
Abstract
Aims:
5-Lipoxygenase (5-LO) is the key enzyme of leukotriene (LT) biosynthesis and is critically involved in a number of inflammatory diseases such as arthritis, gout, bronchial asthma, atherosclerosis, and cancer. Because 5-LO contains critical nucleophilic amino acids, which are sensitive to electrophilic modifications, we determined the consequences of a drug-mediated intracellular release of nitric oxide (NO) on 5-LO product formation by human granulocytes and on 5-LO-dependent pulmonary inflammation in vivo.
Results:
Clinically relevant concentrations of NO-releasing nonsteroidal anti-inflammatory drugs and other agents releasing NO intracellularly suppress 5-LO product synthesis in isolated human granulocytes via direct S-nitrosylation of 5-LO at the catalytically important cysteines 416 and 418. Furthermore, suppression of 5-LO product formation was observed in ionophore-stimulated human whole blood and in an animal model of pulmonary inflammation.
Innovation:
Here, we report for the first time that drugs releasing NO intracellularly are efficient 5-LO inhibitors in vitro and in vivo at least equivalent to approved 5-LO inhibitors.
Conclusion:
Our findings provide a novel mechanistic strategy for the development of a new class of drugs suppressing LT biosynthesis by site-directed nitrosylation. The results may also help to better understand the well-recognized anti-inflammatory clinically relevant actions of NO-releasing drugs. Furthermore, our study describes in detail a novel molecular mode of action of NO.
Rebound Track:
This work was rejected during standard peer review and rescued by Rebound Peer Review (Antioxid Redox Signal 16: 293–296, 2012) with the following serving as open reviewers: Angel Lanas, Hartmut Kühn, Joan Clària, Orina Belton. Antioxid. Redox Signal. 28, 1265–1285.
Introduction
5-
Considerable efforts failed in the past to develop safe and efficient 5-lipoxygenase (5-LO) inhibitors although the 5-LO pathway is of high pathophysiological relevance. Here, we report for the first time that clinically relevant concentrations of nitric oxide-nonsteroidal anti-inflammatory drugs (NO-NSAIDs) lead to direct suppression of 5-LO enzyme activity in vitro and in vivo by targeting cysteines 416 and 418. Our finding provides a novel mechanistic strategy for the development of a new class of direct 5-LO inhibitors. Furthermore, the present results may help to better understand the well-recognized clinically relevant anti-inflammatory actions of nitric oxide, NO-NSAIDs, and other NO-releasing drugs.
We have shown that two approved nonsteroidal anti-inflammatory drugs (NSAIDs), celecoxib and sulindac sulfide, are also able to suppress 5-LO activity at clinically relevant concentrations (40, 63). In addition, we recently found that electrophilic nitro-fatty acids (NFAs) generated endogenously in inflamed tissues are direct 5-LO inhibitors with an in vivo efficacy superior to that of zileuton (4). NFAs were able to irreversibly nitroalkylate 5-LO on the solvent-accessible surface of the enzyme at catalytically relevant cysteine residues (Cys416 and Cys418) (4, 30). In recent, successfully completed phase 1 trials, the pharmacokinetics and safety of NFAs were evaluated following oral and intravenous administration to humans (e.g., ClinicalTrials.gov Identifier: NCT02460146). Subsequent to these studies, the same company is preparing phase 2 trials, with a focus on focal segmental glomerulosclerosis and pulmonary arterial hypertension (1).
Nitric oxide (NO), generated by exposure of macrophages to lipopolysaccharide (LPS), inhibits 5-LO product formation by an as yet unclear mechanism (8, 18). It was demonstrated, however, that suppression of LT synthesis by electrophilic NO might be associated with covalent nitrosylation of different tyrosine and cysteine residues, although these modifications could only be detected in a cell-free system (18) and could not be assigned to specific amino acids. Later on, Coffey et al. found that NO is also able to suppress cellular 5-LO activity via activation of the cytosolic guanylyl cyclase (cGC) and protein kinase G pathway (17). Thus, due to its electrophilic nature and cysteine reactivity, we hypothesized that pharmacologically donated NO may lead to suppression of 5-LO product formation in leukocytes via molecular mechanisms potentially similar to NFAs.
This work was rejected during standard peer review and rescued by Rebound Peer Review (Antioxid Redox Signal 16: 293–296, 2012) with the following serving as open reviewers: Angel Lanas, Hartmut Kühn, Joan Clària, and Orina Belton. The comments by these reviewers supporting the rescue are listed below:
Nitric-oxide-donating nonsteroidal anti-inflammatory drugs (NO-NSAIDs) are anti-inflammatory and antitumorigenic compounds that are able to pharmacologically release NO. These drugs form a class of NSAID derivatives possessing an NO-donor moiety covalently linked, either directly or through an aliphatic/aromatic linker, to the parent NSAID moiety (14, 56). NO-NSAIDs were initially developed to reduce NSAID-induced gastrointestinal toxicity, as NSAIDs also decrease the biosynthesis of protective prostaglandins in the stomach, leading to gastrointestinal bleeding and ulceration events (41).
Several clinical trials have been performed with naproxcinod, the NO-donating derivative of naproxen. This compound received an European Orphan Drug Designation (ODD) in 2013, followed by an ODD from the United States Food and Drug Administration (FDA) in 2015, both for the treatment of Duchenne muscular dystrophy (2).
Because NO is a critical mediator of gastrointestinal mucosal defense (37, 71) and possesses protective properties within gastric mucosa similar to that of prostaglandins (36), the pharmacological donation of NO should counteract NSAID-mediated gastropathy. Subsequent studies have found that NO-NSAIDs also possess anticancer activities against cultured tumor cells and are better at impairing tumor growth in animal models than their respective classical NSAIDs (61, 65, 72). However, the molecular mechanisms underlying these anticarcinogenic effects remain unclear. Here, we report that clinically relevant concentrations of NO-NSAIDs suppress LT formation in vitro and in vivo via direct S-nitrosylation of 5-LO, providing novel mechanistic strategies for the development of future anti-LT drugs.
Results
Clinically relevant concentrations of NO-donating aspirin suppress the formation of 5-LO products in human whole blood
Human venous whole blood from healthy volunteers was supplemented with clinically relevant concentrations of NO-releasing aspirin (NO-ASA, “SML1669”; Sigma-Aldrich, St. Louis, MO) as a model agent for NO-donating drugs. Next, LPS/N-formylmethionyl-leucyl-phenylalanine (fMLP) (“L2630” and “F3506”; Sigma-Aldrich) was added to trigger the biosynthesis of 5-LO products. A highly sensitive and specific liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) methodology was subsequently applied for selective detection of the 5-LO products 5-HETE and LTB4, as previously described (62). NO-ASA inhibited the formation of the 5-LO products LTB4 and 5-HETE in a concentration-dependent manner, with IC50 values ≈39 and 105.6 μM, respectively (Fig. 1A, B).

Aspirin (“A2093”; Sigma-Aldrich) at doses up to 200 μM had no effect on 5-LO product formation. Zileuton, the only approved inhibitor of 5-LO in the U.S. market, which acts by chelating the catalytically essential iron (12), was used as positive control and led to significant reduction of LTB4 as well as 5-HETE production (Fig. 1C, D) at 10 μM. Plasma levels of salicylic acid, the directly and very rapidly formed NO-ASA metabolite, in rats orally dosed once with NO-ASA reached up to 600 μM (10). In patients, maximum plasma levels of salicylic acid reached 160 μM following a single oral dose of 1600 mg NO-ASA (11). Taken together, the data show that clinically relevant concentrations of NO-ASA suppress 5-LO product formation in LPS-treated whole blood.
Effect of NO-ASA and further NO-releasing agents on LO product formation in intact human granulocytes and platelets
Effect of NO-ASA, NO-sulindac, and NO-naproxen
To further investigate the effects of NO-donating drugs on cellular LT biosynthesis, the efficacy of NO-ASA was assessed in A23187 (“C7522”; Sigma-Aldrich)-stimulated human polymorphonuclear leukocytes (PMNLs), a frequently used model for evaluating 5-LO inhibitors (67).
Pretreatment (15 min) of PMNLs with NO-ASA caused a concentration-dependent inhibition of 5-LO product formation, with an IC50 ≈ 12.5 μM (Fig. 2B). To exclude the effects of NO-ASA on the supply of endogenous AA as a substrate, and thus to circumvent cPLA2 and FLAP activity, exogenous AA (20 μM; “506-32-1”; Cayman Chemicals, Ann-Harbor, MI) was provided. However, no change was observed in the inhibitory potency of NO-ASA (Fig. 2B). Consistent with the results of the whole blood assay, ASA at concentrations up to 100 μM failed to suppress 5-LO product synthesis in A23187-activated PMNLs, irrespective of the presence of exogenous AA (Fig. 2B).
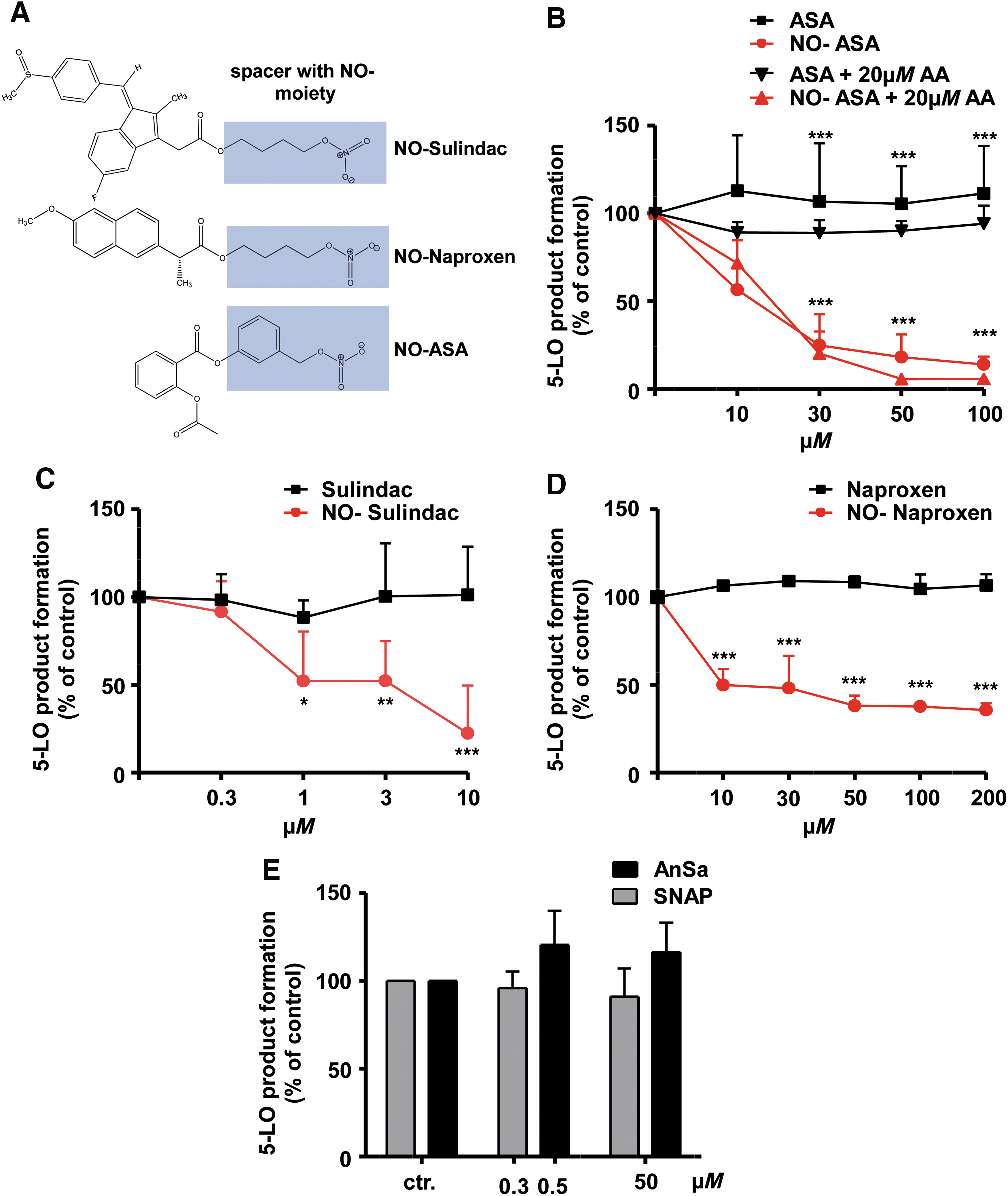
Furthermore, we observed a significant and concentration-dependent suppression of 5-LO product formation by two other NO-NSAIDs, NO-sulindac and NO-naproxen (Fig. 2C, D, structures in A), used at pharmacologically relevant concentrations [NO-naproxen is rapidly hydrolyzed to naproxen, plasma levels of naproxen reach 15.7 μM in rats orally dosed with 6 mg/kg NO-naproxen (60)], whereas sulindac and naproxen were ineffective (Fig. 2C, D). Notably, LTB4 and 5-HETE were suppressed equipotently by NO-ASA and NO-naproxen in PMNL (data not shown). Surprisingly, two different NO donors, 1-amino-2-naphthol-4-sulfonic acid (Angeli's Salt [AnSa], “08751”; Sigma-Aldrich) and S-nitroso-N-acetyl-
Selectivity of suppression of 5-LO product formation by NO-ASA
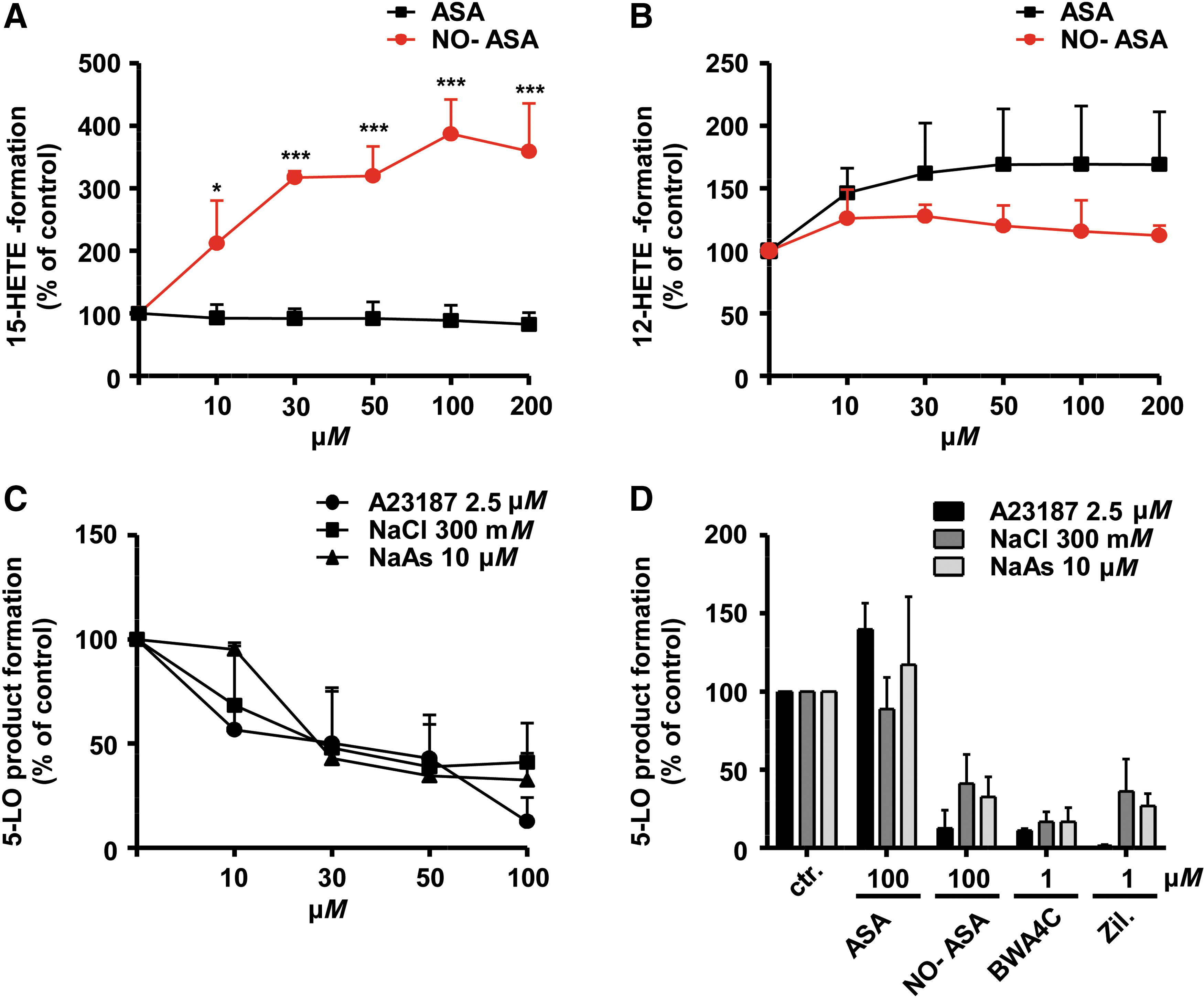
Because PMNL preparations (purity > 95%) contain eosinophils expressing 15-LO-1, we also analyzed the effects of NO-ASA on the concomitant formation of the 15-LO-1 product 15-H(P)ETE (Fig. 3A). NO-ASA significantly increased the formation of 15-H(P)ETE up to fourfold compared to vehicle control in a concentration-dependent manner, whereas ASA had no effect. In addition, we investigated the effect of increasing concentrations of NO-ASA on platelet-type 12-LO activity. 12-H(P)ETE product formation in platelets was triggered by 10 μM AA, which was moderately increased by NO-ASA. ASA at concentrations up to 200 μM was ineffective (Fig. 3B).
Suppression of 5-LO product formation under different stimulatory conditions
Previous studies have shown that the type of stimulus and different signal transduction pathways triggering 5-LO activation can lead to different potencies of 5-LO inhibitors in PMNLs (23). Furthermore, a number of different cellular stimuli are considered to contribute to the increased LT biosynthesis in inflamed tissues (23, 67). Thus, we compared the inhibitory potency of NO-ASA in PMNLs stimulated with the ionophore A23187, hyperosmotic shock (0.3 M NaCl), or genotoxic stress using sodium arsenite (67). All incubations received exogenous AA (20 μM) to assure sufficient and equal substrate supply. As can be seen in Figure 3C, only minor differences in potency were evident. However, as observed with many 5-LO inhibitors, NO-ASA failed to fully reduce 5-LO product synthesis in PMNLs challenged by genotoxic or osmotic stress, with a remaining plateau of ∼30% of 5-LO product synthesis. BWA4C and zileuton (“B7559” and “Z4277”; Sigma-Aldrich) were used as positive controls, suppressing 5-LO product synthesis under all stimulatory conditions (Fig. 3D). Collectively, different NO-NSAIDs led to selective and stimulus-independent suppression of cellular 5-LO, but not 12-LO or 15-LO product formation.
Reversibility of the suppression of 5-LO product formation by NO-ASA in intact PMNLs
Finally, to determine whether the NO-ASA-mediated inhibition of 5-LO was irreversible, PMNLs were treated with NO-ASA, the competitive 5-LO inhibitor BWA4C, or the irreversible, covalent 5-LO inhibitor U73122 (“662035”; Sigma-Aldrich) (21). While 5-LO product formation was restored after threefold washing of cells treated with BWA4C, there was only partial recovery of 5-LO activity in NO-ASA-treated cells and no recovery in U73122-treated cells (Supplementary Fig. S1; Supplementary Data are available online at
Effect of NO-ASA on 5-LO activity in cell-free assays
To circumvent upstream activation pathways, cellular 5-LO regulatory components or mechanisms such as FLAP, mitogen-activated protein kinases (MAPKs), Ca2+ mobilization, nuclear membrane association (51), or interaction with coactosin-like protein (CLP) (55), and to confirm a possible direct action of pharmacologically donated NO on 5-LO, the effects of NO-ASA on 5-LO activity in cell-free assays were tested. NO-ASA only moderately suppressed 5-LO activity in PMNL homogenates and cytosolic fractions (S100) (Fig. 4A), and likewise showed moderate suppression of the activity of partially (adenosine triphosphate [ATP]-affinity) purified recombinant 5-LO supplemented with 20 μM AA (Fig. 4B).
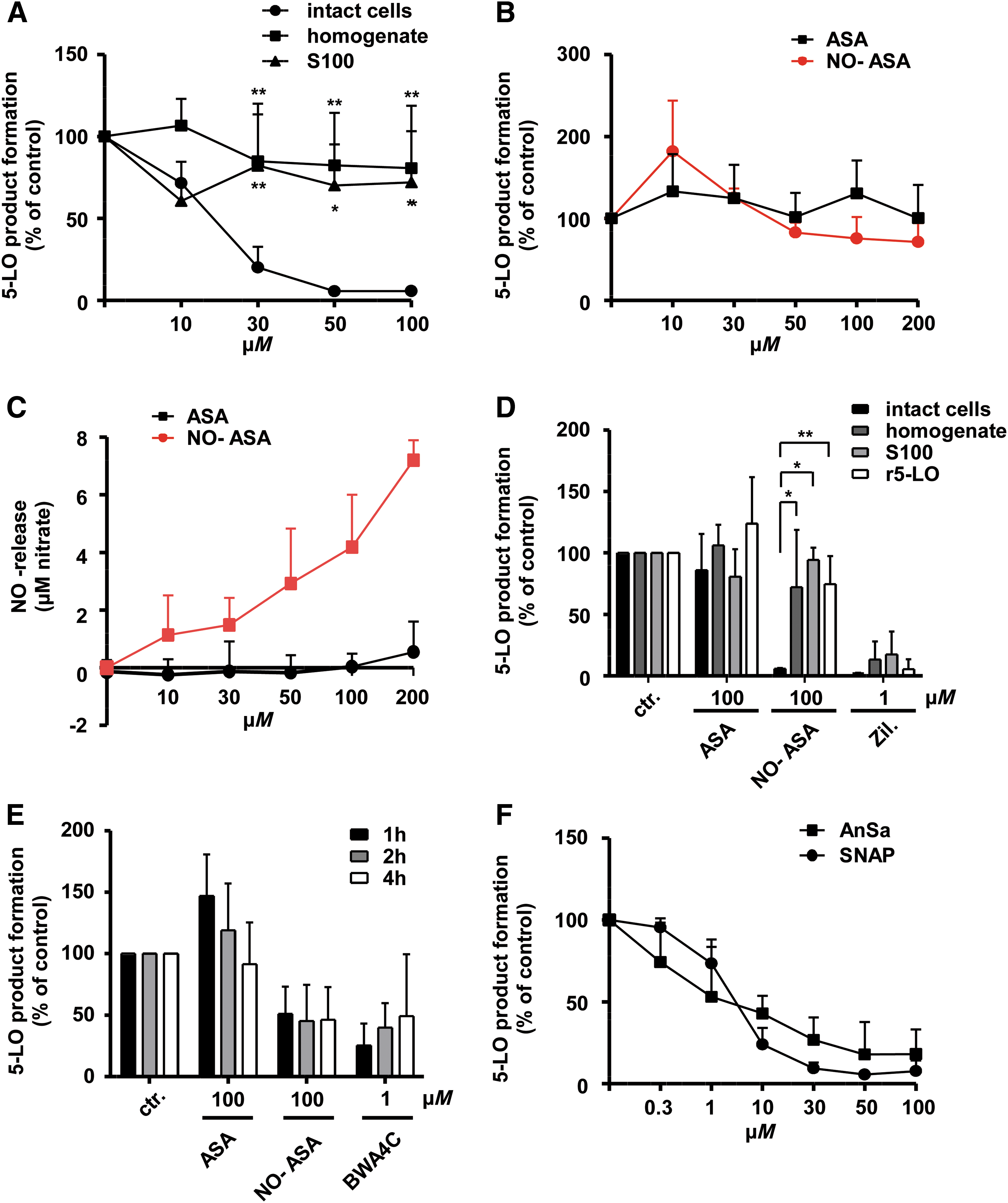
To exclude lack of NO release as a cause for the weak inhibitory effects of NO-ASA in cell-free assays, we next confirmed the concentration-dependent release of NO by NO-ASA in the reaction buffer of incubations with r5-LO using a nitric oxide assay kit (“780051”; Cayman Chemicals) (Fig. 4C). As a control, the direct 5-LO inhibitor zileuton inhibited 5-LO activity in all the cell-free assays, whereas ASA at 100 μM was ineffective (Fig. 4D). Even prolonged incubation times of up to 4 h failed to produce efficient inhibition of r5-LO by NO-ASA (Fig. 4E). However, concentration-dependent and efficient suppression of r5-LO activity was observed when the NO donors SNAP and AnSa were incubated for prolonged time periods of up to 2 h (Fig. 4F).
To summarize, NO-ASA efficiently suppressed cellular 5-LO product formation, but only weakly suppressed the activity of r5-LO in a cell-free environment, whereas the other NO donors, SNAP and AnSa, failed to inhibit cellular 5-LO activity but led to efficient and direct inhibition of the recombinant enzyme. We therefore conclude that an intact cellular environment is probably a requirement for potent and efficient inhibition of 5-LO product formation by NO-ASA in PMNLs.
Role of cellular guanylate cyclase in effect of NO-ASA
We next considered whether NO-mediated activation of cellular guanylate cyclase (GC) by NO-ASA could be the mechanism behind the increased inhibitory potency and efficacy in intact PMNLs, since suppression of cellular 5-LO activity by NO via GC has been previously reported (17). Therefore, we assessed the effects of NO-ASA on 5-LO product formation in PMNLs, in the absence and presence of the established GC inhibitor 1H-(1,2,4) oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, “O3636”; Sigma-Aldrich), preincubated 10 min before addition of NO-ASA. However, ODQ failed to abolish the suppression of 5-LO product formation by NO-ASA (Supplementary Fig. S2A). Similar GC-independent effects were seen with NO-sulindac and zileuton (Supplementary Fig. S2B).
To confirm that the concentration of ODQ was sufficient to inhibit cGC, we analyzed the cyclic guanosine monophosphate (cGMP) level in PMNL, as read-out for cGC activity (Supplementary Fig. S2C). For this, 106 cells were treated with NO-ASA, ASA or vehicle control in the presence or absence of ODQ. BAY-418543 (“256498-66-5”; Cayman Chemicals), as a direct cGC activator, or sodium nitroprusside (SNP, “1614501”; Sigma-Aldrich), as a cell permeable NO donor, were used as positive controls (Supplementary Fig. S2C).
As one can see in Supplementary Figure S2C, activation of cGC by BAY-418543 (100 μM) was reduced by ∼80% when cells were preincubated with 1 μM ODQ, confirming that ODQ is able to largely inhibit cGC activity at the concentration used. However, a complete inhibition of cGC by 1 μM ODQ could not be achieved. Interestingly, NO-ASA was not able to increase the cellular cGMP level. Also, higher concentrations of NO-ASA (300 μM) did not induce formation of detectable cGMP levels in PMNL, indicating that the temporally higher intracellular NO concentrations induced by NO-ASA were not capable of activating cGC. Thus, an inhibition of 5-LO by NO-ASA is unlikely to result from activation of the cGC pathway.
NO-ASA penetrates into PMNLs, triggering a drug concentration-dependent intracellular release of NO
We hypothesized that NO-ASA might penetrate into cells, triggering the metabolic cleavage of the prodrug and producing transiently high intracellular concentrations of NO. In a first series of experiments, we determined the NO release in PMNLs treated with increasing concentrations of NO-ASA (Fig. 5A). As can be seen, suppression of cellular 5-LO product formation (Fig. 5B) was well correlated with a concentration-dependent increase in NO release by NO-ASA in intact PMNL preparations (Fig. 5A). To investigate the extent to which an intracellular release of NO contributes to the total NO formation by NO-ASA, increasing amounts of PMNLs (2.5 × 106 up to 5 × 107) were incubated with 100 μM NO-ASA, 100 μM SNAP, or 100 μM ASA as control (Fig. 5C).

Whereas the release of NO by SNAP was similar in all cell amounts and therefore, independent of intracellular metabolism, NO release by NO-ASA was significantly elevated in the presence of increasing amounts of PMNLs. Release of NO in the absence of cells produced nitrate concentrations of ∼6 μM, whereas under the conditions used in the activity assays (∼5 × 106 PMNLs), nitrate concentration in the supernatant was ∼11.3 μM. This suggests that under the conditions used for the cellular 5-LO assays, ∼40% of the total NO released by NO-ASA derives from intracellular sources. This clearly differs from SNAP, which releases NO nonenzymatically into the aqueous buffer solution, independently of the number of cells.
To confirm these results, we analyzed the amount of NO stored in the pellet of PMNL (30 × 106 cells) after treatment with 100 μM NO-ASA, ASA, SNAP, or vehicle control (Fig. 5D). The amount of intracellular NO formed after treatment of cells with NO-ASA was significantly higher compared to the situation where cells were treated with SNAP, whereas the NO concentration in the supernatant of the cells was similar between both NO donors. In a cell-free setting, only SNAP showed a significant NO release into the aqueous solution, supporting the important role of an intracellular release of NO by NO-ASA.
To further validate the importance of the penetration of the intact NO-ASA molecule into cells and subsequent intracellular cleavage, PMNLs were treated with increasing concentrations of a spacer molecule containing the NO-releasing moiety of NO-ASA, which was synthesized in-house (see Supplementary Experimental Procedures and Supplementary Fig. S3). Lack of the ASA pharmacophore considerably impaired the potency of this derivative (IC50 > 50 μM) compared to NO-ASA (Fig. 5E), suggesting that the ASA pharmacophore facilitates penetration of the NO-releasing moiety into cells and favorably adjusts the subcellular localization to allow direct interaction with 5-LO. Of course, it cannot be fully excluded that the ASA pharmacophore of intact NO-ASA chemically stabilizes the NO-releasing moiety preventing a possible degradation.
Cysteines 416 and 418 in 5-LO are nitrosylated by NO-donating drugs and contribute critically to the NO-mediated suppression of cellular 5-LO activity
Mass spectrometric analysis of isolated r5-LO incubated with NO donors
The posttranslational modification of susceptible, nucleophilic amino acid residues in 5-LO by NO donors was assessed using a combination of mutagenesis and proteomic studies. Incubation of human recombinant 5-LO with the NO donor SNAP for 60 min resulted in the adduction of NO to the cysteines at positions 264, 416 and/or 418, and 449, as confirmed by mass spectrometric analysis (see Fig. 6 and Supplementary Figs. S4 and S5). Notably, we identified an additional mass corresponding to NO within peptide 412–423.
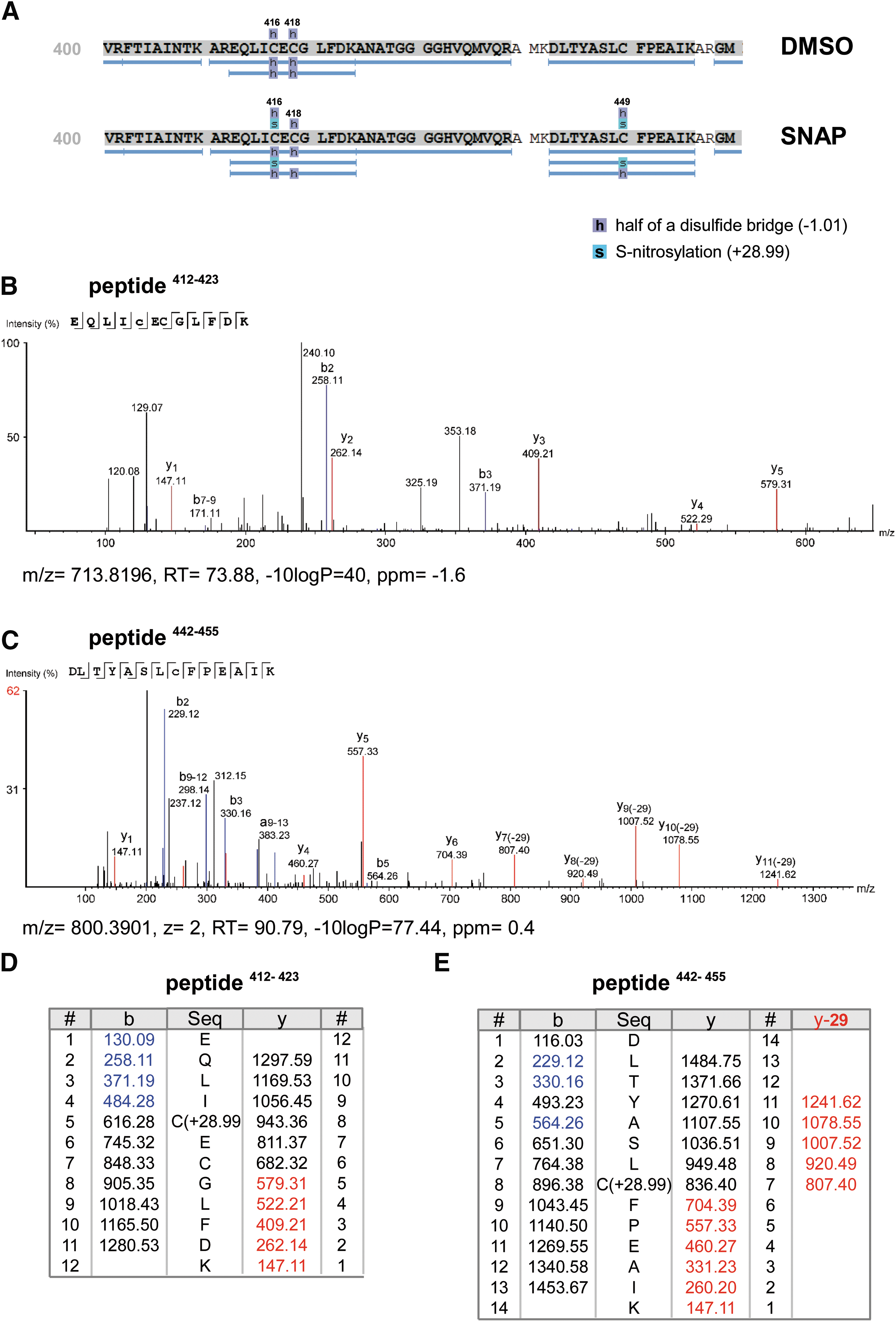
Interestingly, the S-nitrosylation at the closely localized cysteines 416 and 418, forming a double Cys motif (CXC), seems to be considerably more stable than nitrosylation in peptides containing single cysteines, as observed by the absence of characteristic neutral loss fragments, which indicates a more stable modification. The cysteines 159, 300, 416, and 418 are located at the solvent interface of the enzyme and have been shown to be critical for enzymatic activity (34). Thus, treatment with NO donors led to a 5-LO nitrosylation pattern similar to the pattern of nitroalkylation seen with NFAs (4).
Effect of NO-ASA on 5-LO product synthesis and expression in transfected HEK 293T cells
To further address the role of cysteine adduction in the inhibition of 5-LO catalysis by NO-ASA in intact cells, human embryonic kidney (HEK) 293T cells (“ACC-635”; DSMZ, Braunschweig, Germany) were transfected with either 5-LO wild type (wt) or a mutated 5-LO in which surface cysteine residues 159, 300, 416, and 418 were substituted with serine (5-LO 4C).
Whereas concentration-dependent suppression of 5-LO product formation by NO-ASA was observed in cells transfected with 5-LO wt, cells transfected with 5-LO 4C were resistant to any inhibition by NO-ASA (Fig. 7A). ASA failed to suppress 5-LO product formation both in 5-LO wt and 4C transfected cells, whereas suppression of 5-LO product synthesis by BWA4C was similar in both cell types and was independent of the cysteines (Fig. 7B). As a control, and consistent with previous studies, inhibition of 5-LO product formation by the covalently reacting 5-LO inhibitor U73122 was partly abolished in 5-LO 4C transfected cells compared to 5-LO wt transfected cells, as cysteine 416, among others, is crucial for U73122-mediated suppression of 5-LO enzyme activity (34).

Single amino acid mutation studies using 293T cells identified Cys416, Cys418, and to a less extent Cys300, as the amino acids that define sensitivity to NO-ASA (Fig. 7C). Successful transfection of the different 5-LO mutants was confirmed using Western blot analysis (Fig. 7D). We also checked for the expression level of 5-LO in stably transfected 293T cells after treatment with 100 μM NO-ASA at different time points. However, we could not observe any changes in the 5-LO expression level up to 24 h treatment with NO-ASA, excluding that nitrosylated 5-LO gets rapidly degraded (data not shown).
NO-ASA suppresses LPS-induced pulmonary 5-LO product synthesis and airway inflammation
To determine whether the NO-ASA-mediated inhibition of 5-LO also applies to an animal model in vivo, the effects of NO-ASA on 5-LO activation and LTB4-dependent neutrophil infiltration, as a marker of airway inflammation, were studied in the mouse lung after intranasal application of LPS, as previously described (46, 57) (Fig. 8A). Administration of LPS triggered neutrophil infiltration into the lungs and considerably increased pulmonary levels of the 5-LO products LTB4 and 5-HETE (Fig. 8B–D).

Pretreatment of the mice with NO-ASA at an oral dose of 100 mg/kg significantly attenuated LPS-induced neutrophil cell counts as well as 5-HETE levels in bronchoalveolar lavage fluid (BALF). A reduction of LTB4 levels could also be observed. Notably, NO-ASA suppressed neutrophil infiltration better than zileuton and produced 5-LO inhibition similar to the drug. Treatment with ASA had no effect on either neutrophil cell counts or BALF 5-LO product levels.
Discussion
5-LO products derived from the 5-LO pathway play crucial roles in the immune response and in the pathogenesis of inflammatory diseases such as arthritis, asthma, allergic rhinitis, and other chronic inflammatory disorders (29, 48). Furthermore, 5-LO products contribute to atherosclerosis (49) and may regulate the proliferation and survival of cancer cells (27). Considerable effort has been made in the past to develop safe and efficient 5-LO inhibitors. However, many compounds failed under in vivo conditions, either due to adverse effects (interference with other biological processes or the production of reactive radicals) (24, 33) or to a loss of efficacy caused by an elevated oxidative state and/or increased phosphorylation of 5-LO in inflamed tissues (69).
Currently, the only approved drugs with proven 5-LO suppressive efficacy are the NSAIDs sulindac sulfide (63) and celecoxib (40), as well as the iron ligand inhibitor zileuton. However, poor efficacy, unfavorable dosage regimens, and possible hepatic toxicity limit the clinical use of zileuton. In the present study, we demonstrate for the first time that clinically relevant concentrations of several NO-donating NSAIDs suppress 5-LO product synthesis in human isolated PMNLs via direct S-nitrosylation of 5-LO. Furthermore, suppression of 5-LO product synthesis was observed in ionophore-stimulated human whole blood (HWB) as well as in an animal model of pulmonary inflammation.
5-LO is a tightly regulated enzyme with a number of regulatory nodes involved in the control of LT biosynthesis. Thus, suppression of cellular product formation by NO-NSAIDs might result from targeting any of the multiple regulatory components or processes involved, such as cPLA2, FLAP, MAPKs, Ca2+ mobilization, interaction with CLP, or nuclear membrane association (50). However, we found that NO-ASA suppressed 5-LO product synthesis in PMNLs equipotently, regardless of whether AA was provided from endogenous sources via cPLA2 or FLAP or exogenously, excluding interference of NO-NSAIDs with the provision of AA by these proteins.
Interference with upstream 5-LO-activating MAPK signaling by NO-ASA is unlikely, since PMNL activation by calcium ionophore A23187 widely circumvents receptor-coupled signaling and MAPK/phosphorylation-dependent 5-LO activation (67). Furthermore, NO-ASA suppressed 5-LO product formation in PMNLs irrespective of the stimulus used to trigger 5-LO activation, pointing to a direct effect on the 5-LO enzyme. Direct interaction of NO released from NO-ASA with the 5-LO enzyme was confirmed by transfection studies, recombinant 5-LO enzyme activity assays, and the detection of S-nitrosylation at catalytically relevant cysteines in 5-LO using mass spectrometry. Among the amino acids nitrosylated by NO donors, Cys 416 and Cys 418 play an essential role in the catalytic activity of 5-LO (4, 34) and represent the most likely molecular targets of the NO-NSAIDs. However, we cannot fully exclude the possibility that binding to other thiol-reactive targets contributes to the suppression of 5-LO product formation in intact cells.
This reactivity of NO with 5-LO is similar to the results of our recent study, in which electrophilic NFAs suppressed LT biosynthesis in vitro and in vivo, mainly via nitroalkylation of 5-LO at cysteine 418 (4). Thus, selective inhibition of 5-LO activity by NO-ASA in human PMNLs and their lack of effect on 12-LO in human platelets or on 15-LO-1 in human eosinophils may again be a consequence of the targeting of Cys416 and Cys418, which are unique to 5-LO and are not conserved in the human, rat, or murine platelet-type 12-LO or 15-LO-1 isoforms (34). The fate of nitrosylated 5-LO still remains unclear and needs further investigation.
Furthermore, we found that cellular penetration and intracellular cleavage of NO-ASA leading to temporarily high intracellular concentrations of NO are of major importance for the 5-LO-suppressive effects of NO-NSAIDs. However, suppression of 5-LO product formation is most likely not a class effect of all NO-donating drugs but is probably restricted to drugs with certain structural properties, including an NO-releasing moiety as well as a pharmacophore to trigger cellular penetration and, potentially, favorable intracellular localization.
The literature provides supporting evidence for our finding of the inhibition of 5-LO by NO, as the present results fit well with the current view of the clinical and preclinical effects of NO-NSAIDs. NO-ASA has already been described to impair Cys-LT levels in a study screening the effect of different NSAIDs on COX and 5-LO activity in whole blood from aspirin-sensitive asthmatics (26). In addition, the NO-releasing derivative of paracetamol has been shown to be considerably more potent and efficient than the parent drug with regard to analgesic and anti-inflammatory effects (3). Furthermore, an NO-releasing derivative of prednisolone (NCX-1015) was found to be clearly more potent than the parent drug in reducing neutrophil extravasation and cytokine and chemokine synthesis in a murine model of acute inflammation (45). Superior anti-inflammatory efficacy of NO-ASA compared to ASA was also seen in a mouse model of carrageenan-induced rat paw edema (15). As a number of studies have demonstrated the therapeutic efficacy of 5-LO inhibitors in such models of inflammation (70), it is reasonable to speculate that the augmented anti-inflammatory potency of NO-NSAIDs may derive, at least partly, from concomitant inhibition of LT biosynthesis.
Many studies have shown that NO-NSAIDs have a therapeutic effect on cancer growth, both in vivo and in vitro. In an in vivo rat model of colonic adenocarcinoma, treatment with NO-ASA reduced the number of aberrant crypt foci induced by trinitrobenzene/sulfonic acid more effectively than equivalent doses of ASA (5). Similarly, NO-sulindac exerted considerably stronger antiproliferative and apoptotic effects in colorectal cancer cells than sulindac (38). The authors of almost all these studies concluded that the NO-releasing moiety of NO-NSAIDs exerts an extraordinary antitumorigenic activity independent of the parent NSAID moiety. Increasing evidence in the literature implicates 5-LO as a player in the growth of several solid tumor types, including pancreatic, colorectal, prostate, and breast cancers (64). Recently, a role for 5-LO in hemato-oncological tumors such as chronic myeloid leukemia (16) and acute myeloid leukemia (59) was reported. As 5-LO products participate in tumor development (42) and pharmacological inhibition of 5-LO has been shown to suppress the growth of different tumors in a number of animal models (64), the antitumorigenic effects of NO-NSAIDS may, at least partly, be due to NO-mediated inhibition of 5-LO activity and product synthesis. Correspondingly, NO-ASA and NO-naproxen S-nitrosylated the p65 subunit of transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in vitro and in vivo, a result that was considered to be a mechanistic explanation for the chemopreventive effects of NO-NSAIDs (14). In addition, NO-ASA modulated the Keap1 (Kelch-like ECH-associated protein 1)-nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway, leading to translocation of Nrf2 into the nucleus of colorectal cancer cells. This is consistent with findings that electrophilic NFAs suppress both 5-LO and the NF-κB p65 subunit and activate Nrf2 by covalent nitroalkylation of cysteine residues (19, 35). Thus, we hypothesize that NFAs and NO may target the same set of proteins involved in inflammation and tumorigenesis via electrophilic reaction with surface cysteines. Cysteine reactivity is not an obstacle to the development of clinically effective drugs, as already shown by an emerging number of agents containing cysteine-reactive moieties, which have already been approved or are now entering clinical trials. For example, Neratinib, a tyrosine kinase inhibitor in clinical phase 3 for the treatment of breast cancer and other solid tumors (13), acts by covalently binding to specific cysteine residues (22). Furthermore, several other cysteine-reactive drugs have already been successfully approved, that is, afatinib (Gilotrif®) for treatment of patients with squamous cell carcinoma of the lung (39) or dimethyl fumarate (Tecfidera®) as first-line monotherapy for multiple sclerosis (6). As can be seen from the recent approvals, specific molecule scaffolds are required to deliver the reactive Michael acceptor moiety in close proximity to its target structure.
NO-NSAIDs demonstrate considerably less gastrointestinal toxicity than conventional NSAIDs. For example, a single dose of naproxen (80 mg/kg) produced lesions in the rat stomach within 24 h of administration, whereas an equimolar dose of NO-naproxen was completely devoid of any ulcerogenic activity (20). Notably, it was shown that increased LTC4 production in gastrointestinal tissues was a determinant for NSAID-induced ulcerogenicity, and concomitant application of a 5-LO inhibitor abolished the indomethacin-induced gastrotoxicity in pigs (53). Further studies confirmed these findings and it was therefore concluded that 5-LO products play a major role in acute gastric and intestinal damage triggered by NSAIDs (54).
Taken together, the results indicate that suppression of 5-LO product formation by NO-NSAIDs may significantly contribute to the pharmacodynamic profile of NO-NSAIDs in animals and to the clinical effects observed in humans.
Beside these known clinical effects, it is reasonable to speculate that NO-ASA or other NO-NSAIDs may be safer in patients suffering from aspirin-induced asthma (20% of all asthmatics), which is essentially triggered by elevated levels of bronchoconstrictory cysteinyl LTs (32). Our study provides evidence that drugs which release NO intracellularly lead to suppression of 5-LO activity and LT biosynthesis.
Compounds fulfilling the necessary structural requirements could be interesting candidate drugs for the treatment of diseases in which inhibition of 5-LO signaling is of additional therapeutic value. For instance, the combination of cytostatics with NO-donating moieties could not only trigger sensitization to the chemotherapeutic drug (7) but also significantly improve treatment regimens for acute myeloid leukemia, due to the elimination of 5-LO-dependent leukemic stem cells, which often remain after chemotherapy and cause disease relapse (58).
Based on the findings that two naturally occurring mediators, NO and NFA, suppress 5-LO activity under in vivo conditions via the targeting of regulatory cysteines 416 and 418, this mechanism, on the one hand, may be a novel endogenous mechanism for regulating cellular 5-LO activity and thus, decreasing inflammation, on the other hand, could be a promising strategy for the development of future anti-LT therapeutics. Drugs selectively targeting cysteines 416 and 418 in 5-LO by nitrosylation, nitroalkylation, or other covalent modifications may represent a novel class of potent, efficient, and safe 5-LO inhibitors, which may ultimately overcome the limitations of previously developed anti-LT drugs and may allow sustained and efficient suppression of LT biosynthesis under in vivo conditions.
Materials and Methods
Plasmids, cell lines, and chemicals
The expression vector pcDNA3.1-5LO, as well as the 5-LO cysteine mutants (Cys159Ser, Cys300Ser, Cys416Ser, Cys418Ser, or all four), has been described previously (30, 31, 34). The plasmid pT3-5-LO was obtained from Dr. Olof Rådmark (Stockholm, Sweden). 293T (human embryonal kidney cell line, ACC-635) cells were obtained from DSMZ (German Collection of Microorganisms and Cell Cultures GmbH). Cells were cultured in Dulbecco's modified Eagle's medium (“11965-092”; Thermo Fisher Scientific, Waltham, MA), supplemented with 10% FCS (fetal calf serum; “10270106”; Thermo Fisher Scientific), 100 U/mL penicillin, and 100 μg/mL streptomycin (“P7539”; Sigma-Aldrich).
NO-sulindac, NO-naproxen, and NO-ASA spacer were synthesized in-house (see Supplementary Experimental Procedures). AnSa, ASA, calcium ionophore A23187, BWA4C, dimethyl sulfoxide (DMSO), fMLP, IBMX (3-isobutyl-1-methylxanthine, “I5879”), LPS, naproxen, NO-ASA, ODQ, SNP, sulindac, SNAP, and zileuton were purchased from Sigma-Aldrich. AA and Bay-418543 were purchased from Cayman Chemicals.
Isolation of human PMNLs and platelets from venous blood
Human PMNLs were freshly isolated from leukocyte concentrates obtained from the German Red Cross Blood Donor Service (Frankfurt, Germany), as described previously (67). The leukocyte concentrates were sedimented on a dextran gradient. After 20–30 min, the supernatant was collected and retained for platelet isolation and the remaining cell pellet (PMNL) was resuspended in phosphate-buffered saline (PBS). To remove the erythrocytes, hypotonic lysis was carried out. The PMNLs (5 × 106 cells/mL; purity >95%) were resuspended in PBS (pH 7.4) with the addition of 1 mg/mL glucose and 1 mM CaCl2 (PGC buffer). The upper phase of the Ficoll gradient (platelet-rich plasma) was further centrifuged (800 g for 7 min) to obtain platelets, which were then suspended in PBS containing 1 mM CaCl2 (100 × 106 cells/mL).
Preparation of cell homogenates and cytosolic fractions (S100) from PMNLs
To obtain cell homogenates, human PMNLs were resuspended in PBS containing 1 mM ethylenediaminetetraacetic acid (EDTA) sonicated (3 × 10 s) at 4°C, and 1 mM ATP was added. To remove unbroken cells, nuclei, and mitochondria, this cell preparation was centrifuged at 10,000 g for 10 min. The supernatant (cell homogenate) was further centrifuged at 100,000 g for 70 min to obtain the cytosolic fraction (100,000 g supernatant; S100). These cell preparations were used for further in vitro activity assays.
Expression and purification of recombinant 5-LO
Recombinant 5-LO wt was expressed in Escherichia coli BL21 (DE3) cells. Protein expression was started by addition of 0.2 mM IPTG (isopropyl-thio-β-
Determination of 5-LO product formation in intact cells, cell homogenates, and S100 supernatant
For determination in intact cells, 5 × 106 freshly isolated PMNLs were resuspended in 1 mL PGC buffer. After preincubation with the test compounds or vehicle control (0.01% DMSO) at 37°C for either 15 min or 1 h, 5-LO product formation was initiated by the addition of 2.5 μM A23187 (Ca2+ ionophore) or 10 μM sodium arsenite (NaAs) or 300 mM sodium chloride (NaCl), with or without AA. After 10 min at 37°C, the reaction was stopped by addition of 1 mL ice-cold methanol and the 5-LO metabolites formed (5-HETE, LTB4, trans/epi-trans LTB4) were extracted and analyzed by high-performance liquid chromatography (HPLC), as described previously (68).
In addition, cell homogenates and cytosolic fractions (S100) of human PMNLs were used for the testing of the 5-LO inhibitors. Therefore, equal amounts of cellular preparations were incubated with solvent (0.01% DMSO) or test compounds for 15 min at 4°C in the presence of 1 mM EDTA and 1 mM ATP. Thereafter, the reaction was started by prewarming (37°C) as well as by the addition of CaCl2 (2 mM) and 20 μM AA, and stopped after 10 min by the addition of 1 mL ice-cold methanol.
Reversibility of 5-LO inhibition in human PMNLs
To determine the reversibility of the effects of the test compounds, a “washout” assay was performed. Freshly isolated human PMNLs were incubated with test compounds or vehicle control (DMSO) for 15 min and then washed three times with reaction buffer. Next, 2.5 μM A23187 and 2 μM AA were added; the reversible 5-LO inhibitor BWA4C (1 μM) and the irreversible 5-LO inhibitor U73122 (10 μM) were used as controls.
Determination of 15-LO activities in intact human PMNLs
PMNL preparations from human buffy coats exhibit a purity of ∼95%. The remaining 5% of cells are mainly eosinophils expressing 15-LO-1. The formation of the 15-LO-1 product 15-HETE was started by the addition of 2.5 μM Ca2+ ionophore to PMNLs after preincubation with the test compounds or vehicle (0.01% DMSO) at 37°C for 15 min. The reaction was stopped after 10 min by the addition of 1 mL ice-cold methanol, and 15-HETE was extracted and analyzed by HPLC, as described for intact cells.
Determination of 12-HETE formation in human platelets
Freshly isolated platelets (108 cells/mL in PBS containing 1 mM CaCl2) were preincubated with the test compounds for 15 min at 37°C. After addition of 2.5 μM A23187 and 10 μM AA and further incubation for 10 min at 37°C, the reaction was stopped by addition of 1 mL ice-cold methanol, and 12-HETE was extracted and analyzed by HPLC, as described for 5-LO products in PMNL.
Determination of 5-LO product formation under the influence of cGC inhibition
For determination of the influence of cGC on the inhibition of 5-LO product formation, 5 × 106 freshly isolated PMNLs, resuspended in 1 mL PGC, were preincubated with 1 μM ODQ (cGC inhibitor) or vehicle control (0.01% DMSO) for 10 min, followed by incubation with test compounds for 15 min at 37°C. Afterward, 5-LO product formation was initiated by the addition of 2.5 μM A23187 and 20 μM AA. After 10 min at 37°C, the reaction was stopped by addition of 1 mL ice-cold methanol and the 5-LO metabolites formed (5-HETE, LTB4, trans/epi-trans LTB4) were extracted and analyzed by HPLC, as described previously (68).
Determination of product formation of recombinant 5-LO
For determination of recombinant 5-LO activity, 3 μg of partially purified 5-LO was added to 1 mL of a 5-LO reaction mix (PBS, pH 7.4, 1 mM EDTA, 1 mM ATP). After incubation with the test compounds or vehicle control (0.01% DMSO) for 1, 2, or 4 h at 37°C or for 15 min at 4°C, 5-LO product formation was started by adding 2 mM CaCl2 and 20 μM AA. The reaction was stopped after 10 min by addition of 1 mL ice-cold methanol. The 5-LO products (5-HETE, trans/epi-trans LTB4) formed were analyzed by HPLC, as described for PMNL.
Determination of 5-LO product formation in HWB
For assays in whole blood, freshly withdrawn blood from adult and healthy volunteers was obtained with consent by venipuncture and collected in monovettes containing 16 IE heparin/ml. The subjects had no apparent inflammatory conditions and had not taken anti-inflammatory drugs for at least 10 days before blood collection. Aliquots of 3 mL blood were primed with 1 μg/mL LPS for 1 h at 37°C. The formation of 5-LO products (5-HETE, LTB4, and its trans isomers) was initiated by the addition of 1 μM fMLP. Ten minutes before fMLP addition, test compounds or vehicle (DMSO) was added and incubated at 37°C. The reaction was stopped after 15 min by placing on ice and the samples were centrifuged at 600 g for 10 min at 4°C. 5-LO products in the plasma supernatant were analyzed using LC-MS/MS on an API 4000 triple quadrupole mass spectrometer (Applied Biosystems, Darmstadt, Germany).
Determination of cGMP levels in intact PMNL
For the determination of cGMP levels in PMNL, 1 × 106 cells were preincubated with 500 μM IBMX (phosphodiesterase inhibitor) in the presence or absence of 1 μM ODQ (cGC inhibitor) at 37°C. After 30 min, cells were treated with 100 or 300 μM ASA and NO-ASA, 30 or 100 μM Bay-418543 (cGC activator), 10 μM SNP (NO-donating drug) or vehicle control (0.01% DMSO) for 1 h at 37°C. The reaction was stopped on ice, cells were lysed with 0.1 N HCl and 0.5% Triton × 100, and cGMP concentrations were measured via cGMP-ELISA (acetylated format, “ADI-900-014”; Enzo Life Science, Farmingdale, NY) according to the manufacturer's instructions.
Determination of NO
For determination of the total amount of NO in intact PMNL, 5 × 106 cells, 30 × 106 cells, or an increased number (2.5–50 × 106 cells) of freshly isolated PMNLs were resuspended in 1 mL PGC buffer. After preincubation with the test compounds or vehicle control (0.1% DMSO) at 37°C for either 15 min or 1 h, 2 mM CaCl2 and 20 μM AA were added. After 10 min at 37°C, the reaction was stopped on ice and the samples were centrifuged at 10,000 g for 10 min at 4°C. The total amount of NO was assessed indirectly by measuring the levels of its oxidized forms (nitrites and nitrates) in the supernatant or, after three washing steps, resuspension in buffer and sonication, in the pellet (experiment: extra- and intracellular NO level) of these cells using a Nitrite/Nitrate Fluorimetric Assay Kit (Cayman Chemicals) according to the manufacturer's instructions.
Determination of 5-LO product formation and Western blot analysis of proteins in transfected 293T cells
293T cells were transiently transfected with pcDNA3.1-5LOwt, pcDNA-5LO cysteine mutants (4C, C159S, C300S, C416S, C418S) or empty vector and/or pSG5-FLAP by the calcium-phosphate precipitation method, according to widely established protocols. After 48 h, cells were harvested and assayed for 5-LO activity. 2.7–3 × 106 cells/mL were resuspended in PBS containing 1 mM CaCl2. After preincubation with the test compounds or vehicle control (0.01% DMSO) at 37°C for 15 min, 5-LO product formation was initiated by the addition of 2.5 μM A23187 and 20 μM AA. After 10 min at 37°C, the reaction was stopped by the addition of 1 mL ice-cold methanol. The 5-LO metabolites formed were extracted and analyzed by HPLC, as described above.
As a transfection control, a part of the transfected 293T cells were lysed with hot sodium dodecyl sulfate (SDS)-lysis buffer (50 mM Tris pH 6.8, 2% SDS, 2% glycerol) and sonicated for 3 × 10 s. Protein concentrations were determined using the BCA assay kit, according to the manufacturer's protocol (Thermo Fisher Scientific). Equal quantities of protein extracts were separated by 8% SDS-polyacrylamide gel electrophoresis.
For immunoblot analysis, proteins were electrophoretically blotted onto a nitrocellulose membrane (Hybond-C Extra; Amersham Biosciences Ltd., Amersham, United Kingdom) and transfected 5-LO was visualized using a monoclonal mouse anti-5LO antibody (“610694”; BD Biosciences, NJ; 1/1000 dilution, overnight at 4°C). For loading control, blots were incubated with a polyclonal goat anti-actin antibody (“sc-1616”; Santa Cruz Biotechnology, Dallas, TX; 1/2000 dilution, 1 h at RT).
Membranes were blocked at room temperature for 1 h with Odyssey blocking reagent (“P/N 927-40100”; LI-COR Biosciences, Lincoln, NE) and all antibodies were diluted in Odyssey blocking reagent as well. Membranes were washed with PBS containing 0.2% Tween 20 and were then incubated with an IRDye680- or IRDye800-conjugated secondary antibody (“P/N 926-68072” or “P/N 925-32214”; LI-COR Biosciences) in Odyssey blocking reagent. The protein/antibody complexes were visualized on the Odyssey Infrared Imaging System (LI-COR Biosciences).
Sample preparation for mass spectrometric analysis
Three micrograms of recombinant 5-LO was incubated with 100 μM SNAP or DMSO for 60 min at 37°C in PBS pH 7.4 containing 1 mM EDTA and 1 mM ATP, followed by addition of 2 mM CaCl2 and 20 μM AA. After 10 min, the reaction was stopped and proteins were precipitated by addition of ice-cold 80% acetone. Protein pellets were reconstituted in 90 μL of 50 mM ammoniumbicarbonate, 0.1 mM EDTA, and 0.1 mM neocuproine, and digested with 1 μg trypsin (sequencing grade, “V5111”; Promega, Madison, WI) overnight in a chamber filled with argon in a light protected cabinet at 37°C. Samples were acidified with 0.1% trifluoric acid, transferred to a microtiter plate, and sealed in argon atmosphere.
Liquid chromatography/mass spectrometry
Liquid chromatography/mass spectrometry (LC/MS) was performed on a Thermo Scientific™ Q Exactive Plus instrument equipped with an ultrahigh-performance liquid chromatography unit (Thermo Scientific Dionex Ultimate 3000) and a Nanospray Flex Ion-Source (Thermo Scientific, Waltham, MA).
Peptides were loaded onto a C18 reverse-phase precolumn (Thermo Scientific) with loading buffer A/NO (1% acetonitrile, 0.1 mM neocoproine) followed by separation on a 2.4 μm Reprosil C18 resin (Dr. Maisch GmbH, Ammerbuch-Entringen, Germany) in house-packed picotip emitter tip (diameter 100 μm, length 30 cm from New Objectives) using a gradient from 100% mobile phase A (4% acetonitrile, 0.1% formic acid) to 30% mobile phase B (80% acetonitrile, 0.1% formic acid) for 60 min followed by a second gradient to 60% B for 30 min at a flow rate 200 nL/min. Each run was finished by washout with 99% mobile phase B for 5 min and re-equilibration of the column in mobile phase A.
MS data were recorded by the data-dependent acquisition Top10 method, selecting the most abundant precursor ions in positive mode for higher-energy collisional dissociation fragmentation. Lock mass option (44) was enabled to ensure high mass accuracy. The full MS scan range was from 300 to 2000 m/z, with a resolution of 70,000 and an automatic gain control (AGC) value of 3 × 106 total ion counts with a maximal ion injection time of 160 ms. Only more highly charged ions (2+) were selected for MS/MS scanning, with a resolution of 17,500, an isolation window of 2 m/z, and an AGC value set to 105 ions with a maximal ion injection time of 150 ms. Selected ions were excluded for the time frame of 5 s following fragmentation. Full scan data were acquired in profile and fragments in centroid mode by Xcalibur software.
Peaks Studio 7.0 was used for MS data analysis, together with a hybrid database from humans (Uniprot, April 2015, 68506 entries) and Echerichia coli (Uniprot, August 2014, 4251 entries). Configuration of posttranslational modification in the Peaks software was supplemented to identify neutral loss of thiol nitrosylation in the fragment spectra (−28.990464 Da) (66). For database search, the following variable modifications were selected: methionine oxidation (+15.99), deamidation of asparagine and glutamine (+0.98), S-nitrosylation of cysteine (28.99), and half of disulfide bridge (−1.01). The false discovery rate was set to 1%. The enzyme specificity was set to trypsin, and missed cleavages were limited to 3. Initial monoisotopic precursor mass error tolerance was set to 8 ppm and fragment ion tolerance was set to 0.05 Da.
Animals
Female BALB/c mice (Charles River, Sulzfeld, Germany), aged 5–6 weeks, were used after being acclimated to the animal facility for 14 days before the experiments. The animals had access to food and water ad libitum. All experimental procedures were approved by the Animal Ethics Committee at Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, Germany.
Induction of acute lung inflammation and treatment with NO-ASA
Chromatographically purified E. coli-LPS (28) was used to induce acute lung inflammation in mice. Mice were briefly anesthetized with isoflurane, and 2.5 μg of LPS was administered intranasally in a volume of 50 μL. Animals were treated prophylactically 1 h before LPS application with the following substances: NO-ASA given at 100 mg/kg, ASA given at 54 mg/kg, and zileuton at 50 mg/kg. All substances were solubilized in polyethylene glycol (PEG400) and given via gavage in a volume of 100 μL.
Collection of serum and BALF
Sixteen hours after administration of LPS, mice were sacrificed and lungs were lavaged via a tracheal cannula with 2 × 1 mL PBS. Subsequently, leukocytes in the BALF were counted in a Neubauer chamber. After centrifugation, the BALF was frozen at −80°C for further analysis. Cytospin slides of BALF cells were stained with a fast staining procedure (HAEME-Schnellfärbung, Labor+Technik Eberhard Lehmann, Berlin, Germany), according to the manufacturer's instructions. The percentages of granulocytes, lymphocytes, and macrophages in the BALF samples were determined by light microscopy. At least 300 cells per sample were differentiated by a blinded investigator.
Statistics
All data are presented as mean ± SD. GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA) was used for statistical analysis. Data were subjected to two-way analysis of variance (ANOVA) coupled with Bonferroni posttest for multiple comparisons and Student's t-test. A sigmoidal dose–response curve fitting model with a variable slope was used to calculate the IC50 values.
Footnotes
Acknowledgments
This work was funded by the German Research Foundation (DFG projects MA-5825/1-1), the DFG Sonderforschungsbereich SFB 1039/projects A01, A07, and B01, the Else Kröner-Fresenius-Foundation (EKFS), and the Graduate School TRIP (Translational Research Innovation-Pharma). This project was supported by the LOEWE Research Centre for Translational Medicine and Pharmacology, financed by the State of Hessen, the Fraunhofer Institute for Molecular Biology and Applied Ecology IME, Project Group for Translational Medicine and Pharmacology TMP and the Aarhus University Research Foundation (AUFF). T.J.M. is recipient of a Heisenberg fellowship from the German Research Foundation (DFG-MA-5825/2-1). M. Piesche, is supported by PMI Oncologìa (UCM1301, MECESUP grant). The authors thank Sandra Busse for excellent technical assistance and Ann-Katrin Ball for providing stably transfected HEK 293T cells.
Author Disclosure Statement
M.J.P. is a consultant to Leo Pharma a/s and Xellia Pharmaceuticals ApS. All authors declare that there are no conflicts of interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.