
Abstract
Aims:
Our previous clinical trial indicated that the flavonoid dihydromyricetin (DHM) could improve hepatic steatosis in patients with nonalcoholic fatty liver disease (NAFLD), altough the potential mechanisms of these effects remained elusive. Here, we investigated the hepatoprotective role of DHM on high-fat diet (HFD)-induced NAFLD.
Results:
DHM supplementation could effectively ameliorate the development of NAFLD by inhibiting hepatic lipid accumulation both in HFD-fed wild-type mice and in palmitic acid-induced hepatocytes. We reveal for the first time that mitochondrial dysfunction characterized by ATP depletion and augmented oxidative stress could be reversed by DHM treatment. Moreover, DHM enhanced the mitochondrial respiratory capacity by increasing the expression and enzymatic activities of mitochondrial complexes and increased mitochondrial reactive oxygen species scavenging by restoring manganese superoxide dismutase (SOD2) activity. Interestingly, the benefits of DHM were abrogated in SIRT3 knockout (SIRT3KO) mice and in hepatocytes transfected with SIRT3 siRNA or treated with an SIRT3-specific inhibitor. We further showed that DHM could increase SIRT3 expression by activating the adenosine monophosphate-activated protein kinase (AMPK)-peroxisome proliferator-activated receptor-γ coactivator-1 alpha (PGC1α)/estrogen-related receptor-α (ERRα) signaling pathway.
Innovation:
Our work indicates that SIRT3 plays a critical role in the DHM-mediated beneficial effects that include ameliorating mitochondrial dysfunction and oxidative stress in a nutritional NAFLD model both in vivo and in vitro.
Conclusion:
Our results suggest that DHM prevents NAFLD by improving mitochondrial respiratory capacity and redox homeostasis in hepatocytes through a SIRT3-dependent mechanism. These results could provide a foundation to identify new DHM-based preventive and therapeutic strategies for NAFLD.
Introduction
N
Medicinal tea (vine tea) made from Ampelopsis grossedentata has been consumed to treat liver disease. Dihydromyricetin (DHM) is the most prominent flavonoid present in vine tea. Although DHM has been shown to have beneficial effects for nonalcoholic fatty liver disease (NAFLD), the mechanisms by which these effects are mediated were unclear. The present study highlights a key role for SIRT3 in the protective effect of DHM against mitochondrial dysfunction and oxidative stress in a nutritional NAFLD model. Results from this study provide a reasonable mechanism by which DHM ameliorates NAFLD. Our data also provide a potential therapeutic strategy and target for the prevention of NAFLD.
NAFLD is characterized by excessive fat accumulation in the liver and subsequently increased susceptibility to liver damage. The underlying mechanism for the development and progression of NAFLD is complex and multifactorial. Accumulating evidence indicates that abnormalities in mitochondrial function, typified by decreased energy production capacity and impaired redox homeostasis, have a key role in NAFLD pathogenesis (6, 33). Mitochondria are the main cellular sites for lipid oxidation and ATP production. Notably, defective mitochondrial oxidative phosphorylation (OXPHOS) features prominently in patients with NAFLD (23, 50), whereas augmented hepatic OXPHOS promotes fatty acid oxidation and protects against NAFLD (2). Identification of the underlying causes of mitochondrial dysfunction, as well as potential target molecules that could protect cells from mitochondrial dysfunction, will be crucial for treating mitochondria-mediated diseases (15).
Sirtuins are an evolutionarily conserved NAD+-dependent deacetylase family. Of the seven mammalian sirtuins, three (SIRT3, SIRT4, and SIRT5) are mainly localized to mitochondria. SIRT3 is the only sirtuin that has robust deacetylase activity in mitochondria (1) and in hepatocytes is a crucial gatekeeper of redox status, the epigenetic landscape, and lipid homeostasis (46). A previous study found that SIRT3 deficiency led to reduced intracellular ATP level, accompanied by increased reactive oxygen species (ROS) levels (40). Furthermore, SIRT3 knockout (SIRT3KO) mice fed a high-fat diet (HFD) exhibited aggravated metabolic syndrome (25). We previously found that mitochondrial SIRT3 (silent mating-type information regulation 2 homologue 3) enrichment can promote expression of mitochondria-encoded genes, leading to increased ATP synthesis and Complex I activity as well as decreased mitochondrial reactive oxygen species (mtROS) generation (57). However, a recent study by Li et al. (36) demonstrated that SIRT3 inhibition protected against palmitate-induced lipotoxicity, whereas SIRT3 overexpression sensitized hepatocytes to such lipotoxicity, which was in contrast to the previous findings (4, 28). These differing results raise the question of whether SIRT3 could be a critical regulator of mitochondrial dysfunction and redox homeostasis in NAFLD.
Over the past decades, several pharmacological treatments, including vitamin E, metformin, lipid-lowering agents, and insulin sensitizers, have been used in NAFLD treatment (51). Unfortunately, none of these treatments has shown significant efficacy or long-term safety in animal experiments and cellular studies. Recently, increasing evidence suggests that natural polyphenols can exert several beneficial effects on many features of metabolic disorder (48). Such compounds are readily available through the diet and are readily absorbable while having limited side effects. Dihydromyricetin (DHM) is the most abundant flavonoid in Ampelopsis grossedentata, a plant used to make vine tea, comprising over 30% of the dry weight of the leaves and stems of vine tea (20). Previous studies demonstrated that DHM has anti-inflammatory, antioxidative, and antihypertension properties, hepatoprotective, lipid regulatory, and antitumoral effect, and can be used to treat alcoholism (35, 55, 58).
Our previous clinical trial showed that DHM supplementation improves lipid and glucose metabolism in addition to some biochemical parameters in NAFLD patients (12). Nevertheless, little is known about the underlying mechanisms. Therefore, we aim to identify whether dietary DHM attenuates hepatic steatosis and to explore the potential molecular mechanisms involved.
Results
DHM inhibits palmitic acid-induced lipid accumulation in hepatocytes
Primary hepatocytes isolated from 129 wild-type (WT) mice and HepG2 cells were exposed to different doses of palmitic acid (PA) for 16 h to mimic hepatic steatosis in vivo, as shown in previous studies (27). No obvious changes in cell viability were observed in hepatocytes exposed to PA at concentrations of less than 0.2 mM (Fig. 1A, B). After treatment of hepatocytes with different concentrations of PA, an increase in intracellular lipid accumulation was observed (Fig. 1C–E). Notably, PA-induced (0.2 mM) loss of cell viability was significantly inhibited by the pretreatment with DHM at ≥20 μM (Fig. 1F, G). DHM pretreatment also inhibited PA-induced increases in intracellular lipid accumulation in hepatocytes (Fig. 1H–J).
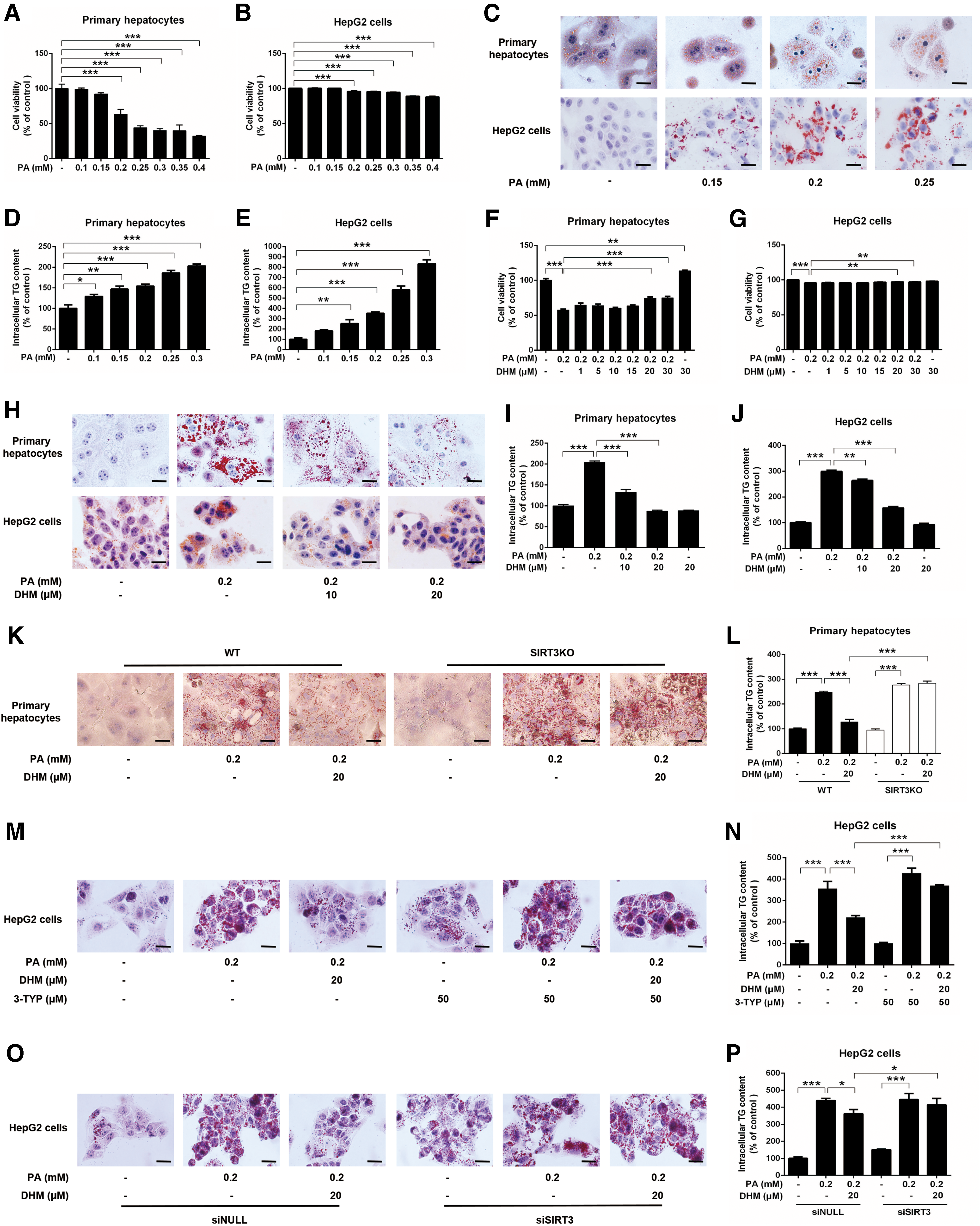
To determine whether the antisteatotic effects of DHM in hepatocytes were mediated via SIRT3, we isolated primary hepatocytes from SIRT3KO and WT mice, and treated them with PA followed by DHM treatment. PA exposure resulted in markedly increased intracellular triglyceride (TG) levels in primary hepatocytes from WT mice, which was inhibited by DHM treatment. However, the PA-induced increases in TG content in hepatocytes from SIRT3KO mice could not be ameliorated by DHM treatment, indicating that SIRT3 deficiency abolished the beneficial effects induced by DHM (Fig. 1K, L). Concomitantly, similar experiments were performed in HepG2 cells that were incubated with an SIRT3-specific inhibitor [3-(1H-1,2,3-triazol-4-yl) pyridine (3-TYP); Supplementary Fig. S1; Supplementary Data are available online at
SIRT3 contributes to the beneficial effect of DHM on HFD-induced hepatic steatosis and inflammatory injury
To assess the potential effect of DHM on NAFLD progression, WT mice and SIRT3KO mice were fed different diets for 12 weeks. After 8 weeks, the body weight was notably increased in the WT mice fed HFD compared with those fed a standard diet (SD), whereas DHM inhibited the HFD-induced body weight increase in HFD-fed WT mice (Fig. 2A). Despite the significant differences in body weight, the amount of food consumption by the different groups was similar throughout the 12-week experimental period (Supplementary Fig. S2). In addition, there was no statistically significant difference in the body weight of SIRT3KO mice fed different diets (Fig. 2B). HFD also increased hepatic lipid accumulation in mice (Fig. 2C–F). Interestingly, DHM significantly attenuated HFD-induced hepatic lipid accumulation only in WT mice and not SIRT3KO mice. Measurement of serum lipid profiles and liver function of WT mice showed that HFD induced notable changes in serum total cholesterol (TC) (Fig. 2G), low-density lipoprotein cholesterol (LDL-C) (Fig. 2H), TG (Fig. 2J), and serum alanine transaminase (ALT) (Fig. 2K), but no difference was seen for serum high-density lipoprotein cholesterol (HDL-C) (Fig. 2I). DHM-induced changes in serum LDL-C, TG, and ALT were abrogated in the SIRT3KO mice.

Meanwhile, there were obvious pathological changes in liver tissue of both HFD-fed WT mice and SIRT3KO mice, as evidenced by increased hepatocyte hypertrophy, vacuolization, inflammatory cell infiltration (Fig. 2 L), and NAFLD activity score (Supplementary Fig. S3), whereas DHM treatment remarkably ameliorated HFD-induced hepatic inflammatory injuries. Together, these observations indicated that genetic deletion of SIRT3 abrogated the inhibitive effects of hepatic lipid accumulation and inflammation by DHM administration. In addition, no significant differences in the abovementioned measurements were found in WT mice treated with DHM alone compared with SD-fed WT mice, indicating that few adverse effects were associated with DHM administration. These results suggest that DHM administration can significantly suppress HFD-induced hepatic lipid accumulation and inflammatory injury, and that SIRT3 contributes to the protective effect of DHM.
DHM increases SIRT3 expression and activity in hepatocyte mitochondria
Based on the findings that SIRT3 might play a crucial role in DHM-mediated amelioration of hepatic steatosis, we next evaluated the effect of DHM on the expression and activity of SIRT3 in liver and hepatocytes. In mitochondria from HFD-fed WT hepatic tissue, we observed increased global protein hyperacetylation, which was suppressed by DHM supplementation (Fig. 3A, B). However, DHM did not affect protein hyperacetylation in mitochondria from SIRT3KO mice. Moreover, in liver samples from WT mice, HFD reduced SIRT3 mRNA and protein expression levels, which were restored by DHM supplementation (Fig. 3C–E). These data implied that DHM could enhance mitochondrial deacetylation via activation of SIRT3 expression.
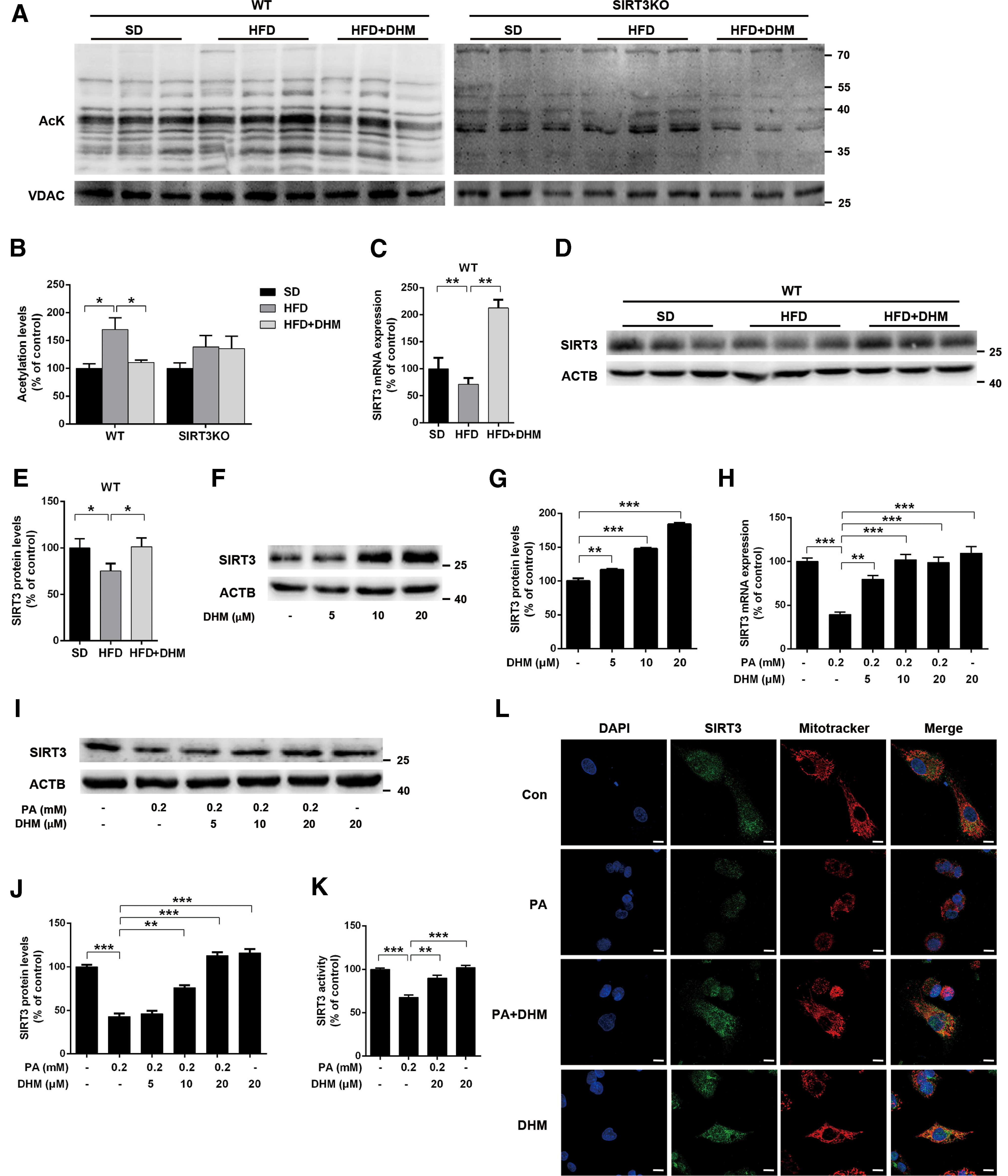
Thus, we further examined the effects of DHM on the expression and activity of SIRT3 in hepatocytes. SIRT3 protein expression was substantially and dose dependently upregulated by DHM (Fig. 3F, G; Supplementary Fig. S4A, B). DHM treatment also inhibited both PA-induced downregulation of SIRT3 expression (Fig. 3H–J; Supplementary Fig. S4C, D) and SIRT3 activity (Fig. 3K). Furthermore, an immunofluorescence assay showed that PA-treated HepG2 cells had reduced mitochondrial SIRT3 expression, which was significantly rescued by DHM (Fig. 3L). Collectively, these results indicate that DHM can activate mitochondrial SIRT3 in the hepatic tissue and hepatocytes.
DHM improves hepatic mitochondrial respiratory capacity through a SIRT3-dependent mechanism
Mitochondria are an important site for nutrient metabolism and play critical roles in lipid metabolism. We next detected whether DHM inhibited hepatic lipid accumulation by regulating mitochondrial respiratory capacity. Hepatic ATP content was decreased in HFD-fed WT mice, which was ameliorated by DHM administration, implying that DHM could have beneficial effects on mitochondrial respiratory capacity (Fig. 4A). Next, we investigated the enzymatic activities and protein expression of mitochondrial respiratory chain (MRC) complexes. As expected, in HFD-fed WT mice, mitochondrial complex I and IV activities were decreased, which was notably attenuated by DHM (Fig. 4B). Moreover, protein expression of mitochondrial DNA (mtDNA)-encoded cytochrome c oxidase I (MT-CO1) and mitochondrially encoded ATP synthase 6 (MT-ATP6), the representative subunit of complex IV and V, respectively, was increased after DHM feeding (Fig. 4C, D; Supplementary Fig. S5A, B). However, the protein expression of nuclear DNA (ncDNA)-encoded subunits of other MRC complexes, including NDUFB8 (NADH: ubiquinone oxidoreductase subunit 8), SDHB (succinate dehydrogenase complex iron/sulfur subunit B), UQCRC (ubiquinol-cytochrome c reductase complex III subunit VII), and ATP5A (ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit 1, cardiac muscle), was unchanged (Fig. 4C, D). Assessment of mRNA expression of MRC-associated mtDNA- or ncDNA-encoded genes showed that Pgc1α, a major transcriptional coactivator associated with mitochondrial biogenesis, had increased mRNA expression in DHM-fed WT mice (Fig. 4E). The downstream targets of peroxisome proliferator-activated receptor-γ coactivator-1 alpha (Pgc1α), nuclear respiratory factor 1 (Nrf1), and mitochondrial transcription factor A (Tfam), which regulate mitochondrial content, were likewise significantly upregulated by DHM administration. Apart from these ncDNA-encoded genes, mRNA expression of mtDNA-encoded MRC complex subunits was upregulated by DHM administration (Fig. 4E). These findings show that DHM recovers the coordinated synthesis of mtDNA- and ncDNA-encoded proteins that are concurrently assembled into OXPHOS complexes.

Analysis of mitochondrial respiration capacity in HepG2 cells showed that PA treatment alone significantly reduced the mitochondrial respiration oxygen consumption rate (OCR) (Fig. 4F), ATP production (Fig. 4G), and spare respiratory capacity (Fig. 4H) in mitochondria, which were all markedly rescued by DHM treatment. However, pretreatment with the SIRT3 inhibitor 3-TYP significantly impaired DHM-induced mitochondrial respiration recovery in PA-treated HepG2 cells (Fig. 4F–H), implying a potential role for SIRT3 in modulating mitochondrial respiratory function. Accordingly, we examined whether DHM-induced regulation of mitochondrial respiratory capacity was dependent on SIRT3. We first detected the enzymatic activities of the MRC complexes and found that DHM treatment rescued the remarkable loss of activities, and these effects were inhibited by 3-TYP (Fig. 4I). DHM treatment also inhibited PA-induced decreases in MT-CO1 and MT-ATP6 expression, which were abrogated by the addition of 3-TYP (Fig. 4J, K; Supplementary Fig. S5C, D). These effects were accompanied by expression of mtDNA-encoded genes, including MT-CO1 and MT-ATP6 (Fig. 4L). These results clearly indicate that DHM improves mitochondrial respiratory capacity through an SIRT3-dependent mechanism.
DHM suppresses hepatic mitochondrial oxidative stress
Mitochondria are considered to be a major source of intracellular ROS, and low levels of superoxide are constitutively produced as a by-product of MRC during oxidative metabolism (38). As an important regulator of mitochondrial function, SIRT3 modulates oxidative processes in mitochondria by activating antioxidant enzymes, leading to a decrease in ROS levels (21). To determine whether HFD-induced mitochondrial dysfunction leads to oxidative stress, we analyzed hepatic samples for several oxidative damage markers. HFD feeding elicited aggravated oxidative stress in hepatocytes as seen by elevated levels of thiobarbituric acid reactive substances (TBARS) (Fig. 5A) and 4-hydroxynonenal (4-HNE) (Fig. 5B, C). These increases were dominantly inhibited by DHM in WT but not in SIRT3KO mice. The results implied that genetic deletion of SIRT3 significantly abrogated DHM-induced inhibition of oxidative stress (Fig. 5A–C).

Concomitantly, there was evident oxidative stress injury in HepG2 cells exposed to PA, whereas DHM treatment effectively inhibited PA-induced oxidative stress (Fig. 5D–F). Pretreatment with 3-TYP notably blocked the capacity of DHM to inhibit oxidative stress (Fig. 5D–F). We found that mitochondrial-derived superoxide (O2 •−) was notably increased in PA-treated HepG2 cells, and this increase could be abrogated by DHM pretreatment. However, DHM-induced inhibition of mtROS was abolished by 3-TYP treatment (Fig. 5G). Collectively, these data demonstrate that DHM suppresses hepatic mitochondrial oxidative stress through SIRT3-mediated signaling.
DHM facilitates the mitochondrial antioxidant defenses through SIRT3-mediated manganese superoxide dismutase deacetylation
Manganese superoxide dismutase (SOD2) is the primary antioxidant enzyme in mitochondria and plays an essential role in mtROS scavenging by catalyzing O2 •− conversion to H2O2 (52). Several recent reports showed that SIRT3 directly activates SOD2 via protein/lysine deacetylation (31). Thus, we analyzed whether DHM could activate SOD2 through SIRT3-mediated deacetylation. In WT mice, HFD feeding resulted in decreased SOD2 activity and increased SOD2 acetylation, which were restored by DHM administration (Fig. 6A–C). However, the action of DHM on the enzymatic activity and deacetylation of SOD2 was abrogated when SIRT3 was genetically deleted in vivo.

Moreover, DHM treatment could restore PA-induced decreases in SOD2 activity (Fig. 6D) and increases in SOD2 acetylation (Fig. 6E–H) in HepG2 cells, and these effects were abolished in the presence of 3-TYP (Fig. 6E, F) or SIRT3 siRNA transfection (Fig. 6G, H). We further examined whether SIRT3 deacetylase activity is required for elimination of PA-induced mtROS. Overexpression of the catalytically inactive mutant SIRT3H248Y (Supplementary Fig. S6) failed to restore SOD2 activity and eliminate mtROS (Supplementary Fig. S7), indicating that SIRT3 deacetylase activity is required for the protective effect of DHM. Collectively, our results demonstrate that DHM enhances mitochondrial antioxidant defenses through SIRT3-mediated deacetylation of SOD2.
DHM activates SIRT3 via stimulation of the adenosine monophosphate-activated protein kinase-PGC1α/ERRα signaling pathway
To investigate the mechanism of enhanced mitochondrial function in mice or hepatocytes treated with DHM, we examined the protein expression of key mediators involved in mitochondrial biogenesis and function. The activity of adenosine monophosphate-activated protein kinase (AMPK), defined as the ratio of phosphorylated AMPKα (pAMPKα) to total AMPKα, was decreased in hepatic tissues from WT and SIRT3KO mice fed HFD, and this decrease could be abolished by DHM administration (Fig. 7A, B). Furthermore, since SIRT3 expression was previously shown to be modulated by AMPK through PGC-1α (26), we evaluated whether the observed AMPK activation was accompanied by upregulation of PGC1α and SIRT3 expression. As expected, DHM administration suppressed HFD-induced decreases in PGC1α expression in hepatic tissues (Fig. 7A, B).
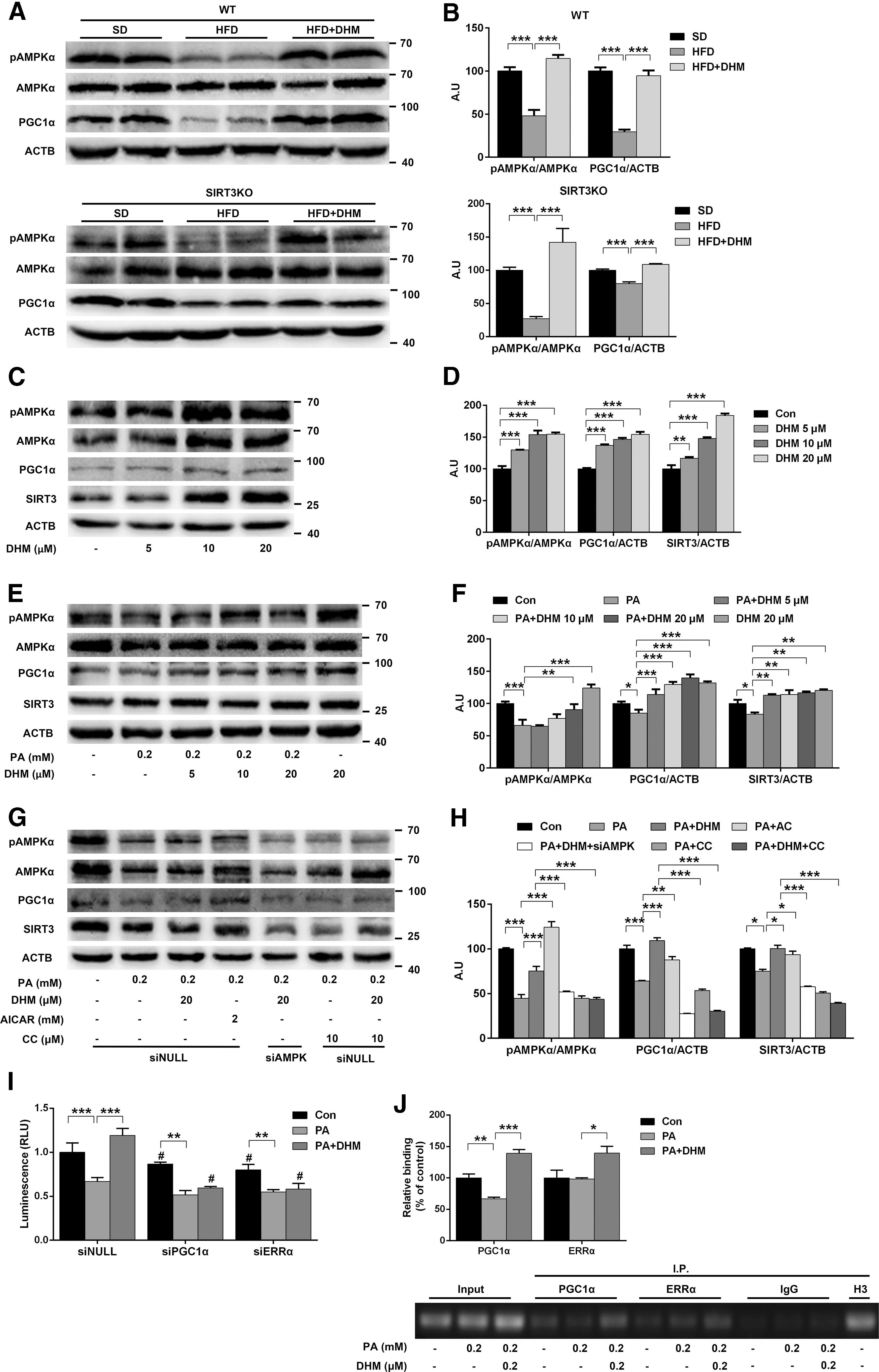
In both untreated and PA-induced HepG2 cells, DHM treatment significantly and dose dependently increased AMPK phosphorylation as well as PGC1α and SIRT3 expression (Fig. 7C–F). The effect of the DHM treatment was similar to that of the AMPK agonist 5-aminoimidazole-4-carboxamide 1-β-D-ribofuranoside (AICAR). However, when HepG2 cells were pretreated with the AMPK-specific inhibitor compound C (CC) or transfected with AMPK siRNA, the inhibitive effects of DHM against PA-induced reductions in AMPK phosphorylation as well as decreases in PGC1α and SIRT3 expression were abrogated (Fig. 7G, H). Our present findings indicated that AMPK activation has a key role in DHM-mediated modulation of SIRT3 expression.
The transcriptional coactivator PGC1α was shown to induce SIRT3 gene transcription through coactivation of estrogen-related receptor-α (ERRα), which could bind to the estrogen receptor response element (ERRE) on the proximal SIRT3 gene promoter region (22). We thus used a luciferase assay to investigate whether DHM-induced SIRT3 activation was dependent on PGC1α/ERRα binding to the SIRT3 promoter in HepG2 cells. SIRT3 promoter-dependent expression of luciferase was dramatically increased after DHM treatment and diminished by transfection with PGC1α siRNA or ERRα siRNA (Fig. 7I). These results suggest that DHM promotes SIRT3 gene transcription through a PGC1α-dependent ERRα-mediated signaling pathway. Indeed, chromatin immunoprecipitation (ChIP) assays performed on HepG2 cells showed enhanced binding activity of PGC1α/ERRα to the SIRT3 promoter on stimulation with DHM (Fig. 7J). The findings thus confirm the involvement of PGC1α/ERRα in mediating DHM-induced SIRT3 expression. Collectively, we conclude that AMPK-PGC1α/ERRα-SIRT3 signaling is likely to be required for DHM-induced regulation of mitochondrial function and activation of SIRT3.
Discussion
Lipid accumulation in the liver is a hallmark and an initial pathological process in NAFLD. At present, several pharmacological treatments for NAFLD have been proposed and tested, but few have exhibited significant efficacy and long-term safety for NAFLD prevention and therapy (47). Accumulating evidence indicates that several polyphenols could alleviate hepatic steatosis in experimental models and clinical trials (48). Naturally occurring polyphenols are abundant and available in the daily diet, and are particularly rich in vegetables. Polyphenol consumption is thought to bring a variety of benefits for health, and thus could be used to prevent or treat different metabolic disorders.
In this study, we showed that the natural flavonoid DHM could effectively ameliorate NAFLD development in a mouse NAFLD model and PA-induced steatosis in hepatocytes. Our results also indicated that genetic deletion of SIRT3 abrogates DHM-induced hepatoprotective effects. Furthermore, we found that DHM triggers the AMPK-PGC1α/ERRα signaling pathway, resulting in SIRT3 enrichment within mitochondria. SIRT3 mediates DHM-induced amelioration of hepatic steatosis and oxidative injury by normalizing mitochondrial functions, including promoting expression of mtDNA-encoded genes, restoring enzymatic activities of MRCs, and increasing mtROS scavenging capacity. Taken together, we conclude that SIRT3 has a master role in the protective effect of DHM on NAFLD in vivo and in vitro, and our results provide new insights into whether SIRT3 could be a therapeutic target for NAFLD. We also define for the first time a reasonable mechanism to explain how DHM ameliorates hepatic steatosis and oxidative injury in an NAFLD model.
A previous report indicated that SIRT3 expression was reduced in HFD-induced metabolic syndrome, which involves insulin resistance, hyperlipidemia, hyperglycemia, and hypertension (25). In agreement with these observations, we found that in both HFD-fed mice and fatty acid-overloaded hepatocytes, SIRT3 expression was notably reduced. Kendrick et al. demonstrated that HFD led to marked hyperacetylation of critical mitochondrial proteins involved in oxidative metabolism and that SIRT3-deficient mice had disrupted activity of MRC complexes (29). Collectively, these results support the notion that a reduction in SIRT3 levels is associated with diminished mitochondrial respiration, reduced energy generation, and elevated levels of ROS that promote mitochondrial dysfunction.
Importantly, our findings also support that SIRT3 expression can be induced by DHM. As a key regulator of mitochondria, SIRT3 enrichment within the mitochondria could afford a variety of benefits. Indeed, we found that DHM attenuates severe mitochondrial dysfunction, characterized by reduced ATP production and MRC enzymatic activities. Assessment of mitochondrial respiration via measurement of the OCR (30) showed that SIRT3KO mice exhibited decreased OCR and exacerbated oxidative stress (9). On the contrary, SIRT3 overexpression gives rise to increased OCR and reduced oxidative stress (8). Consistent with previous reports, we observed increased OCR, ATP production, and spare respiratory capacity in HepG2 cells treated with DHM, but not in SIRT3-inhibited HepG2 cells.
To characterize the mechanisms associated with DHM-mediated improvements in ATP content and MRC enzymatic activities, we assessed mitochondrial biogenesis and several aspects of mitochondrial function. Our previous study further confirmed that SIRT3-dependent deacetylation of FOXO3A promotes mtDNA transcription (57). In addition, SIRT3 was recently shown to regulate several factors associated with mitochondrial integrity, including mtDNA transcription and translation (24). The present study indicated that the potent effects of DHM on mitochondrial function were dependent on SIRT3 induction of mitochondrial biogenesis and mtDNA transcription. An interesting finding that only levels of MT-CO1 and MT-ATP6 were upregulated after DHM feeding was similar to a previous report that linked the beneficial effects of SIRT3 induction with mitonuclear protein imbalance (43), in reference to the altered balance between ncDNA- and mtDNA-encoded OXPHOS subunits. This finding suggests that ncDNA-encoded protein synthesis is not matched by mtDNA-encoded protein synthesis, and that the mitochondrial unfolded protein response and mitophagy might be activated to provide protective capacity in response to a stress state of mitochondria (41). All of these findings strongly indicate that the benefits afforded by DHM treatment could be mediated through activation of SIRT3.
To determine whether oxidative stress-induced NAFLD was dependent on downregulation of SIRT3 expression and whether this expression change could be prevented by an antioxidant, here we showed that DHM could surprisingly upregulate SIRT3 expression and limit lipid peroxidation. ROS are mitochondrial by-products that result from leakage of electrons from the electron transport chain (ETC). An imbalance between ROS production and clearance produces enhanced lipid peroxidation and oxidative stress (14), which leads to dysfunction or death of hepatocytes and other cells in the liver, and contributes to the pathogenesis of acute and chronic liver diseases (39, 51). Mitochondrial fatty acid oxidation (mtFAO) is known to be stimulated in NAFLD, presumably as a compensatory mechanism to restrain lipid accumulation. However, enhanced mtFAO without a concomitant increase in MRC activity can result in mtROS overproduction due to additional electron leakage from the MRC (7). Our data show that hepatic fatty acid overload impacts mtROS homeostasis, leading to severe oxidative stress and lipid peroxidation. However, DHM supplementation promoted mitochondrial biogenesis and restored MRC activity that likely reduced the amount of electrons leaking from MRC complexes that can form superoxide anion radicals.
On the contrary, mtROS scavenging activity is a key part of mitochondrial homeostasis. One of the most important mitochondrial antioxidases is SOD2. Post-translational modifications can regulate SOD2 activity, and SIRT3 catalyzes reversible lysine acetylation and SOD2 activation (19). Our study found that DHM treatment reduced lipid peroxidation in vivo and in vitro, and this effect was associated not only with reduced mtROS synthesis but also increased mtROS scavenging capacity. The deacetylation level and activity of SOD2 both increased following DHM treatment, and these effects were dependent on the presence of SIRT3, which together give insight into the detoxifying process of mtROS.
AMPK is a critical molecule in energy metabolism and also modulates mitochondrial function. As the ratio of AMP/ATP increases, AMPK is activated and triggers a wide range of energy metabolic pathways, eventually leading to increased ATP production (10). As such, AMPK plays a pivotal role in intracellular metabolism and is a prospective therapeutic target for several metabolic diseases. Further examination of signaling pathways involved in DHM-induced, SIRT3-dependent recovery of mitochondrial function and energy metabolism mediated by SIRT3 showed that HFD-fed mice showed a remarkable decrease in the pAMPK/AMPK ratio, and this decrease was prevented by DHM supplementation.
We also assessed whether AMPK impacted PGC-1α transcription and activation (18) and found that consistent with the ratio of pAMPK/AMPK, levels of PGC1α transcription and protein synthesis were also increased by DHM. PGC1α acts as a master modulator of energy metabolism and mitochondrial biogenesis (3) and controls SIRT3 gene expression through coactivation of ERRα that binds to estrogen-related receptor elements in the SIRT3 promoter (42). Concomitant with the upregulation of PGC1α expression induced by DHM, here we showed that SIRT3 expression was also upregulated by DHM in a dose-dependent manner, and this upregulation was suppressed by an AMPK inhibitor and siRNA, providing evidence that DHM effects on SIRT3 expression are most likely mediated through the AMPK-PGC1α signaling pathway. Our results also demonstrated that PGC1α coactivated ERRα to promote ERRα binding to ERRE in the SIRT3 promoter and subsequently to induce SIRT3 mRNA transcription. Taken together, the AMPK-PGC1α/ERRα signaling pathway could be involved in DHM-induced SIRT3 activation.
Based on the data presented here, we propose a potential mechanism to explain the possible mechanism underlying the amelioration of NAFLD by DHM mediated through SIRT3 (Fig. 8). Given the intense clinical interest in developing novel preventive and therapeutic strategies that can mitigate both hepatic steatosis and the consequences of mitochondrial dysfunction, our findings that DHM has both of these functions indicate that it could have significant potential for development as a novel pharmacological agent to improve NAFLD. Although flavonoids are ubiquitous in plants, Ampelopsis grossedentata has high amounts of DHM and thus could be of commercial interest for the phytopharmaceutical and food markets. Since SIRT3 modulates many aspects of mitochondrial function and oxidative stress and is a major factor in many diseases, we believe that DHM supplementation could be beneficial for the management of a wide variety of metabolic diseases.

Materials and Methods
Reagents
DHM (CAS No. 27200-12-0, HPLC ≥98%) was purchased from Chengdu Mansite Bio-Technology (Chengdu, China). PA, bovine serum albumin (BSA), Oil Red O, and collagenase type IV were obtained from Sigma-Aldrich (St. Louis, MO). William's E medium, Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin, and Hanks’ balanced salt solution (HBSS) were purchased from Gibco (Carlsbad, CA). Lipofectamine™ 2000 transfection reagent and MitoSOX™ Red mitochondrial superoxide indicator were obtained from Invitrogen (Carlsbad, CA). Previously described (45) 3-(1H-1,2,3-triazol-4-yl) pyridine (3-TYP; CAS No. 120241-79-4) was a gift from Huifeng Pi (Department of Occupational Health, Third Military Medical University, Chongqing, China). CC and AICAR were purchased from Selleck (Houston). Antibodies against PGC1α and SOD2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against acetylated-lysine, phospho-AMPKα (Thr172), AMPKα, voltage-dependent anion channel (VDAC), and SIRT3 were purchased from Cell Signaling Technology (Beverly, MA). Antibodies against SOD2 (acetyl K68), estrogen-related receptor alpha (ERRα), total OXPHOS rodent WB antibody cocktail, 4-HNE, and β-actin (ACTB) were obtained from Abcam (Cambridge, MA). Other reagents were purchased from Sigma-Aldrich or as indicated in the specific methods.
Animals and experimental design
Male global SIRT3 knockout (129-Sirt3tm1.1Fwa/J, Stock No. 012755) mice (designated as SIRT3KO) were obtained from Jackson Laboratories (Bar Harbor, ME) and respective WT control (129S1/SvImJ) mice (designated as WT) were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). Mice were housed at a controlled temperature (22°C–25°C) and humidity (50% ± 5%) and maintained on a 12-h light/12-h dark cycle at the Experimental Animal Center of Third Military Medical University (Chongqing, China). Food pellets and water were changed every 3 days and were provided ad libitum. At 8 weeks of age, both WT and SIRT3KO mice were randomly divided into four diet groups (n = 5/group): (i) SD group with 10% kcal from fat (D12450; Research Diets, NJ); (ii) HFD group with 45% kcal from fat (D12451; Research Diets); (iii) HFD+DHM group with HFD containing 0.6% DHM, equivalent to 300 mg/kg·bw/day; (iv) DHM group with SD containing 0.6% DHM. Diets containing DHM were prepared according to Lagouge et al. (34). DHM was thoroughly mixed with either powdered SD or HFD at a concentration of 6 g/kg of food, and the pellets were then reconstituted. All diets were irradiated by gamma irradiation and stored at 4°C in light-protected, airtight containers. Body weight and food consumption were measured and recorded weekly for the duration of the study. After 12 weeks of feeding with the corresponding diets, all mice were fasted overnight, anesthetized by intraperitoneal injection of 30 mg/kg·bw pentobarbital sodium and then sacrificed. Serum was prepared by solidification and centrifugation (4°C, 3000 × g, 10 min) and stored at −80°C until analysis of biochemical parameters. Liver samples were collected and frozen immediately in liquid nitrogen and then stored at −80°C for hepatic TG content measurement, Western blot assay, and quantitative real-time polymerase chain reaction (PCR). Some samples were also frozen after fixation in optimal cutting temperature mounting media (Leica, Germany) for Oil Red O staining, or fixed in paraformaldehyde for histological examination. This study was supervised and approved by the Institutional Animal Care and Use Committee of Third Military Medical University (Chongqing, China). All procedures and protocols involving animals were conducted according to established guidelines for humane treatment set by the National Institutes of Health (NIH).
Cell culture and treatments
Primary mouse hepatocytes were isolated from WT and SIRT3KO mice using a two-step collagenase perfusion technique as previously described (13). In brief, mice were anesthetized by intraperitoneal injection of 8 μL/g·bw chloral hydrate. Following anesthesia, the abdominal cavity was opened, and both the inferior vena cava (IVC) and portal vein were exposed to allow perfusion of mouse livers with prewarmed (37°C) HBSS without calcium (containing 0.5 mM EDTA and 25 mM HEPES, pH 7.4) via the IVC at 5 mL/min for 5 min, while the portal vein was cut open. Liver tissue was digested following perfusion with prewarmed (37°C) calcium-containing HBSS (containing 50 U/mL collagenase type IV and 15 mM HEPES). At the end of perfusion, hepatocytes were removed from the liver by gentle scraping, then filtered through a 100 μm cell strainer, and pelleted by centrifuging for 5 min at 350
TG assay
The TG level of liver samples and the intracellular TG content were determined with an enzymatic assay kit (Applygen Technologies, Inc., Beijing, China). The protein concentrations were determined with a BCA Protein Assay Kit (Beyotime, China). The results were expressed as mmol TG per g protein (mmol/gprot).
Oil Red O staining
Liver sample cryosections (7 μm) were prepared in a cryostat (Leica, Germany). Hepatocytes were fixed in 4% neutral formalin for 30 min. The cryosections and fixed hepatocytes were stained with prewarmed Oil Red O working solution for 30 min. After rinsing with distilled water three times, the dishes were then counterstained with hematoxylin (Sigma-Aldrich) for 3 min and gently washed with distilled water. For each dish, three images were captured at 100 × or 400 × magnification using a light microscopy (Leica, Germany). Representative photomicrographs are shown.
Biochemical parameters
Serum parameters, including TC, LDL-C, HDL-C, and alanine transaminase (ALT), were quantified using enzymatic assays (Roche Applied Science, Germany) according to the manufacturer's instructions.
Histological assessment
Immediately after sacrifice of the mice, liver samples were fixed in 4% paraformaldehyde for at least 24 h and then embedded in paraffin wax, followed by sectioning into 5 μm slices. The liver sections were subsequently deparaffinized and rehydrated through a xylene and alcohol series, and then stained with hematoxylin and eosin using a standard procedure. Fibrosis was evaluated using a Masson's trichrome staining kit (Sigma) following the manufacturer's protocol. The presence, type (micro- and macrovesicular), and distribution of steatosis were noted. Steatohepatitis was defined by the presence of steatosis, inflammation, and hepatocellular ballooning (17), as per the FLIP algorithm (5). The amount of steatosis, hepatocellular ballooning, and the foci of lobular inflammation were scored using the NASH-Clinical Research Network criteria (32). Specifically, the amount of steatosis was determined by the percentage (<5%, 5–33%, >33–66%, and >66%, respectively) of hepatocytes containing fat droplets and scored on a 3-point scale. Hepatocyte ballooning was assessed and scored as grade 0 (none ballooning), 1 (little ballooning), and 2 (prominent ballooning). Foci of lobular inflammation were then identified by calculating the number of foci per field (200 × magnification), and scored as 0 (none), 1 (<2), 2 (2 –4), and 3 (>4). The NAFLD activity score (NAS) was defined as the total score for steatosis, inflammation, and ballooning.
Cell viability
Cell viability was analyzed using a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) following the manufacturer's instructions. Briefly, 1 × 104 cells were seeded into 96-well microplates and then exposed to a series of PA concentrations (0, 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, and 0.4 mM) for 16 h. To detect the effect of DHM on PA-induced hepatic steatosis, cells were pretreated with different DHM concentrations (0, 1, 5, 10, 15, 20, 30, and 40 μM) for 2 h and then exposed to 0.2 mM PA for an additional 16 h. Subsequently, cell counting kit, CCK-8, solution (10 μL/well) was added to the wells, and the microplate was incubated at 37°C for 1 or 2 h. The absorbance was measured using a monochromator microplate reader (Molecular Devices, Sunnyvale, CA) at 450 nm. Cell viability was calculated from the ratio of the optical density of experimental cells to that of control cells (set as 100%).
Protein extraction and immunoblotting
Both total and mitochondrial homogenates were prepared for Western blot analysis. Total protein extracts from liver samples were prepared using T-PER Tissue Protein Extraction Reagent (Invitrogen) containing one protease inhibitor cocktail tablet (Roche Applied Science, Germany) and one PhosSTOP phosphatase inhibitor cocktail tablet (Roche Applied Science, Germany) per 10 mL of protein extraction reagent. Total cell lysates from hepatocytes from the different groups were homogenized in cell lysis buffer for Western and IP (Beyotime, China) containing 1% phenylmethanesulfonyl fluoride (Beyotime, China) and one PhosSTOP phosphatase inhibitor cocktail tablet per 10 mL of lysis buffer. For mitochondrial protein extraction, mitochondria were isolated from liver tissues or HepG2 cells with a Mitochondria Isolation Kit (Beyotime, China) in accordance with the manufacturer's instructions. Mitochondrial fractions were prepared by solubilizing isolated mitochondria in mitochondria lysis buffer (Beyotime, China). All lysates were centrifuged at 14,000 × g for 15 min at 4°C. The protein concentration was determined using a BCA assay (Beyotime, China). Immunoblot analysis was performed as we described previously (54). Equal amounts (40 μg) of proteins were separated on sodium dodecyl sulfate polyacrylamide gel electrophoresis and then electrotransferred to poly(vinylidene fluoride) membranes (Bio-Rad, CA) using a wet transfer method. After blocking with 5% nonfat milk or 5% BSA in TBS supplemented with 0.1% Tween-20 for 1 h at room temperature, membranes were incubated with primary antibodies overnight at 4°C. The primary antibodies (Supplementary Table S1) used were as follows: PGC1α (sc-13067, 1:200; Santa Cruz), SOD2 (sc-30080, 1:1000; Santa Cruz), acetylated-lysine (9441, 1:1000; Cell Signaling Technology), phospho-AMPKα (Thr172) (2531, 1:2000; Cell Signaling Technology), AMPKα (2532, 1:1000; Cell Signaling Technology), VDAC (4661, 1:1000; Cell Signaling Technology), SIRT3 (5490, 1:1000; Cell Signaling Technology), β-actin (ACTB) (4970, 1:5000; Cell Signaling Technology), SOD2 (acetyl K68) (ab137037, 1:1000; Abcam), MT-ATP6 (ab192423, 1:1000; Abcam), 4-HNE (ab46545, 1:1000; Abcam), and total OXPHOS rodent WB antibody cocktail (ab110413, 1:200; Abcam). HRP-conjugated anti-mouse or anti-rabbit secondary antibodies were obtained from Invitrogen. Immune complexes were visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore) and the signal was captured by Fusion FX (Vilber Lourmat, France). Densitometry analysis was quantified using ImageJ software (NIH, MD). See original blots in Supplementary Figs. S9 and S10.
RNA extraction and quantitative real-time polymerase chain reaction
Mouse liver tissue and HepG2 cells were harvested in RNAiso Plus reagent (Takara Bio, Japan), and total RNA was extracted according to the manufacturer's instructions. To remove DNA contamination, the extracted RNA was treated with RNase-free DNase on spin-columns (Qiagen). RNA concentration and purity were measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific). First-strand cDNA was synthesized by reverse transcription with random primers using a PrimeScript RT Master Mix (Takara Bio, Japan). Quantitative real-time PCR (qRT-PCR) was carried out with the qTower 2.2 real-time PCR system (Analytik Jena, Germany) using SYBR Premix Ex Taq II (Tli RNaseH Plus) (Takara Bio, Japan). The primers for the targeted genes were synthesized by Sangon Biotech (Shanghai, China). The primer sequences used for gene expression analysis are listed in Table 1. The amplification profile consisted of denaturation at 95°C for 30 s, followed by 40 cycles of 95°C for 5 s and 60°C for 30 s. A melting curve stage was performed at the end of the amplification. Relative fold changes in gene expression were analyzed by the 2−ΔΔCt method and normalized to the internal control gene ACTB.
Primers Used for Real-Time Reverse Transcription Polymerase Chain Reaction
ACTB (Actb), β-actin; G6p, glucose-6-phosphatase; MT-ATP6 (mt-Atp6), mitochondrially encoded ATP synthase 6; MT-CO1 (mt-Co1), mitochondrially encoded cytochrome c oxidase 1; mt-Cytb, mitochondrial cytochrome b; mt-Nd1, mitochondrially encoded NADH dehydrogenase 1; Nampt, nicotinamide phosphoribosyltransferase; Nrf1, nuclear respiratory factor 1; Pepck (Pck1), phosphoenolpyruvate carboxykinase; PGC1α (Pgc1α), peroxisome proliferator-activated receptor-γ coactivator 1 alpha; SIRT3 (Sirt3), silent mating-type information regulation 2 homologue 3 (Sirtuin 3); Tfam, mitochondrial transcription factor A.
SIRT3 deacetylase activity assay
Mitochondria were freshly isolated as described above, and then, the enzymatic activity of mitochondrial SIRT3 was assayed using an SIRT3 Direct Fluorometric Screening Assay Kit (Cayman chemical) as previously described (44) with minor modifications. In brief, freshly isolated mitochondrial extracts (5 mg) were incubated with 15 μL substrate solution on a shaker for 45 min at 37°C, followed by the addition of 50 μL stop/developing solution to each well and incubation for 30 min at room temperature. After excitation at 360 nm, fluorescence was measured at an emission wavelength of 460 nm using a microplate fluorometer (Molecular Devices).
Immunofluorescence microscopy analysis
HepG2 cells were seeded on glass coverslips (NEST, China) in 24-well plates. Following the indicated treatment, intracellular mitochondria were labeled by addition of 100 nM MitoTracker Red CMXRos (Cell Signaling Technology) solution to living cells and incubated at 37°C for 30 min. The cells were then fixed in 4% paraformaldehyde and followed by permeabilization with 0.25% Triton X-100 (Beyotime, China) for 10 min. Nonspecific binding sites were blocked with 5% goat serum for 1 h. The slides were incubated with primary antibody against SIRT3 at 4°C overnight and then incubated with Alexa Fluor® 488 goat anti-rabbit IgG (H+L) secondary antibody (1:500, A11001; Invitrogen) at 37°C for 2 h. Nuclei were counterstained with DAPI. Samples were analyzed using the laser confocal scanning microscopy (Zeiss, Germany).
ATP detection
The ATP content was determined by a luciferin/luciferase method using an ATP Assay Kit (Beyotime, China) according to the manufacturer's instructions. In brief, liver sample homogenates or cell lysates were centrifuged at 12,000 × g for 5 min at 4°C to prepare supernatants for ATP testing. An ATP concentration standard curve was generated using a series of known concentrations of ATP standard solutions (1 nM–1 μM). Subsequently, 100 μL ATP assay buffer was added to each well, incubated for 3 min before the addition of 20 μL sample to each well, mixed, and luminescence measurement using a luminometer (Molecular Devices).
Mitochondrial respiratory complex activity assay
The activities of mitochondrial respiratory complexes I, II, and IV in mitochondria freshly isolated from mouse liver samples or HepG2 cells were analyzed with the corresponding kits from Abcam Mitosciences (Cambridge, United Kingdom) following the manufacturer's instructions.
Mitochondrial OCR determination
OCR, representing key parameters of mitochondrial function, was evaluated with an XF Cell Mito Stress Test Kit using an XF96 Extracellular Flux Analyzer (Seahorse Bioscience, Massachusetts) as described previously (11). Briefly, HepG2 cells (3 × 103 cells per well) were seeded in XF Cell Culture Microplates. After treatment, the cell culture medium was removed and changed to unbuffered XF base medium supplemented with 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose. The cell culture microplates were placed into a 37°C non-CO2 incubator for 1 h before the assay. Subsequently, three compounds (oligomycin, carbonyl cyanide-ptrifluoromethoxyphenylhydrazone [FCCP], rotenone/antimycin A) were prepared in assay medium, adjusted to pH 7.4, and then loaded into ports of a hydrated sensor cartridge. Oligomycin (1 μM, an inhibitor of ATP synthetase), FCCP (0.5 μM, a mitochondrial inner membrane uncoupler that promotes maximum electron flux through the ETC), and rotenone plus antimycin A (0.5 μM, the inhibitor of complex I and III) were injected successively. The parameters of basal respiration, ATP production, maximal respiration, and nonmitochondrial OCR were then programmatically measured. Data were processed using Wave and Report Generator software.
Thiobarbituric acid reactivity
Determination of malondialdehyde (MDA) in liver samples and HepG2 cells was performed using a lipid peroxidation MDA assay kit (Beyotime, China). MDA is an end product of fatty acid peroxidation and can be measured through a chromogenic reaction between MDA and thiobarbituric acid (TBA). Briefly, 100 μL supernatant of liver homogenates or cell lysates was mixed with 200 μL of MDA working solution, incubated in a 100°C water bath for 15 min, and then cooled to room temperature. The mixture was centrifuged at 1000 × g for 10 min and 200 μL supernatant was used to measure absorbance at 532 nm. The TBARS concentration was calculated from an MDA standard curve.
Mitochondrial O2 •− assessment
Mitochondrial O2 •− levels were assessed using a mitochondria- and superoxide-specific probe (MitoSOX Red; Invitrogen), a selective probe for the detection of O2 •−. HepG2 cells (1 × 104 cells per well) were seeded in a 96-well microplate. After the indicated treatment, MitoSOX Red stock solution was diluted with calcium- and magnesium-containing HBSS buffer to prepare a 5 μM working solution. Then, cells were incubated with 5 μM of MitoSOX Red working solution in the dark at 37°C for 20 min. Cells were gently washed three times with warmed HBSS buffer. After excitation at 492 nm, the fluorescence intensity was analyzed at an emission wavelength of 595 nm using a microplate fluorometer (Molecular Devices).
SOD2 activity
Mitochondria from mouse livers and HepG2 cells were separated as indicated above. SOD2 enzymatic activity was assessed based on its capacity to competitively inhibit a combination of WST-8 and superoxide radicals generated by xanthine oxidase, using a Cu/Zn-SOD and Mn-SOD assay kit with WST-8 (Beyotime, China) according to the manufacturer's instructions. SOD1 inhibitor A and B were successively added to the samples to inhibit residual SOD1 activity, followed by incubation with the WST-8/enzyme working solution at 37°C for 30 min. The absorption was measured at a wavelength of 450 nm. When the inhibition rate of WST-8 formazan was 50%, the enzymatic activity of SOD2 was defined as one unit. The protein concentrations were determined using BCA assay. The results were expressed as unit per mg protein (U/mg prot).
RNA interference
siRNAs for SIRT3 (human, sc-61555), AMPK (human, sc-45312), PGC-1α (human, sc-38884), and estrogen-related receptor α (ERRα; human, sc-44706) along with control siRNA (human, sc-44230) were purchased from Santa Cruz Biotechnology. HepG2 cells were transfected with 100 pmol of siRNA for 5–7 h using Lipofectamine 2000 reagent (Invitrogen) following the manufacturer's protocol. Then, the medium was exchanged with a fresh complete medium. At 24 h after transfection, cells were incubated with 0.2 mM PA for 16 h in the presence or absence of DHM (20 μM) added 2 h before and during PA incubation. After treatment, cells were harvested for further experiments.
Luciferase assay
Activation of PGC1α/ERRα-SIRT3 signaling pathways was assessed in HepG2 cells via a luciferase reporter assay. In brief, HepG2 cells (2 × 104 cells per well) were seeded in 48-well plates and incubated until the cells reached 60–70% confluence. Cells were then transfected with the SIRT3 luciferase reporter vector HPRM24033-PG04 (0.25 μg per well; GeneCopoeia, Rockville; Supplementary Fig. S8A) and control vector pEZX-PG04 (0.05 μg per well; GeneCopoeia, Rockville, USA; Supplementary Fig. S8B) mixed in Lipofectamine 2000 reagent. The HPRM24033-PG04 plasmid was generated in a dual-reporter construct, containing an ERRE. Gaussia luciferase (GLuc) was used as the promoter reporter, and secreted alkaline phosphatase (SEAP) was used as an internal standard control for signal normalization. Luciferase activity was analyzed using a Secrete-Pair™ Dual Luminescence Assay Kit (GeneCopoeia) on a luminometer microplate reader (Molecular Devices) as described in the manufacturer's instructions. The GLuc/SEAP ratio was calculated as luminescence intensities (relative light unit).
ChIP assay
For ChIP assays, a SimpleChIP® Enzymatic Chromatin IP Kit (Magnetic Beads; #9003, Cell Signaling Technology) was used as described previously with slight modifications (49). Briefly, after the indicated treatment, ∼4 × 106 cells for each immunoprecipitation were harvested. Cells were fixed in 1% formaldehyde to preserve the protein-DNA interactions occurring in the cell. After termination of crosslinking by the addition of glycine, cells were washed, scraped into cold buffer, and centrifuged at 2000 × g for 5 min at 4°C. Cell pellets were lysed on ice for 10 min in 1 × buffer A containing DTT and protein inhibitor cocktail (inverting every 3 min), and the nuclei were harvested and fragmented using enzymatic digestion solution (1 × buffer B containing DTT and micrococcal nuclease). After incubation for 20 min at 37°C, chromatin was digested into 150–900 bp DNA/protein fragments. Immunoprecipitation was carried out using appropriate ChIP-grade antibodies against PGC1α (sc-13067; Santa Cruz), ERRα (ab16363; Abcam), rabbit histone H3 (as a positive control; #4620, Cell Signaling Technology), and normal rabbit IgG (as a negative control; #2729, Cell Signaling Technology) overnight at 4°C with rotation, and then, the complex was captured by incubation with ChIP-grade protein G magnetic beads for 2 h at 4°C with rotation. Next, chromatin was eluted from the antibody/protein G magnetic beads by incubation with 1 × ChIP elution buffer for 30 min at 65°C. As crosslinking of DNA and proteins was reversed, the samples were digested with proteinase K for 2 h at 65°C as recommended. DNA was then purified using spin columns before analysis. Purified DNA was analyzed by PCR using primers 5′-AGTAAGACCCGAGGTAGGAG-3′ (forward) and 5′-GGAATAAGAGGGATGACAGA-3′ (reverse) to determine the binding of PGC1α and ERRα to SIRT3 promoter. The control primers for human RPL30 were provided with the kit. Amplified DNA was visualized by electrophoresis on a 1% agarose gel and stained with ethidium bromide. Quantitative real-time PCR was performed using SYBR Premix Ex Taq II (Takara Bio, Japan) on a qTower 2.2 real-time PCR system (Analytik Jena, Germany).
Statistical analysis
Data analysis was performed with SPSS 19.0 software (Chicago, IL). All experimental data are expressed as mean ± SEM. Statistical differences among groups were determined with either Student's t-test (for two groups) or one-way analysis of variance followed by Tukey–Kramer post hoc tests (for multiple group comparisons). p-values less than 0.05 were considered statistically significant. Each experiment was performed a minimum of three times.
Footnotes
Acknowledgments
This work was supported by research grants from the National Natural Science Foundation of China (81673155 and 81573131) and the Chongqing Fundamental and Advanced Research Project (cstc2014jcyjA1734).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.