
Abstract
Aims:
Protein tyrosine phosphatase-1B (PTP1B) is a negative regulator of receptor tyrosine kinase signaling. In this study, we determined the importance of PTP1B expressed in endothelial cells for the vascular response to arterial injury in obesity.
Results:
Morphometric analysis of vascular lesions generated by 10% ferric chloride (FeCl3) revealed that tamoxifen-inducible endothelial PTP1B deletion (Tie2.ERT2-Cre × PTP1Bfl/fl; End.PTP1B knockout, KO) significantly increased neointima formation, and reduced numbers of (endothelial lectin-positive) luminal cells in End.PTP1B-KO mice suggested impaired lesion re-endothelialization. Significantly higher numbers of proliferating cell nuclear antigen (PCNA)-positive proliferating cells as well as smooth muscle actin (SMA)-positive or vascular cell adhesion molecule-1 (VCAM1)-positive activated smooth muscle cells or vimentin-positive myofibroblasts were detected in neointimal lesions of End.PTP1B-KO mice, whereas F4/80-positive macrophage numbers did not differ. Activated receptor tyrosine kinase and transforming growth factor-beta (TGFβ) signaling and oxidative stress markers were also significantly more abundant in End.PTP1B-KO mouse lesions. Genetic knockdown or pharmacological inhibition of PTP1B in endothelial cells resulted in increased expression of caveolin-1 and oxidative stress, and distinct morphological changes, elevated numbers of senescence-associated β-galactosidase-positive cells, and increased expression of tumor suppressor protein 53 (p53) or the cell cycle inhibitor cyclin-dependent kinase inhibitor-2A (p16INK4A) suggested senescence, all of which could be attenuated by small interfering RNA (siRNA)-mediated downregulation of caveolin-1. In vitro, senescence could be prevented and impaired re-endothelialization restored by preincubation with the antioxidant Trolox.
Innovation:
Our results reveal a previously unknown role of PTP1B in endothelial cells and provide mechanistic insights how PTP1B deletion or inhibition may promote endothelial senescence.
Conclusion:
Absence of PTP1B in endothelial cells impairs re-endothelialization, and the failure to induce smooth muscle cell quiescence or to protect from circulating growth factors may result in neointimal hyperplasia.
Introduction
Obesity is a major cardiovascular risk factor; however, the pathomechanisms underlying the link between increased body weight and cardiovascular disease are not completely understood. Among others, obesity and metabolic dysfunction are associated with an increased expression of protein tyrosine phosphatase-1B (PTP1B) in brain and metabolic tissues (e.g., skeletal muscle, adipose tissue) (1). PTP1B is a negative regulator of receptor tyrosine kinase signaling, and previous work suggested that PTP1B overexpression underlies the development of insulin and leptin resistance in obesity (16, 29). Conversely, systemic PTP1B deficiency (16, 26) or deletion of PTP1B specifically in brain (6) or proopiomelanocortin (POMC) neurons (3) was shown to protect against diet-induced obesity and the associated metabolic dysfunctions. Pharmacological PTP1B inhibitors have been suggested as therapeutic option to improve or prevent type 2 diabetes and insulin resistance associated with obesity (34).
Obesity and diabetes mellitus are associated with increased (endothelial) protein tyrosine phosphatase-1B (PTP1B) expression, and PTP1B inhibitors have been explored to treat the metabolic dysfunction associated with obesity. In this study, we show that genetic knockdown of PTP1B in endothelial cells aggravates neointima formation in diet-induced obese mice and is associated with reduced lesion re-endothelialization, increased neointimal cell proliferation and activation, enhanced tyrosine kinase and transforming growth factor-beta (TGFβ) signaling, as well as oxidative stress. Mechanistically, genetic knockdown or pharmacological inhibition of PTP1B increased caveolin-1 expression in murine and human endothelial cells and also induced a senescent phenotype and impaired in vitro re-endothelialization, which could be prevented by caveolin-1 downregulation or the antioxidant Trolox. Our results indicate that absence of PTP1B negatively affects endothelial cell properties during vascular wound healing and that antioxidants could be considered to accompany any therapeutic approaches to inhibit PTP1B.
PTP1B is highly expressed in endothelial cells, and we have previously demonstrated elevated levels of PTP1B in endothelial progenitor cells (EPCs) isolated from obese individuals (24). Importantly, PTP1B expression in EPCs decreased following weight loss to levels observed in lean subjects, and both weight loss and pharmacological inhibition of PTP1B restored EPC responsiveness to the angiogenic activities of the adipokine leptin. Pharmacological inhibition or systemic gene deletion of PTP1B has been shown to protect against endothelial dysfunction associated with type 1 diabetes (2, 25) or heart failure (42). Nevertheless, the contribution of endothelial PTP1B expression to cardiovascular risk in obesity is largely unknown.
In addition to new vessel formation and vascular function regulation, endothelial cells play a major role in the control of vascular smooth muscle cell (SMC) proliferation and migration and the restoration of vascular integrity following injury (23). Clinical studies have shown that obesity is an important risk factor for clinical and angiographic restenosis following coronary stent implantation (33, 35). Vascular lesion re-endothelialization following endothelial denudation involves growth factor and tyrosine kinase receptor signaling (9), however, the causal role of PTP1B expressed in endothelial cells and, in particular, endothelial PTP1B overexpression in obesity, for lesion re-endothelialization and neointima formation have never been examined.
In this study, we examined the effect of endothelial-specific PTP1B deletion on vascular response to injury in mice with diet-induced obesity. Our findings suggest that genetic deletion of PTP1B in endothelial cells impairs lesion re-endothelialization and results in increased neointima formation in mice, and findings in human and murine endothelial cells that deletion or inhibition of PTP1B was associated with morphological signs of senescence, increased expression of cell cycle inhibitors, and impaired re-endothelialization may explain our in vivo observations. Importantly, parallel “treatment” with an antioxidant or downregulation of the PTP1B substrate and endothelial cell membrane protein caveolin-1 was able to prevent the detrimental consequences of PTP1B inhibition on endothelial cell properties.
Results
To investigate the importance of PTP1B expressed in endothelial cells for vascular lesion re-endothelialization and neointima formation following injury, mice with conditional, endothelial-specific PTP1B deletion were examined. Successful PTP1B gene excision after tamoxifen feeding was documented in endothelial cells isolated from lungs of endothelial receptor tyrosine kinase Cre recombinase-estrogen receptor fusion protein (Tie2.ERT2-Cre) transgenic (tg) × PTP1Bfl/fl (End.PTP1B knockout, KO) mice and not observed in their Tie2.ERT2-Cre wild type (WT) × PTP1Bfl/fl (End.PTP1B-WT) counterparts (Supplementary Fig. S1A; Supplementary Data are available online at
Analysis of mice, in which a green fluorescent protein (GFP) construct was expressed under control of the Tie2.ERT2-Cre promoter (Tie2.ERT2-Cre tg × IRG), confirmed endothelial-restricted reporter gene expression in uninjured mouse carotid arteries (Supplementary Fig. S1E). Of note, whole blood cell counts (Table 1) and the number of circulating CD11b-positive cells (Table 1 and Supplementary Fig. S2) did not differ between End.PTP1B-WT and End.PTP1B-KO mice.
Whole Blood Cell Counts and Circulating CD11b-Positive Cell Numbers
Data are given as mean ± SEM. Statistical significance was determined using Student's t-test.
End, endothelial; HFD, high-fat diet; KO, knockout; PTP1B, protein tyrosine phosphatase-1B; RBCs, red blood cells; SEM, standard error of the mean; WBCs, white blood cells; WT, wild type.
Endothelial PTP1B deletion enhances neointima formation in diet-induced obese mice
Arterial injury was induced in End.PTP1B-WT and End.PTP1B-KO mice at the common carotid artery using 10% ferric chloride (FeCl3). In mice fed normal chow (n = 11 End.PTP1B-WT and n = 12 End.PTP1B-KO mice), deletion of PTP1B in endothelial cells did not significantly affect neointima formation (not shown). In contrast, morphometric analysis of Verhoeff's elastic stain (VES)-Masson's trichrome (MTC)-stained cross sections through restenotic lesions of End.PTP1B-WT (n = 20) and End.PTP1B-KO (n = 13) mice fed high-fat diet (HFD) revealed that the absence of PTP1B in endothelial cells of obese mice was associated with an increased neointima area (p < 0.001; Fig. 1A) and intima-to-media ratio (p < 0.001; Fig. 1B) and resulted in a significant difference in the degree of lumen stenosis (p < 0.01; Fig. 1C), whereas the media area (p = 0. 372; Fig. 1D) did not differ compared to End.PTP1B-WT mice. Moreover, an increased outward remodeling was observed in End.PTP1B-KO mice (p < 0.05; Fig. 1E). Representative findings of vascular lesions in both genotypes are shown in Figure 1F.
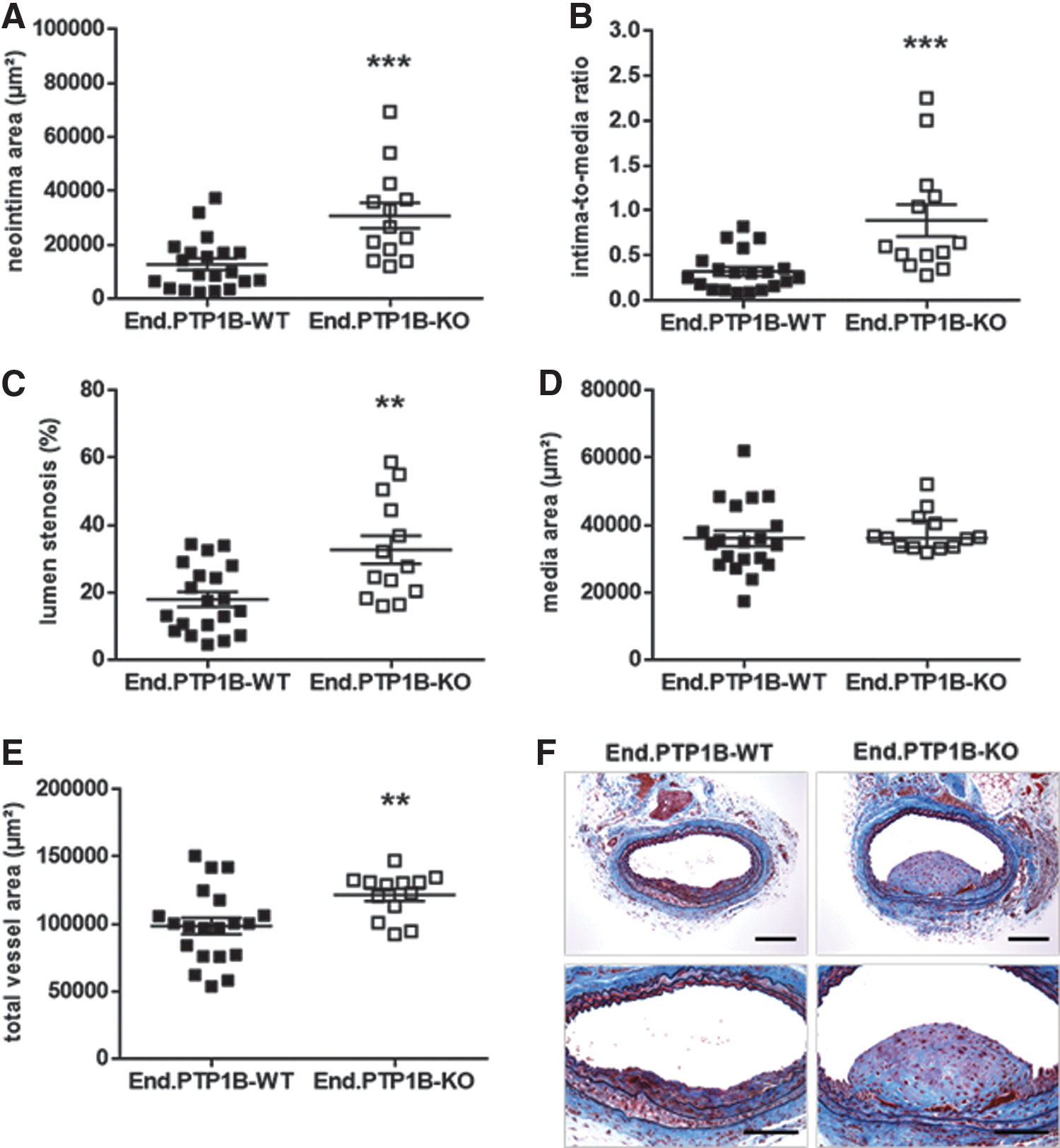
Endothelial PTP1B deletion does not protect against diet-induced obesity
As shown in Table 2, HFD resulted in significantly increased mean body weights, visceral adiposity, and circulating leptin levels in both End.PTP1B-WT and End.PTP1B-KO mice. Nonfasting serum cholesterol, glucose, and insulin levels were higher in obese mice and not affected by endothelial PTP1B deletion, in contrast to previous findings in mice with systemic (16, 26) or brain-specific (6) PTP1B deficiency or deletion of PTP1B in POMC neurons (3). These results suggest that the observed differences in neointima formation between obese End.PTP1B-WT and End.PTP1B-KO mice did not develop secondary to differences in the degree of adiposity or associated metabolic alterations.
Body Weight, Adiposity, and Metabolic Serum Parameter in Nonfasting Mice
Data are given as mean ± SEM.
p < 0.001 and b p < 0.05 versus mice of the same genotype fed NC (determined using one-way analysis of variance).
NC, normal chow; VAT, visceral adipose tissue.
Endothelial PTP1B deletion impairs re-endothelialization in diet-induced obese mice and is associated with neointimal cell proliferation and dedifferentiation
To begin to study the mechanisms underlying our findings of increased neointima formation in the absence of endothelial PTP1B, we began by examining the degree of lesion re-endothelialization. These analyses revealed significantly lower numbers of total cell nuclei (Fig. 2A; representative findings are shown in Fig. 2B) and endothelial lectin-positive cells (Fig. 2C, D) lining the vascular lumen. Of note, endothelial PTP1B deficiency did not affect re-endothelialization in mice fed normal chow (Supplementary Fig. S3). Analysis of human umbilical vein endothelial cells (HUVECs) revealed that inhibition of PTP1B dose dependently impaired their ability to close a wound after scratching of a monolayer (Supplementary Fig. S4A, B).
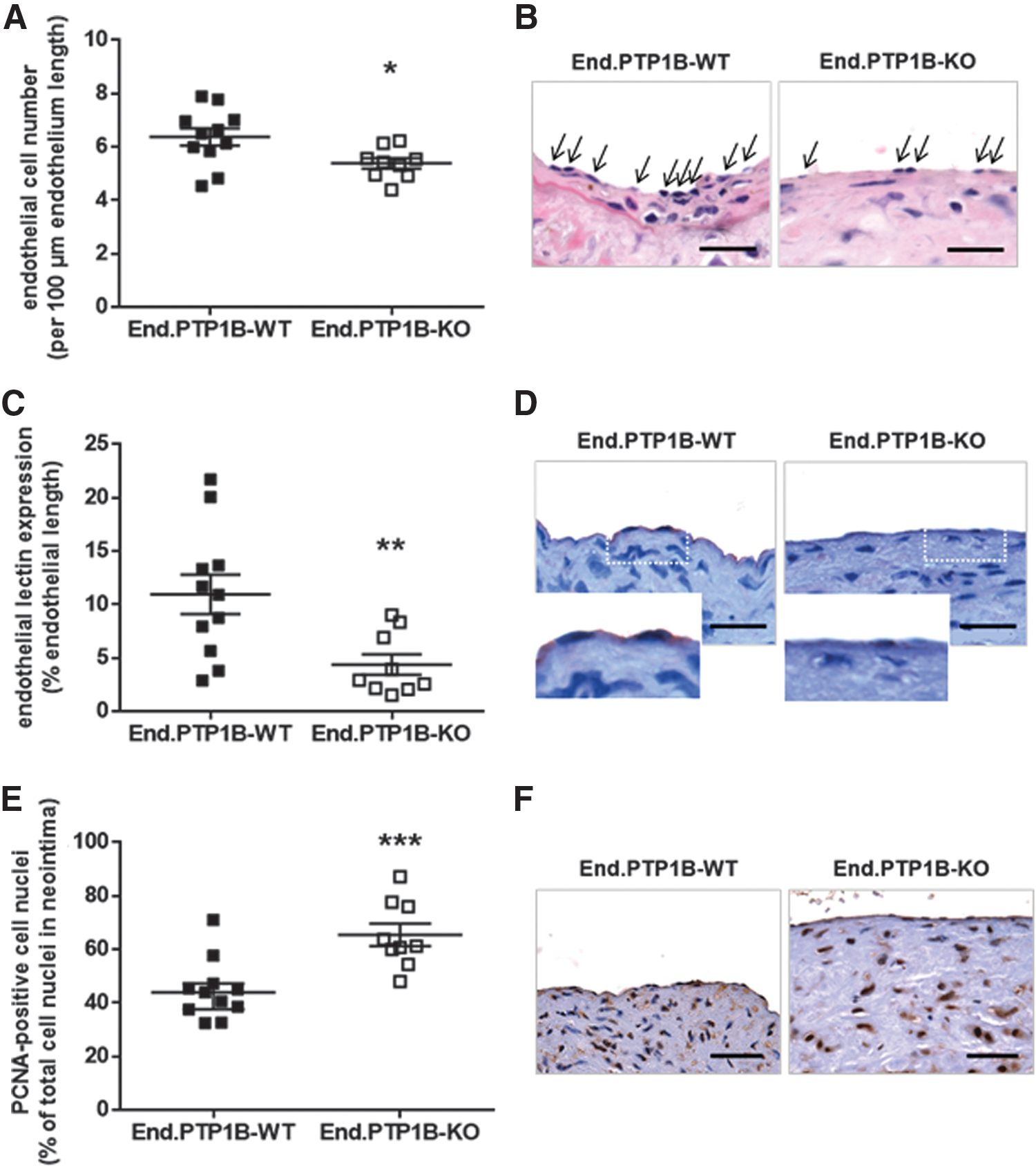
Since the degree of lesion re-endothelialization following vascular injury plays a critical role in controlling the phenotype of intimal SMCs (23), we next examined the cellularity of neointimal lesions. In line with the observed significantly enhanced neointima formation, vascular lesions of End.PTP1B-KO mice exhibited a significantly higher cellularity (178 ± 26 vs. 95 ± 14 cells per neointima; p < 0.01; not shown), and a significantly higher percentage of neointimal cells stained positive for the S-phase-related proliferation marker proliferating cell nuclear antigen (PCNA) (Fig. 2E, F). Moreover, neointimal lesions of End.PTP1B-KO mice exhibited a larger smooth muscle actin (SMA)-immunopositive area (p = 0.06; Fig. 3A, B), which positively correlated with the total number of cell nuclei per neointima (r 2 = 0.669; p < 0.0001; not shown). A significantly higher percentage of cells in the neointima of End.PTP1B-KO mice were immunopositive for vimentin (Fig. 3C, D) or vascular cell adhesion molecule-1 (VCAM1) (Fig. 3E, F), suggesting increased myofibroblast activation in the absence of endothelial PTP1B. Neointimal lesions of End.PTP1B-KO mice also contained significantly higher amounts of interstitial collagen, as detected by Sirius red staining followed by polarization microscopy (Supplementary Fig. S5A, B).

On the contrary, no significant differences were detected with regard to the number of F4/80-positive macrophages within vascular lesions of End.PTP1B-WT and End.PTP1B-KO mice (603 ± 106 [n = 9] vs. 540 ± 134 [n = 9] positive cells per mm2 neointima, respectively; p = 0.721; not shown).
Neointimal lesions of mice with endothelial PTP1B deletion exhibit activated growth factor signaling and increased biomarkers of oxidative stress
Growth factors released from activated platelets, including PDGF and transforming growth factor-beta (TGFβ), have been implicated in neointima formation following vascular injury and thrombosis (9, 38), and defects in endothelial integrity and wound closure in the absence of PTP1B may have exposed neointimal SMCs and other cells to these as well as other mitogens. To examine PDGF and TGFβ signaling activity in the neointima, Src tyrosine kinase and SMAD family member-2/3 (Smad2/3) phosphorylation was examined using immunohistochemistry. As shown in Figure 4, End.PTP1B-KO mice exhibited a significantly increased number of neointimal cells expressing phosphorylated Src kinase (p < 0.01; Fig. 4A, B) or Smad2/3 (p < 0.01; Fig. 4C, D).

Because PDGF and TGFβ are known to signal via the generation of reactive oxygen species (ROS), we compared the degree of oxidative stress in the neointima of End.PTP1B-WT and End.PTP1B-KO mice. These analyses revealed higher amounts of the oxidative stress indicator nitrotyrosine (Fig. 5A, B) and increased numbers of cells immunopositive for the Nox4 type of NADPH oxidase (NOX4; Fig. 5C, D) in vascular lesions of End.PTP1B-KO mice. Immunosignals for endothelial nitric oxide synthase (eNOS) were also significantly higher in neointimal lesions of End.PTP1B-KO mice (Fig. 5E, F), whereas endothelial eNOS expression did not significantly differ between genotypes (Supplementary Fig. S6A, representative findings in Supplementary Fig. S6B). Western blot analysis of HUVECs treated with a PTP1B inhibitor also did not reveal differences in eNOS expression and phosphorylation (at Ser1177; Supplementary Fig. S6C–E).
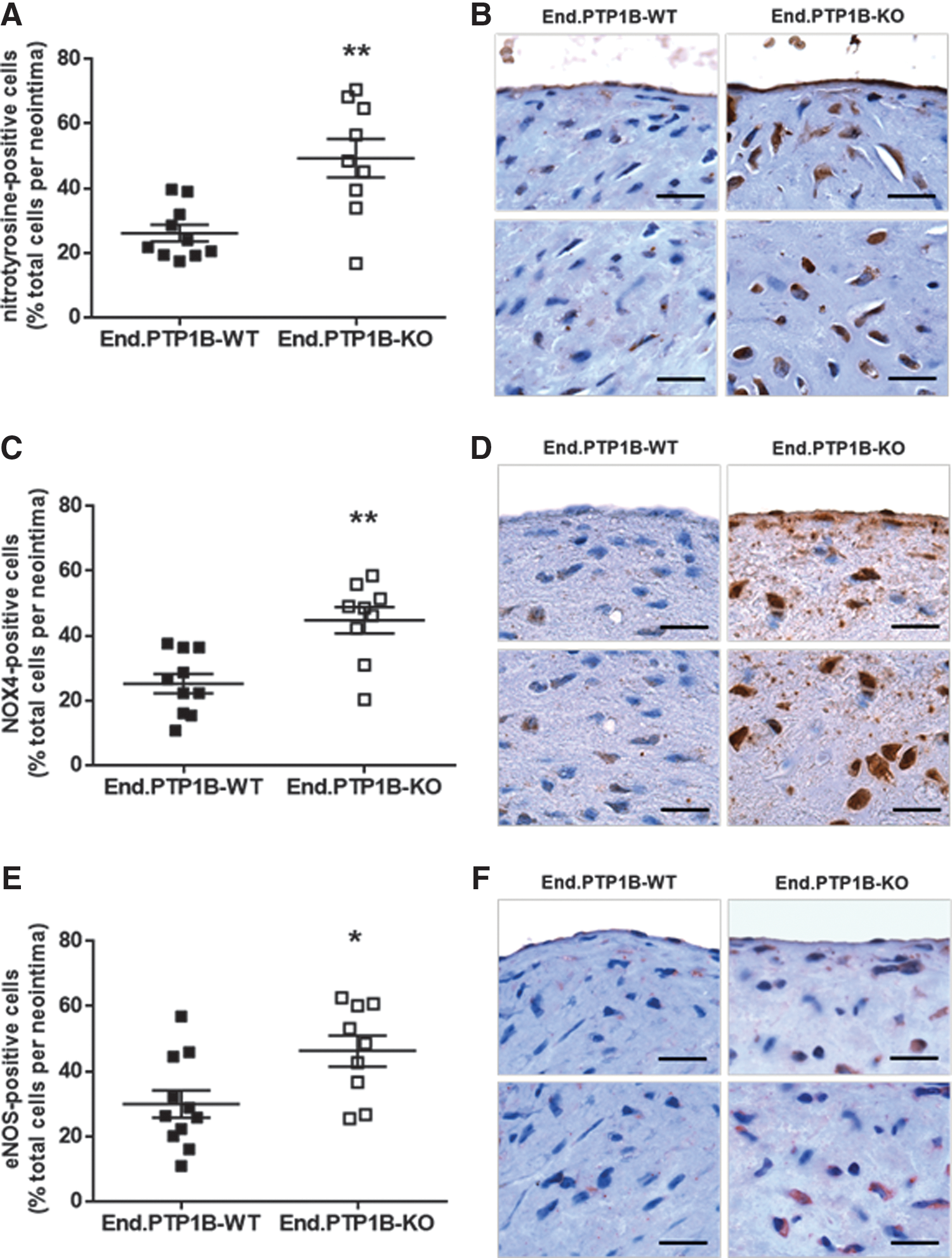
Endothelial PTP1B deletion is associated with increased caveolin-1 expression
PTP1B binds to and colocalizes with caveolin-1 (8), a structural component of cell membrane lipid rafts involved in, among others, inactivation of eNOS (19). Interestingly, vascular lesions of End.PTP1B-KO mice were lined by significantly higher numbers of caveolin-1-positive endothelial cells compared with their End.PTP1B-WT counterparts (p < 0.05; Fig. 6A, B). Caveolin-1 immunosignals were restricted to the endothelium and not observed in other neointimal cells. Western blot analysis confirmed significantly higher phosphorylated (at Tyr14) and total caveolin-1 protein levels in HUVECs incubated with a PTP1B inhibitor, and significantly increased caveolin-1 expression and phosphorylation levels were also observed after incubation of HUVECs with H2O2 (Fig. 6C–E).

Increased total (118% ± 4.8% of controls; p < 0.001) and phosphorylated (208% ± 37% of controls; p < 0.05) caveolin-1 protein levels were also observed in human aortic endothelial cells (HAoECs) following incubation with a PTP1B inhibitor (representative Western blot findings are shown in Fig. 6F). Analysis of primary endothelial cells isolated from mouse lungs revealed a predominant localization of caveolin-1 immunosignals at the cell membrane in End.PTP1B-WT mice, whereas caveolin-1 was found to accumulate at the perinuclear cytoplasm in endothelial cells from End.PTP1B-KO mice (Fig. 6G). Similar expression patterns were observed for VE-cadherin (Fig. 6G).
PTP1B inhibition promotes endothelial senescence: role of oxidative stress
Increased levels of oxidative stress have been implicated in the development of cellular senescence (11), and overexpression of caveolin-1 has been shown to block cellular proliferation and to inhibit cell cycle progression, critical steps in achieving cellular senescence (18, 44). In line with accelerated cellular aging in the absence of PTP1B, a dose-dependent increase in the relative number of senescence-associated β-galactosidase (SA-β-Gal)-positive cells (Fig. 7A) with typical morphological changes such as polynuclear (Fig. 7B) or flattened and enlarged (Fig. 7C) cells was observed in human endothelial cells exposed to a PTP1B inhibitor (representative findings in HUVECs are shown in Fig. 7D). Increased numbers of SA-β-Gal-positive cells were also observed in primary endothelial cells isolated from End.PTP1B-KO compared with End.PTP1B-WT mice (Supplementary Fig. S7). Western blot analysis demonstrated that inhibition of PTP1B was associated with significantly increased protein levels of the tumor suppressor tumor suppressor protein 53 (p53) and the cell cycle inhibitor cyclin-dependent kinase inhibitor-2A (p16INK4A), both in HUVECs and HAoECs (Fig. 7E), and similar findings were observed after prolonged passaging of HUVECs (P13; Fig. 7F). Immunocytochemistry and confocal microscopy also showed that HUVECs treated with a PTP1B inhibitor resembled senescent HUVECs with respect to the predominant cytoplasmic localization of caveolin-1 or the increase in cell size (Supplementary Fig. S8).

To further test the hypothesis that increased oxidative stress is involved in the increased cellular senescence observed in endothelial cells isolated from End.PTP1B-KO mice or in human endothelial cells treated with a PTP1B inhibitor, we examined the effects of the antioxidant Trolox on the above parameters. Pretreatment of HUVECs with Trolox 24 h before addition of the PTP1B inhibitor significantly reduced the number of SA-β-Gal-positive senescent cells to levels observed in control-treated cells (Fig. 8A) and normalized the mean cell area (Fig. 8B). Representative images are shown in Figure 8C. Importantly, Trolox pretreatment restored the impaired capacity of PTP1B inhibitor-treated HUVECs to close a scratch wound injury in vitro (Fig. 8D; representative findings in Fig. 8E). The presence of increased cellular oxidative stress in the absence of PTP1B is also shown after dihydroethidium staining of primary endothelial cells isolated from End.PTP1B-KO mice (three representative images per genotype in Fig. 8F).
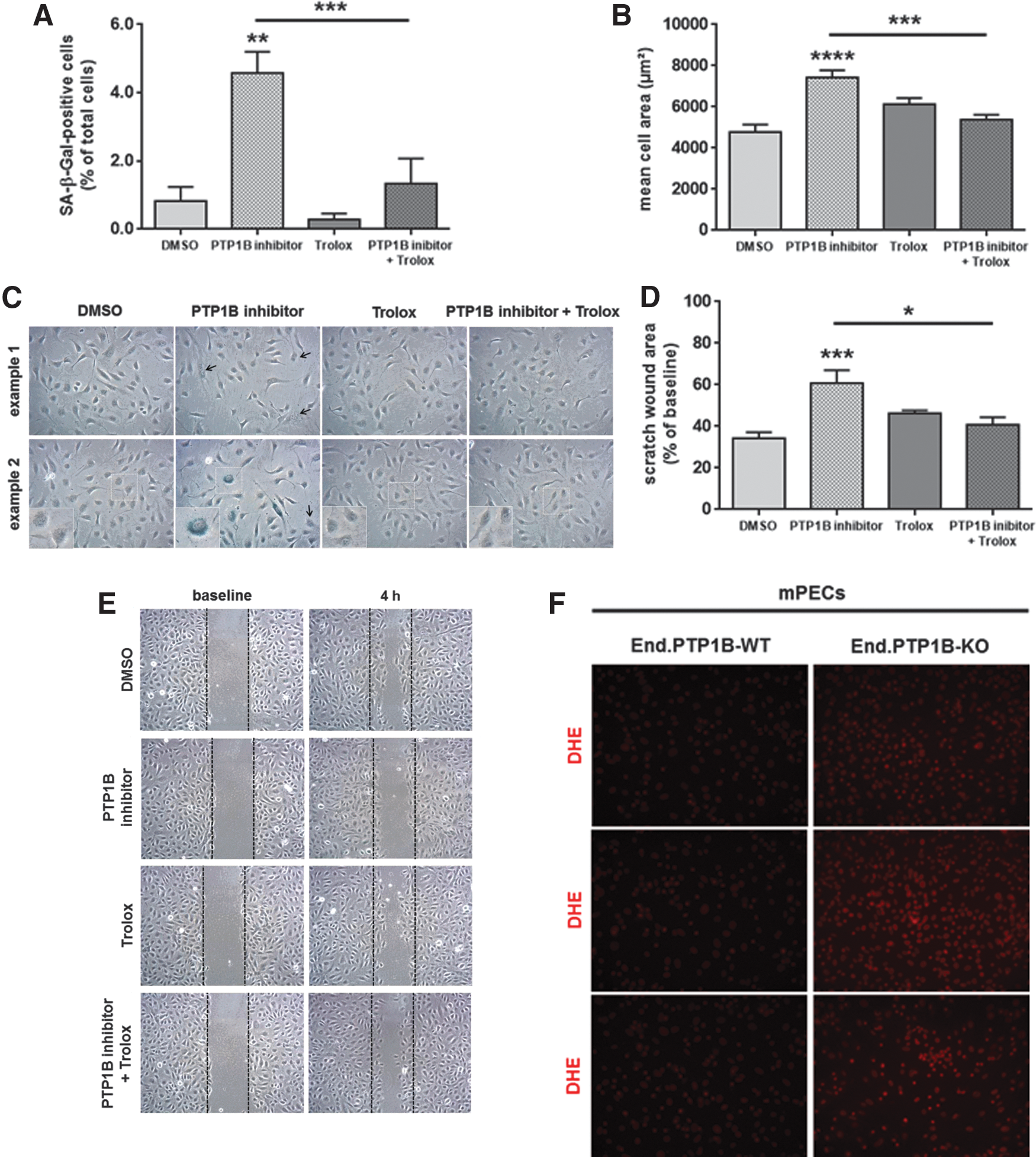
To determine the role of caveolin-1 in the phenotype observed in endothelial cells after genetic deletion or pharmacological inhibition of PTP1B, HAoECs were transiently transfected with caveolin small interfering RNA (siRNA) (Supplementary Fig. S9) followed by treatment with a PTP1B inhibitor 48 h later. As shown in Figure 9, siRNA-mediated downregulation of caveolin-1 significantly reduced the number of SA-β-Gal-positive cells (Fig. 9A, B) and prevented the PTP1B inhibitor-induced increase in p53 (Fig. 9C, E) and p16INK4A (Fig. 9D, E) expression.

Discussion
Endothelial cells play a crucial role during the development of neointima formation following vascular injury as they control lesion re-endothelialization and SMC quiescence. Obesity is associated with an elevated restenosis risk and increased PTP1B expression in endothelial (progenitor) cells, suggesting that endothelial overexpression of PTP1B plays a role in the resistance of endothelial cells to regenerate after endothelial denudation in obesity. In this study, we examined the hypothesis that deletion of PTP1B (and thus removal of the molecular brake of growth factor signaling) in endothelial cells would accelerate the reconstitution of endothelial integrity following injury and reduce intima hyperplasia in mice with diet-induced obesity.
Unexpectedly, we found that endothelial PTP1B deletion was associated with increased neointima formation and more pronounced outward vessel remodeling. The detrimental effects of endothelial PTP1B deletion on neointima formation were evident only in mice with diet-induced obesity, similar to previous studies on the vascular effects of PTP1B (2, 5, 25). End.PTP1B-KO mice exhibited attenuated lesion re-endothelialization and increased endothelial expression of caveolin-1, a cell membrane component involved in eNOS inactivation and NADPH signaling (19), among others. Moreover, we found that vascular lesions of endothelial PTP1B-deficient mice contained higher numbers of activated, proliferating cells and markers of oxidative stress as well as neointimal cells positive for phosphorylated Src tyrosine kinase and Smad2/3, indicative of activated cytokine and growth factor signaling.
Inhibition of PTP1B induced a senescent phenotype (enlarged and multinucleated cells) in human endothelial cells with increased expression of SA-β-Gal, tumor suppressor p53, and the cell cycle inhibitor p16INK4A, which may have caused the impaired re-endothelialization observed in vitro and in vivo. Our findings that the eNOS inhibitor and PTP1B substrate, caveolin-1, is mislocalized in murine and human endothelial cells after genetic or pharmacological deletion of PTP1B, and also after exposure of HUVECs to H2O2 and that the antioxidant Trolox was able to rescue their phenotype strongly suggest a role of increased oxidative stress in the induction of endothelial senescence in the absence of PTP1B, and findings were confirmed in primary endothelial cells isolated from End.PTP1B-KO mice. Furthermore, our results suggest that inhibition of PTP1B may be detrimental in scenarios requiring the functional integrity of endothelial cells, such as following vascular injury and endothelial denudation.
Neointima formation is primarily controlled by the proliferation and migration of vascular SMCs that dedifferentiate and produce abundant extracellular matrix. Earlier studies have shown that SMCs appear only in intima areas not covered by endothelium (23), suggesting that endothelial regeneration determines the extent of intimal SMC accumulation. Conversely, we and others have shown that enhancing the restoration of endothelial integrity by EPC administration reduces neointimal lesion formation (22, 37). Of note, the specific contribution of endothelial progenitors could not be addressed in the present study. The critical importance of lesion re-endothelialization has also been reported in clinical studies. In 1143 randomized patients, incomplete stent re-endothelialization was associated with a greater need of subsequent bypass surgery (41a), and long-term survival rates were higher in patients with complete revascularization after coronary stenting (47).
The endothelial layer is regenerated by cell migration followed by proliferation, cellular processes controlled by growth factors. Many growth factors signal via tyrosine kinase receptor phosphorylation, and signaling is terminated by PTPs. Receptor tyrosine kinases dephosphorylated and thus deactivated by PTP1B include receptors for PDGF, basic FGF and EGF, and several in vitro studies have documented effects of PTP1B overexpression or inhibition on SMC proliferation and migration in response to growth factor stimulation and neointima formation following vascular injury (10, 46).
PTP1B also negatively regulates tyrosine kinase receptors involved in endothelial cell proliferation and migration, such as VEGFR2, Tie1, and Tie2, and pretreatment of cultured endothelial cells with a nonselective PTP inhibitor augmented angiogenic vessel sprouting in response to VEGF or angiopoietin-1 (7). On the contrary, PTP1B substrates also include proteins involved in the regulation of cell adhesion and motility, such as ZO-1 or cortactin, or proliferation, such as p62DOK or p120RasGAP (31). However, the role of endothelial PTP1B for lesion re-endothelialization following vascular injury had not been examined until now.
We have previously shown that obesity is associated with increased PTP1B expression in human EPCs (24). In mice, type 1 diabetes was associated with increased PTP1B expression in the aorta, and similar findings were observed in human arterial endothelial cells exposed to high glucose (25, 50). Importantly, upregulation of PTP1B in states of murine or human obesity was associated with a resistance toward the effects of VEGF or leptin on endothelial proliferation, migration, and tube formation, which could be rescued by weight loss or PTP1B inhibition (24, 50). PTP1B deletion also improved the endothelial dysfunction in mice with streptozotocin-induced type 1 diabetes (25), and PTP1B inhibition ameliorated the resistance to vasorelaxant activities of insulin in type 2 diabetic rats (32). Of note, PTP1B was inhibited or deleted systemically in those studies and may have restored endothelial cell function by improving systemic metabolic dysfunction.
Healthy endothelial cells secrete factors, such as nitric oxide (NO), to induce SMC quiescence (20), whereas endothelial denudation induces SMCs to enter the cell cycle and to proliferate (41). Of note, we did not observe differences between End.PTP1B-WT and End.PTP1B-KO mice with regard to the expression of eNOS in endothelial cells lining the neointima, and PTP1B inhibitors also did not alter the level of phosphorylated and total eNOS in HUVECs. Of note, the eNOS activity is regulated not only by phosphorylation but also by protein–protein interactions. Caveolin-1, an integral cell membrane component highly expressed in endothelial cells, forms an inhibitory complex with eNOS resulting in decreased NO generation (19). Increased endothelial caveolin-1 expression and phosphorylation were evident in primary endothelial cells and vascular lesions of End.PTP1B-KO mice as well as in human endothelial cells treated with a PTP1B inhibitor.
eNOS inactivation due to caveolin-1 overexpression may be one mechanism underlying the observed neointimal hyperplasia and increased neointimal cell proliferation in mice lacking PTP1B in endothelial cells. In addition, caveolin-1 has been shown to negatively regulate endothelial VEGFR signaling and angiogenesis (4, 30), which could also explain the reduced number of endothelial cells covering neointimal lesions in End.PTP1B-KO mice. Similar to the results of the present study, we recently reported elevated caveolin-1 expression in cardiac endothelial cells of End.PTP1B-KO mice (21). Following transverse aortic constriction in mice, the absence of endothelial PTP1B was found to prevent cardiac hypertrophy and fibrosis. It appears that differences in disease mechanisms or the cellular environment determines the net effect of endothelial PTP1B deletion in vivo.
Regarding the underlying mechanisms, PTPs are potential ROS targets and inactivated through reversible oxidation of the catalytic cysteine residue (12). Oxidative stress has been implicated in the development of cellular senescence (11), and reversible oxidation of PTP1B was shown to induce premature senescence in fibroblasts by inactivation of argonaute-2, a member of the RNA-induced silencing complex (49). Caveolin-1 is also a direct binding partner of sirtuin-1, an NAD+-dependent class III histone deacetylase involved in premature senescence, and phosphorylation of caveolin-1 on tyrosine 14 was shown to promote sirtuin-1 sequestration, activate p53 signaling, and induce premature senescence in mouse embryonic fibroblasts (44). Moreover, overexpression of caveolin-1 has been shown to block cellular proliferation and to inhibit cell cycle progression, critical steps in achieving cellular senescence (18, 43). While senescence-induced cell cycle arrest may be beneficial in the setting of cancer progression and help eliminate damaged cells, senescent endothelial cells are known to change their secretory phenotype and to release factors (13), which may stimulate cell proliferation or migration and contribute to neointima formation.
In addition to altered paracrine effects of senescent cells, increased cell death could impair the restoration of an intact endothelial layer that protects SMCs from circulating mitogens, such as PDGF or TGFβ, released from activated platelets following vascular injury. In this regard, genetic deletion of PTP1B in endothelial cells was associated with increased numbers of neointimal cells positive for phosphorylated Smad2/3 indicating activated TGFβ signaling. In addition to the regulation of neointimal cell proliferation, TGFβ promotes the conversion of fibroblasts into pathological myofibroblasts characterized by the expression of α-SMA and the production of extracellular matrix (39), and increased numbers of proliferating and SMA-positive cells as well as higher amounts of interstitial collagens were detected in vascular lesions of End.PTP1B-KO mice. Interestingly, the activity of TGFβ on SMC proliferation (40) or myofibroblast transdifferentiation (14) involves upregulation of NOX4, and elevated levels of oxidative stress were present in lesions of End.PTP1B-KO mice, as demonstrated by the increased neointimal expression of nitrotyrosine and NOX4. Elevated NOX4 mRNA levels have been detected in SMCs isolated from murine atherosclerotic plaques, and NOX4 overexpression in normal SMCs resulted in dedifferentiation, cell cycle arrest, and increased susceptibility to apoptosis (48). Increased exposure to circulating cytokines and growth factors or elevated levels of oxidative stress may also explain the observed increase of phospho-Src-kinase-positive cells in the neointima of End.PTP1B-KO mice. Of note, factors released from activated or dedifferentiated neointimal cells may also have affected endothelial cell phenotype and complicated our findings in vivo.
Taken together, our results demonstrate that genetic deletion and pharmacological inhibition of PTP1B in endothelial cells are associated with increased endothelial expression of caveolin-1 and morphological changes indicating cellular senescence and results in reduced vascular lesion re-endothelialization, increased neointimal proliferation, and enhanced neointima formation in mice with diet-induced obesity. In light of our findings, inhibition of PTP1B as a potential therapeutic target to treat endothelial dysfunction and to prevent the vascular complications of obesity and diabetes should be considered carefully.
Materials and Methods
Animals
Mice with inducible endothelial cell-specific PTP1B deletion (End.PTP1B-KO) were created as described (21). In brief, mice with loxP-flanked (floxed, fl/fl) PTP1B alleles (B6/129SF2/J background; courtesy of Benjamin G. Neel) (6) were crossed with mice expressing a Cre recombinase-estrogen receptor fusion protein (ERT2-Cre) under control of the endothelial receptor tyrosine kinase (Tie2) promoter (DBA2/C57BL/6 background; courtesy of Bernd Arnold) (17). Only littermates were used throughout the study. Cre recombinase activity was induced by feeding adult (i.e., 5- to 6-week-old mice) mice tamoxifen-containing rodent chow (Cat. No. TD.55125; Harlan Laboratories) for 6 weeks (17). Age- and sex-matched Tie2.ERT2-Cre-WT × PTP1Bfl/fl mice fed tamoxifen chow were used as controls.
To visualize Cre recombinase expression, Tie2.ERT2-Cre mice were mated with IRG mice (15), in which Cre-mediated recombination results in green fluorescent protein expression. Obesity was induced by 45% HFD (Cat. No. D12451; Research Diets) for 4 weeks before vascular injury and until sacrifice 3 weeks later. All experiments involving animals were approved by the institutional animal research committee and state authorities and complied with national guidelines for the care and use of laboratory animals.
Serum analysis
Whole blood was collected from anesthetized mice via cardiac puncture. Serum was obtained by centrifugation at 3000 rpm for 10 min and stored at −80°C pending analysis. Serum glucose and cholesterol levels were determined enzymatically using colorimetric assays (Cat. No. EBGL-100 and EHDL-100, respectively; BioAssay Systems, Hayward, CA). Serum leptin and insulin levels were measured using enzyme-linked immunoassays (Cat. No. MOB00; R&D Systems, Minneapolis, MN; and Cat. No. 90080; CrystalChem, Downers Grove, IL; respectively).
Analysis of whole blood
Blood was collected from anesthetized mice by cardiac puncture using ethylenediaminetetraacetic acid (EDTA) as anticoagulant. A 100 μl aliquot was removed and complete blood cell count automatically determined (Sysmex KX21N; Sysmex, Norderstedt, Germany). A second 100 μl aliquot was transferred into a fresh tube and diluted with the same volume of buffer (0.5% bovine serum albumin [BSA]/2 mM EDTA in phosphate-buffered saline [PBS]). FITC-conjugated monoclonal rat antibodies against mouse CD11b (Cat. No. 101205; BioLegend, Koblenz, Germany) were used at 1:100 dilution and incubated for 30 min in the dark. Red blood cells were lysed using FACS lysing solution (Cat. No. 349202; BD Biosciences, Heidelberg, Germany). The solution was centrifuged at 500 × g for 5 min, the supernatant removed, and the pellet suspended in the above buffer and analyzed at an FACS Canto (BD Biosciences). For each analysis, 1 × 106 cells were examined.
Induction of vascular injury
Before the induction of vascular injury and tissue harvest, mice were weighed and anesthetized via intraperitoneal injection of a mixture of 0.5% xylazine (5 mg/kg body weight; Bayer, Leverkusen, Germany) and 2.5% ketamine hydrochloride (100 mg/kg body weight; Hameln Pharma Plus, Hameln, Germany). Vascular injury was induced at the left common carotid artery in 15-week-old male mice using 10% FeCl3 as described (27, 28). Three weeks later, anesthetized mice were perfusion fixed with 4% zinc formalin (Cat. No. Z2902; Sigma-Aldrich, Steinheim, Germany) via the left ventricle, and the injured left and the uninjured, contralateral carotid arteries were harvested followed by embedding in paraffin wax (Cat. No. 39601006; Leica, Wetzlar, Germany) or TissueTek® OCT Compound™ (Cat. No. 4583; Sakura, Staufen, Germany), respectively.
Histochemical and morphometric analysis
Serial cross sections were stained by a combination of VES and MTC stain to simultaneously visualize elastic fibers (black), muscular tissue (red), and extracellular matrix (blue). Five to seven cross sections through the injured arterial segment were morphometrically quantified using image analysis software (Image-Pro Plus; Media Cybernetics, version 7.0), as described (36), and the mean calculated for each animal. For the analysis of lesion re-endothelialization, paraffin-embedded cross sections were stained with hematoxylin and eosin and the number of cell nuclei (dark violet signal) lining the complete neointimal lumen was manually counted. Results are expressed as number of cell nuclei per 100 μm lumen length measured using Image-Pro Plus analysis software and the “measure” function. To visualize interstitial collagen fibers, Sirius red staining was performed and sections photographed under polarized light. The birefringence area was calculated in relation to the total neointimal area. All microscopical analyses were performed using an Olympus BX51 microscope.
Immunohistochemistry
Immunohistochemistry was performed on 4% zinc formalin-fixed paraffin sections. Sections were deparaffinized and rehydrated through a series of graded alcohols. Endogenous peroxidase was quenched using 3% H2O2 (Cat. No. 9681.4; Roth GmbH, Karlsruhe, Germany) in methanol (Cat. No. CP43.1; Roth GmbH). For some antibodies (anti-Smad2/3), sections were permeabilized using 0.05% Triton X-100 (Cat. No. 3051.3; Roth GmbH) at 37°C for 10 min before heat-induced epitope retrieval (in 0.01 M citrate buffer, pH 6.0, or 10 mM Tris/1 mM EDTA, pH 9.0; 800 W for 10 min). Unspecific antigen binding sites were blocked using 10% normal goat serum (Cat. No. ab156046; Abcam, Cambridge, United Kingdom) or avidin/biotin blocking kit (Cat. No. SP-2001; Vector Laboratories, Burlingame, CA; for lectin staining) for 30 min.
Lesion endothelialization was examined using biotinylated isolectin B4 (dilution, 1:50; Cat. No. B-1205; Vector Laboratories). Proliferation was analyzed using rabbit monoclonal antibodies against PCNA (dilution, 1:400; Cat. No. ab92552; Abcam). Neointimal lesion composition was analyzed using rabbit monoclonal antibodies against caveolin-1 (dilution, 1:4000; Cat. No. ab192869; Abcam), α-SMA (dilution, 1:800; Cat. No. A2547; Sigma-Aldrich), vimentin (dilution, 1:1000; Cat. No. NBP1-40730; Novus Biologicals, Littleton, CO), or VCAM1 (dilution, 1:1000; Cat. No. ab134047; Abcam) or rat monoclonal macrophage F4/80 (dilution, 1:200; Cat. No. MCA497GA; Bio-Rad, Puchheim, Germany).
To identify oxidative stress, monoclonal antibodies against eNOS (dilution, 1:50; Cat. No. NB300-500; Novus Biologicals) and NADPH-oxidase subunit 4 (NOX4; dilution, 1:100; Cat. No. ab133303; Abcam) or polyclonal antibodies against nitrotyrosine (dilution, 1:200; Cat. No. 06-284; Millipore, Billerica, MA) were used. To visualize specific signaling events, polyclonal antibodies against p-Src tyrosine kinase (dilution, 1:50; Cat. No. 44-660G; Novex/Thermo Fisher, Waltham, MA) and phospho-SMAD family member-2 (p-Smad2; dilution, 1:500; Cat. No. AB3849; Millipore) were used. GSL I lectin and all primary antibodies were incubated overnight at 4°C.
Secondary antibodies (dilution, 1:1000; Cat. No. B2763, respectively, Cat. No. B2770; Molecular Probes, Eugene, OR) were incubated for 1 h at room temperature (RT). Avidin/biotin peroxidase link (Cat. No. PK-6100; Vector Laboratories) was applied for 30 min and peroxidase substrate (3-amino-9-ethylcarbazole or 3,3′-diaminobenzidine; Cat. No. SK-4200, respectively, Cat. No. SK-4100; Vector Laboratories) until color development. Gill's hematoxylin was used for counterstaining (Cat. No. GHS332-1l; Sigma-Aldrich). Sections were mounted in ImmuMount (Cat. No. 9990412; Thermo Scientific, Waltham, MA) and inspected and photographed on an Olympus BX51 microscope.
Endothelial lectin and caveolin were quantified by measuring the length of the vascular lumen covered with (immuno)-positive cells and expressed per total lumen length. The expression of SMA was expressed as relative SMA-positive area; all other stainings were quantified by relating the number of immunopositive cells to the total number of cells within the neointima. Image analysis software was used for all morphometric analyses (Image-Pro Plus; Media Cybernetics, version 7.0).
Immunofluorescence analysis
To confirm endothelial-specific PTP1B deletion, cryosections through the injured carotid artery were defrosted for 5 min at RT followed by postfixation in acetone for 10 min at −20°C. Unspecific antigen binding sites were blocked using 1% BSA (Cat. No. 2834.2; Roth GmbH)/0.05% Triton X-100 in PBS for 45 min and incubated with rabbit monoclonal antibodies against PTP1B (dilution, 1:50; Cat. No. ab52650; Abcam). Cell nuclei were visualized with 4′,6-diamidine-2-phenylindol (dilution, 1:5000; Cat. No. 6335.1; Roth GmbH). Sections were mounted in fluorescence mounting medium (Cat. No. S2023; Dako/Agilent Technologies, Santa Clara, CA) and photographed on an Olympus BX51 microscope.
Primary murine endothelial cell isolation
Primary mouse endothelial cells were isolated from the lungs of female End.PTP1B-WT and End.PTP1B-KO mice (n = 3 per genotype and isolation). Lungs were washed with 1 × PBS and minced into 1-mm-sized pieces. Enzyme digestion buffer was added containing 1.5 mg/ml collagenase A (Cat. No. LS004154; Worthington, Lakewood, NJ) and incubated for 30 min at 37°C with frequent vortexing. The solution was filtered through 70-μm cell strainers (Cat. No. 352350; Falcon/Becton Dickinson, Franklin Lakes, NJ). To stop digestion, Dulbecco's modified Eagle's medium (containing 20% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin; Cat. No. 319660021; Gibco/Thermo Fisher Scientific, Carlsbad, CA) was used. After centrifugation at 400 × g for 10 min at 4°C, the supernatant was removed and the cell pellet resuspended in 0.5% BSA/2 mM EDTA in 1 × PBS. Following the depletion of hematopoietic cells using CD45-conjugated magnetic MicroBeads (Cat. No. 130-052-301; Miltenyi Biotech, Bergisch Gladbach, Germany), endothelial cells were surface labeled using CD31-conjugated magnetic beads (Cat. No. 130-097-418; Miltenyi Biotech) and isolated using magnetic LS columns and magnetic separators (Cat. No. 130-042-401, respectively, Cat. No. 130-090-976; Miltenyi Biotech).
Isolated cells were cultivated on 0.2% gelatin-coated culture plates in Endothelial Cell Growth Medium MV2 Kit (Cat. No. C-22020; PromoCell, Heidelberg, Germany) at 37°C under 5% CO2. Half of the medium was renewed every other day, and cells were analyzed up to passage 2. The purity of the cell population was examined using flow cytometry and antibodies against CD31 (APC-conjugated; Cat. No. 102510; BioLegend) and ICAM2 (unconjugated; Cat. No. 1925-01; Southern Biotech/Biozol) as endothelial markers as well as against FSP1 (unconjugated; Cat. No. NBP1-89402; Novus Biologicals) to exclude fibroblast contamination (representative dot blots are shown in Supplementary Fig. S10).
Immunocytochemistry
Primary mouse endothelial cells were plated onto coverslips and cultured for 3 days. For immunocytochemistry, cells were fixed at −20°C for 10 min using ice-cold acetone (Cat. No. CP40.1; Roth GmbH), washed five times with PBS, and permeabilized using 0.05% Triton X-100 in PBS. Unspecific antigen binding sites were blocked using 1% BSA in PBS. Primary antibodies against caveolin-1 (Cat. No. 3238; Cell Signaling Technology, Cambridge, United Kingdom), PTP1B (Cat. No. sc1718; Santa Cruz Biotechnology, Dallas, TX), or VE-cadherin (Cdh5; Cat. No. ab33168; Abcam) were incubated for 45 min at RT followed by MFP488- or MFP555-conjugated secondary antibodies (Cat. Nos. MFP A1008, MFP A2428, and MFP A1055, respectively; MoBiTec, Göttingen, Germany). F-actin fibers in the cytoskeleton were detected using rhodamine phalloidin (Cat. No. R415; Life Technologies, Carlsbad, CA). Cell nuclei were detected using DRAQ5 (Cat. No. 65-0880-92; eBioscience, San Diego, CA) and confocal images were collected using a Zeiss LSM710 confocal microscope.
Oxidative stress was visualized using the superoxide indicator dihydroethidium (5 mM in dimethyl sulfoxide [DMSO]; Cat. No. D23107; Life Technologies). Senescent cells were detected using the Senescence Cell Histochemical Staining Kit (Cat. No. CS0030; Sigma), following the instructions of the supplier. Coverslips were washed in PBS, counterstained with nuclear fast red, and mounted using ImmuMount (Thermo Scientific).
Human endothelial cell cultivation
HAoECs (Cat. No. C-12271; PromoCell) and HUVECs (Cat. No. C-12203; PromoCell) were cultured at 37°C under 5% CO2 in endothelial cell growth medium MV2 (Cat. No. C-22022; PromoCell) or endothelial cell growth medium (Cat. No. C-22010; PromoCell), respectively. At 90% confluence, cells were treated with cell-permeable PTP1B inhibitor (Cat. No. 539741-5MG; Calbiochem/Merck Millipore, Darmstadt, Germany) at the indicated concentrations or equal volumes of DMSO for 4 h. The effective concentrations of the inhibitor were determined in preliminary experiments based on its ability to increase VEGFR2 phosphorylation (not shown). In some experiments, cells were incubated with 25 μM of the vitamin E analog and antioxidant Trolox (6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid; Cat. No. 238813; Sigma-Aldrich) for 24 h or with 5 μM H2O2 for 4 h.
HAoECs at 60–70% confluency were transfected with commercially available siRNA targeting human caveolin-1 (60 pmol; Cat. No. sc-29241) or a scrambled siRNA as control (Cat. No. sc-37007; both Santa Cruz Biotechnology) using Lipofectamine RNAi MAX reagent (Cat. No. 13778-030; Invitrogen) according to the manufacturer's protocol and examined 48 h later.
To induce “replicative senescence” in HUVECs, cells were serially passaged until permanent growth arrest. Senescent cells were detected histochemically by SA-β-Gal activity following the manufacturer's protocol (Senescence Detection Kit; Cat. No. ab65351; Abcam). The total number of SA-β-Gal-positive cells or of cells containing more than one nucleus was manually counted and expressed per total number of cells per optical field. The cell surface area was calculated on the same image using TIF images at 20-fold magnification using Image-Pro Plus software and the “area measure” function (at least 5 cells per image). For all 3 parameters, at least 15 optical fields and 5 cells per image per condition were evaluated and results averaged. Cell viability was assessed using the MTS Cell Proliferation Assay kit (Cat. No. G3580; Promega, Fitchburg, MA).
Re-endothelialization in vitro was examined using the scratch wound assay. In brief, HUVECs were seeded on gelatin-coated six-well culture plates. After reaching confluency, the cell monolayer was carefully scratched using a 200-μl pipette tip, any cell debris removed by washing with PBS, and fresh medium with or without PTP1B inhibitor added to the cells. The scratch-induced wound was photographed at baseline and following incubation for 5 h on a phase-contrast microscope (Motic AE31). The mean area between both wound edges was quantified using ImageJ (version 1.46r). Results are expressed as percent of the area at baseline.
Protein isolation and Western blot analysis
HUVECs were resuspended in lysis buffer containing fresh protease and phosphatase inhibitors (Cat. No. 78444; Thermo Scientific). Equal amounts of protein were fractionated by sodium dodecyl sulfate (SDS)/polyacrylamide gel electrophoresis together with molecular weight standards and transferred to nitrocellulose membranes (Protran; Cat. No. 10600002; GE Healthcare, Buckinghamshire, United Kingdom). Membranes were cut horizontally, blocked in 5% BSA (in TBS buffer containing 0.1% Tween-20), followed by incubation with antibodies against phospho- (Tyr14) and total caveolin-1 (; Cat. Nos. 3251 and 3238, respectively; Cell Signaling Technology), phospho- (Ser1177), and total eNOS (Cat. Nos. 9571 and 9586, respectively; Cell Signaling Technology), p16INK4A (Cat. No. 108349; Abcam) or p53 (Cat. No. sc-6243; Santa Cruz Biotechnology).
Protein bands were visualized using horseradish peroxidase-conjugated secondary antibodies (Cat. Nos. NA934V and NA931V, respectively; GE Healthcare) followed by detection with SuperSignal West Pico Chemiluminescent Substrate (Cat. No. 34080; Thermo Scientific). Protein bands were quantified by densitometry and normalized to β-actin (Cat. No. ab8226; Abcam) protein.
Statistical analysis
Results are presented as mean ± standard error of the mean or as median ± interquartile range (Figs. 1D and 2E). Normal distribution was tested using D'Agostino and Pearson omnibus normality test. If two groups were compared, Student's t test for unpaired means was used, if samples were normally distributed, or Mann–Whitney test, if not. If more than two groups were compared, one-way analysis of variance followed by Bonferroni's or Kruskal–Wallis followed by Dunn's multiple comparisons test was used. Statistical significance was assumed, if p reached a value <0.05. For all analysis, GraphPad PRISM data analysis software (version 6.03; GraphPad Software, Inc.) was used.
Footnotes
Acknowledgments
The authors are grateful to Bernd Arnold (German Cancer Research Center, DKFZ, Heidelberg, Germany) for providing Tie2.ERT2-Cre mice and to Benjamin G. Neel (University Health Network, Toronto, Canada) for providing PTP1Bfl/fl mice. IRG mice were a kind gift from Markus Bosmann (Center for Thrombosis and Hemostasis, Mainz, Germany). They acknowledge the expert technical assistance of Anna Kern and Marina Janocha. Results shown in this study are part of the medical doctoral thesis of M.J. This work was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft to K.S.), the Bundesministerium für Bildung und Forschung (BMBF 01E01003; Virchow fellowship to M.L.B.), and the Deutsches Zentrum für Herz-Kreislauf-Forschung (DZHK-Doktorandenstipendium to M.J.).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.