
Abstract
Significance:
Psychosocial stress is associated with alterations in serum glucocorticoids and cytokines, such as interleukin-6 (IL-6) and IL-1β, which functionally interact. However, the molecular mechanisms and physiological relationship between the two systems within the context of stress exposure are not well characterized.
Recent Advances:
Extracellular IL-6, which stimulates the release of cortisol from the zona fasciculata of the adrenal cortex, mediates its intracellular effects by tyrosine phosphorylation of the transcription factor signal transducer and activator of transcription 3 (STAT3). Mitochondrial electron transfer reactions are involved in both STAT3-driven ATP production in oxidative respiration and adrenocortical steroid biosynthesis.
Critical Issues:
The role of STAT3 in oxidative respiration and steroidogenesis suggests that it integrates both nuclear and mitochondrial actions, thereby preserving main steps of glucocorticoid biosynthesis in the adrenal gland under psychosocial stress. This review discusses the notion that these two pathways are together simultaneously involved in protection against chronic stressors.
Future Directions:
Linking the function of cytokines and main components of the hypothalamic–pituitary–adrenal (HPA) axis to molecular mechanisms of mitochondrial redox signaling will be essential for a better understanding of the relevant stress-responsive systems engaged in stress vulnerability. Antioxid. Redox Signal. 28, 760–772.
Introduction
A
In the presence of psychosocial stress stimuli, the nervous, endocrine, and immune systems are engaged in a complex and interacting manner to maintain homeostasis. Acute psychosocial stress transiently activates, in a first wave, the sympathetic nervous system, resulting in the release of the catecholamines epinephrine and norepinephrine from the adrenal medulla as well as of norepinephrine from sympathetic nerve terminals (13). The second wave of stress responses comprises activation of the hypothalamic–pituitary–adrenal (HPA) axis (59), followed by a delayed increase in circulating cytokines such as interleukin-6 (IL-6) and IL-1β (65, 112). Exposure to long-lasting or chronic psychosocial stress is associated with chronic dysfunctions, including HPA axis alterations and elevated cytokine levels (1, 13, 41, 84). In this article, we particularly focus on the HPA axis and cytokines and provide an overview on mechanisms of mitochondrial redox signaling in these stress-responsive systems.
Components and Regulators of the HPA Axis
The HPA axis, regulated by inputs from limbic structures and the prefrontal cortex, is a major part of the neuroendocrine system encompassing interactions among the hypothalamus, the pituitary gland, and the adrenal gland (22, 23). The principal hormones of the HPA axis are corticotropin-releasing factor (CRF), adrenocorticotropic hormone (ACTH, also termed corticotropin), and the major glucocorticoid in humans, cortisol. The 41 amino acid polypeptide CRF has been identified as a key regulator responsible for initiating many of the endocrine, autonomic, and behavioral responses to different kinds of stress, including psychosocial stress (124). CRF derives as a cleavage product from an 196 amino acid precursor and stimulates the release of ACTH into systemic circulation (100). Following exposure to stress stimuli, parvocellular neuroendocrine cells located within the paraventricular nucleus (PVN) of the hypothalamus release CRF into the hypothalamo-hypophyseal portal vessels (14, 101, 103). The blood flow in this capillary plexus transports CRF to the anterior lobe of the pituitary, where it binds to G-protein-coupled CRF receptors expressed on the surface of basophilic cells termed corticotropes (19, 20, 129). Upon binding of CRF to the CRF type I receptor on pituitary corticotropes, the cyclic adenosine monophosphate (cAMP) pathway induces the release of ACTH into the systemic circulation. Corticotropin is derived from the 241 amino acid precursor polypeptide pro-opiomelanocortin (POMC) by proteolytic cleavage through the subtilisin-like prohormone convertase (18, 66). The principal target for circulating ACTH is the zona fasciculata of the human adrenal cortex where it binds to the G-protein-coupled melanocortin type 2 receptor (MC2-R) and subsequently stimulates the synthesis and secretion of glucocorticoids (87).
Electron Transfer Reactions in Steroid Biosynthesis
The action of ACTH in steroidogenesis includes conversion of cholesterol into pregnenolone, which is the initial step of glucocorticoid biosynthesis. The enzyme catalyzing the synthesis of pregnenolone is mitochondrial cytochrome P450scc located at the matrix side of the inner mitochondrial membrane (10, 85). Cholesterol side-chain cleavage involves monooxygenase reactions, including two hydroxylation steps, which generate first 22R-hydroxycholesterol and then 20α,22R-dihydroxycholesterol (53, 113) (Fig. 1). Both hydroxylation reactions utilize electron transfer mechanisms and require molecular oxygen and two electrons per oxidation step (42, 43, 51). The transfer of electrons from nicotinamide adenine dinucleotide phosphate (NADPH) to the heme iron of cytochrome P450scc occurs via two electron transfer proteins, adrenodoxin reductase and adrenodoxin, which are both expressed in the adrenocortical zona fasciculata (24, 49, 56, 57, 136). The flavin adenine dinucleotide coenzyme of adrenodoxin oxidoreductase receives two electrons from NADPH and transfers them to the electron transfer protein adrenodoxin (42, 146). Adrenodoxin, a member of the ferredoxin family of electron-transferring nonheme iron proteins, contains a [2Fe-2S] cluster as redox-active group (39, 115) (Fig. 2). Its primary function is to act as a specific electron carrier between the NADPH-dependent adrenodoxin reductase and cytochrome P450scc, which makes it an irreplaceable redox component of the side-chain cleavage of cholesterol in steroid hormone biosynthesis. Given the essential role of the reductase-adrenodoxin-P450scc system in the conversion of cholesterol to pregnenolone, further research is needed to study changes in the expression level of this electron transfer path in adrenocortical steroid-producing cells under stress exposure.
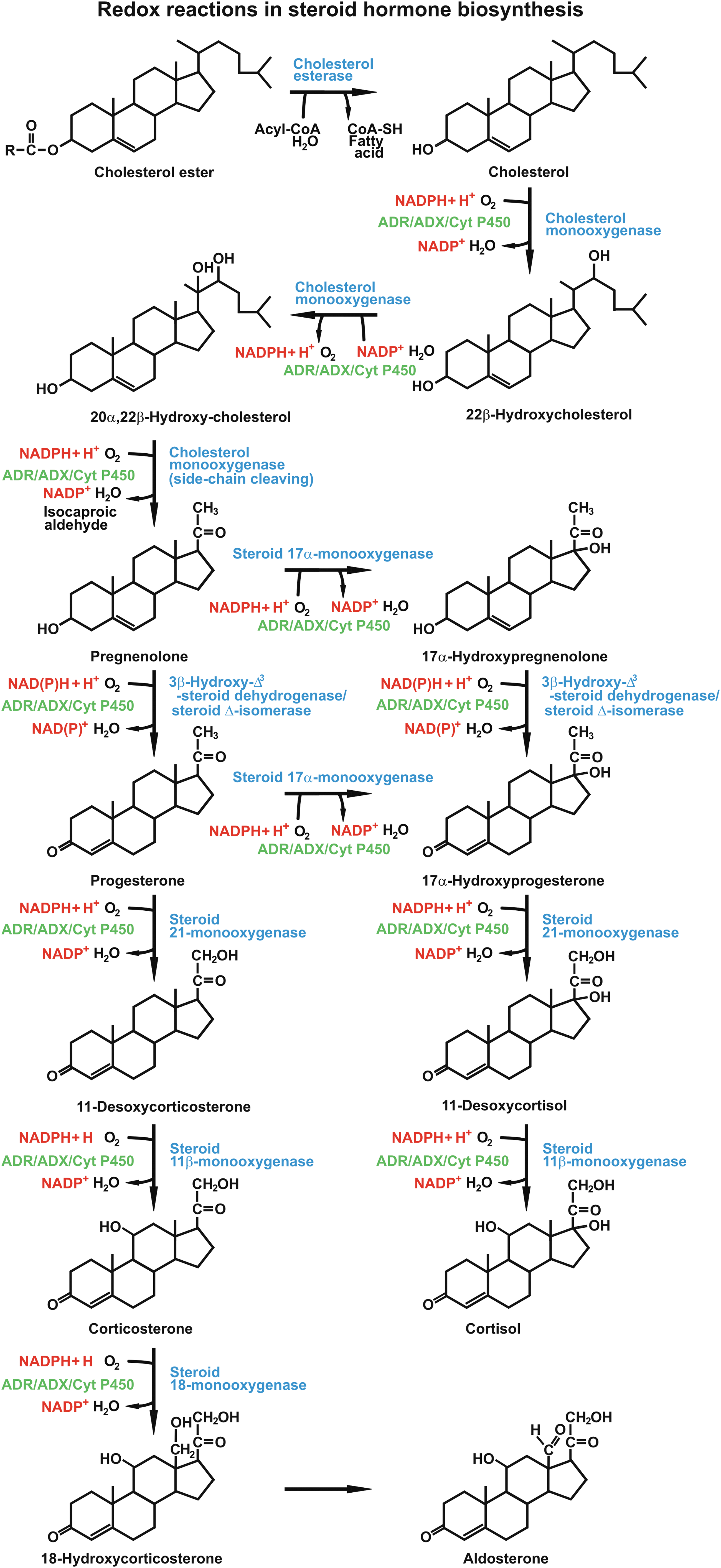
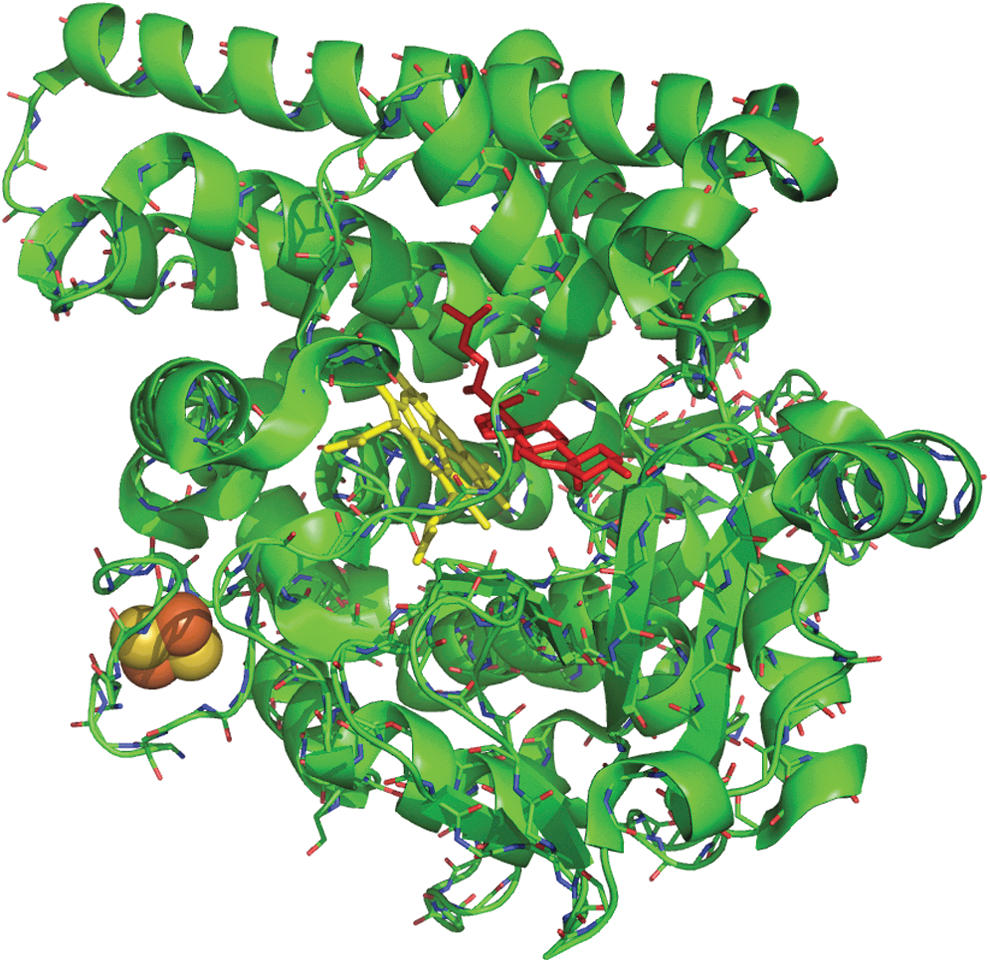
Basal Functioning of the HPA Axis
The basal activity of the HPA axis is characterized by pronounced circadian variability that is mediated by superimposed positive inputs from central pacemakers. Every hour, neurons in the PVN secrete two or three mostly synchronous pulses of CRH and arginine vasopressin into the hypophysial portal system that subsequently stimulate ACTH and cortisol secretion (14). These pulses are at their peak early in the morning and lowest in late evening. Therefore, the circadian rhythm of HPA activity is characterized by highest spontaneous secretory activity during the second half of nocturnal sleep and lowest activity in the late evening (12, 33, 132). Accordingly, the pattern of cortisol secretion shows an early morning maximum, declining levels throughout the day, a quiescent period of minimal secretory activity at night, and an abrupt elevation during late sleep (125). HPA axis activity is regulated in a long-loop negative feedback system (17, 68, 143). The end product of the axis, cortisol, exerts negative feedback effects on ACTH-secreting cells in the pituitary, on CRH-secreting cells in the hypothalamus, and on extrahypothalamic sites that regulate CRH synthesis and secretion (48, 58, 86). The circulating steroids inhibit the synthesis and release of CRF within the hypothalamus and suppress production of POMC-derived neuropeptides in the pituitary gland (54), indicating that downregulation of the HPA axis by cortisol occurs at different levels in classical inhibitory feedback loops.
Stress Reactivity of the HPA Axis and Cytokines
Psychosocial stress is one of the most potent stimuli to activate the HPA axis in humans. The magnitude of HPA axis responses and recovery trajectory depends on characteristics of the stressor. More precisely, situations that include social-evaluative threat, in which others could negatively judge performance, provoke largest cortisol and ACTH increases, particularly if the outcome seems uncontrollable (30). Other situation characteristics proposed to stimulate HPA axis activity include perceived novelty or unpredictability (76). Many current acute stress paradigms use standardized stressors that combine public speaking with cognitive tasks (e.g., mental arithmetic) and thus comprise these characteristics. Indeed, stressors such as the Trier Social Stress Test (TSST) (59) elicit highest cortisol and ACTH increases and slowest recovery (62). In line with Lazarus' transactional definition of stress (67), anticipatory cognitive appraisal processes, including an individual's perceived threat, challenge self-concept of one's own abilities, and control expectancy, predict cortisol and thereby HPA axis stress reactivity to a great extent (35, 138). In addition, a variety of other factors, including noise exposure and psychological parameters such as measures related to personality or affect are also known to modulate an individual's HPA stress reactivity [for review, see e.g., Refs. (21, 89)]. These stressors can relate to mood disorders and sleep fragmentation, which in turn impair the circadian clock and affect stress hormone release (137).
Acute psychosocial stress induces a kinetic response of the HPA axis and elevation of circulating cytokines that as small extracellular, glycosylated signaling molecules execute pleiotropic effects at nano- or picomolar concentrations. In terms of ACTH, the peak release resulting from TSST stress induction is observed immediately after stress cessation, while salivary cortisol peaks 10 to 20 min after stress cessation [e.g., Ref. (59)]. In most healthy subjects, HPA measurements return to baseline levels within an hour of stress cessation. Regarding cytokines, acute stress induces delayed increases in circulating levels of proinflammatory cytokines with robust effects of particular IL-6 and IL-1β (65, 112). Our recent randomized-controlled investigation of the kinetics of cytokine responses to TSST stress induction compared with a nonstress control condition over a 120-min poststress interval revealed peak responses of both plasma IL-6 and IL-1β 90 min after cessation of the stressor (65).
Chronic stress as a cumulative result of repeated or prolonged stress exposure has been associated with changes in both basal HPA axis functioning and HPA responsiveness to stress. While work stress, caregiving to a demented spouse, loneliness, and low economic status are considered to represent chronic psychosocial stressors (41), exhaustion or burnout is conceptualized as a consequence of chronic stress exposure (1, 80, 81). Findings on alterations in basal HPA axis functioning with burnout or exhaustion are divergent and include altered HPA axis activity in terms of both lower and higher circadian cortisol secretion (75, 81, 91, 93) as well as altered cortisol awakening responses (28, 40, 75, 93, 97, 109). Moreover, vital exhaustion or burnout has been associated with stronger HPA axis suppression after dexamethasone administration (8, 84, 109), higher cortisol reactivity to low-dose ACTH stimulation (140), and lower glucocorticoid sensitivity of inflammatory cytokine production (139). Responsiveness of the HPA axis to acute stress induction seems to be blunted with exhaustion or burnout (29, 52, 91). An integrating explanation for these findings is as follows: stress initially induces elevated endogenous glucocorticoid secretion that with ongoing stress exposure shifts into a hypoactive exhausted HPA axis with decreased immunocompetence and increased risk of inflammation (1, 78).
Indeed, chronic psychosocial stress has been associated with elevated levels of circulating inflammatory cytokines, particularly of IL-6 (41). Dementia caregivers showed elevated IL-6 levels compared with controls, both cross-sectionally (72) and prospectively over 6 years (55). Similarly, independent of confounders, socially less well-connected men and women had higher IL-6 levels (69), and several studies found higher IL-6 levels with lower socioeconomic status (37, 45, 61, 69). Finally, higher vital exhaustion was associated with higher circulating IL-6 levels in cardiac patients (50).
Stress-Related Actions of Glucocorticoids
In response to psychosocial stress, glucocorticoid hormones are released from the adrenal gland to regulate a broad variety of metabolic, immune, and behavioral processes, including hyperglycemia, dysproteinemia, electrolyte disturbances, elevated blood pressure, immunomodulatory effects, mood disorders, and sleep fragmentation (Fig. 3) (105). In the liver, cortisol stimulates hyperglycemia-causing gluconeogenesis and facilitates the synthesis of liver glycogen. Most of the circulating cortisol is bound to a specific transport protein, corticosteroid-binding globulin (CBG), also known as transcortin (26, 135). This bound fraction of cortisol is physiologically inactive and functions as a reservoir (122). Approximately 5%–10% of the total cortisol in the circulation is unbound and biologically active (83). Unbound cortisol is metabolized in the liver and kidneys with a biological half-life time of 80–100 min. Notably, cortisol measurements in plasma or serum reflect total cortisol concentrations, that is, bound and unbound cortisol, whereas saliva measurements assess the unbound and biologically active cortisol fraction (58). The hydrophobic unconjugated steroids are secreted into saliva predominantly by passive diffusion through the acinar cells of salivary glands and remain unbound to carrier proteins (128).

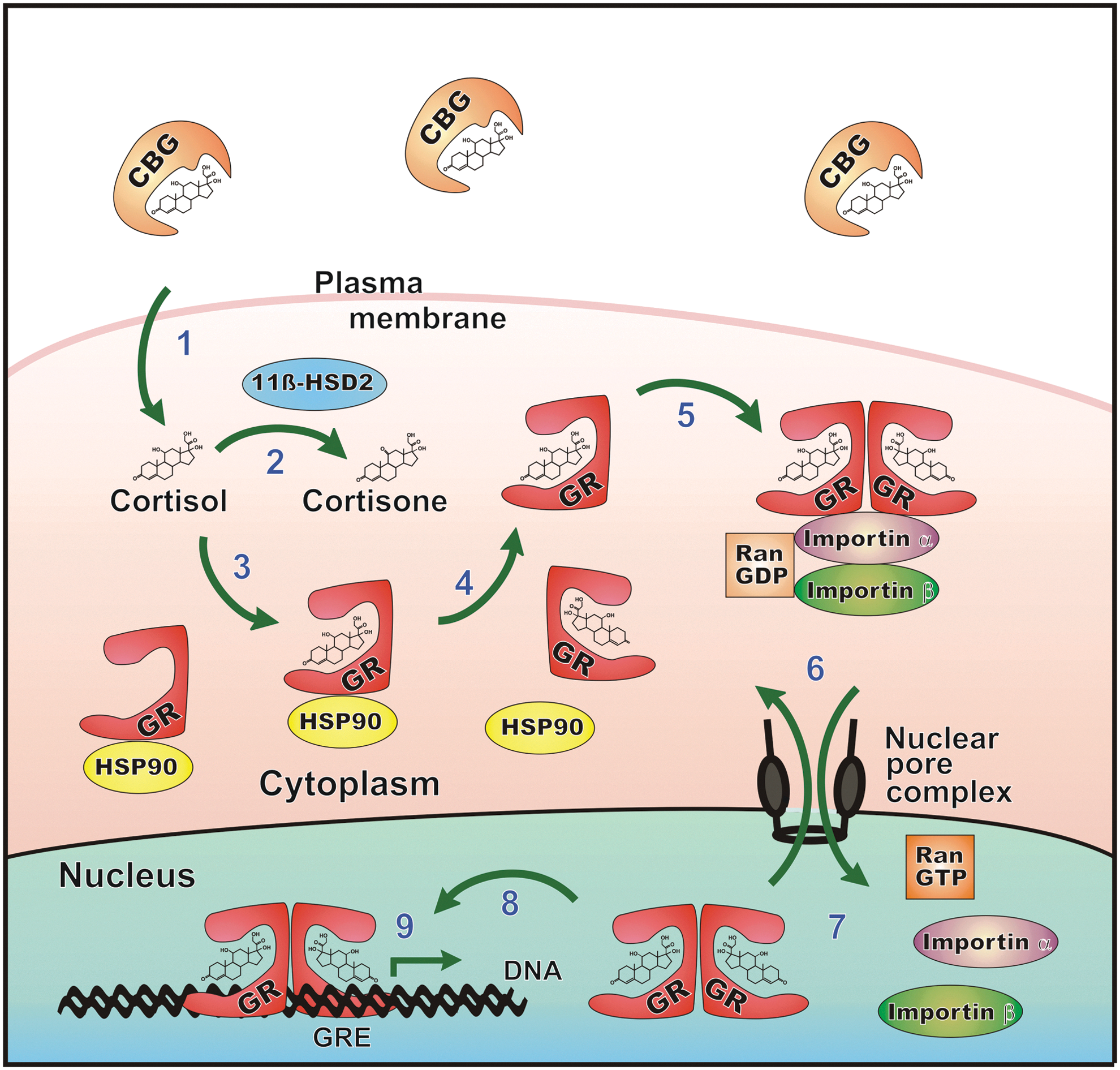
Cortisol exerts its numerous physiological effects via binding to two types of intracellular receptors on target tissues, the mineralocorticoid receptor (MR) and glucocorticoid receptor (GR) (5, 31) (Fig. 4). GRs are ubiquitously expressed, whereas MRs are expressed only in selected tissues (e.g., kidney, colon, heart, and brain) and at lower levels. In sites such as the vascular endothelium, MRs bind aldosterone exclusively because cortisol is excluded by local inactivation via 11β-hydroxysteroid dehydrogenase 2 (11β-HSD2), which converts cortisol to the less active metabolite cortisone (34). In contrast, 11β-HSD1 is predominantly a reductase that regenerates active cortisol from cortisone, thereby amplifying GR activation (130).

GR, encoded by the NR3C1 gene, resides in its unbound state in the cytosol. Binding of cortisol to the GR induces a conformational change resulting in the release of chaperons (including heat shock protein 90) and its subsequent translocation into the nucleus (15, 36). The activated GR complex binds to glucocorticoid response elements and modulates the expression of genes coding for proteins with immunosuppressive properties, including lipocortin-1, IL-10, IL-1 receptor antagonist, and IκBα, which is an important inhibitor of NFκB-mediated inflammation (4, 6, 11, 71, 106) (Fig. 5). In vascular endothelial cells, glucocorticoids inhibit the expression of inducible, but not constitutive, nitric oxide synthase at a transcriptional level, which may contribute to immunosuppressive and blood pressure-elevating effects of cortisol (98). Several studies using cultured cells or experimental animal models have shown that corticosteroids inhibit NADPH oxidase-mediated superoxide anion production and p22 phox mRNA expression [for a review, see Ref. (107)].
Studies investigating the reactivity of oxidative stress parameters to psychosocial stress induction provide diverging findings. In the hypothalamus of socially isolated rats, behavioral changes were associated with increased expression of markers of oxidative stress (8-hydroxy-2-deoxyguanosine and nitrotyrosine) and NOX2 mRNA (25). In addition, brain superoxide has been suggested as a key regulator of cardiovascular response to emotional stress in rabbits (77). Interestingly, in humans, acute psychological stress induction significantly altered plasma levels of cortisol, induced the recruitment of lymphocyte subsets, such as peripheral blood natural killer T cells, and suppressed the production of reactive oxygen species (ROS), as detected using chemiluminescence (2). In line with this, we previously found that in healthy men, acute psychosocial stress increased stress hormone release and reduced stimulated NADPH oxidase-derived superoxide anion production by monocyte-derived macrophages (63, 64). Moreover, in healthy subjects undergoing a brief mental stressor, the circadian rhythm of ROS synthesis was altered and an overall decreased formation of ROS was observed (3).
Functional Interactions Between Cytokines and HPA Signaling
Beside their beneficial metabolic effects following exposure to stressful stimuli in terms of energy preservation, plasma glucocorticoids suppress the production of distinct cytokines, such as interleukin-1 (IL-1) and IL-6, thereby modulating immune reactions (123). An early clinical observation by Späth-Schwalbe et al. suggested a stimulating effect of IL-6 on the activity of the HPA axis (110). Steensberg and colleagues demonstrated that compared with saline infusion, administration of physiological concentrations of recombinant IL-6 to healthy volunteers led to elevated plasma levels of the two anti-inflammatory cytokines, IL-1 receptor agonist (IL-1ra) and IL-10 (111). Furthermore, the authors revealed that IL-6 induced the release of cortisol into the circulation, which resulted in neutrocytosis and lymphopenia. Although in CRF-deficient mice, secretion of glucocorticoids was impaired after having been challenged with most stimuli, Muglia et al. found evidence of a near-normal glucocorticoid response upon IL-6 stimulation, indicating that IL-6 functions as an essential activator of the HPA axis during systemic inflammatory and other stressors in the absence of CRF expression (88).
Interestingly, administration of IL-1β elevates plasma ACTH and corticosterone levels in rats and induces a hyper-responsiveness of the HPA axis. Intracerebroventricular injection of IL-1β in castrated rats increased circulating ACTH and corticosterone levels measured 2 h later, whereas destruction of the PVN completely blocked the increase of HPA axis activity (99). Tumor necrosis factor α acts synergistically on IL-1β-mediated ACTH release (126). Similarly, other cytokines, such as IL-6, leukemia inhibitory factor (LIF), ciliary neurotrophic growth factor, oncostatin M, and cardiotrophin-1, also lead to elevated plasma ACTH concentration in the presence of IL-1β. These so-called IL-6-type cytokines signal through the common transmembrane receptor subunit glycoprotein 130 (gp130), resulting in subsequent activation of the Janus-activated kinase/signal transducer and activator of transcription 3 (JAK-STAT3) pathway (44, 73, 119, 145).
Effects of IL-6 on Adrenocortical Steroidogenesis
It has been suggested that elevated plasma IL-6 concentrations observed in subjects with chronic stress (37, 45, 55, 61, 69) potently activate steroid synthesis in the adrenal gland [for a review, see e.g., Ref. (95)] (Fig. 3). In rat adrenal gland cells, IL-6 and corticotropin act synergistically to stimulate release of corticosterone into the circulation (104). The elevated circulating IL-6 may directly stimulate adrenocortical steroidogenesis and thus maintain high cortisol levels even in the presence of a suppressed HPA axis. By means of RT-PCR and immunohistochemistry, it was demonstrated that the human adrenal gland expressed the IL-6 receptor (9, 94).
In a recent publication, Strickland et al. reported that stimulation of cultured human NCI-H295R adrenocortical cells with IL-6 resulted in increased production of cortisol (114). The release of cortisol from H295R cells was associated with increased expression of the cholesterol side-chain cleavage enzyme and other enzymes involved in steroidogenesis. Stimulation of H295R cells with IL-6 upregulated the expression of steroidogenic acute regulatory protein, which regulates the transfer of cholesterol from the outer to the inner mitochondrial membrane and serves as an important regulator of steroidogenesis (114). In addition, the authors found that IL-6 stimulation induced tyrosine phosphorylation of both STAT1 and STAT3, which was mitigated upon treatment of cells with AG490 or piceatannol. Incubation with these inhibitors significantly decreased expression of steroidogenic factor-1 (SF-1) and activator protein 1 (AP-1) mRNA, suggesting that IL-6 triggers cortisol release via a STAT3-dependent mechanism (114). In summary, IL-6 exerts stimulatory effects on the biosynthesis of cortisol in adrenocortical cells and modulates the suppressed HPA axis at different levels, thereby integrating responses from the immune, endocrine, and nervous systems.
Intracellular Signal Pathways for IL-6
Binding of an IL-6-type cytokine to its heterodimerized receptor activates Janus kinases (JAKs), which are named after the ancient, two-faced Roman God of gates and doorways. JAKs are noncovalently attached to the carboxy termini of the cytokine receptor, and after transphosphorylation, activated JAKs phosphorylate specific receptor tyrosine residues, which then serve as docking sites for STAT3 (145). Once recruited to the receptors, STAT3 molecules become phosphorylated by the JAKs and translocate to the nucleus where they bind to their target promoters as transcriptionally active dimers (27) (Fig. 6). The recruitment to promoters of IL-6-responsive genes is achieved through binding to palindromic target sequences, termed gamma-activated sites (GASs) (27).

The domain architecture of STAT3 is structurally similar to the other six human STAT family members and contains a conserved N-terminal domain, a coiled-coil domain, a DNA-binding domain, an Src homology 2 (SH2) domain for dimerization, a linker domain, and a C-terminal transactivating domain (7). For maximal transcriptional activation, both phosphorylation at a critical serine residue in position 727 and a tyrosine residue in position 705 are required (134). Tyrosine phosphorylation is a prerequisite for high-affinity DNA binding, while phosphorylation of serine 727 is dispensable for binding to GAS elements (133). Previous studies have demonstrated that tyrosine-phosphorylated STAT3 heterodimerizes with other members of this protein family, namely phospho-STAT1 and probably STAT5 (92, 121).
In myocardial tissue, STAT3 is activated under stressful conditions such as pressure overload and myocardial infarction [for a review, see Ref. (141)]. In several important studies, Hilfiker-Kleiner and her colleagues demonstrated that hearts from STAT3 knockout mice produced elevated ROS levels and, in contrast to wild-type animals, were not protected from myocardial damage by ischemic preconditioning (46, 47). Furthermore, it was shown that STAT3 protected against hypoxia/reoxygenation-induced cardiomyocyte injury by inducing the expression of manganese superoxide dismutase, thereby scavenging ROS generation (90).
Noncanonical STAT3 Functions in Oxidative Respiration and Naïve Pluripotency
Besides its role as an inducible nuclear transcription factor, STAT3 elicits nonclassical functions in mitochondria by augmenting the activities of complex I and II of the electron transport chain (ETC; Fig. 7). The mitochondrion is the subcellular compartment for STAT3-regulated changes in oxidative respiration where the main steps of glucocorticoid biosynthesis also take place. In mitochondria from murine STAT3 knockout hearts, the rate of oxygen consumption was decreased when pyruvate or malate was used as a complex I and succinate as a complex II substrate, confirming that STAT3 expression upregulates mitochondrial respiration (38, 131). Furthermore, STAT3 was detected in immunoprecipitates from mitochondrial extracts prepared from liver using a monoclonal antibody against complex I of the ETC (131). Previous studies have demonstrated that STAT3 binds directly to the cell death regulator gene associated with retinoid–interferon-induced mortality 19 (GRIM-19), which inhibits STAT3-dependent gene expression (70, 144). Mutational analysis has revealed that the transactivation domain of STAT3, including its serine 727 residue, is required for interaction with GRIM-19 as the serine-to-alanine substitution mutant S727A has almost completely lost its capacity to bind to the GRIM-19 component of the mitochondrial respiratory chain complex I (144). In the presence of this mutation, the mitochondrial import rate of STAT3 is reduced in a GRIM-19-dependent manner, suggesting that GRIM-19 functions as a chaperone for the recruitment of STAT3 to the mitochondrial inner membrane (120).

Recently, Meier et al. demonstrated that binding of STAT3 to cyclophilin D, a structural component of the mitochondrial permeability transition pore, was important for reducing mitochondrial ROS production after oxidative stress (79). The authors revealed that the amino terminus of STAT3 was required for binding to cyclophilin D (79). Using a transgenic mouse line with cardiomyocyte-specific overexpression of mitochondria-targeted STAT3 harboring the DNA-binding mutation E434A/E435A (termed MLS-STAT3E), Szczepanek and colleagues characterized the cytoprotective effects of mitochondrial STAT3 during ischemia (117). The authors showed that under conditions of ischemia, activities of the ETC complex I (NADH-ubiquinone oxidoreductase) and complex II (succinate–ubiquinone oxidoreductase) were decreased in MLS-STAT3E-expressing hearts compared with hearts from animals expressing wild-type STAT3, whereas complex III (ubiquinol–cytochrome c oxidoreductase) and complex IV (cytochrome c oxidase) activities were unchanged. Furthermore, mitochondrial localization of transcriptionally inactive MLS-STAT3E in cardiomyocytes prevented the release of cytochrome c from mitochondria into the cytosol during ischemia, suggesting that STAT3 attenuated cellular injury during ischemia and reperfusion by regulating mitochondrial respiration (118).
These observations are particularly interesting in the light of previous findings, which demonstrated that STAT3 directed self-renewal of pluripotent embryonic stem cells and induced pluripotent stem cells downstream of the LIF-receptor/gp130 complex (127, 131, 142). Molecular reprogramming of a differentiated cell back to a naïve pluripotent cell is mediated by the synergistic action of NANOG and activated STAT3, both of which are master regulators of naïve pluripotency acquisition (116). The homeoprotein NANOG and tyrosine-phosphorylated STAT3 both cooperate to upregulate the downstream target Krüppel-like factor 4 (KLF4), which is a canonical Yamanaka factor required to induce pluripotent stem cells (116). Recently, Carbognin et al. demonstrated that STAT3 upregulated the expression of mitochondrial transcripts and triggered the assembly of components of the ETC during reprogramming from primed to naïve pluripotency (16).
In summary, cytokine-mediated regulation of mitochondrial activity appears to be critical for changes in the metabolic energy profile and the induction of naïve pluripotency. The LIF/IL-6-driven transcription factor STAT3 plays a pivotal role in integrating nuclear gene expression, mitochondrial energy metabolism, and stem cell expansion. Whether the link between STAT3-driven, enhanced oxidative respiration and the induction and/or maintenance of pluripotency is also important in the context of psychosocial stress remains to be investigated.
Perspective
The surge in glucocorticoid hormone secretion in response to a perceived stressful challenge, including exposure to psychosocial stress, triggers adaptations needed to restore homeostasis in a fight-or-flight situation, whether or not to withstand the stressor or, alternatively, move the subject out of the danger zone. After secretion of catecholamines, the release of glucocorticoids initiates a second wave of physiological responses to prepare the organism for a stress-activated defense mechanism. In this perspective, STAT3-regulated cytokines such as circulating IL-6 constitute a third line of defense by coordinating the multitude of physiological responses that are required for maintaining the responsiveness of the HPA axis which is under negative feedback control by the elevated cortisol levels. The long-lasting consequences of the supposed steroidogenic stress-coping mechanisms exerted by IL-6-type cytokines may include altered immune responses and beneficial changes in energy metabolism.
Based on the abovementioned observations that assign IL-6 protective functions, we assume that IL-6 is a fundamental component of a rescue program that orchestrates survival pathways for the organism in the presence of threatening stimuli rather than simply being a proinflammatory cytokine. Future research is needed to disentangle the canonical and noncanonical pathways for STAT3 activation under psychosocial stress. We speculate that a coordinated interplay between both nuclear and mitochondrial functions is required for the cytoprotective effects mediated by STAT3 and, under mental stress in the absence of an infection, enhanced cortisol production may contribute to the overall beneficial effects of STAT3. Since it is unclear how STAT3 modulates electron transfer reactions in oxidative respiration and, on a molecular level, affects steroidogenesis, further investigations are urgently warranted. This research will hopefully add to our understanding of the relationship between circulating IL-6 and glucocorticoids, both of which are elevated in subjects exposed to psychosocial stress and functionally interact in complex pathways.
Footnotes
Acknowledgments
This work was funded by research grants from the Swiss National Science Foundation (PP00P1_128565/1 to P.H.W.) and the German Research Foundation (INST 38/550-1 to P.H.W. as well as ME_1648/4-3 and ME_1648/7-1 to T.M.). The funding sources had no impact on the writing of the article or the decision to submit the manuscript for publication.