
Abstract
Aims:
Redox active ultrafine particles (UFP, d < 0.2 μm) promote vascular oxidative stress and atherosclerosis. Notch signaling is intimately involved in vascular homeostasis, in which forkhead box O1 (FOXO1) acts as a co-activator of the Notch activation complex. We elucidated the importance of FOXO1/Notch transcriptional activation complex to restore vascular regeneration after UFP exposure.
Results:
In a zebrafish model of tail injury and repair, transgenic Tg(fli1:GFP) embryos developed vascular regeneration at 3 days post amputation (dpa), whereas UFP exposure impaired regeneration (p < 0.05, n = 20 for control, n = 28 for UFP). UFP dose dependently reduced Notch reporter activity and Notch signaling-related genes (Dll4, JAG1, JAG2, Notch1b, Hey2, Hes1; p < 0.05, n = 3). In the transgenic Tg(tp1:GFP; flk1:mCherry) embryos, UFP attenuated endothelial Notch activity at the amputation site (p < 0.05 vs. wild type [WT], n = 20). A disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) inhibitor or dominant negative (DN)-Notch1b messenger RNA (mRNA) disrupted the vascular network, whereas notch intracellular cytoplasmic domain (NICD) mRNA restored the vascular network (p < 0.05 vs. WT, n = 20). UFP reduced FOXO1 expression, but not Master-mind like 1 (MAML1) or NICD (p < 0.05, n = 3). Immunoprecipitation and immunofluorescence demonstrated that UFP attenuated FOXO1-mediated NICD pull-down and FOXO1/NICD co-localization, respectively (p < 0.05, n = 3). Although FOXO1 morpholino oligonucleotides (MOs) attenuated Notch activity, FOXO1 mRNA reversed UFP-mediated reduction in Notch activity to restore vascular regeneration and blood flow (p < 0.05 vs. WT, n = 5).
Innovation and Conclusion:
Our findings indicate the importance of the FOXO1/Notch activation complex to restore vascular regeneration after exposure to the redox active UFP. Antioxid. Redox Signal. 28, 1209–1223.
Introduction
V
Ambient ultrafine particles suppress forkhead box O-subfamily (FOXO1) as an important co-activator with notch intracellular cytoplasmic domain (NICD) to impair vascular regeneration.
The Notch signaling pathway is an evolutionarily conserved intercellular signaling pathway that is critical in cell-fate specification and embryonic development (4, 14, 22, 27, 48, 57). On ligand binding, the Notch receptor undergoes proteolytic cleavages to release notch intracellular cytoplasmic domain (NICD). After translocation to the nuclei, NICD forms a transcriptional activation complex consisting of recombination signal-binding protein for immunoglobulin J region (Rbp-Jκ), suppressor of hairless, Lag-1 (CSL), and master-mind like (MAML) to induce Notch target genes (6).
The Forkhead box O-subfamily (FOXO) protein is a transcription factor that regulates hormonal control, cellular metabolism, and differentiation (1). Analogous to Notch signaling in cell fate and vascular maturation, FOXO1 (FKHR), the dominant isoform of the FOXO, is essential for vascular growth. Conditional deletion of FOXO1 results in abnormal sprout formation and vascular migration, resulting in a hyperplastic vascular network; whereas constitutively active FOXO1 suppresses vascular expansion, resulting in a sparse vascular network (62). Endothelial FOXO1 physically interacts with canonical Notch signaling by binding to CSL, enhancing co-repressor clearance to promote Notch signaling (26). FOXO1 ablation recapitulates the Notch1 knockout phenotype, whereas the FOXO1 interaction with NICD promotes Notch1 activation in vascular, muscular, and neuronal differentiation (26).
In this context, we assessed whether exposure to ambient UFP inhibits Notch signaling via FOXO1/Notch activation complex to impair vascular regeneration. After tail amputation in embryonic zebrafish, vascular regeneration occurred; whereas UFP exposure attenuated Notch activity and impaired regeneration. UFP further down-regulated FOXO1 expression, resulting in reduced FOXO1 and NICD co-localization. Although rescue with NICD messenger RNA (mRNA) partially restored Notch activity, rescue with FOXO1 mRNA reversed UFP-mediated reduction in Notch activity, leading to restored vascular network and blood flow to the amputated site. Thus, the redox-active UFP down-regulates FOXO1-mediated Notch signaling, revealing the essential role of FOXO1/Notch activation complex in vascular regeneration.
Results
UFP exposure impaired vascular regeneration
To assess the effects of UFP on vascular regeneration, we used the transgenic Tg(fli1:GFP) zebrafish embryos to visualize the vascular endothelium in response to tail amputation at 3 days post-fertilization (dpf) (Fig. 1A, B). Embryos in the control group developed a complete loop formation connecting the dorsal aorta (DA) with the dorsal longitudinal anastomotic vessel (DLAV) at 3 days post-amputation (dpa) (Fig. 1C), accompanied with restored blood flow to the amputated site (Supplementary Video S1A; Supplementary Data are available online at
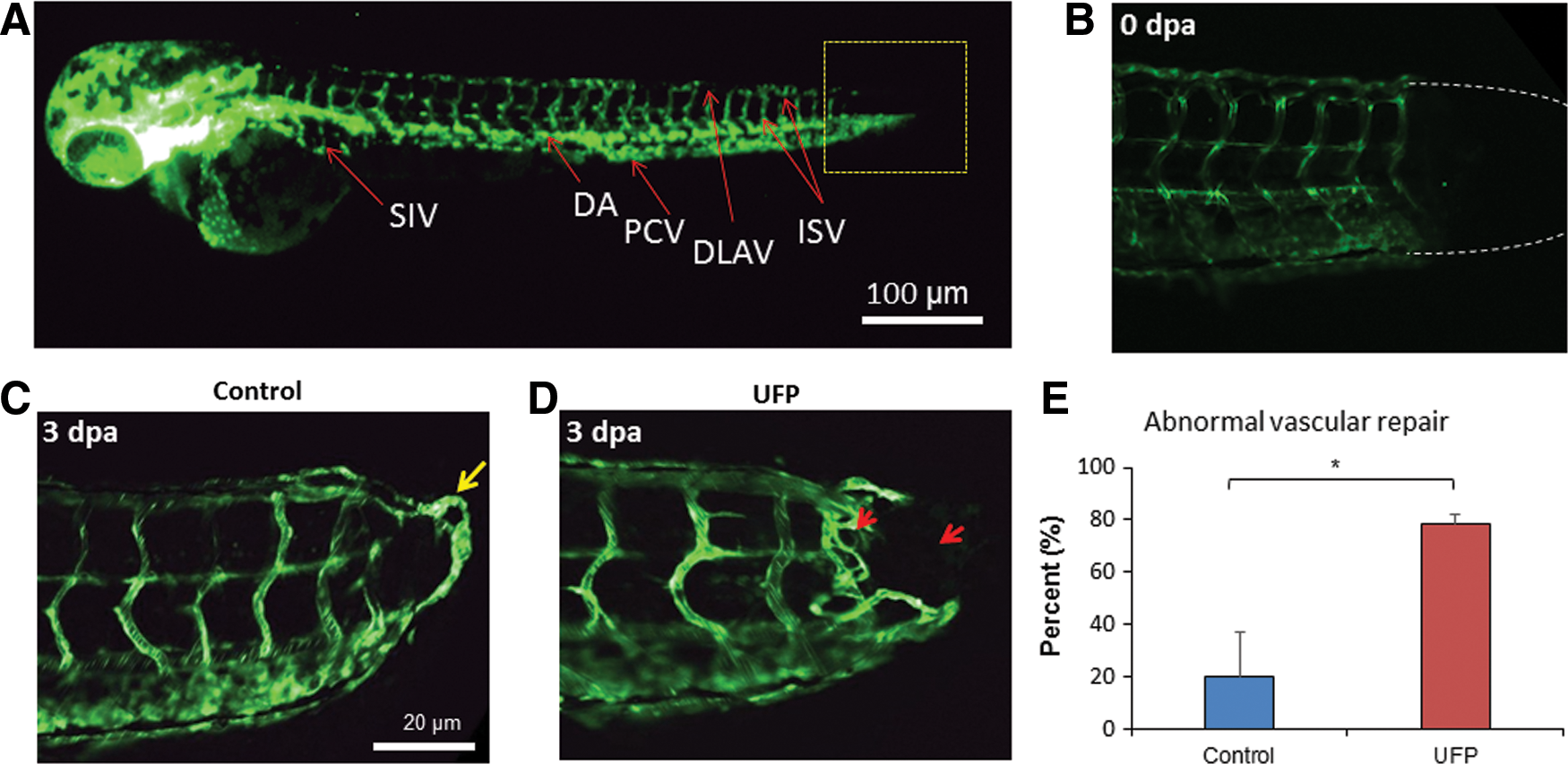
UFP exposure down-regulated Notch-related genes
To assess Notch signaling in response to UFP exposure, we used the pJH26 Notch reporter. UFP significantly reduced Notch activity in human aortic endothelial cells (HAECs) in a dose-dependent manner (*p < 0.05 vs. control, n = 3) (Fig. 2A), accompanied with down-regulation of Notch signaling-related genes, including Notch ligand Dll4 and Notch target Hes1, in a dose- and time-dependent manner (*p < 0.05 vs. control, n = 3) (Fig. 2B, C). In the zebrafish embryos, UFP exposure also down-regulated Notch signaling-related genes, including Notch ligands (JAG1 and JAG2), the Notch receptor (Notch1b), and Notch targets (Hey2 and Hes1) (*p < 0.05 vs. control, n = 3) (Fig. 2D). The UFP-mediated attenuation in Notch signaling was corroborated by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) inhibitor (GI254023X) that inhibits Notch receptor activation (Fig. 2D). Thus, UFP inhibited Notch activity and down-regulated Notch-related mRNA expression in both HAEC and zebrafish embryos.
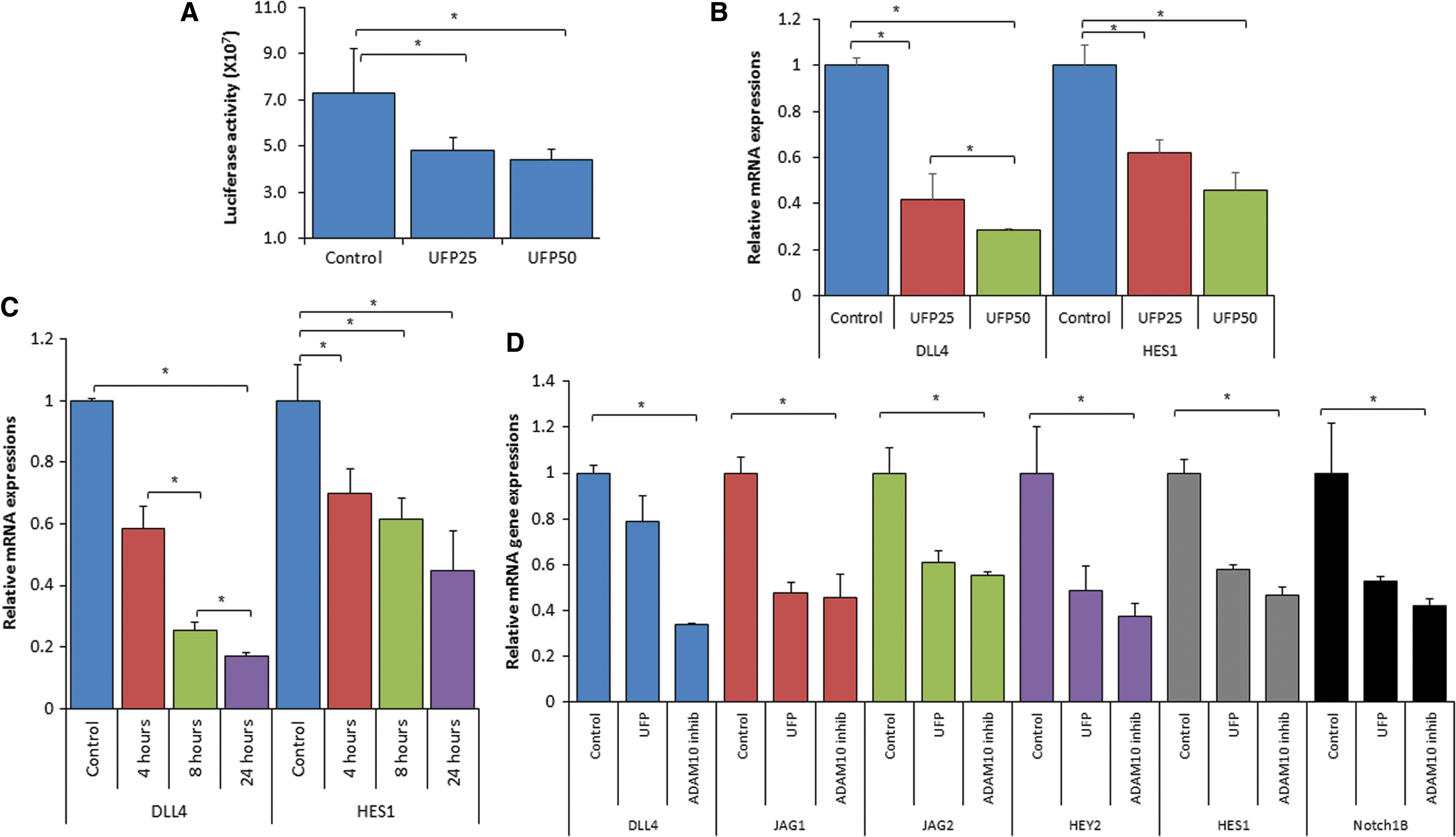
UFP-attenuated Notch signaling impaired vascular regeneration
We further performed gain- and loss-of-function analyses to validate Notch signaling-mediated vascular regeneration in the transgenic Tg(fli1:GFP) embryos. Similar to the effects of UFP, ADAM10 inhibitor, GI254023X, impaired vascular regeneration after tail amputation (*p < 0.05, n = 20) (Fig. 3A, B). However, rescue with NICD mRNA attenuated the proportion of embryos with ADAM10 inhibitor- and UFP-impaired vascular regeneration from 65% to 31% and from 80% to 31%, respectively (*p < 0.05, n = 20) (Fig. 3C). To further assess whether the reduction in Notch signaling impairs vascular regeneration, we constructed a dominant negative Notch 1b (DN-Notch1b) mRNA that inhibits 96% of Notch signaling (Supplementary Fig. S3). Seventy five percent of embryos injected with DN-Notch1b mRNA developed abnormal vascular regeneration and exhibited embryonic lethality (*p < 0.05, n = 20), which was reduced to 50% by NICD mRNA rescue (Fig. 3C). In agreement with DN-Notch1b mRNA, UFP exposure during embryogenesis retarded development and promoted embryonic lethality (Supplementary Fig. S5). Hence, UFP-attenuated Notch signaling is implicated in impaired vascular regeneration.
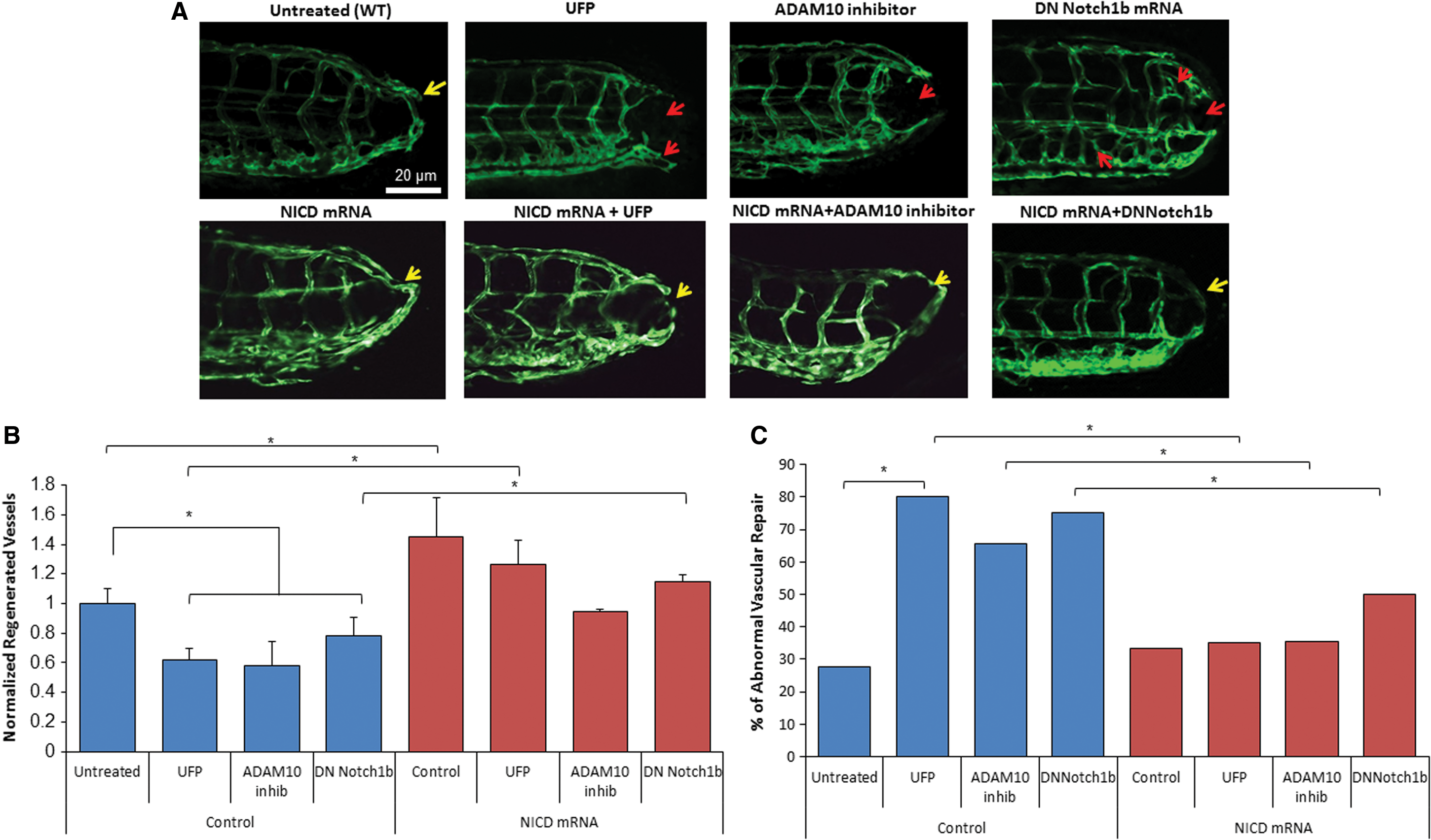
Reduced endothelial Notch activity impaired vascular regeneration
We further crossbred the Notch reporter transgenic fish Tg(tp1:GFP) with Tg(flk1:mCherry) line to demonstrate Notch activity-mediated vascular regeneration. The tp1 (Epstein Barr Virus terminal protein 1) in the Notch reporter line contains two Rbp-Jκ binding sites for NICD, thereby reporting regional Notch1b activation (28). Vascular endothelial Notch activity (as visualized in yellow) was reduced after UFP exposure, ADAM10 inhibitor treatment, or injection of DN-Notch1b mRNA, accompanied with incomplete vascular loop closure (*p < 0.05 vs. control, n = 29 for control, n = 28 for UFP, n = 29 for ADAM10, n = 15 for DN-Notch1b); whereas NICD mRNA injection rescued endothelial Notch activity and restored vascular regeneration (Fig. 4A). Endothelial Notch activity was further quantified by a customized MATLAB algorithm to color-code the accentuation of co-localized Notch activity in the vasculature (Fig. 4B). These findings further support that UFP exposure inhibits endothelial Notch activity to impair vascular network formation.

UFP exposure attenuated FOXO1-mediated Notch activation complex
To elucidate the mechanism underlying UFP-mediated reduction in Notch activity, we analyzed the protein levels of NICD and Notch co-activators to form the activation complex. The total, active, and nuclear NICD protein levels remained unchanged in HAEC after 6 h of UFP exposure (Fig. 5A). Consistently, intact level of NICD protein was observed in response to various UFP exposure time (2–6 h; *p < 0.05 vs. control, n = 3) (Supplementary Fig. S6). In contrast, FOXO1 expression, but not MAML1 expression, was significantly reduced after 6 h post-initial UFP exposure (*p < 0.05 vs. control, n = 3) (Fig. 5B). FOXO1 mRNA expression was also reduced in a dose- and time-dependent manner by UFP treatment (*p < 0.05 vs. control, n = 3) (Fig. 5C). Treatment of the proteasome inhibitor, MG-132, in the presence of UFP restored UFP-attenuated FOXO1 protein expression, suggesting that UFP also stimulate the degradation of FOXO1 via proteasome (Fig. 5D) (*p < 0.05 vs. control, n = 3). To determine whether suppressing FOXO1 expression affects the formation of the Notch activation complex, we performed immunoprecipitation and immunofluorescence against NICD and FOXO1, respectively. Immunoprecipitation with different UFP exposure time (2–6 h) revealed that UFP suppressed FOXO1 expression 4–5 h post-initial exposure, whereas it significantly attenuated FOXO1-mediated NICD pull-down 5 and 6 h after exposure (Fig. 5E). Immunofluorescence further uncovered a reduction in NICD and FOXO1 co-localization (*p < 0.05, n = 3) (Fig. 5F). Silencing FOXO1 with small interfering RNA (siRNA) recapitulated the UFP-mediated reduction in Notch activation complex formation (*p < 0.05, n = 3) (Fig. 5E), and it down-regulated Notch signaling gene expression. These data corroborate FOXO1-mediated Notch signaling (Supplementary Fig. S7), and UFP suppression of FOXO1 expression, leading to a reduction in FOXO1/Notch activation complex.

FOXO1 is an essential co-activator for the Notch activation complex for vascular repair
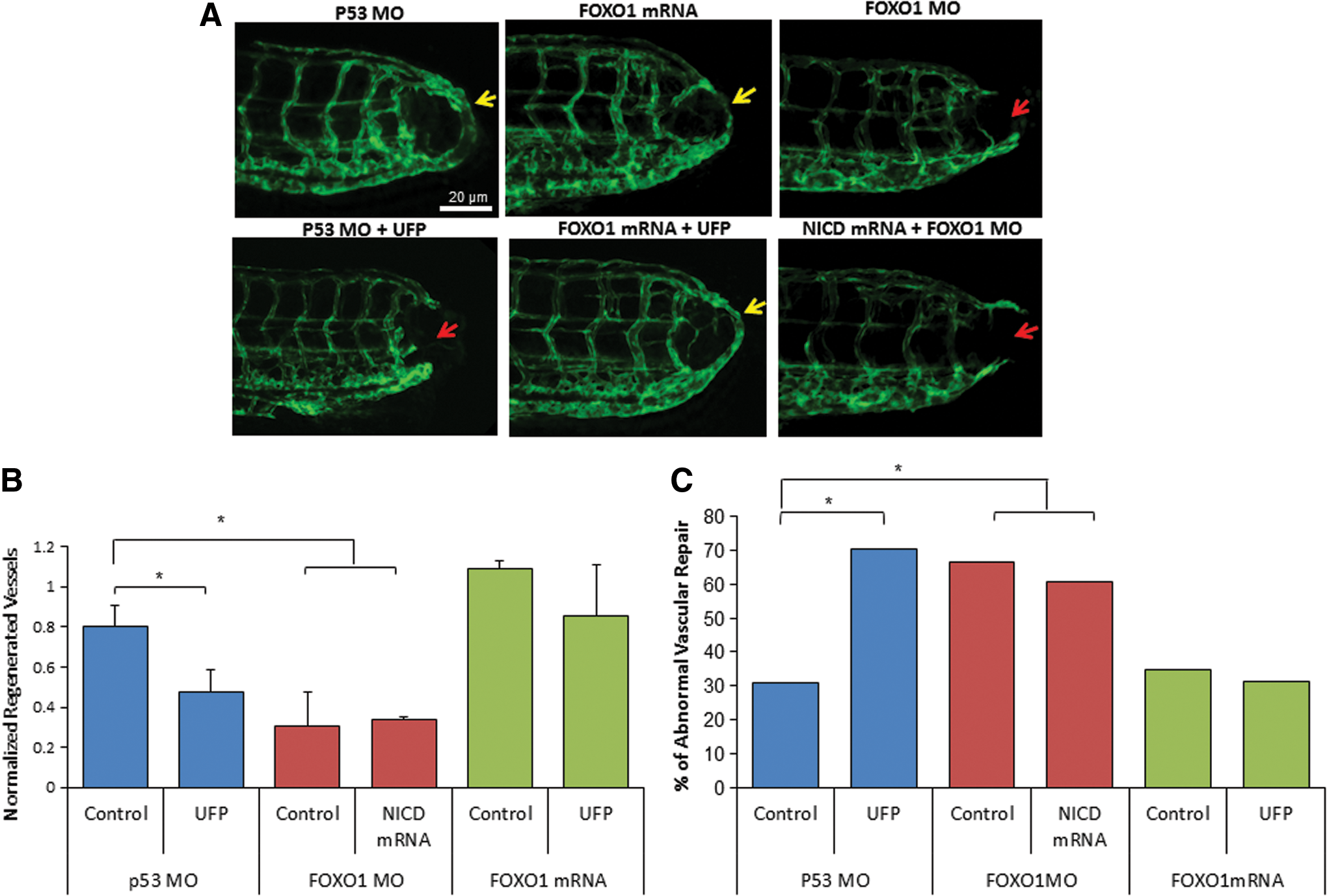
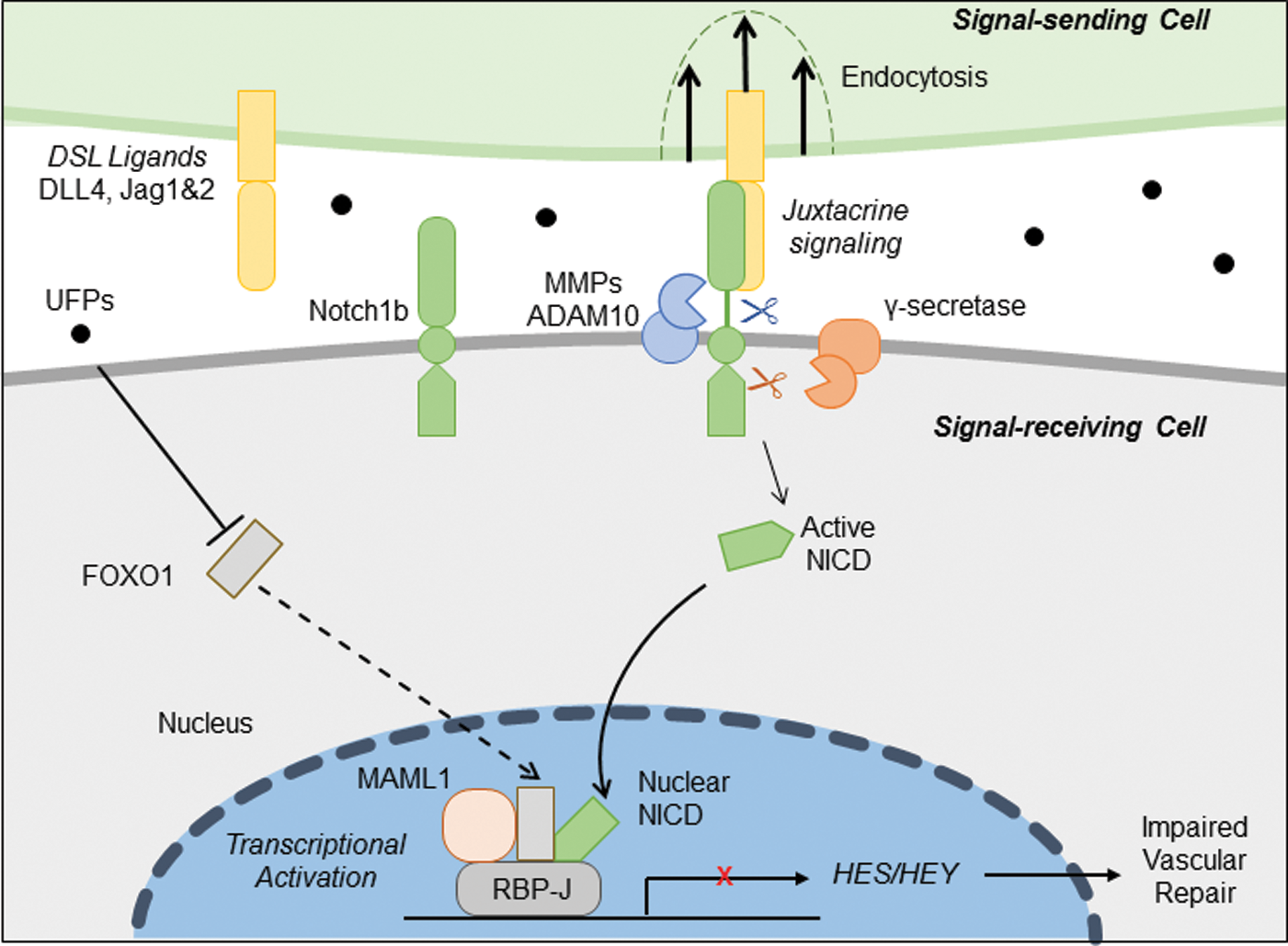
To assess the importance of FOXO1/Notch activation complex for vascular regeneration, we performed gain- and loss-of-function analyses on FOXO1. Wholemount zebrafish immunofluorescence staining with anti-FOXO1 validated the reduction of FOXO1 expression after FOXO1 morpholino oligonucleotide (MO) micro-injection (Supplementary Fig. S7A). Embryos injected with P53 control MO developed normal vascular repair at 3 dpa (*p < 0.05, n = 20), whereas FOXO1 MO impaired vascular regeneration analogous to UFP-mediated effect (Fig. 6A). FOXO1 mRNA rescue restored regeneration despite the presence of UFP (*p < 0.05, n = 25), whereas co-injection of FOXO1 MO with NICD mRNA failed to restore vascular regeneration (*p < 0.05, n = 17) (Fig. 6A–C). FOXO1 MO attenuated Notch activity in the Tg(tp1:GFP, flk1:mCherry) embryos (Fig. 7 and Supplementary Fig. S7B), and co-injection of FOXO1 MO and NICD mRNA partially restored Notch activity (*p < 0.05, n = 15) (Fig. 7A, B). Taken together, FOXO1 is an essential co-activator of the Notch activation complex, and UFP inhibits FOXO1-mediated Notch signaling to impair vascular regeneration after tail amputation (Fig. 8).
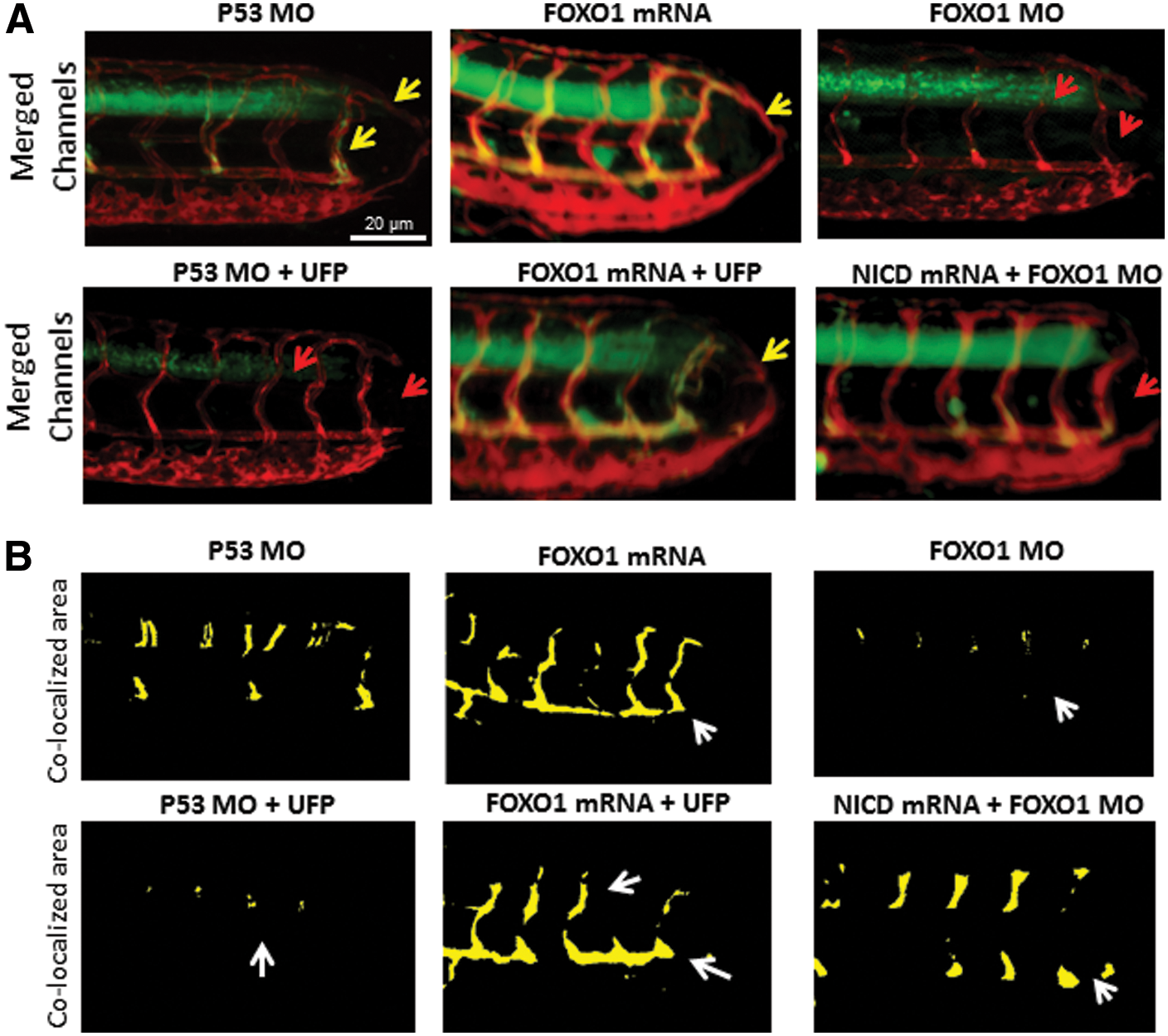
Discussion
The novel contribution of our study lies in the elucidation of UFP-mediated disruption in FOXO1/Notch complex to impair vascular network formation. DN-Notch 1b mRNA injection or ADAM10 inhibitor treatment supports Notch signaling as the mechanism underlying UFP-impaired vascular regeneration. We further uncovered that UFP attenuated FOXO1 expression and FOXO1/NICD co-localization. Although NICD mRNA rescue partially restored Notch activity, FOXO1 mRNA rescue completely restored UFP-attenuated Notch activity and blood flow to the injured site. Hence, FOXO1 is an essential co-activator for the Notch activation complex for Notch signaling-mediated vascular regeneration.
The Notch signaling pathway regulates stem cell differentiation and proliferation (6, 39, 51, 54, 61). Ablation of Notch1 induces developmental retardation, followed by collapsed arterial/venous specification and vascular malformation (27). Dysregulated Notch activity results in abnormal endothelial proliferation, leading to a hyperplastic vascular network that is prone to developing cancer (3). Missense mutation in the Notch3 gene is the underlying cause of the degenerative vascular disease known as Cerebral Autosomal-Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) (23). In addition, Notch signaling is implicated in the initiation of sprouting angiogenesis (16, 37). After UFP exposure, zebrafish embryos developed impaired vascular repair and aberrant appearance in intersomatic space (Figs. 1 and 3), accompanied with reduced Notch activity and down-regulation of Notch target genes (Fig. 2). We further corroborated UFP-mediated reduction in Notch activity by using the transgenic Tg(tp1:GFP) Notch activity reporter line (Figs. 3 and 7). In addition, ADAM10 inhibitor or DN-Notch1b mRNA strengthened the role of Notch signaling in restoring vascular network formation. As a corollary, UFP attenuated vascular endothelial cell migration and tube formation (Supplementary Fig. S2). Thus, UFP attenuates Notch signaling to impair vascular regeneration.
NICD, MAML1, and FOXO1 bind to the CSL domain to form a Notch activation complex. Although UFP inhibited Notch activity, NICD and MAML protein levels remained unchanged. For this reason, we uncovered down-regulation of FOXO1 mRNA and protein expression after UFP exposure (Fig. 5). FOXO1 and Notch cooperation has been reported in myogenic differentiation and neural stem cell differentiation (25, 26). Our data suggest that UFP exposure suppressed FOXO1-mediated Notch activation complex formation, thus attenuating Notch signaling for vascular regeneration.
Exposure to ambient PM2.5 promotes cardiovascular, pulmonary, and gastrointestinal disorders (7). Epidemiological studies associated maternal exposure to air pollutants with increased occurrence of congenital heart diseases (12). Maternal exposure to ozone (O3) at the second-month gestation increases the risk of aortic and pulmonary valve anomalies (52). UFP are the redox-active sub-fraction of PM2.5, harboring elemental carbon and polycyclic aromatic hydrocarbons as products of incomplete combustion from urban environmental sources, including the exhaust from diesel trucks and gasoline vehicles (30). Their large surface-to-volume ratio favors their potential adsorption to or absorption in the pulmonary and cardiovascular systems (13, 44, 56). Long-term epidemiological studies on UFP exposure demonstrate adverse respiratory function (40, 49), exercise-induced cardiac ischemia (47), and arrhythmias (17). At the molecular level, UFP induces JNK-mediated superoxide (O2 ·−) production and NF-κB-mediated monocyte recruitment to prime atherosclerosis (32, 34). UFP exposure via inhalation increases plasma lipid metabolites and reduces high density lipoprotein (HDL) anti-oxidant capacity to accelerate atherosclerosis in low density lipoprotein receptor (LDLR)-null mice (31). UFP inhalation further promotes atherogenic lipid metabolites and macrophage infiltrates in the intestine (30), where microbiota composition was altered to produce atherogenic lipid metabolites (35). In our zebrafish model of vascular injury and repair, we provide the first molecular insights into UFP-suppressed FOXO1 as a co-activator for the Notch activation complex.
The emerging role of redox-sensitive microRNAs (miRNAs), including miR-223 and −375, has been implicated in disrupting FOXO1 signaling (45) and cellular proliferation (63, 65). Further, miR-154 and −379 have been reported to regulate inflammatory responses (21, 46). Although miR-3188 targets the mTOR pathway to suppress p-PI3K/p-AKT/c-JUN signaling (67), miR-132 activates the PI3K/AKT pathway to decrease FOXO1 expression (36). PM2.5 has been reported to modulate the levels of a number of miRNAs, including miR-223 and miR-375 (5, 53, 64). Thus, UFP could down-regulate FOXO1 by modulating miRNAs. In addition, we have previously reported that UFP activates JNK, and JNK activation promotes protein ubiquitination and degradation via proteasome activity (32, 59). In this study, we observed that UFP down-regulated FOXO1 protein levels via proteasome degradation (Fig. 5D), which could be mediated by the JNK pathway (Supplementary Fig. S9). However, the precise mechanism whereby UFP exposure suppresses FOXO1 expression warrants further investigation (19).
Overall, we demonstrate FOXO1 as an important co-activator with NICD for assembling the Notch activation complex for vascular development and regeneration (11). UFP-mediated reduction in FOXO1/NICD complex formation was corroborated by immunoprecipitation of FOXO1-mediated NICD pull-down, immunofluorescence of FOXO1 expression, and FOXO1/NICD co-localization. Thus, exposure to redox-active UFP disrupts Notch activity to impair vascular network formation.
Materials and Methods
UFP collection
UFP were collected at the University of Southern California (USC) campus near downtown Los Angeles, California. UFP are characterized by a mixture of particulate pollutants, including ambient PM from heavy duty diesel trucks, light duty gasoline vehicles, and PM generated by photochemical oxidation of primary organic vapors (60). UFP were collected with High-Volume Particle Sampler, operating at 400 L/min (42), on Zefluor PTFE membrane filters (3 μm; Pall Life Sciences). Collected PM samples were extracted from the filter substrates by soaking and vortexing in ultrapure Milli-Q water followed by sonication and neutralization. Metal and organic content of the UFP samples was quantified by using inductively coupled plasma mass spectrometry and Siever 900 Organic Carbon Analyzer, respectively, as reported (18, 58). Mass fraction of the total organic carbon and major metals (in units of ng/μg UFP) are summarized in Supplementary Figure S1.
Vascular endothelial cell culture and exposure to UFP
HAECs were cultured in endothelial growth medium (Cell Applications, San Diego, CA) supplemented with 5% fetal bovine serum (FBS), 1% penicillin/streptomycin (Life Technologies), and 0.1% fungicide. HAEC were propagated for experiments between passages 4 and 7, and were treated with UFP, ADAM10 inhibitor, GI254023X, or proteasome inhibitor, MG-132 (Sigma-Aldrich, St. Louis, MO) at indicated concentrations diluted in M199 media (Life Technologies).
Preparation of NICD, FOXO1, and DN-Notch1b mRNA
Rat NICD and mouse FOXO1 complementary DNA (cDNA) were amplified from donor plasmids and cloned into the plasmid pCS2+ at the BamHI/EcoRI sites (for NICD) and BamHI/XbaI sites (for FOXO1), respectively. Zebrafish DN-Notch1b cDNA was amplified from zebrafish cDNA with primers excluding the intracellular domain and cloned into pCS2+ at EcoRI/XhoI sites. Clones with insert of interest were selected by polymerase chain reaction (PCR) screening and validated with sequencing. mRNAs were prepared by using the mMessage SP6 kit (Invitrogen) by following the manufacturer's instruction. The in vitro transcribed mRNAs were purified by using a total RNA isolation kit (Bio-Rad) for in vivo rescue experiments.
Notch reporter activity assay
HEK-293 cells were grown to sub-confluence in 24-well plates. The cells were transfected with the Notch reporter plasmid pJH26 with or without control or DN-Notch1b plasmid overnight by using Lipofectamine 2000 (Thermo Fisher Scientific). The cells were treated overnight with UFP at different concentrations in M199 containing 0.1% FBS. The cells were lysed in Passive Lysis Buffer (Promega), and luciferase activities were quantified with a Luminometer using Bright-Glow substrate (Promega).
Quantitative real-time PCR analysis
Notch signaling-related gene mRNA expression patterns, including Notch ligands DLL4, JAG1 and JAG2, Notch receptor Notch1b, downstream target Hey2, and FOXO1, were assessed by quantitative real-time PCR. RNA was isolated by using the Bio-Rad total RNA kit (Bio-Rad), and it was synthesized into cDNA by using the iScript cDNA synthesis kit (Bio-Rad). Synthesized cDNAs were diluted in the molecular biology reagent water (Sigma-Aldrich) for PCR amplification with qPCR master mix (Applied Biological Materials, Inc.). The individual mRNA expression patterns were normalized to actin expression. Sequences of primers and MOs are listed in Table 1.
DN, dominant negative; FOXO1, forkhead box O-subfamily; MO, morpholino oligonucleotide; NICD, notch intracellular cytoplasmic domain.
Western blot analysis and immunoprecipitation
HAEC treated with or without UFP were lysed with standard RIPA buffer supplemented with phosphatase inhibitor as previously described (33). Cytosolic and nuclear lysates were prepared as previously described (29). Protein concentrations of each sample were determined by DCP assay. Western blotting was done as previously described (33) with anti-FOXO1 (H-128; Santa Cruz Biotechnology, Inc.), anti-MAML1 (EMD Millipore, Inc.), and anti-NICD (V1744; Cell Signaling Technology, Inc.). Equal loading was verified with anti-β-tubulin (AA2; Santa Cruz Biotechnology, Inc.). Blot densitometry was performed with the FluorChem FC2 imaging software for chemiluminescence. Immunoprecipitation of FOXO1 and NICD was performed with Pierce Crosslink magnetic IP/CI-IP kit (Thermo Fisher Scientific) by following the manufacturer's instruction. Elution of imunoprecipitation was neutralized with neutralize buffer (Thermo Fisher Scientific) for Western blot analysis.
Assessment of vascular regeneration with the transgenic Tg(fli1:GFP) zebrafish tail amputation model
The transgenic Tg(fli1:GFP) zebrafish line, in which vascular endothelial cells were labeled with GFP under the promoter of Fli1 (also known as ERGB), was used to image the vascular regeneration. Tg(fli1:GFP) fish embryos were injected with or without NICD mRNA, P53 control MO (GeneTools, LLC), FOXO1 MO (GeneTools, LLC), FOXO1 mRNA (2–4 pg), or DN-Notch1b mRNA, and they were cultured in standard E3 medium supplemented with 0.05% methylene blue at 28.5°C for 3 days. At 3 dpf, ∼100 μm of the posterior tail segments was amputated by using a clean razor blade under a phase-contrast microscope (Zeiss). Embryos were then returned to fresh E3 medium, or E3 medium with UFP or ADAM10 inhibitor. At 3 dpa, embryonic zebrafish were randomly picked and immobilized in neutralized 0.02% tricaine solution (Sigma-Aldrich), and they were mounted in 1–2% low-melting agarose (Sigma-Aldrich) on a glass coverslip to image regeneration of blood vessels with confocal microscopy (Zeiss).
Notch activity in double transgenic Tg(tp1:GFP; flk1:mCherry) embryos
The Tg(tp1:GFP) Notch reporter line was crossbred with the Tg(flk1:mCherry) line to localize endothelial Notch signaling. NICD mRNA, FOXO1 MO, and FOXO1 mRNA were micro-injected to modulate Notch signaling and to elucidate FOXO1/Notch cooperation. At 6 dpf (3 dpa), embryos were randomly selected to image Notch activation in the central nervous system and in the amputated tail. Embryos from the remaining group were sorted and treated with and without UFP and ADAM10 inhibitor. After 3 days of treatment, Notch signaling localized in the vasculature was scanned at 3–5 μm intervals in the Z direction by using dual-channel confocal microscopy (Zeiss). Images of Notch activity and the vascular endothelial layer were acquired and superimposed to visualize endothelial Notch activation. Images from each channel are displayed as Supplementary Figure S8. Stacked images from both channels were projected onto a single plane by using ImageJ to visualize three-dimensional (3D) Notch activation.
Immunofluorescence staining
After UFP exposure, HAECs were fixed with 4% paraformaldehyde (PFA) and stained with antibody against NICD and FOXO1 diluted in 2% bovine serum albumin (BSA; Sigma-Aldrich). Images were acquired by using dual-channel confocal microscopy (Zeiss) and superimposed by using ImageJ.
Visualization of FOXO1/NICD co-localization
To accentuate co-localization of NICD and FOXO1, we customized a MATLAB (Mathworks) algorithm. Multi-level image thresholds were applied for segmenting the single slice in each channel. Threshold levels for each channel were manually chosen to generate binary masks of the slice. Pixels defined with binary masks were merged together and visualized as overlapping regions, whereas the remaining regions were regarded as background.
FOXO1 knockdown
Scrambled (Qiagen) and FOXO1 siRNA (Thermo Fisher Scientific) were transfected to HAEC with Lipofectamine 2000 (Invitrogen) as previously described (32). To confirm FOXO1 knockdown, cells were applied to immunofluorescence staining and imaged under fluorescent microscopy (Zeiss) at 48 h post-transfection.
Endothelial cell migration and tube formation assays
For migration assays, HAEC monolayers at confluence were scratched with 200 μL pipette tips. After the scratch, cells were treated with or without UFP at 25 μg/mL in M199 media (Life Technologies). Cell migration was imaged under phase-contrast microscopy (Olympus IX 70) at 4, 8, 12, and 24 h. For the quantification of cell migration, distances between each inner border of cells were assessed by using ImageJ. For tube formation assays, confluent HAEC were pre-treated with or without UFP at 25 μg/mL and re-suspended in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 25 ng/mL VEGF and 5% FBS. Cells were then added to 96-well plates coated with growth factor-reduced Matrigel (BD Biosciences) and were incubated with or without UFP. Tube formation was imaged under phase-contrast microscopy at 4, 8, 12, and 24 h.
Quantification of the regenerated intersegmental vessels
To quantify changes in vascular repair, we used Amira 3D imaging software. The whole vasculature on the posterior tail segment was masked (purple). The area of regenerated vessels was derived manually by designating segmental vessels (pink) connecting the DA and the DLAV (Supplementary Fig. S4).
Imaging blood flow on the tail post-regeneration
Double transgenic Tg(fli1:GFP; gata1:DS-RED) zebrafish underwent tail amputation at 3 dpf, and they were maintained with or without UFP at 25 μg/mL for 3 days. At 3 dpa, fish were immobilized and placed in 1–2% low-melting agarose to image the circulation of erythrocytes through the regenerated vessel. Images of the blood flow and endothelial layer were taken separately with QIcam at 20–30 frames per second. Images were superimposed by using Corel imaging software.
Wholemount zebrafish immunofluorescence staining
WT zebrafish embryos were injected with P53 MO and FOXO1 MO (GeneTools, LLC), respectively, and they were maintained in standard E3 medium at 28.5°C for 3 days. At 3 dpf, embryos were fixed in 4% PFA solution overnight and were subjected to pure acetone for dehydration. Embryos were then rehydrated with 0.2% PBST and were blocked with 2% BSA (Sigma-Aldrich). Zebrafish embryos underwent 24 h of incubation with FOXO1-targeted antibody (C-9; Santa Cruz Biotechnology, Inc.) to assess reduction of FOXO1. Fluorescent images were acquired by using a confocal microscope (Zeiss) by mounting embryos in 1–2% low-melting agarose.
Statistical analysis
Data were expressed as mean ± standard deviation and compared among separate experiments. Unpaired two-tail t test and two-proportion z-test were used for statistical comparisons between two experimental conditions. p Values <0.05 were considered significant. Comparisons of multiple values were made by one-way analysis of variance, and statistical significance for pairwise comparison was determined by using the Tukey test.
Footnotes
Acknowledgments
The authors are grateful to Dr. Weinmaster for providing the NICD plasmid and the JH26 Notch reporter plasmid, and they thank Drs. David Traver at UCSD and Nathan Lawson at the University of Massachusetts Medical School (Worcester, MA) for generously providing the Tg(tp1:GFP) line. This study was supported by National Institutes of Health R01HL083015 (T.K.H.), R01HL111437 (T.K.H.), R01HL129727 (T.K.H.), R01HL118650 (T.K.H.), and the American Heart Association 16SDG30910007 (R.R.S.P.), and it was funded by EPA under STAR program (C.S.) and the South Coast Air Quality Management District Award (C.S.).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.