is very similar to the human enzyme, both being monomeric proteins containing two domains (13, 145) but with the first harboring an additional β-strand at the N-terminal and an extra α-helix at the C-terminal domain (Fig. 7A). Interestingly, in trypanosomatid GlxII, three highly conserved basic residues involved in binding the glycine carboxylate of the thioester in HsGlxII (Arg249, Lys143, and Lys252) (27) are substituted by alanine, tyrosine, and cysteine, respectively (83, 145). These changes contribute to reduce electrostatic repulsion with the positively and hydrophobic backbone of Sp (Fig. 7B, C). Consistently, a double Tyr→Arg and Cys→Lys mutant of LiGlxII conferred this chimeric enzyme the capability to use GSH thioester with a kinetic efficiency similar to that of the human enzyme (13). On the contrary, cocrystallization of LiGlxII with Sp bound close to the active site revealed that polyamine binding is stabilized by hydrophobic interactions with residues that are absent in the human enzyme, such as Ile171 and Ala173 (Cys141 and Lys143 in HsGlxII) (Fig. 7D, E).
TXN, a Trypanothione-Adapted Trx-like Protein
TXN is phylogenetically related to Trx, but forms a distinct molecular clade within this protein superfamily and is exclusive and essential to kinetoplastids (40, 71, 106, 119, 138). The phenotype associated with TXN downregulation is growth arrest and increased sensitivity toward oxidative stress (40). TXN sequences have a conserved WCPPCR motif in the active site and are ∼5 kDa larger than classical Trx due to the presence of several insertions. They reduce protein disulfides using a dithiol mechanism (Fig. 4D) (25, 140) and are poorly active toward GSH-protein mixed disulfides (30, 109). Unlike canonical Trx that is reduced by TrxR, TXN is reduced by T(SH)2 (25, 106, 109, 119). Thus, the oxidative half of TXN catalytic cycle is similar to Trx, while the reductive half resembles that of Grx, which are reduced by a low-molecular-weight thiol (LMWT).
The physicochemical factors that favor an efficient thiol/disulfide exchange between oxidized TXN and T(SH)2 under physiological conditions (Table 3) are (i) the standard redox potential (Eo′) of T(SH)2/TS2 couple (57) that facilitates electron flow from T(SH)2 to TXN (134); (ii) the nucleophilic cysteine of TXN (134) and T(SH)2 cysteines (115) have pKa values that match the cytosolic pH of parasites (63); and (iii) an entropic contribution due to the formation of an intramolecular disulfide in the substrate, a phenomenon reported to enhance thiol/disulfide exchange in five- to six-membered cyclic disulfides (156). Given the similar redox potentials of the TXN/trypanothione couple, the dithiol/disulfide ratio of trypanothione has been shown to exert an important control on TXN activity. For instance, in the presence of 1 mM T(SH)2, TXN activity has been shown to be strongly inhibited (50% inhibition) by 50 μM TS2 (48). Thus, the T(SH)2/TS2 ratio determines the flow of electrons funneled via TXN.
TXN from different trypanosomatid species.
Tested at a 1:2.2, T(SH)2:GSSG ratio.
Tested at a 1:10, T(SH)2:GSSG ratio.
With TrxR from Drosophila melanogaster or E. coli.
Tested against Crithidia fasciculata peroxiredoxin.
With Plasmodium falciparum TrxR.
With human TrxR.
Amino acid sequence of the peptide = SQLWCLSN.
2CGrx1, dithiolic glutaredoxin 1; 2CGrx2, dithiolic glutaredoxin 2; 2-ME, 2-mercapto ethanol; BSA, bovine serum albumin; GPx, glutathione-like tryparedoxin-dependent peroxidase; nd, not determined; Trx, thioredoxin; TrxR, thioredoxin reductase; TXN, tryparedoxin; TXNPx, tryparedoxin-dependent peroxidase.
Kinetic characterization of TXN from different species highlighted the preference of this enzyme for T(SH)2 over GSH (4, 71, 96, 106, 112, 119, 134, 151) (Table 3). Intriguingly, Gsp proved to be similarly efficient as T(SH)2 in delivering electrons to TXN for reduction of RnR (48) but not of a peroxidase (159). TXN from C. fasciculata (151) and T. brucei (134) was also reported to be a target of reduction by TrxR from E. coli and H. sapiens, respectively. The K M of human TrxR for TbTXN (43.5 μM) was similar to the value reported for its physiological substrate Trx. Although TrxR is absent in kinetoplastids, its presence in Euglenids suggests that redundant paths to keep TXN reduced might have coexisted until a T(SH)2-system was fully functional in a kinetoplastid ancestor.
Interestingly, substantial turnover rates were also observed for the reduction of GSSG catalyzed by TXN at the expense of T(SH)2 (106). At first glance, this suggests that TXN can contribute to sustain the pool of GSH in a parasite lineage devoid of GR activity. However, the high K M value for GSSG of CfTXN1 (∼1 mM) (71) indicates that this reaction will become relevant only when GSSG reaches millimolar concentrations. For comparison, TXN attains its maximum reaction velocity (V max) at a 1000-fold lower concentration of two of its cognate substrates, namely a peroxiredoxin (TXNPx K M = 2 μM for CfTXN1) (71) and RnR (K M = 4 μM for TbTXN) (48), than that required for GSSG. Instead, trypanosomal Grx can efficiently complement or take over the reduction of GSSG using T(SH)2 as reducing substrate (30, 39, 107).
At variance with parasite class I Grx (see section Class I Grxs, redox hubs between GSH and trypanothione metabolism), CfTXN2 (151) and TbTXN displayed marginal reductase activity on mixed disulfide of GSH and β-mercaptoethanol (106). Kinetic characterization using glutathionylated albumin as a model protein revealed a catalytic efficiency for TXN 500-fold lower than that of trypanosomal Grx (30) (Table 3). Although this rules out a physiological role for TXN in protein deglutathionylation, the recent identification of trypanothionylated proteins (164) poses the question whether this redoxin can act on these mixed disulfides.
The structure of TXN is constituted by four α-helices and five to seven β-strands, depending on the species. Despite the relatively low sequence identity and larger size of TXN with respect to Trx (i.e., canonical structure composed of three α-helices and four β-strands), two α-helices and four β-strands of TXN are well superimposed to the corresponding core elements of Trx. The main structural insertions in TXN are a β-hairpin at the N-terminus and an ∼20-residue stretch, including α2. Higly conserved residues in the active site of Trx have a very similar spatial organization in TXN (Fig. 8A) (1, 2, 60, 78, 88, 89, 98).

Comparison of the structures available for TXN revealed the following major differences: (i) in TbTXN, the orientation of the tryptophan preceding the active site restricts the access to this site, (ii) CfTXN1 nuclear magnetic resonance (NMR) structure indicates that the N- and C-termini and the region near the active site, as well as α1-β2 hairpin and α2, are relatively dynamic regions. Notably, the active site of CfTXN1 remained almost unperturbed by changes in its redox state, and only minor structural modifications make the sulfur atom of the nucleophilic cysteine slightly more accessible to partners (1). The two characteristic prolines in the active site of TXN do not show a particular conformation nor influence significantly the spatial positions of the adjacent cysteines, which almost overlap those of human Trx (Fig. 8A). However, permutation of TXN active site (CPPC) into a Trx- (CGPC) or Grx-like (CPYC) active site did not result in gain of Trx- or Grx-like activities but reduced its T(SH)2-dependent activity (151).
Although the structure of TXN provides a clue on how T(SH)2 binding to TXN can be affected by Pro43, there is not an obvious rationale on how this proline pair may modulate catalysis. In CfTXN2, a network of hydrogen bonds allows proton shuttling between uncharged internal residues (Tyr35, Thr48, Tyr81) via Ser37 to Cys44 favoring the dissociation of the Cys41 to its thiolate form (78). This activation mechanism differs from that proposed for Trx where buried charged groups contribute to thiol ionization (51). The structure of the catalytically inactive C44S CfTXN2 mutant, bound to Gsp via a disulfide with Cys41, has been solved and provided insights on the interaction with the substrate. Its binding is stabilized by electrostatic interaction between Arg129 and the γ-glutamate carboxylate of Gsp and by hydrogen bonds between the cysteinyl residue of Gsp and with the peptide bond connecting Ile110 with the conserved cis-proline (78). Hydrophobic contacts of Sp with Trp40 and Pro43 also contribute to substrate binding. Unfortunately, the structure of this complex does not provide information on the bound conformation of the Sp tail because Gsp harbors an additional positive charge at its terminal amino group that prevents its correct attachment to TXN. Although several reports emphasize the lower catalytic efficiency of TXN with Gsp as reducing substrate (159, 163), the K M of TXN for Gsp has not yet been assessed to confirm whether catalysis and/or binding are impaired.
Modeling of T(SH)2 binding to the TXN active site suggested electrostatic interaction of the guanidino group of Arg129 with the carboxyl group of N 1-bound glutathionyl moiety and between the secondary amino group of Sp and the carboxylate of Glu73 (78). These two residues are strictly conserved in TXN but not in Trx. The model also proposes that Arg45 side chain interacts with the carboxylate of N 8-bound GSH, hence facilitating the approach of the second thiol of T(SH)2 into a position where it can attack the mixed disulfide between Cys41 and the substrate cysteine (Fig. 8B). Several experimental evidence support this dynamic binding model: (i) the weak competitive inhibition exerted by bis(ophthalmyl)spermidine on TXN is fully abrogated if its nitrogen at position 4 of the polyamine chain is replaced by a carbon (98) and (ii) charge inversion by introducing the mutations Glu73Arg, Arg129Asp, and Arg42Asp rendered CfTXN2 almost redox silent (78, 151).
Trx of Trypanosomatids, an Orphan Redoxin
Despite trypanosomatids being devoid of TrxR, they encode a single copy Trx gene bearing more than ∼70% sequence identity with canonical Trx. Trx is dispensable for both life stages of African trypanosomes, as demonstrated by the lack of phenotype of Trx-knockout parasites (141). Trypanosomatids also encode a second Trx sequence that has an atypical WCEPC active site and is fused to a class II Grx domain (see section 1CGrx1, a Class II Grx with Lineage-Specific Features). TbTrx has a redox potential very similar to that of classical Trx (−270 mV) and its cysteines have pKa values close to that of physiological media (63, 142) (Table 3). TbTrx accepts T(SH)2, but not Gsp, as an electron donor, although with a remarkable lower efficiency than that for its preferred reducing partner: TrxR (135, 142). Nonetheless, compared to Trx from another unicellular parasite (90), the trypanosomal protein is a poor substrate for TrxR (135, 142) (Table 3).
In agreement with the substrate selectivity of TR, TbTrx was not a substrate for this parasite-specific reductase (135). The rate of TbTrx oxidation by TS2 and GSSG was 7- and 1.6-fold, respectively, higher than its reduction by the reduced LMWT (142). However, the relatively high concentration of reduced T(SH)2 (0.23–1.2 mM) and the comparatively low intracellular concentration of Trx (estimated at 50 nM for T. brucei) suggest that Trx is maintained in its reduced state under physiological conditions, avoiding thus a futile redox cycle with T(SH)2 (142). Compared to TXN and other Trx, the trypanosomal Trx does not appear to have evolved as an optimal reducing partner of RnR. This is, in part, explained by the almost one order of magnitude higher apparent K M of TbRnR for TbTrx (K M = 29 μM) with respect to TXN. In contrast, Trx proved to be a highly efficient reductant of a 2-Cys peroxiredoxin (135, 142) but not of a glutathione-like tryparedoxin-dependent peroxidase (GPx) (76) (Table 3).
Overall, the crystal structure of reduced TbTrx does not show significant differences with that of homologous proteins from other eukaryotes (64), except for the active site Trp30. In most Trx, this residue forms a flat surface but in TbTrx is flipped out toward the exterior, with an orientation similar to that observed for the spinach Trx-f (28). Similar structural plasticity of the active site tryptophan has been observed for TXN, as described above, and for yeast protein disulfide isomerase, PDI (161). This indicates that Trp30 can adopt different orientations to optimize the interaction with specific redox partners. Like classical Trx, TbTrx retains hydrophobicity at the area interacting with other proteins but at variance with them, basic residues populate the outer rim of this region, which confers the trypanosomal protein with an unusual positive surface potential (pI = 8.5 for TbTrx vs. pI = 5.4 for LmTrx or pI = 4.5–5.0 for most Trx).
An additional distinctive feature of trypanosomatid Trx is the presence of a Trp (TbTrx and TcTrx) or a Cys (LmTrx) at position 25, which in most Trx is occupied by a strictly conserved aspartate (131, 135), that lowers the pKa of the nucleophilic cysteine and contributes to proton shuttling during catalysis (34, 80, 81). The kinetic data obtained for TbTrx clearly show that an acidic residue at this position is not essential for catalysis. In fact, the structure of TbTrx shows that the indole nitrogen of Trp25 is connected to Cys34 via a hydrogen-bonded water molecule, resembling the hydrogen-bonded network established by aspartate and cysteine in canonical Trx (34). However, although major structural differences are not evident with respect to the active site region of other Trx, the lower electrostatic potential of Trp probably renders Cys less polarized and reactive, which may explain the poor catalytic efficiency of TbTrx toward certain targets.
Grxs, Redoxins That Use GSH as Cofactor
Grxs are small and ubiquitous proteins that represent the most diverse group from the Trx-fold family and, despite its relatively low amino acid conservation (16–37% identity), share a highly conserved fold with all the members of this superfamily (backbone root-mean-square deviation in the range 1.3–2.3 Å) (9, 153). Grxs are divided into several classes based on homology, with class I and class II being the most represented (Fig. 9). Class I comprises single-domain Grxs that harbor one or two cysteine residues in their active sites and catalyze thiol/disulfide exchange reactions between protein cysteines and GSH (i.e., glutathionylation/deglutathionylation, Fig. 4E) (68, 105). The catalytic mechanism used by Grx has been thoroughly studied (14, 15, 45, 67, 68, 104, 110). Class II Grxs are higly diverse in terms of sequence and domain arrangements. Most of them harbor a CGFS active site, are not engaged in the catalysis of thiol/disulfide exchanges but participate in iron/sulfur cluster (FeS) biogenesis facilitating the transfer of FeS to target proteins using GSH as a nonredox cofactor (12, 103, 137, 139).

Class I Grxs, redox hubs between GSH and trypanothione metabolism
Most trypanosomatids harbor genes for two class I Grxs. † Dithiolic glutaredoxin 1 (2CGrx1) has high similarity with typical class I Grxs [35–42% sequence identity and identical active site motif CP(Y/F)C]. Dithiolic glutaredoxin 2 (2CGrx2) possesses an overall low sequence identity with 2CGrx1 (20–25%) and an atypical C(Q/E)(F/Y)C (CQFS in leishmanial proteins) active site motif. Interestingly, T. cruzi lacks sequences for 2CGrx1 but contains two copies encoding for a 2CGrx2. The high level of identitity among trypanosomal 2CGrx2 sequences (∼80%) and the absence of orthologs outside the Kinetoplastidea (30, 39) suggest that this gene evolved exclusively in this lineage. At a mechanistic level, parasite dithiolic glutaredoxin (2CGrx) behaves as classical class I Grxs that use the N-terminal active site Cys (monothiol mechanism) to drive thiol/disulfide exchange reactions with their substrates (109) (Table 3). In African trypanosomes, 2CGrx1 is a cytosolic protein while 2CGrx2 localizes in the parasite mitochondrion (30). In T. cruzi, 2CGrx2 displayed a predominant cytosolic localization (109). Class I Grx seems to play from general to specific roles in the biology of trypanosomatids. T. brucei has been shown to totally dispense with class I Grx, although the cytosolic isoform contributes to protein deglutathionylation and participates in a pathway that modulates parasite's thermotolerance (30, 117). For T. cruzi, overexpression of 2CGrx2 provided resistance toward oxidative damage and promoted the growth of intracellular parasites but it had a proapoptotic effect in noninfective parasites (109).
As highlighted above, trypanosomatids lack GR and convert a large amount of GSH into T(SH)2 (93), raising the question about the physiological reductant of 2CGrx and GSSG in these organisms. T(SH)2 proved 100- to 1000-fold more efficient than GSH in reducing trypanosomal 2CGrx (30) (Table 3). Given that GSH and T(SH)2 concentrations are on the same order of magnitude (high μM range) (93), the kinetic data clearly support a major role for trypanothione in maintaining the pool of reduced 2CGrx (30). Interestingly, the kinetic constants obtained for the reduction of 2CGrx by T(SH)2 are comparable to those reported for the reaction between this dithiol and oxidized TXN (106, 107, 119). In principle, this would suggest a competition between both oxidoreductases for the reducing substrate. Considering that the cytosolic concentration of TXN in the infective form of T. brucei (40) is ≥25-fold higher than the 2 μM estimated for 2CGrx1 (30), this competition will be only possible if the K M of 2CGrx1 for T(SH)2 (not yet determined) is in the low to submicromolar range (Table 3).
Trypanosomatid 2CGrx1 and, to a minor extent, 2CGrx2 displayed a significant T(SH)2-dependent GSSG reductase activity (Table 3). For Li2CGrx1, the forward reaction was ∼80-fold faster than the opposite reaction, whereas for Tc2CGrx2, it exceeded by two orders of magnitude that mediated by Gsp or the reverse reaction (k′ for both reactions = 2–4 × 102 M−1·s−1) (109). Under almost identical assay conditions, TcTXN1 displayed second-order rate constants for each of these reactions similar to those of Tc2CGrx2 (109). The estimated intracellular concentration of Tc2CGrx2 and Li2CGrx1, and K M values of these redoxins for T(SH)2 are missing to estimate the contribution to GSH/GSSG homeostasis of Grx versus TXN. However, the data above suggest that, at least in T. cruzi, GR function can be fulfilled by the joint action of both T(SH)2-dependent redoxins.
Additional studies showed that trypanosomatid 2CGrxs, in particular 2CGrx1, are able to deglutathionylate protein and nonprotein substrates (30) with an efficiency comparable to that of human glutaredoxins (67, 86), but they cannot compete with the protein disulfide reductase activity of TXN (Table 3). Interestingly, while cytosolic 2CGrx1 has been shown to contribute with about 50% of the deglutathionylase activity in African trypanosomes (117), none of the 2CGrxs from T. brucei catalyzed the opposite reaction, protein glutathionylation (30). Given the occurrence of trypanothionylated proteins (164), it deems necessary to investigate the precise contribution of TryS-amidase, 2CGrx, and TXN in the recovery of the unmodified form of the proteins.
Another interesting feature of Tb2CGrx1, not mimicked by Tb2CGrx2, is its ability to bind FeS using GSH or even T(SH)2 as ligands, which leads to protein dimerization (30, 108). The lack of activity displayed by holo-Tb2CGrx1 in the hydroxyethyl disulfide (HED) assay [reduction of a heterodisulfide between β-mercaptoethanol and GSH (15)] and the incapacity of the oxidized protein to bind the inorganic complex clearly demonstrate an involvement of the active site Cys (most probably the N-terminal Cys) in iron coordination. Moreover, the reversibility of this protein/ligand interaction suggests that the cluster regulates the oxidoreductase activity of Tb2CGrx1 (30).
We have recently solved the NMR-structure of Tb2CGrx1 (150), and preliminary analysis shows an almost identical folding with human Grx2 (a class I Grx that binds FeS) and a strict conservation of residues shaping the binding groove of GSH (Fig. 10A–C). The most outstanding feature of trypanosomatid 2CGrx1 is the presence of a Trp at position 18 (30) that substitutes an otherwise conserved Lys—or to a lesser extent, an Arg—that in homologues from other organisms has been shown to interact with the glycine carboxylate of GSH. Because T(SH)2 lacks the free carboxylate of both glycines, an hydrophobic residue at this position is more suited to interact with the aliphatic chain of Sp or the Sp-GSH amide bond (see section: Enzymatic Recycling of Trypanothione by TR and Substrate Selectivity). Interestingly, also 2CGrx2 contains a less bulky yet hydrophobic residue (alanine) at this position (30). Nevertheless, the chemical nature of the residue occupying this position appears not to be fully determinant for substrate discrimination since EcGrx1, which has an Arg, accepts T(SH)2 as substrate (30).
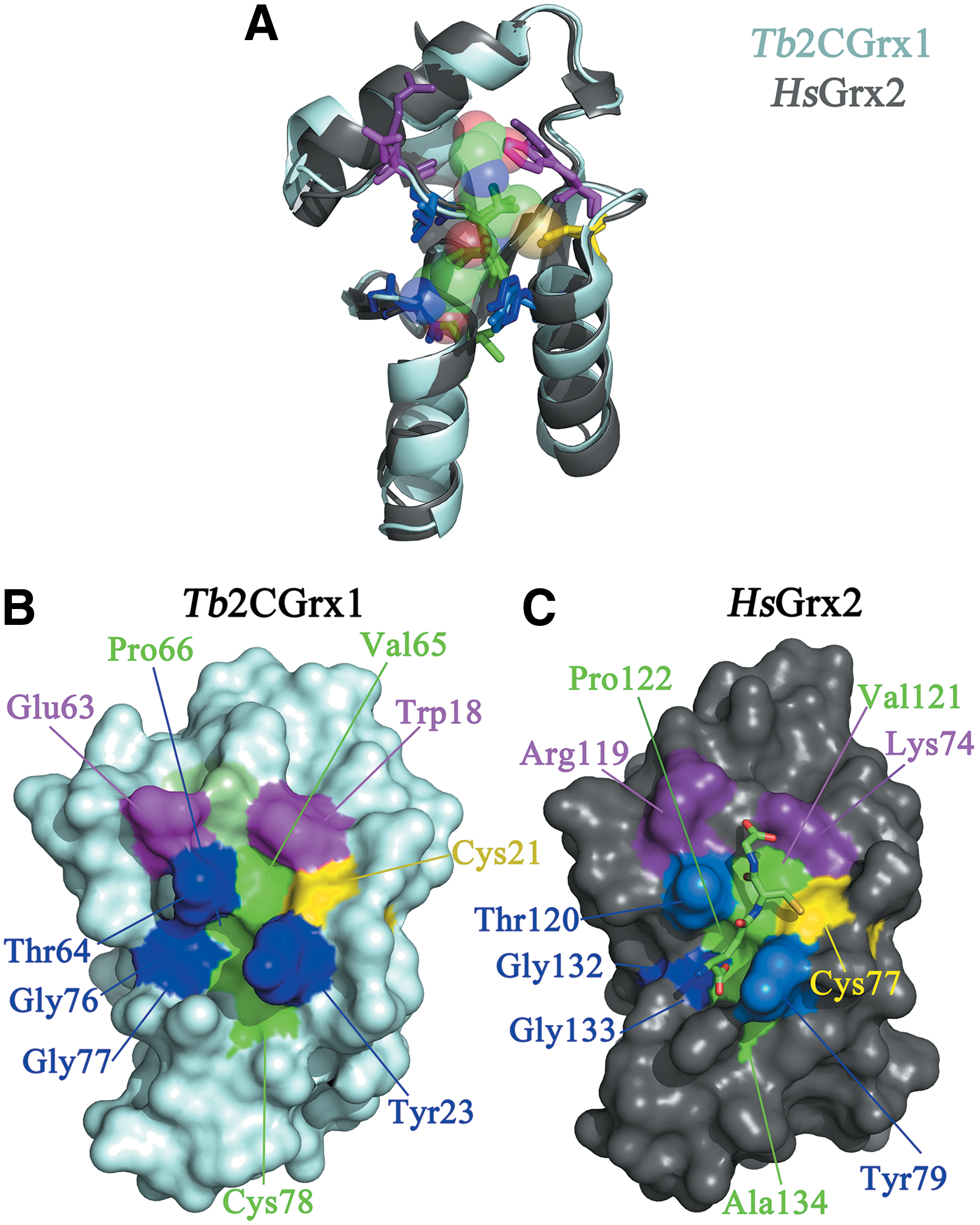
Another interesting characteristic of trypanosomatid 2CGrx1 is the invariable presence of an acidic residue (Glu63 in Tb2CGrx1) preceding the highly conserved TVP motif (or cis-Pro loop), which can modulate redox activity and interaction of Grx with substrates (52, 136). In most 2CGrx1 from other organisms, positively charged amino acids occupy this position, being polar (Thr or Ser) and nonpolar (Gly) substitutions less represented (Fig. 9). Assuming the anchoring of GSH as determined for HsGrx2, Glu63 is in a suitable position to interact with the secondary NH of T(SH)2. Interestingly, Tb2CGrx2, in which this glutamate is substituted by an histidine, displayed a 50-fold lower activity than Tb2CGrx1 in catalyzing the reduction of GSSG by T(SH)2 (30) (Table 3).
1CGrx1, a class II Grx with lineage-specific features
From the three class II Grxs encoded by trypanosomatids, two are single-domain mitochondrial proteins (monothiolic glutaredoxin 1 [1CGrx1] and monothiolic glutaredoxin 2 [1CGrx2]) and the third one is a fusion of a Grx with an N-terminal Trx (monothiolic glutaredoxin 3 [1CGrx3]). While the Grx domains show a relatively low sequence identity between proteins (i.e., 1CGrx1 vs. 1CGrx2 vs. 1CGrx3), each protein is highly conserved among trypanosomatid species (>60% amino acid identity), indicating a common ancestry for these genes. At variance with class II Grx from other organisms, the parasite proteins have a highly variable active site motif C(G>A>R)(F/Y)(T/S) (39). 1CGrx2 is the closest homologue of the typical monodomain class II Grx of prokaryotes and eukaryotes, whereas 1CGrx1 appears to be a lineage-specific Grx derived from 1CGrx2 (39, 108). 1CGrx1 contains an ∼30-mer N-terminal extension, highly conserved within the kinetoplastid genus and lacks two strictly conserved cysteine residues present outside the active site (39, 108). The higher sequence identity of the Grx domain of 1CGrx3 with 1CGrx2 (39%) rather than with 1CGrx1 (26%) suggests that the first likely evolved from a fusion of a 1CGrx2 domain with a Trx domain.
Tb1CGrx1 lacks redox activity (59) and its indispensability in vivo was associated to its capacity to bind an iron-sulfur cluster using the active site cysteine (C104) and different LMWT (108). In vitro, Tb1CGrx1 was a target of glutathionylation or Gsp-lation at its C-terminal cysteine (C181) (41, 59). Despite its high abundance (41), Tb1CGrx1 was not identified among the S-thiolated proteins of African trypanosomes (164), which rules out any biological significance to the modification observed in vitro.
Almost all class II Grxs are monomeric proteins that dimerize on FeS binding and, despite early contradictory results (41, 59) ascribed to the kinetoplastid-specific N-terminal tail of the protein (108), Tb1CGrx1 has been shown not to be an exception (154). Structural analysis of the globular Grx domain of Tb1CGrx1 [deletion mutant lacking the first N-terminal 76 residues (Δ76-)1CGrx1 (108)] shows that most residues involved in GSH binding and iron-sulfur assembly in other monothiolic glutaredoxins (1CGrxs; e.g., Lys96, Cys104, and Asp159) are conserved and have a spatial orientation such as that observed in the holo-form of two 1CGrx (HsGrx5 and EcGrx4) with noncovalently bound GSH (61, 87).
NMR titration experiments revealed that the noncovalent interaction between GSH or T(SH)2 with Δ76- or full-length 1CGrx1 is almost negligible at the estimated physiological concentrations of the ligands (108), supporting the lack of selectivity of the protein for LMWT. This differs from data obtained under similar conditions for HsGrx5 (12) and can be explained by the highly dynamic conformation of the conserved cis-Pro loop that modulates the accessibility of the ligands to the Tb1CGrx1 binding pocket [Sturlese et al., unpublished; (108)]. For EcGrx3 (class I Grx), it has been demonstrated that mutation of a residue located in the helix preceding the cis-Pro loop and with side chain pointing toward the cis-Pro does not affect protein folding but enhances the intrinsic motion of this element allowing for increased specificity toward larger substrates (52, 136).
Another unique feature of the trypanosomal protein is the presence of a negatively charged surface on the opposite side of the active site. Whether this region and the N-terminal tail of 1CGrx1 are involved in interactions with partner proteins is unknown, but the high conservation of these elements among trypanosomatid homologues and the partial capacity of Tb1CGrx1 to surrogate the function of the yeast ortholog (59) support the idea that they are lineage-specific adaptations.
Thiol Transferases, Proteins with Functions Far Beyond Electrophile Detoxification
Thiol transferases catalyze the conjugation of electrophilic molecules to LMWT and, by doing so, neutralize highly reactive and cytotoxic groups present in endobiotics (e.g., lipoperoxides, aldehydes) and xenobiotics (e.g., heavy metals, drugs).
Trypanosomatids were reported to contain thiol transferase activity toward a variety of electrophiles (Fig. 4G). For Leishmania and Crithidia, this activity was associated with the ribosomal eukaryotic elongation factor 1B (eEF1B) and depended on T(SH)2 or Gsp but not on GSH (e.g., catalytic constant [k cat]/K M for 1-choro-2,4-dinitrobenzene (CDNB)-conjugation with Gsp, T(SH)2, and GSH was 0.6, 1.2, and 0.003 × 106 M−1·s−1, respectively, for CfeEF1B complex) (165, 167). The LmeEF1B complex was also shown to decompose lipoperoxides at the expense of T(SH)2 with high efficiency (k cat/K M = 3 × 103 M−1·s−1 for cumene hydroperoxide), compared to other conjugation/addition reactions, but not high enough to compete with GPx-like trypanothione-dependent peroxidase (k cat/K M = 106−107 M−1·s−1 (47, 77). However, the higher K M (mM) for and refractoriness to inactivation by peroxides of eEF1B (165, 167) with respect to classical peroxidases, suggests that the first might protect membranes under heavy oxidative stress. The structural determinants conferring selectivity for T(SH)2 to kinetoplastid eEF1Bγ remain unknown as well as the relevance of this protein for parasite survival.
Glutathione S-transferases (GST)-like proteins with significant similarity to ω and τ classes were identified in T. cruzi and L. infantum (66). The T. cruzi protein (Trypanosoma cruzi protein of 52 kDa [Tc52]) lacked GST and trypanohione-S-transferase activity toward different electrophiles but, surprisingly, presented a remarkable “T(SH)2:GSSG thiol transferase” activity (k cat/K M = 4 × 105 M−1·s−1 for GSSG reduction) (114, 116). The structural organization of Tc52 suggests that, upon gene duplication, each domain of the protein evolved separate specificities for LMWT (114).
“Thiol-dependent reductase 1” (TDR1) is the leishmanial homologue of Tc52 (66). Despite the high amino acid identity between both proteins (47%), they differ in their selectivity for the oxidized targets. TDR1 has GSH-dependent reductase activity toward dehydroascorbate and HED but a comparable modest GST activity and proved inactive in the reduction of protein disulfides with GSH or T(SH)2 (44, 66). Similarly to Tc52, LiTDR1 presented T(SH)2-dependent GSSG reductase activity (66) (Fig. 4). LiTDR1 also showed deglutathionylase activity (k cat/K M = 1.5 × 104 M−1·s−1). Importantly, LiTDR1 has been shown to catalyze the GSH-dependent reduction of a clinically used antimony (Sb)(V)-drug into its metalloid active Sb(III) species (44).
LiTDR1 is a homo-trimer, where the two GST domains of each subunit consist of N-terminal Grx-like and C-terminal α-helical subdomains. The Grx subdomains contain each a dithiol CPFC or a monothiol CPFV active site motif, with several residues involved in GSH binding or catalysis being conserved. However, they differ in the binding region of the glycyl moiety of GSH, where a glutamine (position 267) replaces an arginine (position 39) in the C-terminal Grx-like domain. Similar to classical GST, the ligand binding domain of TDR1 is located near (∼10 Å) the GSH-binding site but at variance with them it is smaller, partially occluded, and with a different shape and charge. The nonhydrophobic and positively charged nature of the TDR1 pocket favor the interaction with acidic substrates, which may, in part, explain the lower deglutahionylating activity when a noncharged, half-hydrophobic, half-polar glutathionylated SQLWCLSN peptide is used as substrate.
A common feature of the Grx-like domain of TDR1 and Tc52 with that of trypanosomatid class I Grx is the substitution of a highly conserved basic residue (K34 in human Grx), involved in recognition of the glycyl carboxylate of GSH in classical Grx, by noncharged ones (Asp/Ser or Asn in LiTDR1 and Tc52, and Trp or Ala in trypanosomatid's 2CGrx). This may partly explain the capability of TDR1 and Tc52 to utilize T(SH)2 as substrate. On the contrary, the different efficiencies of these proteins to reduce protein disulfides were ascribed to an amino acid change (long branched Arg39 of TDR1 replaced by a Gly in Tc52) that likely affects the accessibility to the active site of TDR1.
For its crucial role in modulation of mammalian host immune response, Tc52 has been rated as a virulence factor and a vaccine candidate for T. cruzi (69, 111). Specific peptide sequences present in Tc52 but not redox-mediated mechanisms, as originally proposed (122), are responsible for its immunoactivity (21). LiTDR1 is immunologically silent (144) but, as noted above, it likely participates in the reductive activation of anti-leishmanial prodrugs (44).
Concluding Remarks
From the analysis presented above, it emerges that on the acquisition of polyamine-GSH synthesizing activity by the incorporation of a bacterial GspS-encoding gene, a kinetoplastid ancestor began to develop a new redox system prompted by the higher reactivity of the novel thiol(s). Given the net positive charge of Gsp/T(SH)2 that favors the interaction with negatively charged bases, an initial selective advantage provided by these polyamine thiols may have been providing protection against radical damage of DNA. The occurrence of genes encoding for components of classical redox systems in kinetoplastids (Trx, Grx) and phylogenetically related organisms (TrxR and GR in E. gracilis) that, in some cases, conserve selectivity for their canonical substrates suggest that refurbishing of the redox system likely occurred in a gradual manner. Nonetheless, the emergence of TryS from GspS, followed by that of TR from GR and TXN from Trx, were likely key steps toward rewiring LMWT pathways in trypanosomatids. The performance of the T(SH)2-dependent system was so successful in meeting the high metabolic demands of trypanosomatids for reducing power that several important genes (GR, TrxR, and catalase) were eliminated during genomic scale-down. In this respect, the T(SH)2:GSSG reductase activity of Grx, thiol-transferases, and TXN provided an efficient way to maintain the pool of GSH in the absence of GR, and the pair TXN/T(SH)2 could fully take over the TrxR/Trx functions.
The physicochemical properties of Gsp and T(SH)2 shaped subtle but specific modifications on the interacting proteins to achieve selectivity. The charge seems to be the major discriminating factor but an increased hydrophobicity at the ligand binding pocket also enhanced the affinity for the larger positive/aliphatic glycyl-spermidine amide (TR, TXN, GlxII, TDR1). Thus, the ligand binding site of trypanothione-dependent proteins contains hydrophobic residues (often a Trp) that substitute the otherwise highly conserved basic amino acids occupying similar positions in the homologue/ortholog proteins that rely on GSH as ligand. Substrate selectivity was also driven by a specific steric demand of the polyaminic ligand to accommodate in the interacting pocket. This, in turn, required structural rearrangements that involved small variation of the domain orientation in multidomain proteins (TR, GlxI) and/or by insertions/deletions or even single mutations in specific regions (GlxI/II, TryS). The intrinsic flexibility of trypanothione also contributes to its promiscuity to fit into different active sites and/or induce conformational changes in the enzymes to facilitate binding. These structural adaptations are more evident when they result in a mutually exclusive specificity for GSH or T(SH)2, as in the case of TR or Glx. Structural adaptations are more elusive when the level of selectivity is low, as observed for Grx, which can react with both LMWT.
Such uniqueness of the redox system of trypanosomatids is at the same time their Achilles heel for the development of highly specific therapeutic approaches against these pathogens. In this regard, the single components of the triad TryS/TR/TXN are attractive drug targets, given their indispensability for parasite survival and the absence of homologue sequences in mammals.
Footnotes
Acknowledgments
M.A.C., M.Be., and G.S. acknowledge the support of ICGEB grant CRP/URU14-01. M.A.C. also acknowledges the financial support from FOCEM (MERCOSUR Structural Convergence Fund [COF 03/11]). M.Be. and M.S. acknowledge the Università degli Studi di Padova (UNIPD) for the support (PRAT project, code CPDA137397/13). Financial support by Access to Research Infrastructures activity in the Seventh Framework Programme of the EC (Project No. 261863, Bio-NMR) is gratefully acknowledged for providing access to NMR spectrometers. B.M. acknowledges the international cooperation program UNIPD, “Coimbra Group Scholarship Programme for Young Professors and Researchers from Latin American Universities.” B.M. and M.A.C. thank the uninterested collaborations of Dr. Michael W. Gray (Dalhousie University, Halifax, Canada), Dr. Ellis C. O'Neill (Oxford University, Oxford, United Kingdom), Dr. Mark C. Field (University of Cambridge, Cambridge, United Kingdom), Dr. Gertraud Burger (University of Montreal, Montreal, Canada), and Dr. Julius Lukes (Biology Centre, České Budějovice, Czech Republic) for providing unpublished sequences from Euglenozoa organisms.