
Abstract
Significance:
In 2003, structural studies revealed that eukaryotic 2-Cys peroxiredoxins (Prx) have evolved to be sensitive to inactivation of their thioredoxin peroxidase activity by hyperoxidation (sulfinylation) of their peroxide-reacting catalytic cysteine. This was accompanied by the unexpected discovery, that the sulfinylation of this cysteine was reversible in vivo and the identification of a new enzyme, sulfiredoxin, that had apparently co-evolved specifically to reduce hyperoxidized 2-Cys Prx, restoring their peroxidase activity. Together, these findings have provided the impetus for multiple studies investigating the purpose of this reversible, Prx hyperoxidation.
Recent Advances:
It has been suggested that inhibition of the thioredoxin peroxidase activity by hyperoxidation can both promote and inhibit peroxide signal transduction, depending on the context. Prx hyperoxidation has also been proposed to protect cells against reactive oxygen species (ROS)-induced damage, by preserving reduced thioredoxin and/or by increasing non-peroxidase chaperone or signaling activities of Prx.
Critical Issues:
Here, we will review the evidence in support of each of these proposed functions, in view of the in vivo contexts in which Prx hyperoxidation occurs, and the role of sulfiredoxin. Thus, we will attempt to explain the basis for seemingly contradictory roles for Prx hyperoxidation in redox signaling.
Future Directions:
We provide a rationale, based on modeling and experimental studies, for why Prx hyperoxidation should be considered a suitable, early biomarker for damaging levels of ROS. We discuss the implications that this has for the role of Prx in aging and the detection of hyperoxidized Prx as a conserved feature of circadian rhythms. Antioxid. Redox Signal. 28, 574–590.
Introduction
B
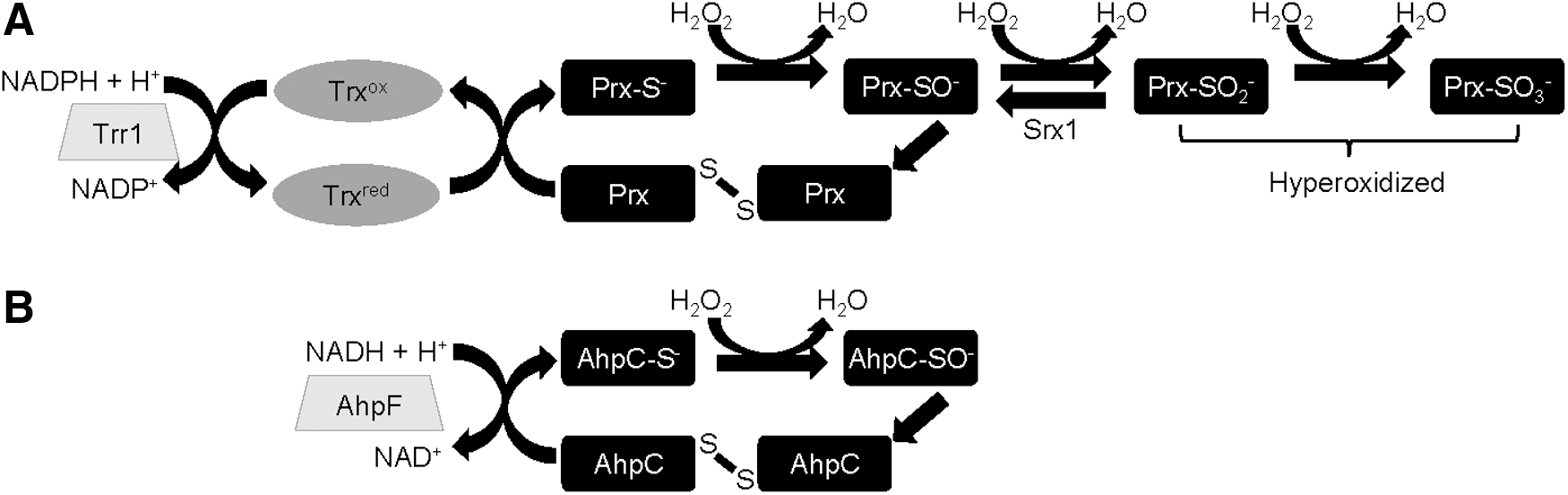
What Makes Some Prx More Sensitive to Sulfinylation of the Peroxidatic Cysteine?
Although typical 2-Cys Prxs are practically ubiquitously expressed, seminal studies by Wood et al. (2003) revealed that 2-Cys Prx that are sensitive to hyperoxidation share GGLG and YF motifs, which are not present in their prokaryotic peroxide-resistant counterparts (Fig. 2). By impeding the unfolding of the Prx to its locally unfolded state, these GGLG and YF motifs serve to delay the formation of the disulfide between the sulfenylated peroxidatic cysteine and the resolving cysteine, thus greatly increasing the probability that the sulfenylated cysteine will encounter a second peroxide molecule and become further oxidized to the sulfinylated form (Fig. 1). Subsequent work has identified that some eukaryotic 2-Cys Prx are more sensitive to hyperoxidation than others. For example, the peroxidatic cysteine of the mitochondrial 2-Cys Prx, Prx3, is more resistant to hyperoxidation than its cytoplasmic counterparts, Prx1 and Prx2, due to its higher rate of disulfide formation (23, 32, 70).

Other factors have also been shown to influence the sensitivity of Prx to hyperoxidation. Although the minimal catalytic unit of 2-Cys Prx is dimeric, under physiological conditions these dimers are cooperatively associated into oligomeric complexes, usually decamers, in which the reactivity of the peroxidatic cysteine (and thus the thioredoxin peroxidase activity) is increased, apart from the susceptibility to hyperoxidation (3, 67, 78). The rate of reduction of Prx disulfides also influences the extent to which Prx will become hyperoxidized in vivo. For instance, the lack of thioredoxin reductase activity in the endoplasmic reticulum (ER) limits the recycling of Prx4 disulfide dimers to the thiol state and thus the rate at which Prx4 can become hyperoxidized (15). The presence of other peroxide-removing activities has also been shown to protect Prx from hyperoxidation both in vitro and in vivo (21, 70, 84). Thus, the extent to which any 2-Cys Prx becomes hyperoxidized in vivo (Table 1) depends on a number of things, including the local concentration of peroxide and rate at which the sulfenylated form is able to locally unfold to allow disulfide formation, but also the concentrations of 2-Cys Prx and availability of thioredoxin.
Examples of studies investigating the extent of hyperoxidation of Prx in response to physiological changes and exogenous sources of reactive oxygen species. The minimum level of stress and earliest time point at which Prx hyperoxidation was detected are indicated, together with the extent of hyperoxidation and the methodology by which this was estimated.
1D, one-dimensional; 2D, two-dimensional; ACTH, adrencorticotrophic hormone; eCG, equine chorionic gonadotropin; hCG, human chorionic gonadotropin; IAM, iodoacetamide; IEF, isoelectric focusing; PAGE, polyacrylamide gel electrophoresis; ROS, reactive oxygen species; UVB, ultraviolet B.
How Is Hyperoxidation of Prx Reversed?
Although glutathione/glutaredoxins and thioredoxins can reduce oxidized cysteine derivatives, such as sulfenic, sulfenylamide, and disulfides, cysteine sulfinylation is resistant to reduction by any of these enzymes/redox couples. Consequently, cysteine sulfinylation has generally been considered irreversible. However, Woo et al. made the remarkable discovery that the hyperoxidation of the peroxide-reacting cysteine in human Prx1 to the sulfinylated derivative was slowly reversed in vivo, actively regenerating the thioredoxin peroxidase active form (94). At the same time, the enzyme responsible for reducing hyperoxidized 2-Cys Prx in yeast was identified as a 13 kDa H2O2-induced protein of previously unknown function and thus named “sulfiredoxin” (8). Significantly, homologues of sulfiredoxin were found to be encoded in the genomes of almost all the eukaryotes and prokaryotes where hyperoxidation-sensitive Prx are present, suggesting that this mechanism for reduction of hyperoxidized Prx is conserved (69). Although sulfiredoxin-mediated reduction of cysteine sulfinate still appears to be limited to the sulfinylated peroxidatic cysteine of 2-Cys Prx (95), human sulfiredoxin has also been shown to deglutathionylate actin and PTP1B (29) and denitrosylate the peroxidatic cysteine of Prx2 (81), raising the possibility that these activities also contribute to its in vivo function. Interestingly, sulfiredoxin shares some homology with bacterial ParB, a DNA-binding protein that is required for chromosomal partitioning, from which it appears to have evolved (4). However, importantly, the catalytic cysteine 99 that is essential for all known enzymatic activities of sulfiredoxin is not present in ParB.
Shortly after the discovery of sulfiredoxin, it was proposed that a second eukaryotic protein, sestrin, also catalyzed the reduction of hyperoxidized Prx (13). This function was proposed, in part, based on homology between sestrin and mycobacterial AhpD, an enzyme that reduces alkyl hydroperoxides and also disulfides in the Prx AhpC. However, subsequent studies have been unable to detect any in vitro or in vivo activity of sestrin in reducing hyperoxidized Prx (93). This is even the case in the nematode worm Caenorhabditis elegans, which lacks a sulfiredoxin encoding gene but expresses a sestrin homologue (61, 83). Further, other roles for sestrin have been identified that suggest that the lower levels of hyperoxidized Prx in cells in which sestrin is overexpressed may be due to indirect effects on the expression of sulfiredoxin and even other ROS-metabolizing enzymes [reviewed in Ho et al. (35)].
In contrast to sestrin, there is substantial evidence that sulfiredoxin is important for the reduction of hyperoxidized 2-Cys Prx under stress conditions in diverse phyla, including cyanobacteria (10), plants (75, 76), mammals (17), and the divergent yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe (8, 11, 88). Interestingly, the levels of sulfiredoxin mRNA have been found to be undetectably low in many organisms under normal growth conditions (11, 76, 88). Indeed, consistent with sulfiredoxin's functions being restricted to stress conditions, the phenotypes associated with loss of sulfiredoxin in mice, yeast, and plants have generally been found to relate to stress or toxin-induced conditions where its expression is induced (8, 11, 41, 43, 72, 76, 88, 91, 100). Moreover, although Prx are found in multiple cell compartments, organisms generally express a single sulfiredoxin gene, encoding proteins that have been shown to be largely confined to the cytoplasm in mammalian cells (57) or chloroplast in plants (76). These findings have raised questions as to whether reduction of hyperoxidized Prx has any significant role under normal physiological conditions, particularly in cell compartments that lack sulfiredoxin. Indeed, the absence of sulfiredoxin, or any evidence that hyperoxidized Prx become reduced, in C. elegans suggests that reducing hyperoxidized Prx is not of major importance, even in organisms where the Prx is sensitive to hyperoxidation (61, 83). However, in evolutionarily divergent yeast and in mammalian cells, sulfiredoxin gene expression is rapidly induced in response to various stress conditions. Moreover, sulfiredoxin is also imported into the mitochondria under stress conditions where it is important for reducing hyperoxidized Prx3 (44, 57). Nevertheless, although it is widely accepted that sulfiredoxin is important for the reduction of hyperoxidized Prx, it is notable that transcriptional, post-transcriptional, and post-translational mechanisms are present to ensure that, under optimal growth conditions, sulfiredoxin levels are kept very low and restricted to specific compartments (8, 11, 41, 44, 53). Indeed, the evolved sensitivity of eukaryotic 2-Cys Prx to hyperoxidation of the peroxidatic cysteine, and the tightly regulated expression and localization of sulfiredoxin, provides strong circumstantial evidence to suggest there are advantages to be gained from hyperoxidation of 2-Cys Prx in response to rises in intracellular H2O2 levels. In the remainder of this review, we will explore the various possible advantages that Prx hyperoxidation has been proposed to confer and the evidence in support of each hypothesis.
Loss of Function: Advantages of Inhibiting Thioredoxin Peroxidase Activity
The co-evolution of sensitivity to hyperoxidation with the utilization of H2O2 as a signaling molecule led to the original proposal of “The Floodgate Model” (98). In this model, it was suggested that the abundance and reactivity of 2-Cys Prx with H2O2 meant that they presented a significant barrier to the utilization of H2O2 as a signaling molecule. Accordingly, it was proposed that, for H2O2 to encounter and react with a cysteine on a “signaling” protein, it was important to first overcome this Prx barrier. By inactivating the thioredoxin peroxidase activity, Prx hyperoxidation would thus allow H2O2 to accumulate and react with target H2O2-sensitive signaling proteins (Fig. 3A). The Floodgate model has certainly provided a plausible explanation for why Prx might have evolved to be sensitive to hyperoxidation, potentially solving the conundrum as to how H2O2-regulated target signaling proteins ever become oxidized in the presence of more abundant and more reactive 2-Cys Prx. In support of the principle underlying this hypothesis, phosphorylation has been shown to reduce the thioredoxin peroxidase activity of mammalian Prx1 and thus promote H2O2-dependent signaling in response to growth factors (16, 19, 97). However, there is no evidence that 2-Cys Prx become hyperoxidized to any significant extent by H2O2 generated in response to growth factors or most other physiological stimuli (19, 25, 97) (Table 1). Moreover, although Prx hyperoxidation has now been detected in many biological situations (Tables 1 and 2), there remain few examples demonstrating that Prx hyperoxidation is important for downstream increases in signaling. Perhaps the best evidence to date in support of the Floodgate model has come from studies that have detected increased hyperoxidation of mitochondrial Prx3 in the adrenal cortex in response to adrenocorticotrophic hormone (ACTH). This Prx3 hyperoxidation is caused by increased levels of mitochondrial H2O2 generated by cytochrome P450s during corticosteroid production. Importantly, ACTH-induced Prx3 hyperoxidation has been shown to be required for the associated increase in cytoplasmic activation of the p38 mitogen activated protein kinase (MAPK) (Fig. 3A) (43). However, the activation of p38 MAPK has been shown to require cytosolic Prx (22, 87) and involve the formation of disulfides between Prx1 and the mitogen activated protein kinase kinase kinase (MAPKKK) Ask1 (40). Thus, the hyperoxidation of Prx3 detected by Kil et al. may actually serve to increase the H2O2 available to oxidize another Prx, Prx1, rather than, as suggested by the Floodgate model, a less peroxide-reactive signaling protein. Moreover, despite the attention that this model has received, there is still remarkably little evidence that inactivation of the thioredoxin peroxidase activity of 2-Cys Prx by hyperoxidation is responsible for in vivo increases in H2O2. For example, even in the case of the significant ACTH-induced increases in Prx3 hyperoxidation, described earlier, it is unclear as to whether the extent to which Prx3 becomes hyperoxidized (<30%) would limit the removal of mitochondrial H2O2 sufficiently to cause an increase in cytoplasmic H2O2 (43).

Examples of the physiological situations in which increases in hyperoxidized Prx have been detected, indicating the point at which peak levels of hyperoxidized Prx were detected and the method of detection. Immunoblots using anti-PrxSO2/3 antibodies (96) to detect hyperoxidized Prx were normalized to levels of the indicated Prx, using appropriate anti-Prx antibodies, unless otherwise indicated (*).
PBMC, proliferating blood monocytes.
Certainly the levels and duration of H2O2 exposure required to hyperoxidize the bulk of 2-Cys Prx activity seem to be far in excess of that produced in response to growth factors or other endogenous “non-stressful” H2O2-generating events (Table 1). It is possible that the inactivation of the thioredoxin peroxidase activity of 2-Cys Prx leads to local increases in H2O2 available for signaling, as proposed for growth factor-induced Prx phosphorylation and for ACTH-induced increases in p38 activity (19, 43, 97). However, it remains challenging to detect such proposed local increases, and even more difficult to determine whether any increases in H2O2 are actually caused by, rather than the cause of, Prx hyperoxidation. Moreover, recent in silico investigations have suggested that even localized increases in H2O2 are unlikely to be sufficient to promote rapid oxidation of signaling proteins, such as protein tyrosine phosphatase (PTP) (85). Indeed, there is increasing evidence that, rather than acting as barriers, Prx play direct, positive roles in promoting the H2O2-induced oxidation of, at least some, signaling proteins [for a review, see Netto and Antunes (55)]. For instance, in the fission yeast S. pombe, increases in exogenous H2O2 that are sufficient to cause H2O2-induced hyperoxidation of the 2-Cys Prx Tpx1 actually prevent the H2O2-induced oxidation and activation of the AP-1-like transcription factor Pap1, in complete contradiction to the predictions of the Floodgate model (11, 88).
Indeed, our further work in S. pombe has suggested an alternative advantage of inhibiting the thioredoxin peroxidase activity of Prx. Similar to the premise of the Floodgate model, we also proposed that the high abundance and H2O2 reactivity of 2-Cys Prx means that there are conditions under which loss of the thioredoxin peroxidase activity by hyperoxidation of Prx is important. However, in contrast to Wood et al.'s proposal that loss of Prx activity increases the H2O2 available for signaling functions, our work suggests that hyperoxidation is important to limit the oxidation of thioredoxin, the other substrate of Prx activity, thus increasing the amount of reduced thioredoxin available to reduce other oxidized proteins under stress conditions (Fig. 3B) (27). This alternative hypothesis arose, in part, from our observation that the sensitivity of 2-Cys Prx to hyperoxidation had co-evolved in organisms in which Prx disulfides, generated during the catalytic reduction of peroxides, were reduced by thioredoxin, rather than by a dedicated Prx reductase, AhpF (Fig. 1). Thioredoxin is a relatively broad specificity oxidoreductase that not only is an essential cofactor for many enzymes that involve a redox step in their catalytic mechanism but also functions to reduce disulfides in signaling proteins, such as PTPs, the Ask1 MAPKKK or yeast AP-1-like transcription factors, and oxidatively “damaged” proteins under oxidative stress conditions [for reviews, see Refs. (30, 50)]. Accordingly, as demonstrated in S. pombe, when thioredoxin reductase activity is limiting, the consequence of coupling the peroxidase activity of the single 2-Cys Prx, Tpx1, to thioredoxin is that, in cells exposed to H2O2, Prx disulfides can dramatically deplete the amount of reduced thioredoxin available to reduce other substrates and repair oxidative protein damage (27). Indeed, we have shown that this ability of Tpx1 disulfides to drive thioredoxin oxidation, and thus inhibit the reduction of other oxidized proteins, is vital for stable H2O2-induced oxidation of the Pap1 transcription factor, which in its oxidized form activates a stress-protective gene expression program (12). These findings suggest that the coupling of Prx to thioredoxin may have evolved to provide a means for low levels of H2O2 to promote thioredoxin oxidation, and thus regulate the activity of downstream thioredoxin/thioredoxin family substrate proteins (Fig. 3B).
As Prx acquired this new H2O2-signaling function, as H2O2-dependent regulators of thioredoxin activity, we speculated that this may have generated the selection pressure for increased sensitivity to hyperoxidation; to uncouple Prx activity from thioredoxin under acute stress conditions and thus divert thioredoxin toward repair pathways (Fig. 3B). Indeed, we demonstrated that both thioredoxin and the thioredoxin-dependent repair enzyme, methionine sulfoxide reductase A (Mxr1), were inhibited under oxidative stress conditions in S. pombe cells expressing a hyperoxidation-resistant Tpx1 mutant (Tpx11-181) (27). Notably, although new gene expression is inhibited, preventing cells from adapting by increasing the expression of ROS defense proteins, both wild-type and Δtpx1 mutant yeast are able to recover after prolonged exposure to very high (25 mM) concentrations of H2O2 (27). However, ectopic expression of sulfiredoxin under these acute stress conditions, where it is not usually expressed, maintains the levels of Tpx1 disulfides, increasing the inhibition of thioredoxin and resulting in a Tpx1-dependent loss of cell viability (27). Thus, we demonstrated that hyperoxidation of Tpx1 is important for cell survival under acute stress conditions. This suggests that inactivation of the thioredoxin peroxidase activity of Tpx1 is vital to allow thioredoxin-dependent maintenance of essential activities when ROS levels inhibit new gene expression/protein synthesis.
Moreover, our data suggest that the inactivation of Tpx1 by hyperoxidation also protects S. pombe from damage at lower, sub-lethal concentrations. Notably, thioredoxin is not only important for repairing oxidative protein damage, and supporting the activity of methionine sulfoxide reductase, but also required for ribonucleotide reductase, which is important for DNA synthesis and repair. Indeed, consistent with the prolonged inhibition of thioredoxin in cells expressing hyperoxidation-resistant Tpx11-181 (27), the cell division cycle of these cells is de-regulated, and nuclear damage increased, when these cells are exposed to non-lethal levels of H2O2 at which Tpx1 is usually hyperoxidized [Brown, Morgan, and Veal, unpublished observations and Day et al. (27)].
Apart from revealing that inactivation of the thioredoxin peroxidase activity of Prx is important to repair damage and allow cell survival under acute stress conditions, these yeast studies suggest that the coupling of Prx to thioredoxins may have evolved as a key mechanism to allow H2O2 signaling. For instance, we have subsequently demonstrated that the thioredoxin peroxidase activity of Tpx1 is required for the H2O2-induced oxidation of the Pap1 transcription factor and oxidative stress resistance, not because it is required to remove peroxides, but because it promotes the H2O2-induced oxidation of the thioredoxin-like protein, Txl1 that reduces Pap1 (12). Accordingly, in cells expressing a thioredoxin peroxidase-defective Tpx1 mutant, Tpx1C169S, the thioredoxin-like protein Txl1 is not oxidized in response to H2O2, Pap1 oxidation is inhibited, and cells are unable to mount Pap1-dependent transcriptional responses (12). Although the effect of Prx hyperoxidation on thioredoxin activity has yet to be investigated in other systems, Prx have been shown to act as important Trx regulators in plants and budding yeast, where they promote thioredoxin oxidation in response to light-stimulated increases in H2O2 (9, 26). Moreover, the observation that the facultative anaerobe, Vibrio vulnificus, possesses both a thioredoxin-coupled, hyperoxidation-sensitive 2-Cys Prx and a stress-induced, AhpF-coupled 2-Cys Prx suggests that 2-Cys Prx may also have evolved distinct functions in response to different levels of H2O2 in Vibrio and other prokaryotes (Enterobacter sp., Pseudomonas sp., Pseudoalteromonas sp., Salmonella sp., Shewanella sp.) where both robust and hyperoxidation-sensitive 2-Cys Prx are present (2, 33) (Fig. 2).
Thus, we propose that the coupling of thioredoxin to the reduction of 2-Cys Prx may have evolved originally in some bacteria as an effective means to regulate the host of cellular thioredoxin-dependent enzymatic and signaling activities in response to rises in H2O2. However, inevitably with this advantage came the risk that, under acute stress conditions, the continuous formation of Prx disulfides could lead to prolonged thioredoxin oxidation, preventing maintenance of vital cellular activities and repair of oxidatively damaged proteins or DNA. Moreover, if thioredoxin reductase activity is not limiting then, unchecked, the thioredoxin peroxidase activity of Prx could potentially deplete NADPH and, consequently, inhibit the glutathione system too. Thus, given the importance of NADPH, thioredoxin, and glutathione for survival under oxidative stress conditions, this connection of the peroxidase activity of 2-Cys Prx to the thioredoxin system would be predicted to generate a strong selective pressure to de-couple thioredoxin from Prx reduction in cells exposed to high levels of oxidants.
Gain of Function: Increased Non-Peroxidase Activities of Hyperoxidized Prx
Both the models discussed earlier consider advantages of Prx hyperoxidation in terms of “loss of function” of the thioredoxin peroxidase activity, either to allow H2O2 signaling or to preserve thioredoxin in times of acute stress (Fig. 3). In addition to these proposals, Prx have been shown to be multifunctional proteins, with a chaperone activity that protects against protein aggregation (18, 34). Accordingly, although hyperoxidation inhibits the thioredoxin peroxidase activity of Prx, it has been proposed that it increases other chaperone and signaling activities of Prx that protect against oxidative damage (Fig. 3C). Here, we will review the evidence that although hyperoxidation inactivates the thioredoxin peroxidase activity of Prx, it may lead to a “gain of function” in other activities.
Chaperone Activity
The identification that 2-Cys Prx also have a chaperone function came from studies in the budding yeast S. cerevisiae. Investigations into the reduced thermotolerance of S. cerevisiae lacking the predominant 2-Cys Prx, Tsa1, lead to the discovery that 2-Cys Prx can protect against thermal aggregation of proteins in vitro and in vivo independent from their peroxidase activity by acting as molecular chaperones (38). This chaperone activity is promoted by heat or H2O2-induced formation of higher order Prx complexes (38, 82). Although the chaperone activity of Prx does not appear to require either of the cysteines that are essential for thioredoxin peroxidase activity, the reversible, H2O2-induced formation of higher order Prx complexes does require the peroxide-reacting cysteine, as well as the cycling activity of thioredoxin. Moreover, in cells lacking sulfiredoxin, these H2O2-induced higher molecular weight forms with increased chaperone activity persist for longer (38). Together, these findings suggest that hyperoxidized Prx more readily participates in higher molecular weight forms with increased chaperone activity. Thus, it has been proposed that, although hyperoxidation inactivates the peroxidase activity of Prx, it promotes this chaperone activity, reducing protein aggregation and, consequently, promotes the survival of cells exposed to thermal stress (38). Moreover, further oxidation of the peroxide-reacting cysteine to a sulfonyl derivative has been shown to further increase the chaperone activity of Tsa1 (48).
In vitro assays suggest that chaperone activity is likely to be a general feature of 2-Cys Prx, for example shared by human Prx1 and Prx2 (37, 54), and Prx from plants (45), protozoa (82), and bacteria (20). Other studies have also identified that other post-translational modifications (37), mutations, and changes in pH that promote formation of higher molecular weight oligomeric forms of Prx also increase this chaperone activity (20, 37). However, given that hyperoxidation is not essential for this chaperone activity (82), yet completely inhibits the thioredoxin peroxidase activity, it seems unlikely that increased chaperone activity alone could provide enough selective pressure to drive the evolution of peroxide-sensitive Prx. Indeed, it is possible that some of the in vivo protection against thermal protein aggregation associated with Prx hyperoxidation could also be due to increased availability of thioredoxin to reduce oxidized proteins (27). Recently, further light was cast on this issue by the discovery that hyperoxidation of S. cerevisiae Tsa1 is important for the in vivo recruitment of ATP-dependent chaperones (heat shock proteins) to aggregates that form under oxidative stress conditions (31). This suggests that hyperoxidized Prx can also facilitate the action of heat shock proteins (e.g., Hsp70) in actively removing or refolding damaged proteins. Accordingly, this may provide an important advantage under oxidative stress conditions (31) (Fig. 4).

Other Activities for Hyperoxidized Prx
Prx hyperoxidation has also been proposed to increase or promote other peroxidase-independent signaling activities. In many cases, this is also proposed to be due to an increase in the stability of oligomeric forms. For instance, hyperoxidation of human Prx2 has been shown to result in the formation of filamentous oligomers. Interestingly, the presence of these forms is associated with cell cycle arrest (71). Although it is currently unclear how these structural changes serve to regulate cell responses, the potential for altering the interaction partners of Prx has been revealed by more recent studies. For example, in contrast with the predictions of the Floodgate model, and with the increased p38 activation associated with hyperoxidation of Prx3 (43) (Fig. 3A), hyperoxidation of human Prx1 increases its association with the phosphatase mitogen-activated kinase phosphatase (MKP)-5, thus preventing ROS-induced inactivation of MKP-5 and maintaining low p38MAPKα activity (86). By stabilizing the decameric form, hyperoxidation has also been shown to promote the non-covalent interaction of human Prx2 with a thioredoxin-like protein, Erp46 (64). Intriguingly, this latter study is consistent with the view that hyperoxidation may also serve to regulate the activity of thioredoxin/PDI-like proteins, which could also lead to downstream effects on protein folding/homeostasis or signaling under oxidative stress conditions.
Although Prx hyperoxidation leads to a loss of peroxidase activity, the recent identification that at least some eukaryotic Prx, including human Prdx1, also have catalase activity (2 H2O2→2H2O + O2), even when exposed to concentrations of H2O2 at which they are usually hyperoxidized, suggests that hyperoxidized Prx may retain some catalytic peroxide-removing activity (80, 101). Although the functional significance of this catalase activity has yet to be established, the dependence of this catalase activity on Fe3+ and a conserved GVL motif found in the majority of hyperoxidation-sensitive Prx (Fig. 2) raises the intriguing possibility that these Prx may have a bifunctional activity profile; with thioredoxin-dependent peroxidase and signaling activities at low H2O2 concentrations and catalase and chaperone/other signaling activities that are retained, or gained, at higher H2O2 concentrations.
When Does Prx Hyperoxidation Occur In Vivo and to What Extent?
Detecting Prx hyperoxidation
In assessing the relative contributions of each of the proposed functions for Prx hyperoxidation in vivo, it is important to understand the circumstances under which Prx become hyperoxidized. The generation of antibodies that are specific to the hyperoxidized, sulfinylated or sulfonylated, forms of Prx (αPrxSO2/3) has provided an important tool for detection of hyperoxidized Prx, circumventing the need for electrophoretic or mass spectrometric methods to distinguish hyperoxidized from reduced Prx monomers (48, 96). This has led to a dramatic increase over recent years in the number of conditions under which Prx hyperoxidation has been detected (Table 2). In most cases, comparisons with the signal from an antibody that detects all Prx redox states is used as a control to ensure that changes in the level of hyperoxidized Prx are not due to changes in total Prx levels. These studies have provided important information as to the circumstances under which there is a detectable increase in hyperoxidized Prx. However, use of αPrxSO2/3 antibodies alone to detect Prx hyperoxidation does have some limitations. Most importantly, this approach gives no indication of the extent to which Prx are hyperoxidized, preventing any assessment of whether a tiny fraction or the vast majority of peroxide-reacting Prx cysteines are hyperoxidized. Moreover, although sulfonylation is unlikely to occur to any significant extent in physiological conditions, the αPrxSO2/3 antibodies, most frequently used, recognize both sulfinylated Prx, which is a substrate for sulfiredoxin, and sulfonylated Prx, which is not. Further, although the sequence identity of the sulfinylated peptide against which they are raised means these antibodies are effective at detecting hyperoxidation of 2-Cys Prx from a broad range of species, this also prevents them from distinguishing between different hyperoxidized 2-Cys Prx in the same sample, unless the Prx are sufficiently different in size to allow electrophoretic separation from other Prx, for example, Prx3 (Fig. 2). To examine whether specific Prx are hyperoxidized, and whether this hyperoxidized Prx represents a significant proportion of the total Prx, it is necessary to separate hyperoxidized Prx from other redox forms, before immunoblotting and analysis with antibodies to Prx that are not specific to a particular redox state. This separation can be effected in a number of ways; for example, by covalent modification of the reduced cysteines in protein samples with agents that allow electrophoretic separation of reduced and hyperoxidized Prx forms based on differences in mobility, for example, AMS, or by using isoelectric focusing gels that allow separation on the basis of charge differences (8, 14, 24, 90). Notably, the sensitivity of the peroxidatic thiol to oxidation means that, unless care is taken to prevent oxidation occurring in vitro during protein extraction, the relative amounts of reduced and disulfide-bonded Prx will not reflect the in vivo distribution (24). However, using these methods, it is possible to quantitatively determine the extent to which a specific Prx become hyperoxidized (14, 24, 27, 84) (Table 1). Nevertheless, although there are now a vast number of studies reporting in vivo hyperoxidation of Prx, in few cases has the percentage of Prx that remains in its catalytically active form also been determined (Tables 1 and 2). This is important for assessing the effect of hyperoxidation. For instance, even if hyperoxidized Prx is detected, if this only represents a tiny fraction of the total Prx, then it is unlikely that this will have a significant downstream, general effect on either H2O2 levels or the availability of reduced thioredoxin. It is possible that localized Prx hyperoxidation might have a localized effect on levels of H2O2 or reduced thioredoxin, particularly if there are physical constraints on the diffusion of H2O2 from the site at which it is generated in vivo [as discussed by Heppner et al. (34)]. However, if the bulk of the Prx remains in active cycling forms, we speculate that it is perhaps more likely that any biological effect of hyperoxidation is due to an increase in a Prx's chaperone or signaling activities (Fig. 3C).
Is Prx hyperoxidation a cause or consequence of oxidative stress?
In addition to providing invaluable insight into the structure and redox biochemistry of Prx, in vitro studies have demonstrated that some eukaryotic Prx are intrinsically more sensitive to hyperoxidation than others (23, 70). However, it is impossible for in vitro studies to simulate the complex cellular environment. The potential influence of other cell components on the sensitivity of Prx to hyperoxidation is illustrated by an in vitro study from the Winterbourn lab that demonstrated that the presence of other peroxide-removing enzymes, such as catalase, protects Prx against hyperoxidation (70). Indeed, the in vitro and in vivo sensitivity of Prx to hyperoxidation can be quite different. For instance, although Prx4 is as intrinsically sensitive to hyperoxidation as Prx2 in vitro, the absence of an efficient ER recycling system for Prx4 disulfides means that much less Prx4 becomes hyperoxidized in vivo, even in cells exposed to very high (10 mM) concentrations of H2O2 (15) (Table 1). Similarly, Prx2 has been shown to be less susceptible to extensive hyperoxidation in cells where thioredoxin activity is limiting (49). Therefore, to understand the physiological causes of Prx hyperoxidation, it is important to take into account the relative in vivo levels of Prx, thioredoxin, and other peroxide-metabolizing systems.
Over recent years, computational models, incorporating both reaction rate parameters, determined from biochemical studies of purified proteins, and in vivo considerations, such as cell H2O2 permeability and the relative concentrations of different enzymes, have started to be utilized to address how cells respond to oxidants (1, 6, 84, 85). In our recent study, we took this approach to investigate how the in vivo environment affects the oxidation state of Prx. Taking advantage of the presence of a single S. pombe Prx, Tpx1, the availability of quantitative proteomic data for the concentration of each S. pombe protein (52), and established redox Western blotting methods (27), we obtained quantitative time course data for the distribution of Tpx1 between different oxidation states in cells exposed to different concentrations of H2O2. Using these data, we were able to construct a mathematical model representing the redox reactions of Tpx1 that was able to simulate the in vivo data (84). We learned a number of things from this model. Notably, rather than a linear increase in the amount of Tpx1 that became hyperoxidized in response to exogenous H2O2, we discovered that Tpx1 was protected from hyperoxidation by an in vivo peroxide-removing activity, such that Tpx1 hyperoxidation was only detected when this activity was overcome and intracellular H2O2 concentrations began to increase (Fig. 4). Notably, this observation that intracellular H2O2 levels undergo a biphasic response to increasing doses of extracellular H2O2 was also found to be true for a human kidney cell line (HEK293) (84). Moreover, although the amount of exogenous H2O2 required to breach the cellular peroxide-buffering capacity was much lower, nevertheless, as in S. pombe, hyperoxidation of Prx was only detected in kidney cells once the intracellular H2O2 concentrations began to rise more rapidly. Indeed, a perusal of the literature suggests that Prx hyperoxidation is also correlated with increases in intracellular H2O2 in other cells (Table 1). Of course, as Prx are very efficient at removing H2O2, it might be expected that inactivation of Prx by hyperoxidation would be correlated with a rise in intracellular H2O2. However, we were able to show that there was no change in the threshold extracellular H2O2 bolus required to cause intracellular H2O2 concentrations to rise in cells expressing a hyperoxidation-resistant Tpx1 mutant (84). Moreover, loss of Tpx1, the single glutathione peroxidase, Gpx1, or catalase did not affect the bi-phasic response of S. pombe. This suggests that none of these peroxidases makes a significant contribution to the cell's ability to buffer the internal environment against an external bolus of H2O2. In contrast, thioredoxin was found to be critical for this bi-phasic response. It is likely that other reductants, such as glutathione and methionine, also contribute to the cellular H2O2-buffering capacity, but a key role for thioredoxin is consistent with findings from a model constructed by the Kemp group that suggested that the thiol proteome makes an important contribution to the H2O2-buffering capacity of the cell (1). Although relatively limited protein thiol oxidation has generally been observed in mammalian cells treated with sufficient H2O2 to cause some Prx hyperoxidation (5), to our knowledge, the effect/s of sustained Prx activity in these circumstances on thioredoxin activity and the thiol proteome have not been investigated. Therefore, it remains possible that rapid thioredoxin-mediated reduction of protein disulfides, enhanced by diversion of thioredoxin from Prx toward other substrates, plays a widespread role in protecting proteins against sustained oxidation (Fig. 4). In any case, regardless of the nature of the H2O2-removing activity, our data imply that Prx hyperoxidation only occurs in vivo once this peroxide-buffering capacity is saturated and the intracellular H2O2 levels start to rise more rapidly (Fig. 4).
Intriguingly, our model and in vivo data also suggest that the main route by which Prx become hyperoxidized involves the sulfenylation of both peroxidatic cysteines in a Prx dimer, before formation of the disulfide bond between the Prx partners. The remaining sulfenylated peroxidatic cysteine in the disulfide-bonded dimer subsequently becomes sulfinylated by reacting with a second H2O2 molecule. Although other routes may also contribute, this reaction sequence is consistent with the detection of hyperoxidized disulfides before hyperoxidized monomer (27, 39, 84). Moreover, it also fits with our finding that hyperoxidized Prx were only detected once the cellular H2O2-buffering capacity was breached, as the chances of a second sulfenylation event taking place before disulfide formation would be greatly increased once intracellular H2O2 levels began to increase more substantially (84).
The finding that the trigger for Prx hyperoxidation is a rise in H2O2 after saturation of cellular H2O2-buffering capacity has important implications for the various situations in which hyperoxidized Prx are detected in vivo. For example, this implies that increases in Prx hyperoxidation, detected as a conserved feature of many circadian rhythms (Table 2 and references within), are preceded by an increase in the levels of H2O2 above the cell's H2O2-buffering capacity. Moreover, our data raise the intriguing possibility that differences between the intrinsic sensitivity of different Prx to hyperoxidation in different organisms or organelles may reflect the H2O2-buffering capacity of the specific Prx's in vivo environment.
Implications for the physiological roles of Prx
As discussed earlier, the simple view that, like their more robust bacterial counterparts, the main function of eukaryotic 2-Cys Prx is to remove H2O2 has been challenged by a number of discoveries. These include the increasing evidence that these 2-Cys Prx are multifunctional proteins, with chaperone and signaling activities that do not require their thioredoxin peroxidase activity, but that are increased by post-translational modifications, including hyperoxidation. In addition to peroxidase-independent functions of Prx, in their thioredoxin peroxidase-active form, Prx have been shown to actively promote redox signaling, both by acting as peroxide transducers to initiate the oxidation of redox-regulated proteins (40, 79) and by promoting the oxidation of thioredoxin family proteins (12) [for a review, see Netto and Antunes (55); Fig. 3B]. Both these functions are inhibited by Prx hyperoxidation, along with any ROS-detoxifying function of the thioredoxin peroxidase activity.
Based on these findings, it is possible that the increased ROS levels, oxidative damage, and/or activation of stress-activated signaling pathways frequently observed in cells lacking Prx could partly reflect loss of one or more of Prx's other activities, rather than rises in H2O2 directly caused by loss of the thioredoxin peroxidase activity. Indeed, as our studies suggest, although Prx may provide an effective means to reduce the low levels of peroxides generated by normal metabolism, in organisms where they are coupled with thioredoxin, this peroxidase activity is not well suited to deal with an increase in H2O2 above these levels, particularly when thioredoxin reductase activity is limiting (12, 84). Hence, we propose that Prx hyperoxidation should be viewed as a consequence rather than a cause of the associated rises in ROS. This is in accordance with others that have proposed that hyperoxidized Prx are a suitable early biomarker for oxidative stress (74).
Increases in hyperoxidized Prx have now been detected in response to many stimuli and, most intriguingly, as a conserved feature of many circadian rhythms (Table 2). In most of these studies, the anti-PrxSO2/3 signal is normalized to the signal from an antibody that detects both reduced and hyperoxidized forms of the Prx in question (Table 2). Although, as discussed earlier, this is sufficient to demonstrate that the changes in hyperoxidized Prx do not reflect changes in total Prx levels, there remain few studies in which the % of Prx that is hyperoxidized has been accurately determined. Filling this gap will be important if we are to discern whether the main function of hyperoxidation in a given situation is to limit either the removal of H2O2 and/or oxidation of thioredoxin, which would most likely require the bulk of Prx to become hyperoxidized, or as a post-translational modification promoting the formation of stable oligomeric forms and interactions with signaling or chaperone/misfolded proteins. For example, formation of a small or localized amount of hyperoxidized Prx, such as has been frequently observed (Table 1), may be unlikely to impact rates of peroxide removal or thioredoxin oxidation, but they might be sufficient to promote alternative chaperone or signaling functions (Figs. 3C and 4). Indeed, although Prx have been demonstrated to have localized ROS-protective activities, in some cases these do not require the resolving cysteine, which is essential for thioredoxin peroxidase activity. Hence, it seems likely that, in at least some cases, chaperone or signaling activities of Prx are responsible for protecting against rises in ROS (12, 51, 92).
Role of Prx hyperoxidation in aging
Increases in hyperoxidized Prx (Tsa1) have been detected during the replicative aging of budding yeast (53) (Table 1) that recent work suggests are important to protect against the aggregation of proteins (31). Accordingly, this raises the possibility that it is this increased chaperone function that may be responsible for the lower ROS levels and extended replicative lifespan of cells expressing sulfiredoxin, rather than the increased thioredoxin peroxidase activity of Tsa1 (53). This raises the possibility that hyperoxidized Prx may also contribute to the pro-longevity function of Prx in other models of aging (58).
We have used our modeling approach to examine how chronological aging might be predicted to affect the sensitivity of the S. pombe Prx, Tpx1, to hyperoxidation. Quiescent cells are an established model of chronological aging, with the chronological lifespan determined as the length of time quiescent cells retain the capacity to re-enter the cell cycle and divide. S. pombe cells undergo extensive changes in gene expression as they enter quiescence that promote their survival in stressful conditions (52). When our model is repopulated with the concentrations of proteins found in quiescent cells, it predicts that the H2O2-buffering capacity of quiescent S. pombe is much lower than that of exponentially growing cells (Fig. 4B). Although these predictions require experimental confirmation, this would be expected to increase the sensitivity of Tpx1 to hyperoxidation, consistent with the possibility that Prx hyperoxidation might also have a protective life-extending role in aging S. pombe. This fits with the observation that Prx2 is more susceptible to hyperoxidation in chondrocytes from older human subjects (21). However, it remains to be determined whether a chaperone activity for hyperoxidized Prx is important for the pro-longevity functions of Prx in animal models (47, 56, 61, 62). Although hyperoxidized PRDX-2 has been shown to extend C. elegans survival of a thermal stress (61), increases in hyperoxidized Prx are not detected during C. elegans aging, despite the lack of any sulfiredoxin activity (83). Notably, all somatic cells in adult C. elegans are post-mitotic, so together these studies raise the possibility that hyperoxidized Prx may be specifically important for maintaining the replicative capacity of aging mitotic cells (53).
Conclusions and Future Directions
Although, as discussed, the thioredoxin peroxidase activity of Prx may be more important for maintaining redox homeostasis and promoting ROS signal transduction than directly counteracting damaging rises in ROS, Prx, undoubtedly, play central roles in protective ROS responses. It is, therefore, not surprising that deregulation of their activity should have been linked to many diseases and aging [for reviews, see Refs. (58, 66)]. It is currently difficult to discern whether ROS-protective effects of Prx are due to removal of H2O2, positive H2O2-signaling functions, chaperone activity, or a combination of all three. Accordingly, a full understanding of how the different activities of Prx contribute to cellular defenses against rises in ROS will require a better spatial resolution of (i) where ROS levels increase, (ii) where hyperoxidized Prx are detected, (iii) correlation with the localized distribution of Prx between dimers and decamers, and (iv) the availability of thioredoxin/thioredoxin reductase and sulfiredoxin. We predict that new methods to measure intracellular changes in H2O2 or NADPH in vivo and mathematical models, incorporating in vitro kinetic analysis of the association and disassociation of Prx oligomers, and interactions with thioredoxin, sulfiredoxin, and other partner proteins, will together provide valuable tools toward a more complete understanding of how cells initiate appropriate responses to different levels of ROS and the key role/s that Prx play in these responses.
Footnotes
Acknowledgments
The authors thank the reviewers and many other colleagues in the peroxiredoxin field for their insightful comments and helpful discussions that have informed this article. Z.E.U. and L.E.T. were funded by the BBSRC (BB/F015895/1, BB/F016980/1, BB/F023065/1). Discussions between E.A.V. and C.S.P. were facilitated by an NRF Travel grant. They are grateful to the many contributors in this field whose work has informed this review and apologize to any person whose work they may have inadvertently overlooked.