
Abstract
Significance:
Ion channels play an important role in the regulation of organelle function within the cell, as proven by increasing evidence pointing to a link between altered function of intracellular ion channels and different pathologies ranging from cancer to neurodegenerative diseases, ischemic damage, and lysosomal storage diseases.
Recent Advances:
A link between these pathologies and redox state as well as lipid homeostasis and ion channel function is in the focus of current research.
Critical Issues:
Ion channels are target of modulation by lipids and lipid messengers, although in most cases the mechanistic details have not been clarified yet. Ion channel function importantly impacts production of reactive oxygen species (ROS), especially in the case of mitochondria and lysosomes. ROS, in turn, may modulate the function of intracellular channels triggering thereby a feedback control under physiological conditions. If produced in excess, ROS can be harmful to lipids and may produce oxidized forms of these membrane constituents that ultimately affect ion channel function by triggering a “circulus vitiosus.”
Future Directions:
The present review summarizes our current knowledge about the contribution of intracellular channels to oxidative stress and gives examples of how these channels are modulated by lipids and how this modulation may affect ROS production in ROS-related diseases. Future studies need to address the importance of the regulation of intracellular ion channels and related oxidative stress by lipids in various physiological and pathological contexts. Antioxid. Redox Signal. 28, 949–972.
Introduction
I
Expanding our knowledge on factors that regulate ion channel functions under physiological and pathological conditions is therefore of utmost importance. In addition to classical regulatory stimuli such as transmembrane voltage, membrane tension, and ligands (92), lipids and reactive oxygen species (ROS) are emerging as key molecules able to influence channel activity. In fact, these integral membrane proteins are surrounded by a complex lipid environment that plays an active role in the structural and functional modulation of channels (163). In addition, lipid-derived signaling molecules also importantly impact channel function in many cases.
Ample evidence for the modulation of ion channels by oxidative stress is also available and, vice-versa, many ion channels can modulate ROS production within the cells. In the present review we focus our attention only on intracellular ion channels as they primarily impact intracellular ROS production. We summarize the current knowledge about the contribution of these proteins to oxidative stress, also in relation to intracellular signaling involving lipids.
Oxidative Stress and Consequences for Cell Function
Oxidative stress is an increase in the concentration of ROS, which include superoxide anion (O2
However, under physiological conditions, as more than 90% of the total intracellular ATP is generated by mitochondria, this organelle produces ROS as a normal consequence of electron leak in the respiratory chain associated with oxidative phosphorylation and ATP production (116) (see the Regulation of ROS Release by Mitochondrial Ion Channels section).
Oxidative stress is known to lead to lipid, protein, and DNA damage due to peroxidation and might result in cell death, but it may also directly regulate molecules such as ion channels and numerous protein kinases that are critical for cellular signaling pathways. Indeed, redox signaling is now recognized to mediate different tissue-specific cellular functions, including muscle contraction/relaxation, metabolic cycles, insulin secretion from pancreatic β cells, T cell homeostasis, self-renewal and differentiation in stem cells, diabetes, and metabolic syndrome [for review see, e.g., O-Uchi et al. (157)].
Today it is clear that ROS are related to cellular toxicity (at high ROS levels) and also to fine-regulation of physiological processes (at lower ROS concentrations). ROS in general can influence redox-sensitive processes within the cell in several systems, and H2O2 specifically modifies the function of various proteins, including transcription factors, kinases, and phosphatases (94).
In the present review therefore we bring attention to ion channels that modulate ROS production within the physiological range and to a few examples of ion flux-modulated oxidative stress in response to lipid signaling molecules that act on intracellular channels. Although to date there are only few concrete examples that directly link modulation of intracellular ion channels by lipids to downstream signaling events involving ROS, hopefully the possible connections described in this review will prompt the field of signaling and of intracellular channels to gain a more in-depth insight into this relevant topic.
Modulation of Intracellular Ion Channel Function by Lipids and Lipid Signaling Molecules
There are different kind of lipids that can affect ion channel function in various contexts, including fatty acids, polyunsaturated fatty acids (PUFAs), phospholipids, phosphoinositides, ceramide, sphingolipids, cholesterol and lipoxins. The exact molecular mechanisms by which they exert their regulatory function on channel activity have not fully been elucidated yet in detail for each class of these lipids. In general, membrane surface potential importantly contributes to the membrane potential acting on the channel pore; thus, agents modifying the surface potential may exert their action indirectly, via changes of this parameter, but direct interactions with transmembrane domains or specific binding sites have also been proposed in several cases [(234), for review, see Poveda et al. (163)].
The effect of fatty acids was extensively studied on intracellular channels directly in the organelle membrane especially in the cases of mitochondria and lysosome. In the former case, this was achieved both using classical bioenergetic approach [for reviews see, e.g., Di Paola and Lorusso (56) and Schonfeld et al. (188)] and electrophysiological experiments to directly visualize the effect of fatty acids/lipids on the channel behavior, including kinetics of closure and openings and the probability of being open for a given channel.
Figure 1 summarizes intracellular ion channels residing in different organelles and highlights their regulation by different kinds of lipids. The present review focuses prevalently on the modulation of these channels, while only briefly mentioning their intracellular location, main characteristics, and function. For recent reviews providing a more detailed description of intracellular ion channel function, see, for example, Checchetto et al. (43), Kiselyov and Muallem (112), Xiong and Zhu (226), and Xu et al. (227). Although the concentration of lipid signaling molecules varies depending on the stimuli and cell type, the concentrations used in most articles for studying channel function are within the physiological range.

Regulation by saturated, unsaturated fatty acids, and PUFAs
The most studied examples regarding fatty acid regulation refer to channels in the mitochondria, the “powerhouse” of the cells, being the prevalent site of ATP synthesis. Table 1 summarizes the effect of the lipid molecules discussed in the Modulation of Intracellular Ion Channel Function by Lipids and Lipid Signaling Molecules section on various ion channels that display intracellular location.
The general structure of different classes of lipid molecules is shown.
5-HD, 5-hydroxydecanoate; AA, arachidonic acid; DAG, diacylglycerol; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; FA, fatty acids; IMAC, inner membrane anion channel; IMM, inner mitochondrial membrane; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3-gated calcium release channels; Kv, voltage-gated potassium; LCFA, long-chain fatty acids; mitoBKCa, mitochondrial big-conductance calcium-dependent potassium channel; mitoKATP, mitochondrial ATP-dependent potassium channel; naChR, nicotinic acetylcholine receptor; n.d., non-determined; PI(3,5)P2, phosphatidylinositol 3,5-bisphosphate; PI(4,5)P2, phosphatidylinositol 4,5-bisphosphate; PIP2, phosphatidylinositol bisphosphate; PM, plasma membrane; PTP, permeability transition pore; PUFAs, polyunsaturated fatty acids; RyR, ryanodine receptor; S1P, sphingosine 1-phosphate; Sph, sphingosine; TASK-3, two-pore acid-sensitive potassium channel 3; TPC1, two-pore channel 1; TRIC, trimeric intracellular cation; TRP, transient receptor potential; UCP1, uncoupling protein 1; VDAC, voltage-dependent anion channel.
It has been clarified more than a decade ago that in the presence of long-chain fatty acids (LCFA), the mitochondrial uncoupling protein 1 (UCP1) increases the permeability of the inner membrane for H+ [for review, see Bertholet and Kirichok (21)]. UCP1 plays an important role in the thermogenesis in brown adipose tissue, since by uncoupling proton transport from oxidative phosphorylation, it generates heat. Using patch clamping of the IMM, in a seminal work, native UCP1 was shown to operate as a LCFA/H+ symporter (64). The authors provided evidence that chloride current through UCP1 is very small compared with the currents obtained using LCFA and concluded that passage of chloride, previously thought to be the major anion transported by UCP1, is unlikely to significantly contribute to UCP1-mediated uncoupling.
Other mitochondrial channels are also affected by fatty acids. The inner membrane anion channel (IMAC) has been reported to be activated by low, nonlytic concentrations (≤30 μM) of LCFA as well as by myristic acid (187). LCFA concentration in the serum under physiological concentration ranges between 200 and 600 μM (145). Opening of the mitochondrial permeability transition pore (PTP) is also triggered by fatty acids and LCFA (162). PTP is a large channel with conductance values ranging up to 1.5 nS (239) possibly formed by the dimeric form of ATP synthase (22, 78). Long-lasting opening of PTP is linked to loss of membrane potential, swelling of mitochondria, and cell death (19, 20).
Finally, 5-hydroxydecanoate (5-HD), a member of the family of fatty acids, is widely considered as a selective inhibitor of the mitochondrial ATP-dependent potassium channel (mitoKATP), although in the absence of a definitive molecular identification of the mitoKATP, the question of selectivity is still debated [for recent review, see Laskowski et al. (122)]. mitoKATP is inhibited by ATP and its function has been linked to cardioprotection (see the Regulation of ROS Release by Mitochondrial Ion Channels section). The recent work that proposed the renal outer medullary K+ channel ROMK2 (Kir1.1b) as the channel-forming subunit of mitoKATP (69) did not address by direct, electrophysiological experiments the inhibition by 5-HD of mitoKATP toward the plasma membrane (PM) KATP.
Among fatty acids, some of the PUFAs play an important regulatory role on ion channel function. PUFAs are integral, structural components of the phospholipid bilayer of cell membranes. However, specific conditions such as brain ischemia can increase the level of both extracellular and intracellular PUFAs and can trigger the hydrolysis of membrane phospholipids with consequent release of free fatty acids, particularly of arachidonic acid (AA), a 20-carbon ω-6 PUFA (76). Intracellular AA concentration can reach 200 μM, for example, in pancreatic β cells when these cells are exposed to high extracellular glucose (224) and increases to similar levels in other cell types as well in response to physical exercise, tissue ischemia, or inflammation. In mitochondria, Szewczyk and colleagues reported that AA, docosahexaenoic acid (DHA), and eicosapentaenoic acid (EPA) were all able to increase the open probability of the mitochondrial big-conductance calcium-dependent potassium channel (mitoBKCa) in the 10–30 μM range (159).
Opening of the mitochondrial PTP has also been shown to be triggered by AA, via an indirect mechanism involving oxidized pyridine nucleotides arising from the increased oxygen consumption and oxidative stress induced by AA (39). Subsequent thiol oxidation triggers opening of PTP, which may have a deleterious effect on mitochondrial function and on the fate of the whole cell, being PTP opening linked to various forms of cell death (169). It has also been demonstrated that a rise in intracellular calcium concentration leads to activation of cytosolic phospholipase A2 with generation of AA, which causes apoptosis exclusively through the mitochondrial pathway involving PTP (162). Opening of this pore is facilitated by fatty acid acyl-CoAs such as palmitoyl-CoA as well (71). Interestingly, dietary intake of PUFA, in particular DHA and EPA, has been shown to exert cardioprotective effects by suppressing Ca2+-induced opening of the PTP (109), although a PUFA-rich diet did not prolong survival and cardiac function of cardiomyopathic hamsters (72).
Whether other mitochondria-specific channels such as the mitochondrial calcium uniporter (MCU) (51) are directly modulated by PUFA and AA is still an open question, to our knowledge. Likewise, modulation of endoplasmic reticulum (ER)-resident trimeric intracellular cation (TRIC) channels (231), whose pore structure has just been resolved (230), by fatty acids and PUFA has not been studied so far. This appears to be the case also for the first identified protein underlying the lysosomal/endosomal potassium conductance, TMEM175 (36). Instead, AA has been shown to activate ryanodine receptor (RyR) in pancreatic β cells (225) and seems to be able to induce calcium release via inositol 1,4,5-trisphosphate (IP3)-gated calcium release channels (IP3R) and two-pore channel 1 (TPC1) in endothelial cells (241).
PUFA inhibits most of the voltage-gated ion channels, apparently by binding to the open state of the channel [for recent review, see Moreno et al. (151)] and exerting a direct effect on ion channels through domains that do not involve the voltage sensor module (83). Voltage-gated channels with selectivity for potassium, sodium, or calcium are crucial for both neuronal and cardiac excitability. PUFA-mediated channel modulation has been investigated mostly for plasma membrane-located channels. However, in view of the fact that organelles also harbor members of the voltage-gated families (227) (see also Fig. 1), such modulation by lipids may well occur also in intracellular channels. In particular, the voltage-gated potassium channels Kv1.3 (201), Kv1.5 (125), and Kv7.4 (208) are present in a functional form in the IMM, while Kv1.3, Kv1.1, Kv1.2, Kv2.2 (102), and Kv10.1 have been described in the inner nuclear membrane (44, 199). Kv1.3 was found also in the cis-Golgi compartment (237) while the ER harbors Kv1.6 (237).
Basically, all these channels have a counterpart in the PM with a well-defined function and the mechanisms targeting them to the different organelles are mostly unclear. Likewise, the function of these organellar voltage-gated Kv-type channels is still largely unknown, except for mitoKv1.3 that emerged as a key player in apoptosis (124), mitoKv7.4 that seems to be related to cardioprotection (208), and mitoSKCa that regulates respiration and calcium uptake (95). Instead, nuclear Kv10.1 and Kv1.3 are thought to impact gene expression regulation (102, 167). Among the above-listed channels, to our knowledge, for none of them their modulation by PUFA or AA has been studied directly in the intracellular membranes where they are located, but information regarding their PM counterparts might be of relevance.
An early work showed that μM doses of AA (EC50 1.55 μM), by acting on both the channel protein and its lipid environment, depressed outward-rectifying K+ channels exhibiting features compatible with those of the Kv1.3 channel (213). It has also been illustrated that incorporation of fatty acids with different degrees of unsaturation and carbon chain lengths into the PM influences the function of Kv1.3 channels of lymphocytes (204). PUFAs such as linoleic acid, AA, and DHA decreased the activation and inactivation time constants of the Kv1.3 channel gating without affecting the voltage dependence. Instead, saturated and monounsaturated fatty acids did not cause significant changes in the channel kinetics (204). Since Kv1.3 of the PM is crucial for immune response (34), it has been hypothesized by the authors that a diet rich in PUFAs and the PUFA-induced modification of Kv1.3 kinetics might have important consequences on activation of T cells under pathophysiological conditions.
A possible contribution of Kv1.3 modulation to the beneficial effects of dietary PUFA supplement in autoimmune reactions and/or during chronic inflammation has been hypothesized. Since mitochondria are emerging as crucial players in chronic inflammation, a modification of mtKv1.3 by, for example, AA and other PUFAs might be an important aspect to be investigated in future studies. Likewise, the finding that lipoxins, which are endogenous eicosanoids released during the resolution phase of inflammation, inhibit PM Kv1.3 activity in lipopolysaccharide-activated macrophages at physiologically relevant concentrations (500 nM) is of possible importance in the context of the known anti-inflammatory actions of these lipids (152). Beside PM Kv1.3, PM Kv1.5 has also been shown to be inhibited by PUFA, including α-linolenic acid, suggesting a link between the antiarrhythmic potential of PUFA and the block of the atrial PM channel, Kv1.5 (83).
Regulation of intracellular ion channels by phospholipids, phosphoinositides, and inositol-trisphosphate
Membrane phospholipids, which might affect channel activity, have been proposed to alter the function of voltage-gated ion channels, characterized by the presence of positively charged, voltage-sensing residues in the transmembrane regions: the negatively charged phosphate groups in the membrane lipids were proposed to stabilize the positively charged residues during the voltage-sensor gating process (228).
However, concrete examples of intracellular ion channel modulation by membrane phospholipids are restricted to cardiolipin (CL), the signature phospholipid of mitochondrial membranes. CL indirectly regulates oxidative phosphorylation (101), the release of cytochrome c in its oxidized form (and thus, apoptosis), and the assembly and function of various mitochondrial membrane proteins [for review, see Monteiro et al. (149)]. CL can also regulate mitochondrial ion channels, for example, the gating kinetics of the outer mitochondria membrane porin (named also voltage-dependent anion channel, VDAC) are affected by CL (182) and the conformational state giving rise to functional UCP1 is stabilized by CL (126).
Another class of lipids with known ion channel modulatory function are phosphoinositides, messengers able to regulate most cellular processes and to regulate ion channel function (10, 91, 137). Phosphatidylinositol can be phosphorylated at various positions on the inositol ring, giving rise to seven different phosphoinositide species. Their effects on intracellular channels are shown in Figure 2. Interestingly, these different signaling molecules are not evenly distributed in the various intracellular membranes, but are abundant in the Golgi complex, in early and late endosomes. They play important roles in the membrane/protein trafficking at the endosomal system (25).

Among these phosphoinositides, phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] is emerging as an important molecule able to provide a unique layer of specificity at intracellular compartment-mediated signaling (205). Phosphatidylinositol bisphosphate (PIP2), a minor acidic membrane lipid found primarily in the inner leaflet of the PM, has been reported to either activate or inhibit various PM channels and also to modulate intracellular ones. PIP2 concentration has been estimated to vary between 2 and 10 μM (146).
PIP2 acts as an activator of the intracellular ryanodine-sensitive calcium release channels (RyR) (163); instead, it inhibits PM cyclic nucleotide-gated channels (CNG), transient receptor potential-like (TRPL) channels, capsaicin-activated transient receptor potential (TRP) channels, and the ER-located IP3R. In the case of voltage-gated calcium, some voltage-gated potassium (Kv) and hyperpolarization activated cyclic nucleotide-gated (HCN) channels, PIP2 has a dual effect on ion channel function, acting both as an activator and as an inhibitor (177).
PIP2 affects the function of PM Kv7.4 (97, 131), of PM Kv1.3 (144), and of PM two-pore acid-sensitive potassium channel 3 (TASK-3) (138), all found to be active also in the IMM (Fig. 1). In particular, while PIP2 increased the open probability of PM Kv7.2-Kv7.4 homomultimers, it inhibited PM Kv1.3. Whether such effects can be observed also in the case of the mitochondrial channels is still unknown. Interestingly, PI(4,5)P2 was recently identified in mitochondria and its removal was shown to be associated with increased mitochondrial fragmentation and autophagy (181), a process that is part of the “integrated stress response” (115).
By directly patch-clamping isolated lysosomes, Xu and colleagues described in a seminal work that lysosomal but not PM-localized mucolipin (TRPML1) is activated by phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2], a lysosome-specific PIP2. Instead, the channel is negatively regulated by PI(4,5)P(2). TRPML1 is an endolysosomal ion channel permeable to mono- and divalent cations, including Ca2+, Fe2+, and Zn2+, that has emerged as a major regulator of the endolysosomal function (234). Mammalian two-pore channel proteins (TPC1, TPC2; TPCN1, TPCN2) that encode ion channels in intracellular endosomes and lysosomes were also proposed to mediate endolysosomal calcium release. By directly recording TPCs in endolysosomes, they were found to be activated by PI(3,5)P2 (218).
Altogether, the idea proposed by these authors is that phosphoinositide regulation sets compartment-specific activity of membrane channels and transporters. A recent study discovered, by structural and functional analyses, a central role for PIP2 in stabilizing the cytoplasmic gate of the ion permeation pathway in TRIC channels (230). Among the several nonselective cation channels, including TRPV1, TRPM8, mitsugumin23, and pannexin channels, present in the ER of different cells types and possibly function as Ca2+ leak channels, the function of TRPV1 (178, 210) and TRPM8 (133, 179) is directly modulated by PIP2. TRPC3 (65, 217), found also in the IMM, is activated by PIP2 (127).
PIP2 hydrolysis in the PM, which occurs by receptor-activated phospholipase C (PLC), generates the soluble second messenger, IP3 and 1,2-diacylglycerol (DAG). Soluble IP3 binds to the cytosolic side of the IP3 receptor located in the ER, induces its opening and causes Ca2+ release. Inositol 1,4,5 trisphosphate receptors (IP3R) are the primary intracellular Ca2+ release channels in nonexcitable cells and are present with three isoforms that are differently distributed in different tissues and able to form homo- and hetero-tetramers (106, 223). IP3-triggered opening of IP3R results in the activation of multiple downstream signaling cascades [for recent reviews see, e.g., Levine and Patel (128) and Chang and Liou (41)].
The presence of PLC-δ1 has been described in liver mitochondria and was proposed to be linked to the regulation of calcium uptake into this organelle (113). Whether the PIP2 products IP3 and DAG can directly regulate the calcium uniporter and other mitochondrial calcium uptake/release pathways such as the sodium/calcium exchanger (161), LETM1 (103), RyR (23, 24, 100) [for recent reviews see, e.g., Foskett and Philipson (68), Rizzuto et al. (176), and Wagner et al. (215)], and TRPC3 is an interesting but still open question.
A low level of RyR1 is detectable in the IMM of heart mitochondria and provides a ruthenium red-insensitive, rapid transport pathway for Ca2+. Likewise, a small fraction of TRPC3 is located in mitochondria. TRPC3 can be activated by DAG-containing oleic acid (37, 98), but again, whether the same regulation occurs at mitoTRPC3 is still unknown. Finally, IP3 may also have an indirect regulatory effect on organellar ion channels, for example, by “directing” Ca2+ to specific intracellular targets harboring calcium-permeable or calcium-activated channels (as well as IP3 receptors), such as mitochondria and lysosomes (206).
Ion channel modulation by sphingolipids, bioactive lipids, and cholesterol
Sphingolipids comprise a large class of structural and bioactive lipids whose metabolism is regulated by a large family of enzymes (195). Sphingosine (Sph) and other small sphingolipids such as ceramide (Cer) and sphingosine 1-phosphate (S1P) are emerging as important regulators of immune cell function and play critical roles in inflammation, cellular growth, differentiation, proliferation, apoptosis, infection, metabolism, and related pathologies [see, e.g., Bartke and Hannun (13), Becker et al. (17), Espaillat et al. (62), Gulbins and Li (84), Hannun and Obeid (87), and Kornhuber et al. (114)].
Sph can be generated by hydrolysis of ceramide via the action of ceramidases in lysosomes, the PM, the ER, as well as the Golgi complex (209), and levels of free Sph are in the range of 10–100 pmol/106 cells (196). S1P concentration within the cell reaches sub-μM concentrations (197), while ceramide level can reach even 20 μM under certain conditions. A small change in ceramide concentration can drastically increase the levels of Sph or S1P (13).
Sph inhibits itself the activity of the skeletal muscle Ca2+ release channel RyR (192) and has recently been shown to act as a positive regulator of calcium release from acidic stores by a mechanism involving specifically the TPC1 lysosomal channel (93). Sph directly reduces the single-channel activity of RyR channels [for review see, e.g., Sabbadini et al. (184)]. Lysosomal accumulation of sphingomyelins and cholesterol, caused by alteration in sphingomyelinase (SMase), inhibits TRPML1 channel activity and therefore lysosomal exocytosis and trafficking (221). In cultured human umbilical vein endothelial cells, S1P was also shown to mobilize intracellular calcium, by inducing release of this ion via IP3-gated ER channels (190).
Whether the regulation observed for the PM calcium-dependent channel by S1P, that is, enhancement of BKCa channel activity by increasing its Ca2+ sensitivity (111), occurs also in the mitoBKCa, still awaits clarification. Likewise, sphingosylphosphorylcholine (SPC) but not S1P shifted the activation midpoint of PM Kv1.3 by about 20 mV toward positive membrane potentials (207) but whether this occurs also for mitoKv1.3 is still unknown.
Ceramide and GD3 ganglioside are two known mediators of apoptosis: during apoptosis, GD3 translocation takes place from PM lipid rafts to mitochondria, and an increase in ceramide content occurs (149). There is compelling evidence that ceramide plays a key role in different diseases [for reviews see, e.g., Kornhuber et al. (114) and Schenck et al. (186)], including the neurodegeneration and amyloidogenesis occurring in the brain in Alzheimer's disease (40).
Short-chain C2 ceramide sensitizes mitochondria to Ca2+-induced PTP opening, resulting in ceramide-induced enhancement of the loss of mitochondrial membrane potential and of calcium uptake into the matrix (203). In fact, ceramide targets protein phosphatase 2A (PP2A) in a mitochondria-associated signaling complex to induce dephosphorylation of the BH3-only protein Bad. Dephosphorylated Bad sensitizes PTP to Ca2+ through a VDAC-mediated process (183). In sharp contrast, long-chain ceramides inhibited the opening of the mitochondrial PTP induced by oxidative stress, sulphydryl (SH) group crosslinking, or high Ca2+ load, suggesting that ceramide affects a major, common PTP regulatory pathway (155).
Ceramide (C6) potently inhibits the PM Kv1.3 channel (27, 55, 85), but again, to our knowledge, an effect on mitoKv1.3 has not been demonstrated, even though ceramide is clearly present in mitochondria at physiologically relevant concentrations (89). It has also been proposed that ceramide itself can form channels in the outer mitochondrial membrane and thus contributes to cytochrome c release. Although both C2 and C16 ceramides were shown to form large and stable channels in artificial membranes (194), the hypothesis that ceramide channels mediate cytochrome c release during apoptosis needs additional experimental evidence.
In addition to the above lipids with signaling function, cholesterol, a structural membrane lipid, also alters ion channel function of the PM. Indeed, lipid modulation of ion channels can be mediated by their segregation into specific lipid domains, for example, into lipid rafts, characterized by high cholesterol and sphingolipid content. In some cases, cholesterol potentiates the activity of the channel, as it is the case of γ-aminobutyric acid (GABA)-gated channel or the nicotinic acetylcholine receptor (nAChR). In other cases, as for the voltage- and Ca2+-gated K+ potassium BKCa and Kir channels, cholesterol diminishes their activity (163).
Cholesterol, being present in tiny quantities in organellar membranes, is generally not considered to crucially impact on intracellular channel function. This view is currently changing, as active (not raft-localized or bulk) cholesterol was found to be present in various organelles. The mitochondrial 18-kDa translocator protein (TSPO) is a core element of cholesterol trafficking between cytosol and mitochondria (75) and the mitochondrial inner membrane is the site of conversion of cholesterol to steroid hormones, oxysterols, bile acids, vitamin D, and other derivatives (121). Late endosomes/lysosomes are responsible for dispatching sterols obtained from endocytosed low-density lipoproteins to other organelles. Among mitochondrial and lysosomal channels, porin (VDAC) binds cholesterol (220), and vice versa, VDAC function affects mitochondrial cholesterol distribution and function (35).
Intracellular Ion Channel Function and ROS Production
Although ion channels and transporters do not contribute directly to ROS production, their opening or closures frequently lead to modification of organelle function that promotes ROS release. This is a particularly important aspect when studying intracellular channels, since ROS are emerging as key players in the context of a number of pathologies.
Regulation of ROS release by mitochondrial ion channels
As mentioned in the Introduction section, under physiological conditions, most of the ROS within the cells are produced at the level of mitochondria, and growing evidence points to the important contribution of mitochondrial channels to the regulation of ion homeostasis imbalances that profoundly affect bioenergetic efficiency, ROS production, and mitochondrial integrity (202). Ion channels in this organelle modulate the mitochondrial membrane potential, generated thanks to oxidative phosphorylation. Figure 3 illustrates the cross talk between ROS release and mitochondrial channel function.
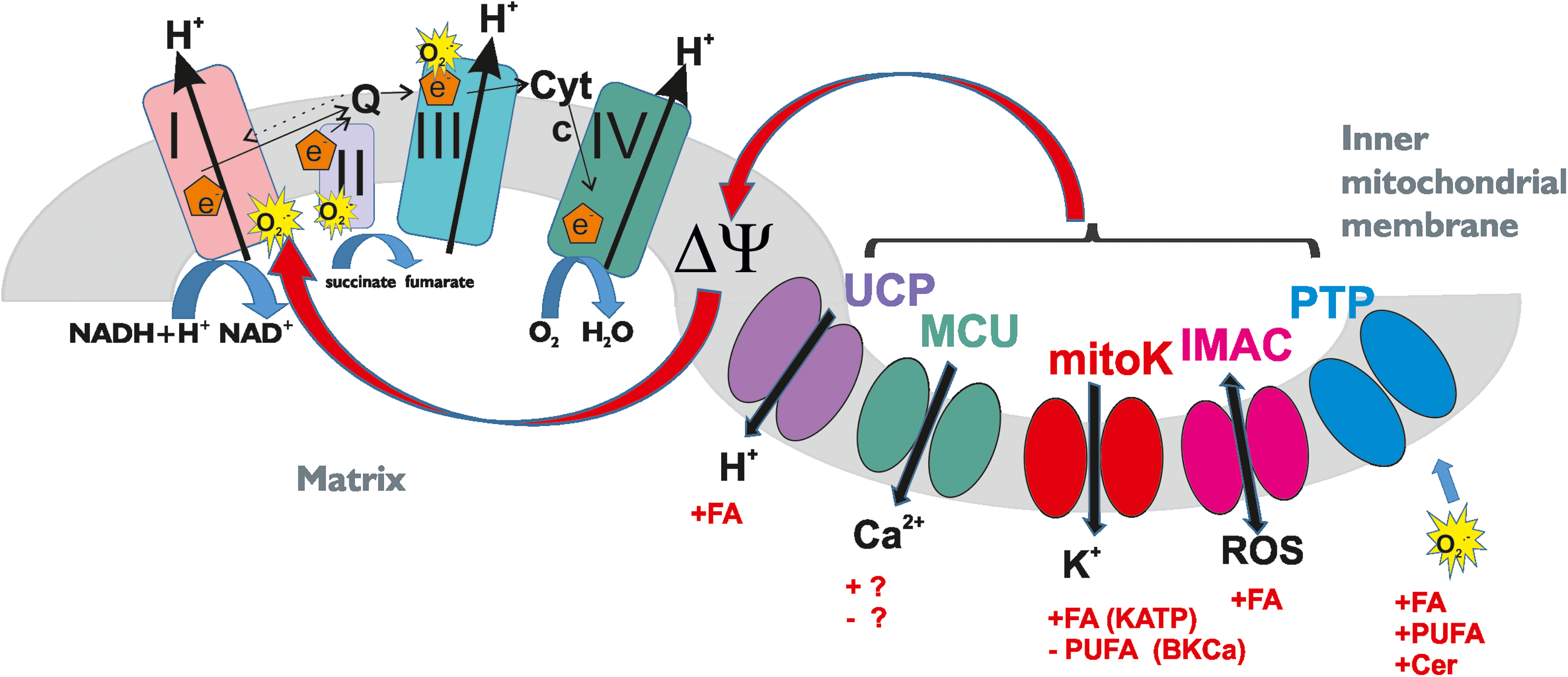
Both depolarization and hyperpolarization have consequences on the rate of superoxide formation [for reviews see, e.g., Malinska et al. (143), Murphy (153), and Zorov et al. (240)]. Excellent recent reviews summarize the mechanism of mitochondrial ROS production (157), and therefore, we only briefly mention here the main connection between membrane potential change and ROS release. Hyperpolarization lowers the efficacy of complex IV (cytochrome c oxidase), induces the reduction of respiratory chain complexes and of intermediates, and as a consequence, it increases the probability of a one-electron transfer to oxygen at respiratory complexes I (nicotinamide adenine dinucleotide [NADH] dehydrogenase) and III (CoenzymeQ-cytochrome c oxidoreductase). Thus, enhanced O2
Complex-III-dependent ROS formation can also be significant at high respiration rates (depolarized conditions) because of a high concentration of semiquinone at the bc1 complex (143). Recent data highlighted that complex II (succinate dehydrogenase) also generates ROS when electrons are supplied by either the reduced ubiquinone pool or succinate (165). Independently of the site of production, which might depend on the substrates being oxidized, the short-lived O2
The majority of information regarding modulation of ROS release by mitochondrial ion channels has been obtained using pharmacological tools. Genetic proofs are still limited, since still to date a large part of the observed channel activities are “orphan” channels, that is, without molecular identity. A combination of different techniques, including subtractive proteomics and whole-genome screening, has recently allowed to identify proteins possibly underlying channel activities in this organelle. Application of electrophysiology either using recombinant proteins or performed on genetically manipulated mitochondria lacking putative channels finally led to the identification of several genes encoding for mitochondrial ion channels.
Thanks to the efforts of numerous groups, components of the long-sought mitochondrial Ca2+ uniporter complex (MCUC) (16, 52) of the PTP (78) and the mitoKATP (69) have finally emerged. In addition to these mitochondria-specific activities, as mentioned above, numerous channels found in mitochondria, have multiple localizations within the cells [for reviews see, e.g., Laskowski et al. (122) and Szabo and Zoratti (202)].
The mitochondrial potassium channels mitoKATP, mitochondrial calcium-dependent potassium channel (mitoKCa), and mitoKv1.3 were among the first organellar channels to be recognized to modulate the fate of the cells via regulation of ROS released from mitochondria: the former two channels modulate ROS generation in pathophysiological conditions such as ischemic/reperfusion (I/R) in the heart. Their opening confers protection against I/R-induced damage, since influx of potassium into the matrix via these channels following the electrochemical gradient for K+ results in depolarization, which in turn reduces matrix Ca2+ overload and ROS overgeneration [for reviews see, e.g., Aon et al. (3), Duchen (60), Garlid et al. (74), and Malinska et al. (143)].
In isolated mitochondria from the heart and brain (as well as in isolated heart), activation of mitoBKCa with pharmacological modulators NS1619 and CGS7184 was shown to reduce ROS production (88, 117, 229). Importantly, a functional coupling has been recently described between mitoBKCa and the respiratory chain complexes (18). In agreement, overexpression of mitochondrial SK2 channels resulted in reduced mitochondrial ROS formation (95). Inhibition of Kv1.3 has instead been linked to apoptosis, since it causes hyperpolarization of the mitochondrial inner membrane with consequent increase in ROS release. This in turn triggers PTP opening, which results in swelling and cytochrome c release, finally causing apoptosis (123, 200).
PTP opening also induces ROS production in vitro and in vivo (240) by triggering a specific conformational change of complex I that dramatically increases ROS production (15, 119). In turn, ROS are potent PTP inducers through several mechanisms: oxidants increase intracellular Ca2+ levels and above a certain threshold, mitochondrial Ca2+ or membrane hyperpolarization might also produce ROS through inhibition of respiratory complexes or displacement of cytochrome c from CL in the IMM (19, 20). These events would induce ROS generation by decreasing the rate of respiration and by PTP induction, which completely stops electron flow along the respiratory chain and causes release of mitochondrial glutathione.
Transient PTP inductions might prompt short bursts of ROS and propagating waves of short PTP openings in the surrounding mitochondria (240). Experimental evidence shows that mitochondrial depolarization following an ischemic insult can propagate as a wave across tissues during global ischemia (139). Mitochondria can behave as a “coupled oscillatory network”: the depolarization of a few mitochondria can cause oscillations of Δψm throughout the cell.
While Zorov and colleagues identified the cyclosporine A-sensitive PTP as the mediator of mostly irreversible dissipations, Aon et al. (3) found that the IMAC is the ion channel that opens transiently in response to an elevation of O2
The proposed chain of events taking place envision first an ROS-induced opening of the IMAC, which partially dissipates ΔΨm and releases O2
Since ROS production is critically dependent on the proton motive force, proton leak is expected to limit oxidative damage. The uncoupling protein 2 (UCP2), which mediates proton leak, has been proposed to regulate cell survival specifically by decreasing formation of mitochondrial ROS, as partial dissipation of the membrane potential via the flux of protons through UCP2 may diminish mitochondrial superoxide production (9, 54). A prominent role for UCP2 in pathological states associated with oxidative stress, including degenerative diseases, atherosclerosis, stroke, aging, cancer, and diabetes, has been envisioned by various groups. In fact, UCP2 overexpression was shown to prevent oxidative injury, while knockout or pharmacological inhibition of UCP2 promotes ROS release in various cell types.
The role of UCP2 in multiple diseases seems to be ascribable to the fact that, similarly to the above-discussed channels, it is able to modulate mitochondrial membrane potential and to induce a protective, mild uncoupling—consequently its open state decreases ROS release. Indeed, an increased ROS production was observed in UCP2 knockout mice (6). Interestingly, UCP2 overexpression has been proposed to directly contribute to the Warburg phenotype (185) and in an orthotopic model of breast cancer, overexpression of UCP2 led to development of tumors (8).
As to the mitochondrial calcium uniporter, the hypothesis has been forwarded that MCU mediates uptake of both Fe2+ and Fe3+, causing mitochondrial depolarization and increased ROS production in brain mitochondria (198). Rapid, acute accumulation of Zn2+ in addition to Ca2+ in the mitochondrial matrix can apparently occur via MCU and trigger ROS generation. The pathological importance of this finding is illustrated by the fact that during ischemia, cytosolic Zn2+ buffering is impaired due to acidosis and oxidative stress. Therefore, high concentration of this ion is present in the cytosol that may contribute to neurodegeneration (47, 147). It has to be mentioned, however, that direct, electrophysiological proof in favor of MCU mediating metal ion flux beside calcium flux is still missing.
A further proof that links matrix calcium increase via MCU to ROS production comes from a study where the dominant negative form of MCU (MCUb) was expressed in hearts, resulting in preserved ΔΨm and reduced ROS during ischemia/reperfusion. Interestingly, no protection occurred from I/R, suggesting that compensatory mechanisms come into play (168). A synergistic effect between MCU and elevated basal ROS production in triggering superoxide flashes has also been suggested to take place in intact cells. Either knockdown of MCU or scavenging mitochondrial basal ROS diminished the flash response. The authors suggest that by this mechanism, physiological levels of mitochondrial Ca2+ and ROS synergistically regulate stochastic PTP opening and tiny quantity of ROS production in intact cells (96).
Interestingly, a recent work highlights that not only ROS but also nitric oxide production can be controlled by mitochondrial channels. In particular, it was found that reduction of mitochondrial Ca2+ uptake by knockdown of MCU or of the essential MCU regulator (EMRE) yielded a significant decrease of the Ca2+-triggered nitric oxide increase, independently of global cytosolic calcium signals. Since knockdown of the MCU gatekeeper, MICU1, instead increased both mitochondrial Ca2+ accumulation and Ca2+-induced nitric oxide signals, the authors suggested that manipulation of mitochondrial Ca2+ uptake represents a novel strategy to control nitric oxide synthase-mediated nitric oxide production (42). In turn, nitric oxide was proposed to induce mitochondrial Ca2+ accumulation due to the blockade of PTP, which may act as calcium-release channel (1).
Finally, it has to be mentioned that the ROS species, especially the O2
ROS production regulation by non-mitochondrial ion channels
As to the contribution of non-mitochondrial channels to global intracellular ROS production, an important role is played by IP3R at the ER via a mechanism involving ER-mitochondria cross talk and by lysosomal channels.
As many of the intracellular channels, IP3R itself is triggered by ROS (82), resulting in Ca2+ release from the ER. This in turn may cause a mitochondrial Ca2+ overload, given the physical contact existing between the two organelles at the level of the MAMs (mitochondria-associated membranes) (175), ultimately boosting mitochondrial ROS production and mitochondrial damage. Released mitochondrial ROS then further trigger IP3R opening, creating a “circulus vitiosus.”
A recent work gives an elegant demonstration of the fact that indeed not only elevated calcium microdomains exist at the level of MAMs but also dynamic nanodomains of H2O2 are present, which function to sensitize ER Ca2+ channels (29) (see the Calcium-ROS Interplay at Mitochondria-ER Contact Sites Involving IP3-Regulated Calcium Channels section). Such a feedback loop is proposed to be linked to the cardiotoxic effects of some drugs as well as to I/R damage. In endothelial cells, it has been suggested that superoxide produced upon macrophage activation facilitates IP3R-linked apoptotic cascade (142).
In the endolysosomal compartment, TRPML1 has emerged as a major regulator of oxidative stress (48). As mentioned in the Introduction section, lysosomes are not only digestive organelles and energy sensors but they are also involved in cytoprotection by autophagic degradation of damaged organelles and by absorbing transition metals otherwise contributing to cytoplasmic oxidative stress. Endolysosomes are the main Fe2+ entry pathway into the cells and also the main Fe2+ releasing organelle, through the TRPML1 (also named MCOLN1) channel, as demonstrated in an elegant work (58). Fe2+ accumulation in the lysosomes has been linked to oxidative stress observed in lysosomal storage diseases and in aging (80, 112, 118). A strong oxidative stress in the brain of the mouse model for mucolipidosis type IV, a lysosomal storage disease caused by the loss of TRPML1 function, further supports a role for TRPML1 in oxidative stress (80).
Impact of ROS on intracellular ion channel function
Not only can ion channel function alter ROS production but also a feedback mechanism that involves ROS-induced modifications of mitochondrial ion channels as well as of the respiratory chain components ensures a further layer of spatiotemporal control of mitochondrial ROS production. As already illustrated for some intracellular channels (e.g., VDAC and PTP), ROS can alter their function by posttranslational modification of cysteine residues.
For example, in the case of one of the isoforms of VDAC, namely VDAC3, our recent study (171) provides evidence for different oxidation-linked Cys modifications by mass spectrometry analysis: Cys residues were identified either in the completely reduced state with SH groups, or in sulfinic and sulfonic oxidation states as well as in the form of disulfide bridges. Most importantly, the oxidative state of VDAC3 has been linked to its functional properties such as the ability to conduct ions, similarly to other channels, for example, RyR (148). The change in ion conductance depending on the oxidation state in the case of VDAC3 most probably reflects also the ability of this channel to allow the flux of metabolites such as ATP, but studies to prove this hypothesis have not been undertaken yet.
Cys as well as methionine residues in general can undergo also other kinds of posttranslational modifications, for example, glutathionylation, surhydration, and nitrosylation [for review see, e.g., O-Uchi et al. (157)]. Indeed, oxidative stress has been shown to induce RyR glutathionylation and consequent Ca2+release from the sarcoplasmic reticulum (135), while ROS-caused sulfonylation of a Cys residue of UCP1 sensitized the protein to adrenergic stimulation (46). In addition, channel activity can be modulated via ROS also in an indirect way, through redox-sensitive kinases. For example, both MCU and PTP have been reported to be activated by the redox-sensitive Ca2+/calmodulin-dependent protein kinase II [(104), but see Fieni et al. (67)].
Another example of ROS-regulated channel is the lysosomal TRPML1, which results in calcium release from this organelle and also in activation of autophagy and lysosome biogenesis [(232) and see below in the Lysosome Ion Channel Modulation by ROS: A Feedback Mechanism section]. The reader is suggested to consult excellent, recent reviews on the modulation of intracellular ion channels by oxidative stress (112, 157) for a detailed description of this issue. Obviously, such a regulation by ROS of the channels that are themselves implicated in the regulation of ROS release is important for generating a feedback control, which in most cases potently amplifies signaling (see, e.g., for ROS-induced ROS release in the case of mitochondria, discussed in the Regulation of ROS release by mitochondrial ion channels section).
Interplay Between Lipid-Mediated Regulation of Intracellular Channels and ROS Production
As already mentioned in the Introduction section, our current knowledge regarding processes involving lipid-mediated modulation of ion channels that lead to alteration of ROS production is rather limited. Here, we give a few examples of physiologically relevant lipid-ion channel-ROS cross talk in different contexts. The first example refers to the most well-studied interplay between calcium channels regulated by IP3 at the ER-mitochondrium interaction surface.
Calcium-ROS interplay at mitochondria-ER contact sites involving IP3-regulated calcium channels
As briefly mentioned above, calcium signaling pathways interact with other cellular signaling systems, among them also with ROS. Importantly, this interaction is bidirectional: ROS can regulate cellular calcium signaling and calcium signaling is important for ROS production (79). Ca2+ and ROS share a common double-face behavior. Perhaps the most distinctive property of the Ca2+ signal is its ambivalence: while essential to the correct functioning of cells, Ca2+ becomes an agent that mediates cell distress, or even (toxic) cell death, if its concentration and movements inside cells are not carefully controlled (32). Similarly, high ROS levels exceeding the cellular antioxidant capacity cause detrimental effects with pathological consequences, however as mentioned above, it is also evident that a rise in intracellular ROS plays important physiological roles in many organs (94).
Ca2+ and ROS cross talk is likely to occur in localized microdomains (Fig. 4). ER-mitochondria interface appears to be a preferential location since both these organelles are involved in ROS production and Ca2+ signal generation. ER lumen is endowed with high Ca2+ concentration (in the mM range), which is maintained by the active transport of the SERCA ATPase and acts as the main intracellular store for the ion, making it available for the control of the intracellular processes. Furthermore, its highly oxidizing environment guarantees the process of oxidative protein folding and the formation of disulfide bond among cysteines (5). The ER is also the major site for the synthesis of sterols and phospholipids that constitute the bulk of the lipid components of all biological membranes, many enzymes and regulatory proteins involved in lipid metabolism reside in the ER. The ER, therefore, plays a fundamental role in controlling membrane lipid composition and membrane lipid homeostasis in all cell types (63).
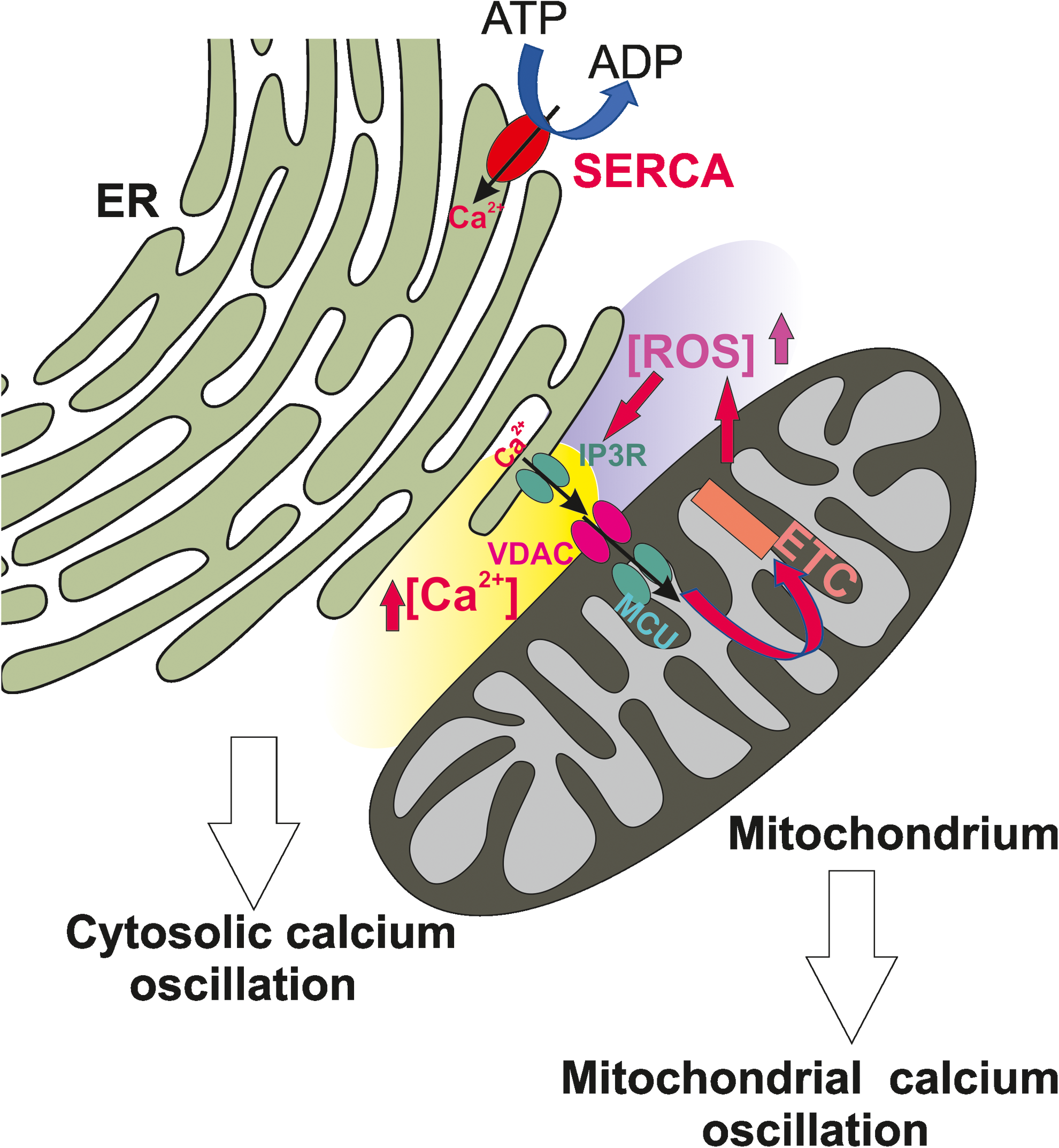
As briefly mentioned in the previous sessions, mitochondria are essential for ensuring numerous fundamental physiological processes such as cellular energy, redox balance, modulation of Ca2+ signaling and important biosynthetic pathways. They also govern the cell fate by participating in the apoptosis pathway. Their main system to take up Ca2+ is the MCU complex, a macromolecular structure that guarantees Ca2+ accumulation inside mitochondrial matrix on increases in cytosolic Ca2+. Ca2+ uptake is driven by the mitochondrial membrane potential (negative inside the mitochondrial matrix) generated by the respiratory chain, and the attainment of the electrochemical equilibrium is prevented by the activity of mitochondrial Ca2+ extrusion systems, that is, the H+/Ca2+ and the Na+/Ca2+ exchangers (53).
Local Ca2+ delivery from ER to mitochondria is responsible for mitochondrial depolarization and ETC stimulation, and thus, ROS production activation. At the same time, ROS produced at mitochondrial levels impinge the activity of the SERCA pump thus limiting Ca2+ reuptake in the ER lumen (49, 193, 238). Higher amounts of ROS can further target ER-based Ca2+ channels, that is, the RyR and IP3R, leading to increased Ca2+ release, thus apparently triggering a vicious cycle.
However, it is now clear that both Ca2+ and ROS act as local second messengers and that their action is mutually interconnected. This aspect has clearly emerged in cardiac and neuronal cells, where it has been shown that antioxidant treatment attenuates cell excitability (2, 219). Indeed, ROS have been shown to directly modulate RyR activity by oxidizing redox-sensing thiol groups (238). As far as the action on IP3R, it has been suggested that O2
High Ca2+ concentration microdomains are generated at the ER-mitochondria contact site level, where IP3Rs are in a close proximity with the MCU (173, 174). The uptake of Ca2+ by the MCU is crucial to ATP production through the activation of the pyruvate dehydrogenase and two Krebs cycle dehydrogenases. Ca2+ influx into the mitochondrial matrix is also responsible for mild mitochondrial depolarization. Both ETC stimulation and mitochondrial depolarization lead to electron leak from complex I/II of the respiratory chain and increased ROS generation, which, as mentioned before, at physiological levels appears to contribute to the generation of Ca2+ signaling.
ER-mitochondria contact sites are determinant for both Ca2+ signaling and lipid biosynthesis, and it has recently emerged that impairment in the communication between the two organelles is linked to a number of pathological conditions since they represent an important check point for cell function [for review see, e.g., Lam and Galione (120)]. Thus, in the last years, the molecular understanding of signaling events occurring at this site has attracted great interest. Similarly to Ca2+-mediated signaling, ROS-mediated signaling has recently emerged to be characterized by the presence of nanodomains (Regulation of ROS Release by Mitochondrial Ion Channels section) at elevated concentrations that may selectively target proteins specifically localized in their proximity, such as IP3Rs.
A role for ROS in regulating IP3Rs activity has been shown by several groups in different cellular models (107, 172) as well as it has been reported that also H2O2 has a sensitizing effect on IP3Rs (170, 236). It is known that the cell exposure to extracellular oxidants, such as thimerosal, stimulated the IP3-mediated Ca2+ release (30, 33, 110) and that the degree of sensitization of IP3Rs is sufficient to trigger Ca2+ oscillation in unstimulated cells (105).
Seminal studies to understand this regulation have shown that the conformational state of IP3Rs is dependent on oxidation state. Disulfide bridge formation has been described at the ER luminal domain of the IP3 (90). ER luminal redox state, together with luminal Ca2+ and pH, contributes to IP3R regulation by the interaction with protein partners, among which ERp44, an ER lumenal protein that belongs to the thioredoxin protein family, which also includes protein disulfide isomerase (PDI). The ERp44-IP3R interaction is dependent on the presence of reduced cysteine residues in the IP3R protein: hyperoxidation of the ER lumen could disrupt the interaction causing IP3R hypersensitivity, Ca2+ release, and apoptosis (129).
ER lumen presents a highly oxidizing environment, and thus, it is unlikely that physiologically generated mitochondrial ROS are able to modify this ambient and impact on IP3R activity by acting at the intraluminal side. Thus, the mechanism by which IP3Rs activity is controlled by the luminal side appears to act mainly under stress conditions, but the molecular detail is still not completely elucidated. The mechanisms operating at the cytosolic side have been better investigated in the last few years, because they could have functional relevance in the regular activity of the receptor.
In 2014, Hajnoczky and coworkers provided evidence for the biological meaning of the sensitization of IP3 to oxidants showing that physiologically relevant ROS can control both the cytosolic and mitochondrial Ca2+ signaling (12). Intriguingly, they have demonstrated that cytosolic Ca2+ transients generated by cell exposure to oxidants are generated by Ca2+ mobilization from the ER through the IP3 receptors and that IP3-mediated Ca2+ release was greatly enhanced by local O2
As far as the mechanism for sensitization, the authors postulate that mechanisms other than disulfide bridge formation, described to occur only at the ER luminal side (90), should take place since O2
Hajnoczky and coworkers have demonstrated the existence of H2O2 nanodomains at the ER-mitochondria interface, and shown their action on IP3Rs, proposing that H2O2 may have a physiological role in mitochondrial Ca2+ signal modulation (29). Intriguingly, they have obtained indication that H2O2 is continuously produced in resting conditions at the mitochondrial IMS and in the matrix, but H2O2 accumulation at the ER-mitochondria interface was dependent on cristae remodeling as a consequence of mitochondrial Ca2+ uptake, rather than on Ca2+-mediated stimulation of the respiratory chain. Accordingly, H2O2 concentration in the cytosol or in the mitochondrial matrix is essentially undetectable, further demonstrating its localized accumulation.
On cell stimulation, ER Ca2+ release events increase matrix Ca2+, activate K+ uptake through the mitoBKCa channels, and increase matrix volume, thus inducing the cristae to open versus the ER-mitochondria interface and release H2O2. Considering that the regions of Ca2+ transfer between the ER and the mitochondria are those where IP3Rs have the major density, it is easy to understand that the same regions experience consistent H2O2 accumulation, and the proteins participating in their constitution are those principally exposed to H2O2 action. IP3R is among them: thus, Ca2+ signals are both the cause and the consequence of ROS-mediated signaling. This model, together with previous observations [for review see, e.g., Szabo and Zoratti (202)], also points to a dual role of mitoBKCa in I/R protection, via both regulation of ROS production and of mitochondrial volume.
In addition to the regulation of IP3R-mediated Ca2+ signaling by ROS, it has also been proposed that ROS can stimulate Ca2+ release from the ER by activating PLC (191, 211). In 2014, Abramov and coworkers showed that PLC preferentially cleaves oxidized lipids, thus suggesting that ROS-mediated lipid peroxidation further contributes to ER Ca2+ release (57). The study was performed in astrocytes where vitamin E or Trolox incubation was shown to inhibit ROS production without affecting the intracellular Ca2+ store content, but evoking reduced Ca2+ response after activation of the ATP receptor coupled to the IP3 generation. Conversely, the application of H2O2 stimulated Ca2+ oscillations, and pharmacological PLC inhibition completely prevented them.
Altogether these results suggested antioxidant action on PLC activity. In contrast, other studies have shown that ROS can affect PLC function causing a depletion of the intracellular Ca2+ stores (77, 214). Thus, the precise mechanism of this regulation is still obscure, but it underlines the complexity of the Ca2+ and ROS signaling cross talk (57).
In summary, it is evident that excess ROS overwhelm antioxidant defenses, leading to oxidative stress, and that this plays an important role in the pathogenesis of numerous diseases, including cardiovascular disease, metabolic syndrome, neurodegenerative diseases, inflammation, and cancer. For a long period, ROS have been considered the toxic side-products of metabolic reactions, but it is now quite clear that ROS also represent signaling molecules for the proper functioning of the organisms and that the therapeutic application of antioxidant compounds must be reconsidered thinking about the use of ROS-forming enzymes or ROS donors that may locally act. Indeed, this point of view is supported by the fact that numerous therapeutic approaches based on the broad use of antioxidants not only were ineffective but had detrimental consequences as well (38).
Interplay between mitochondrial lipid content and ROS production
Mitochondrial lipids are able to influence mitochondrial ROS production by multiple ways, by directly interacting with respiratory chain complexes and also by interaction with channels. First, diet can profoundly affect mitochondrial membrane lipid composition [see e.g., Barzanti et al. (14), for review see, e.g., Monteiro et al. (149)]. As a consequence, altered respiration and ROS production can occur, even in vivo (150). In two elegant works, the group of Monteiro investigated the molecular mechanism of the cardioprotective effects of dietary ω-3 PUFAs, in particular DHA and EPA. DHA intake was reported to increase both DHA and EPA and decrease AA content (a potent activator of PTP) in mitochondria. Both PUFAs (DHA and EPA) suppressed Ca2+-induced opening of the PTP. As mentioned above, opening of PTP can trigger a high ROS production by ROS-induced ROS release mechanism with deleterious consequences for the cell.
In summary, dietary supplementation with DHA, but not EPA, profoundly altered mitochondrial phospholipid fatty acid composition and delayed Ca2+-induced PTP opening. However, this occurred only in noninfarcted hearts, even though AA concentration decreased also in infarcted hearts (109, 156). The authors interpreted their data as the phospholipid composition was not the only factor responsible for the delay in PTP opening in healthy rats.
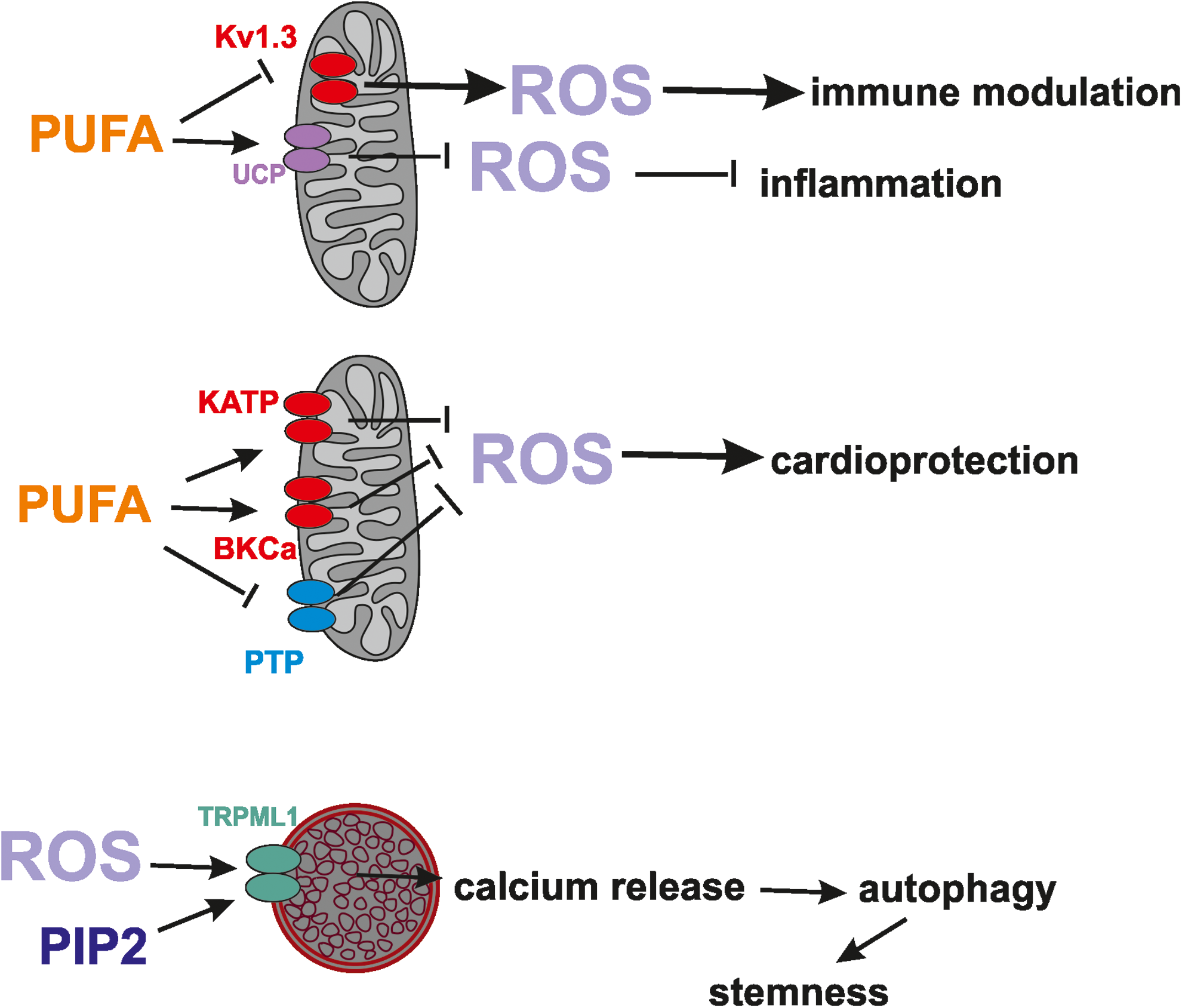
The cardioprotective effects of PUFA might, a priori, occur also by modulation of other ion channels in mitochondria that can decrease ROS production and delay the opening of PTP this way. PUFA, in particular AA, has been shown to inhibit ROMK1 (141), however, it does not affect function of the splice variant ROMK2, which was proposed to be a component of mitoKATP that has a documented role in modulation of ROS release and cardioprotection (4). Epoxyeicosatrienoic acids (EETs)—cytochrome P450 metabolites of AA—activate PM KATP in ventricular myocytes and may play an important part in preconditioning as well, for example (28), possibly also via activation of mitoKATP.
In addition, since pharmacological activation of BKCa mitigates ROS production, and AA, DHA, and EPA all activate mitoBKCa (159), it is more than likely that these PUFAs exert a cardioprotective effect also by modulation of the mitochondrial calcium-dependent potassium channel BKCa.
Another mitochondrial channel possibly modulated by AA is mitoKv1.3, whose inhibition triggers ROS release from mitochondria causing PTP opening and subsequent apoptosis (201). The PM Kv1.3 is indeed inhibited by AA as well as by ceramide (see the Ion channel modulation by sphingolipids, bioactive lipids, and cholesterol section), but whether the mitochondrial channel is also affected by these lipids awaits experimental validation. Ceramides alter membrane permeability and promote oxidative stress, possibly via direct inhibition of respiratory chain complexes and/or by channel formation (see the Ion channel modulation by sphingolipids, bioactive lipids, and cholesterol section), or by triggering PTP opening (149). In addition, it is more than plausible to suppose that ceramide can trigger ROS release also via inhibition of mitoKv1.3, but this hypothesis has still to be experimentally tested.
Lysosome ion channel modulation by ROS: a feedback mechanism
An exciting, emerging area in the field of redox biology is understanding of the contribution of lysosomes to ROS homeostasis within the cells. A recent, milestone work in this respect shows for the first time that mitochondria-produced ROS can directly activate TRPML1 in the lysosomes, causing calcium release from this organelle to promote autophagy (234). In particular, the released calcium triggers PPP3/calcineurin-dependent dephosphorylation of the transcription factor EB, which can then enter the nucleus to promote transcription of autophagy-related genes. As a final output, autophagy mediated by the lysosomes will be enhanced and will help the removal of damaged mitochondria that produce excessive ROS (Fig. 5). Thus, as in the case of the ER-mitochondria cross talk, also in this case a “reciprocal feedback” takes place among two organelles involving ROS and calcium (235).

As is the case of ER, also lysosomes can be found physically proximal to mitochondria (130). TRPML1 has been previously shown to be activated by PI(3,5)P2 (59, 66, 234) and this event controls intracellular membrane trafficking (59). On the contrary, TRPML1 is inhibited by accumulation of sphingomyelins and cholesterol (221). An interesting question is whether modulation of TRPML1 by these lipid molecules is also able to trigger the same series of events, that is, enhancement of autophagy, independently of an increased ROS level. It is of note that another PIP2, PI(4,5)P(2), together with clathrin, seems to be a crucial player for maintenance of lysosome homeostasis and lysosome reformation (180).
Another example of cross talk between mitochondria and lysosomes is illustrated by the observation that during ischemia, lysosomes release Fe2+ (most probably via TRPML1), which is taken up into mitochondria presumably by MCU. Increased mitochondrial iron then triggers an increased ROS production, which in turn opens PTP and kills the cells after reperfusion (233).
Possible Links of the Lipid-Channel-ROS Axis to Pathologies and Future Perspectives
As illustrated by a few examples in the previous section, modulation of redox state and of ROS production within the cells might be more complex than originally thought, due to feedback controls involving different organelles and possibly also lipid-regulated ion channels. Exciting novel results point to mitochondrial ROS as crucial regulators of different pathology-related processes, besides the already mentioned ischemic damage and tumorigenesis. For example, mitochondrial ROS have an essential role in the lipid dysfunction-linked impaired anticancer immunosurveillance, as recently demonstrated in a seminal article where the authors showed that in the nonalcoholic fatty liver disease, accumulation of fatty acid, especially of linoleic acid in helper T lymphocytes, induced a strong mitochondrial ROS release and consequent cell death of the immune CD4+ T cells (140). In turn, the lack of these helper cells in the hepatic tissue promoted hepatocarcinogenesis.
In general, it is becoming clear that mitochondrial ROS serve as an important signaling mechanism for T and B cell-mediated immunity and for the correct development of the immune system subpopulations [for reviews see, e.g., Wahl et al. (216)]. T cell receptor-triggered mitochondrial ROS generation by complex I was shown to be necessary for activation-induced interleukin-2 and interleukin-4 production and secretion in resting and preactivated human T cells (108), but ROS produced by complex III also seem to sustain interleukin-2 expression during T cell receptor activation (189).
Given the important role of some mitochondrial ion channels in the fine-regulation of membrane potential and of ROS production, it would not be surprising if these channels also played a role in immunity. In fact, the UCP2 function has been shown to control immune cell activation by modulating the production of mitochondrial ROS and the MAPK pathway [for review see, e.g., Emre and Nubel (61)]. A clear relationship between UCP2 and inflammasome activation has been found, reporting that UCP2 impacts on the expression and activation of NLRP3 (NACHT, LRR, and PYD domain-containing protein 3) inflammasome in human macrophages. In particular, UCP2 overexpression enhanced the expression levels of NLRP3, while the UCP2 inhibitor genipin suppressed it. Genipin also affected ATP and H2O2-mediated interleukin-1β secretion (166). Similarly, miR-133a-1 was reported to reduce inflammasome activation via the suppression of UCP2 expression (11).
A similar relationship might be expected for the other mitochondrial channels whose activity impacts ROS release, as mitochondria represent a platform for inflammasome assembly and mitochondrial ROS trigger NLRP3 oligomerization (81, 212). Interestingly, dietary PUFA was shown to decrease body lipid accumulation by upregulation of UCP2 and this was associated with anti-inflammatory effects in rodents (132). Indeed, it is emerging that fatty acid-derived mediators are crucial molecules for directing inflammation, in part, also by modulation of various ion channel activities (45).
Another interesting recent article shows that mitochondria-derived ROS-induced sensitization of UCP1 to adrenergic stimulation in brown adipose tissue supports the UCP1-dependent thermogenesis and energy expenditure in the whole body (46). Given that ceramide accumulation has been linked to obesity, in addition to insulin resistance and metabolic disorders (70), it would be interesting to investigate whether this lipidic regulator can directly affect UCP1 and UCP2 function.
Finally, an elegant study provides evidence that the normal function of autophagy in muscle stem cells is that of removing the damaged mitochondria that would otherwise release substantial amount of ROS. Thus, autophagy prevents the mitochondrial ROS-induced senescence of muscle stem cells (73). On the basis of the results mentioned in the Lysosome Ion Channel Modulation by ROS: A Feedback Mechanism section, it is possible to hypothesize that lysosomal TRPML1 channels might play a crucial role also in the context of stemness maintenance.
In summary, there is an ever increasing list of relevant pathologies that are ultimately linked to the dysregulation of intracellular ROS levels. Given that mitochondria and lysosomes are important contributors to overall ROS production, there is a special focus on these organelles to clarify how they can orchestrate normal cell function and how they contribute to the development of pathologies on dysregulation. Given that several different ion channels in these organelles orchestrate ROS release, identification of physiological factors that modulate ion channel activities is of importance.
Among the “naturally occurring” modulators are dietary lipids, whose intake may indeed change the lipid composition of the organelle membranes and possibly result in the formation of intracellular lipid signaling molecules (Fig. 6). Of course, these lipids are likely to have pleiotropic effects within the cells, but whether the lipid-modulated channel activity impacts a given process can be checked with relatively high confidence, using cells/organisms lacking a given channel protein. This approach, and so the dissection of specific processes linked to modulation of intracellular channels by lipids, has now finally become possible for many organellar channels, thanks to the increasing success in the identification of proteins giving rise to these channel activities. As stated in this review, still many questions are unanswered, but we foresee an exciting era where studies addressing the lipid-ion channel-ROS axis will gain further importance and will be also connected to upstream and downstream signaling pathways.
Footnotes
Acknowledgments
The authors are grateful for financial support by the Italian Association for Cancer Research (15544 to I.S.) and the Italian Ministry (Progetti di Rilevanza Nazionale PRIN 2015795S5W to I.S.), BIRD162511 del 2016 to L.L. and CPDA153402 to M.B. from the University of Padova.