
Abstract
Significance:
The nicotinamide adenine dinucleotide (NAD+)/reduced NAD+ (NADH) and NADP+/reduced NADP+ (NADPH) redox couples are essential for maintaining cellular redox homeostasis and for modulating numerous biological events, including cellular metabolism. Deficiency or imbalance of these two redox couples has been associated with many pathological disorders.
Recent Advances:
Newly identified biosynthetic enzymes and newly developed genetically encoded biosensors enable us to understand better how cells maintain compartmentalized NAD(H) and NADP(H) pools. The concept of redox stress (oxidative and reductive stress) reflected by changes in NAD(H)/NADP(H) has increasingly gained attention. The emerging roles of NAD+-consuming proteins in regulating cellular redox and metabolic homeostasis are active research topics.
Critical Issues:
The biosynthesis and distribution of cellular NAD(H) and NADP(H) are highly compartmentalized. It is critical to understand how cells maintain the steady levels of these redox couple pools to ensure their normal functions and simultaneously avoid inducing redox stress. In addition, it is essential to understand how NAD(H)- and NADP(H)-utilizing enzymes interact with other signaling pathways, such as those regulated by hypoxia-inducible factor, to maintain cellular redox homeostasis and energy metabolism.
Future Directions:
Additional studies are needed to investigate the inter-relationships among compartmentalized NAD(H)/NADP(H) pools and how these two dinucleotide redox couples collaboratively regulate cellular redox states and cellular metabolism under normal and pathological conditions. Furthermore, recent studies suggest the utility of using pharmacological interventions or nutrient-based bioactive NAD+ precursors as therapeutic interventions for metabolic diseases. Thus, a better understanding of the cellular functions of NAD(H) and NADP(H) may facilitate efforts to address a host of pathological disorders effectively. Antioxid. Redox Signal. 28, 251–272.
Introduction
N
In this review, we will examine the biosynthesis of NAD+ and NADP+ with an emphasis on recent discoveries into the extracellular sources of NAD+, the newly identified synthetic enzymes, and the role of nicotinamide nucleotide transhydrogenase-mediated NADPH production. We will also discuss the subcellular distribution of NAD(H) and NADP(H) and their intercompartmental trafficking. In addition, we will extensively discuss the paradoxical functions of these two redox couples in maintaining cellular redox homeostasis as well as their regulation of cellular metabolism under various physiological and pathological conditions.
Biosynthesis of NAD+ in Mammals
In mammalian cells, NAD+ is synthesized from four different precursors (Fig. 1), tryptophan (Trp), nicotinic acid (NA), nicotinamide (NAM), and nicotinamide riboside (NR), through three pathways: the de novo pathway, the Preiss–Handler pathway, and the salvage pathway (24, 66, 88, 141) (Fig. 2). The salvage pathway predominates in most cell types.

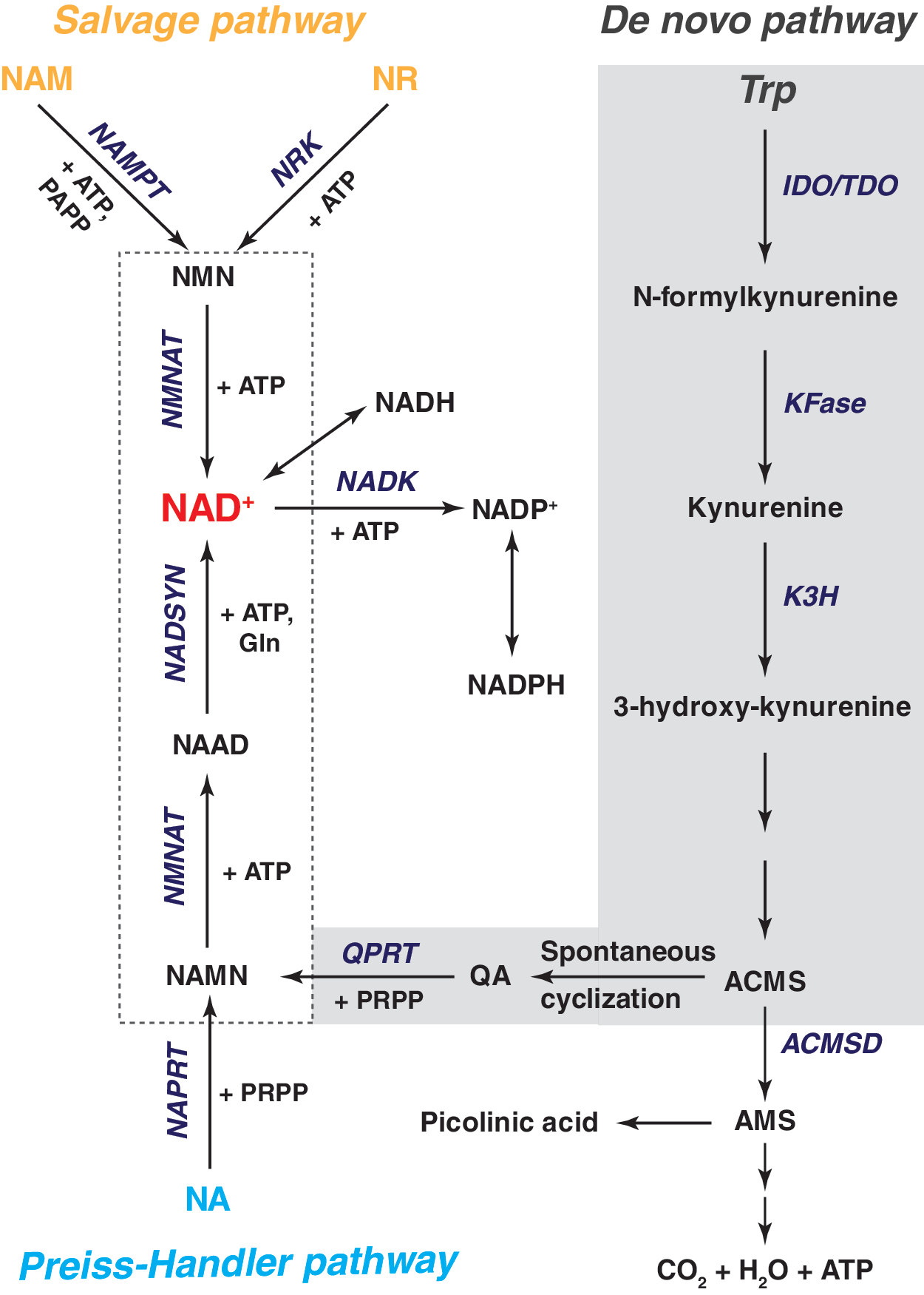
De novo synthesis of NAD+
De novo NAD+ synthesis from L-Trp is mediated by enzymes in the kynurenine pathway via an eight-step process (Fig. 2). Conversion of L-Trp to N-formylkynurenine is the first and rate-limiting step, which is catalyzed by indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO) (88). TDO is primarily expressed in the liver (114). IDO expression has been detected in extrahepatic cells, including human vascular endothelial and smooth muscle cells (15, 30), dermal fibroblasts (47), macrophages (52), neurons, microglia, and astrocytes (53). Human lung, placenta, and small intestine show relatively higher IDO activity than neuronal tissue (137). Since the enzymatic activity of IDO and TDO requires oxygen, their activity is anticipated to be attenuated under low oxygen tension. Surprisingly, current literature shows that hypoxia enhances IDO and TDO activity in vitro and in vivo (40, 63). This paradoxical phenomenon could be explained by the findings that IDO can utilize superoxide anion (O2 •−) as its substrate (88), and O2 •− production is increased under hypoxia (98). Future studies are needed to investigate the precise effects of hypoxia on TDO/IDO activity.
In the next steps of this pathway, kynurenine formamidase (KFase) converts N-formylkynurenine into kynurenine, which is further hydroxylated into 3-hydroxy-kynurenine by kynurenine-3-hydroxylase (K3H). K3H, localized to the mitochondrial outer membrane, is a NADPH-dependent and flavin-adenine dinucleotide (FAD)-containing monooxygenase (88). K3H expression in human tissues is abundant in the liver and placenta, with lower levels in the kidney (5).
Once formed, hydroxylated kynurenine next undergoes two additional enzymatic reactions forming an unstable intermediate, 2-amino-3-carboxy-muconate-semialdehyde (ACMS). ACMS can be removed from the NAD+ synthetic pathway by its decarboxylation into 2-amino-3-muconate-semialdehyde (AMS) catalyzed by ACMS decarboxylase (ACMSD), a process that ultimately leads to the formation of picolinic acid or CO2 and H2O. ACMS can also undergo spontaneous cyclization to form quinolinic acid (QA), which is then converted to nicotinic acid mononucleotide (NAMN) by quinolinate phosphoribosyltransferase (QPRT) using phosphoribosyl pyrophosphate (PRPP) as a cosubstrate. Of note, the QPRT reaction is not efficient and occurs only when the level of QA exceeds the enzymatic capacity of ACMSD, which renders this reaction as a second rate-limiting step in de novo NAD+ synthesis (88) and helps to explain why Trp-dependent synthesis is less efficient than the other two NAD+ biosynthetic pathways.
NAMN is then adenylated to form NA adenine dinucleotide (NAAD) with the catalysis of ATP by NMN adenylyltransferases (NMNATs). In mammals, three isoforms of NMNATs exist with a distinct tissue- and organelle-specific distribution (36, 37, 106, 149). NMNAT1, an exclusively nuclear enzyme, is ubiquitously expressed in human tissues. It is highly abundant in the heart and skeletal muscle, expressed to a lesser extent in the liver and kidney, and barely detectable in the brain and small intestine (11, 36). NMNAT2 is located in cytosol and Golgi apparatus, and its expression in human tissues is found principally in the brain and weakly in the heart and skeletal muscle (14, 106). In contrast, NMNAT3 is found in cytosol and mitochondria (14, 149). Tissue distribution shows that this enzyme is mostly present in human lung and spleen, where the other two isoforms are barely detectable (14, 149). The tissue- and organelle-specific expression pattern implies that the function of NMNATs is not redundant and also explains the cellular compartmentalization of NAD+.
The final step in the de novo pathway is the conversion of NAAD into NAD+ by ATP-dependent NAD+ synthetases (NADSYNs), which catalyze an amidation reaction using glutamine or ammonia as an amide donor. Two NADSYN isoforms (NADSYN1 and 2) have been identified in humans. NADSYN1 expression is enriched in the liver, kidney, small intestine, and testis, where the expression of NADSYN2 is weak (62). Interestingly, NADSYN1 utilizes both glutamine and ammonia as amide donors, whereas NADSYN2 only catalyzes ammonia-dependent NAD+ synthesis (62).
The Preiss–Handler pathway
This pathway was discovered by Preiss and Handler in human erythrocytes and rat liver, where they found NA could be converted into NAD+ in a three-step process that produces NAMN and NAAD as intermediate metabolites (103, 104).
In mammals, NA is first metabolized into NAMN by NA phosphoribosyltransferase (NAPRT) at the expense of PRPP (Fig. 2). NAPRT expression is widespread and its messenger RNA (mRNA) has been detected in almost all human tissues tested thus far (35). ATP is a dual allosteric modulator that can stimulate or inhibit NAPRT activity at low (<100 μM) or high (100–640 μM) concentrations, respectively (43). Intriguingly, metabolites of glucose and fatty acids have distinct effects on NAPRT activity, as well. For instance, dihydroxyacetone phosphate (DHAP) and pyruvate stimulate its activity, whereas glyceraldehyde-3-phosphate (G3P), phosphoenolpyruvate, fructose-1,6-biphosphate (F-1,6-BP), acetyl-CoA, succinyl-CoA, and CoA inhibit its activity (43). To date, the mechanisms for these differential regulatory effects are unknown.
NAMN serves as the converging point for the Preiss–Handler pathway and the de novo pathway, with NAMN undergoing the same reactions for NAD+ synthesis as described in the aforementioned section. NA seems to be a more efficient precursor for NAD+ synthesis than Trp, as 1 mg NA is equivalent to ∼60 mg dietary Trp (12).
The salvage pathway
NAM, a product of niacin and SIRT enzymes, can be used to synthesize NAD+ through the salvage pathway (24, 141). In mammals, NAM phosphoribosyltransferase (NAMPT), a rate-limiting enzyme, catalyzes the conversion of NAM and PRPP into NMN, which is then converted into NAD+ by NMNATs that catalyze the formation of NAAD from NAMN in the de novo pathway (Fig. 2) (24, 141). NAMPT mRNA is ubiquitously expressed in all tissues tested, with higher levels present in bone marrow, liver, and muscle than other tissues (41, 115). Immunocytochemical staining demonstrates that NAMPT protein is found in the nucleus and cytoplasm with a change in its intracellular distribution according to the proliferation state of the cells (72).
Notably, in addition to intracellular NAMPT (iNAMPT) protein, an extracellular form of NAMPT (eNAMPT) has also been identified. The eNAMPT, also denoted pre-B cell colony-enhancing factor (PBEF), was initially recognized as a secreted cytokine, which synergizes cell colony formation induced by stem cell factor and interleukin 7 (115). Later, Rongvaux et al. (112) showed that PBEF is a cytosolic NAMPT involved in NAD+ biosynthesis in mouse liver. Furthermore, eNAMPT was found in human circulation, and leukocytes were also identified as a source for eNAMPT (41). Revollo and colleagues (109) reported that mouse adipocytes also secreted eNAMPT, which exhibits even higher NAD+ biosynthetic activity than the intracellular iNAMPT. Consistent with these observations, a high concentration of NMN is present in mouse plasma, and plasma eNAMPT and NMN levels are reduced in NAMPT heterozygous knockout females (109). Recently, Yoon et al. (145) demonstrated that deacetylation of iNAMPT by the SIRT1 deacetylase enhances eNAMPT secretion and activity in murine adipocytes, and that adipocyte-specific knock out or knock in of NAMPT systemically affected plasma eNAMPT levels, hypothalamic NAD+ biosynthesis, SIRT1 function, and exercise capacity.
NR is a newly discovered NAD+ precursor that feeds into the salvage pathway (16). NR is first phosphorylated into NMN by NR kinases (NRK1-2), after which NMNATs catalyze the production of NAD+ (Fig. 2). NRKs are highly conserved in eukaryotes (16). NRK1 is ubiquitously expressed in mammalian tissues, whereas NRK2 expression is restricted to the heart, brain, and skeletal muscle and not present in the liver, kidney, lung, or pancreas (19). Overexpression of NRK1 in NIH3T3 cells and hepatocytes elevates cellular NAD+ levels in response to NR addition (108). In mice, injection of NR augmented NAD+ levels in the muscle, liver, brain, and brown adipose tissue, an effect that was abrogated in NRK1 knockout mice (108).
Importantly, NR has been detected in cow's milk as a natural nutrient (16, 130). It has been estimated that cow's milk typically contains about 12 μmol NAD+ vitamin precursor/L, of which 60% is present as NAM and 40% as NR (130). A recent study reported that oral NR supplementation is bioavailable for NAD+ biosynthesis in healthy volunteers and that NR exhibits unique and superior pharmacokinetics compared to NA and NAM in mice (128). These lines of evidence highlight the potential use of NR as an NAD+ source and therapeutic agent to treat metabolic diseases.
A systematic regulatory network, denoted “NAD+ World,” has been developed to describe the critical roles of NAMPT-mediated NAD+ biosynthesis and NAD+-dependent SIRT1 in the regulation of metabolism and aging in mammals (70), providing important insights into the systems-level regulatory mechanisms that connect metabolism and aging in mammals. The NAD+ World 2.0 emphasizes the importance of intertissue communication among organs and provides a systems biology approach for modeling and controlling the aging process and longevity in mammals (69). This NAD+-dependent control system consists of two feedback loops involving eNAMPT and iNAMPT, and the NAD+-dependent SIRT1 enzyme.
Figueiredo and colleagues (31) constructed a metabolic network describing NAD+ biosynthesis and degradation, and used the concept of elementary flux modes to explore the potential routes in the network of NAD+ generation, which include the three known biosynthetic pathways mentioned above. Such a systematic analysis and comparison of metabolic networks specific for yeast and humans highlight the differences across species regarding the use of precursor biosynthetic routes and NAD+-dependent signaling. A network of functional associations among the NAD+-related proteins mentioned above can be derived from String v10 (125), providing a systems-level view of their functional connections (Fig. 3).

Biosynthesis of NADP+
NADP+, a structural analogue of NAD+, is synthesized by transferring a phosphate group from ATP to the 2′-hydroxyl group of the adenosine ribose moiety of NAD+ (Fig. 2) (2, 144). This reaction is catalyzed by NADKs, the sole enzymes responsible for de novo NADP+ synthesis in both prokaryotic and eukaryotic cells (144), suggesting that NADK is a determinant of cellular NADP+ levels. NADK mRNA was found to be equally expressed in most human tissues with the exception of skeletal muscle and small intestine (79). In Escherichia coli and Salmonella enterica, the NADK enzyme is highly selective for its substrates NAD+ and ATP, and its activity requires divalent cations, such as Mg2+, Ca2+, and Mn2+ (50, 71). These features are conserved in human NADK enzyme (79, 134). Interestingly, bacterial NADK activity is strongly inhibited by NADPH and NADH, and less strongly by NADP+ (50, 71), suggesting the existence of a negative feedback mechanism. To date, it is unknown whether these allosteric factors also regulate human NADK activity.
For many years, humans were believed to have only one cytosolic NADK protein (102); however, this dogma was challenged by the discovery of a mitochondrial NADK (MNADK) by two different groups (97, 148). Ohashi et al. (97) discovered that a human protein, C5orf33, catalyzes the formation of NADP+ from NAD+ and ATP, and is localized to mitochondria in HEK293 cells. The MNADK mRNA was detected in all tissues tested, and its levels were found to be much more abundant relative to the cytosolic NADK (97). A separate study confirmed the finding that C5orf33 is a MNADK, and showed that MNADK is evolutionarily conserved in multicellular organisms (148). In mice, its mRNA is expressed most highly in the liver, followed by mitochondrial-rich tissues, for example, heart and skeletal muscle (148).
In addition, MNADK mutation was recently discovered in patients with dienoyl-CoA reductase (DECR) deficiency with hyperlysinemia, a rare disorder affecting the metabolism of polyunsaturated fatty acids and lysine (65). Fibroblasts from MNADK mutant patients had decreased DECR activity and reduced mitochondrial NADP(H) levels with no change in cytosolic NADP(H) levels (65). Overexpression of the wild-type MNADK restored DECR activity in patient fibroblasts (65), suggesting that MNADK might be an appealing therapeutic target for this disorder.
Metabolic Sources and Cellular Compartmentalization of NAD(H) and NADP(H) Couples
The distribution of the NAD+/NADH and NADP+/NADPH redox couples is highly compartmentalized due to specific localization of NAD+ and NADP+ biosynthetic enzymes and the bioavailability of NAD+ precursors (Fig. 4). Importantly, since the mitochondrial inner membrane is impermeable to NAD(H) and NADP(H) (24, 102, 141), the mitochondrial and cytosolic NAD(H) and NADP(H) pools are regulated by multiple shuttles, such as the malate–aspartate shuttle for the NAD(H) pools and the isocitrate-α-ketoglutarate (α-KG) shuttle for NADP(H) pools (66). These shuttle mechanisms enable cells to maintain redox and energy homeostasis in normal and stressed states.

NAD(H) in the extracellular milieu
Historically, NAD+ and NADH have been viewed as unable to be transported across membranes (73, 143). This concept has been challenged by recent evidence. First, NAD+ and its precursors have been detected at nanomolar concentrations in plasma and other body fluids (96, 109, 120). Moreover, NAD+ can be released into superfusates of mouse urinary bladder and gastrointestinal smooth muscle, as well as canine mesenteric artery, under both basal and stimulated conditions (92, 120). Finally, addition of exogenous NAD+ or NADH to culture medium augmented intracellular and mitochondrial-specific NAD+ and NADH levels in several experimental systems; however, the mechanisms by which NAD(H) is transported across membranes remain elusive.
It has been proposed that extracellular NAD(H) could be released from dying cells; however, recent observations suggest that transmembrane transporters in living cells may mediate active exo/endocytosis of NAD(H) (55). In support of this concept, addition of exogenous NAD+ to culture medium was found to increase intracellular NAD+ and NADH levels in HeLa cells, a process that was abrogated when cells were cultured at 4 °C, suggesting the involvement of a specific and active transporter (101). In addition, connexin 43 hemichannels were reported to mediate transmembrane NAD+ fluxes in a Ca2+-dependent manner in 3T3 fibroblasts (20), and P2X7 receptors were shown to transport NADH across the plasma membranes of astrocytes (86).
Alternatively, extracellular NAD+ could be metabolized into its cell-permeable precursors, which then enter cells via specific carrier proteins (Fig. 4). CD38, an NAD+-consuming enzyme, is an ectoenzyme tethered to the outer surface of plasma membrane that converts NAD+ to cyclic ADP ribose (cADPR) and NAM by an ADP-ribosyl cyclase reaction (32). As discussed in the section of Biosynthesis of NAD+ in mammals, NAM can be metabolized by eNAMPT into cell-permeable NMN (41). Moreover, CD73, an ectoenzyme that was previously characterized as a 5′-AMP nucleotidase, can also dephosphorylate NMN to NR (46). The latter, in turn, can be transported into cells through dipyridamole-sensitive nucleoside transporters (94). Furthermore, NAM, NMN, and NR have been found in milk and plasma (109, 130).
Once these precursors are transported into cells, they can be utilized to synthesize NAD+ through the pathways described above. Indeed, exogenous addition of NAD+ precursors (NAM, NR, and NMN) to cells grown in culture increases intracellular NAD+ levels and protects against cell death (51, 101). Thus, although additional studies are necessary to determine the precise mechanisms involved, evidence suggests that extracellular NAD(H) can directly or indirectly affect intracellular NAD(H) levels to regulate cellular functions.
NAD(H) and NADP(H) in tissues and subcellular organelles
In general, NAD(H) and NADP(H) are predominantly bound to intracellular proteins, with free NAD(H) and NADP(H) accounting for only a small proportion of these dinucleotide pools (66). The intracellular content of NAD(H) and NADP(H) differs markedly among tissues and cell types. In rat liver, total NAD(H) (free and bound) and total NADP(H) were reported to be 3166 and 1788 nmol/g dry weight, respectively (126). In rat heart, the amount of total NAD+ was estimated to be 500 nmol/g wet weight (33). A total NAD+ concentration of 368 μM was reported for mouse erythrocytes (136). By contrast, in human erythrocytes from healthy adults, the concentrations of NAD+, NADH, NADP+, and NADPH were found to be 48, 1.4, 26, and 16 μM, respectively (124).
Notably, recently developed genetically encoded fluorescent biosensors provide new and accurate measures of cellular NAD(H) and NADP(H) levels and their compartmental pools. Using the Peredox-mCherry fluorescent biosensor, Hung and colleagues (68) found a higher cytosolic NADH/NAD+ ratio in primary astrocytes than primary neurons, suggesting that the redox state in primary astrocytes is more reduced than that in neurons. We recently developed a ratiometric fluorescent biosensor, SoNar, that demonstrates greater variability in cytosolic NADH/NAD+ and greater stability of mitochondrial NADH/NAD+ in live cells in various stress states (150). Another group utilized the RexYFP biosensor and showed that mitochondria contain much higher NADH levels than cytoplasm in HEK293 cells (17). Furthermore, fluorescent lifetime imaging, which can differentiate NADH and NADPH fluorescence, reveals that the enzyme-bound NADPH/NADH ratio is higher (2.2:1) in glia-like outer pillar supporting cells and substantially lower (0.4:1) in outer hair cells of rat cochlea (18).
The intracellular distribution of NAD(H) and NADP(H) is highly compartmentalized. For example, in rat liver, ∼40% of total NAD(H) and 59% of total NADP(H) are found in mitochondria (126). In cultured HEK293 cells, the estimated total cellular NAD+ was ∼365 μM and mitochondrial NAD+ is ∼246 μM, suggesting that the majority of intracellular NAD+ is present in the mitochondria (138). While nuclear membranes may be freely permeable to NAD+ and NADP+, the mitochondrial inner membrane is generally impermeable to both dinucleotides [NAD(P)+] (66, 143). Therefore, while cytosolic and nuclear NAD(P)+ can be maintained through the pathways mentioned in the Biosynthesis of NAD+ in Mammals and Biosynthesis of NADP+ sections, mitochondrial NAD+ and NADP+ levels are maintained through additional organelle-specific mechanisms (Fig. 4). The presence of mitochondrial NMNAT (NMNAT3) suggests that NAD+ can be synthesized from NMN in this organelle (24). The recent identification of MNADK suggests another means by which cells maintain mitochondrial NADP+ levels (97, 148). In addition, SIRT3-5 and PARP1, mitochondrial NAD+-consuming proteins, degrade NAD+ into NAM, which can replenish the NAD+ pool through the salvage pathway in this organelle (24).
It is interesting to note that cellular NAD(H) and NADP(H) pools possess differential sensitivity in response to diverse stimuli. Cytoplasmic and nuclear total NAD(H) is susceptible to changes in cellular nutrient levels (e.g., glucose, lactate, and pyruvate), whereas total mitochondrial NAD(H) is relatively well maintained and required for cell survival in response to toxic stresses (138, 151).
Metabolic sources of NAD(P)H and cytosolic/mitochondrial shuttles
The interconversion between NAD(P)+ and their reduced forms NAD(P)H can occur in cellular energy metabolic pathways such as glycolysis, the pentose phosphate pathway (PPP), the tricarboxylic acid (TCA) cycle, and mitochondrial oxidative phosphorylation (Figs. 5 and 6).
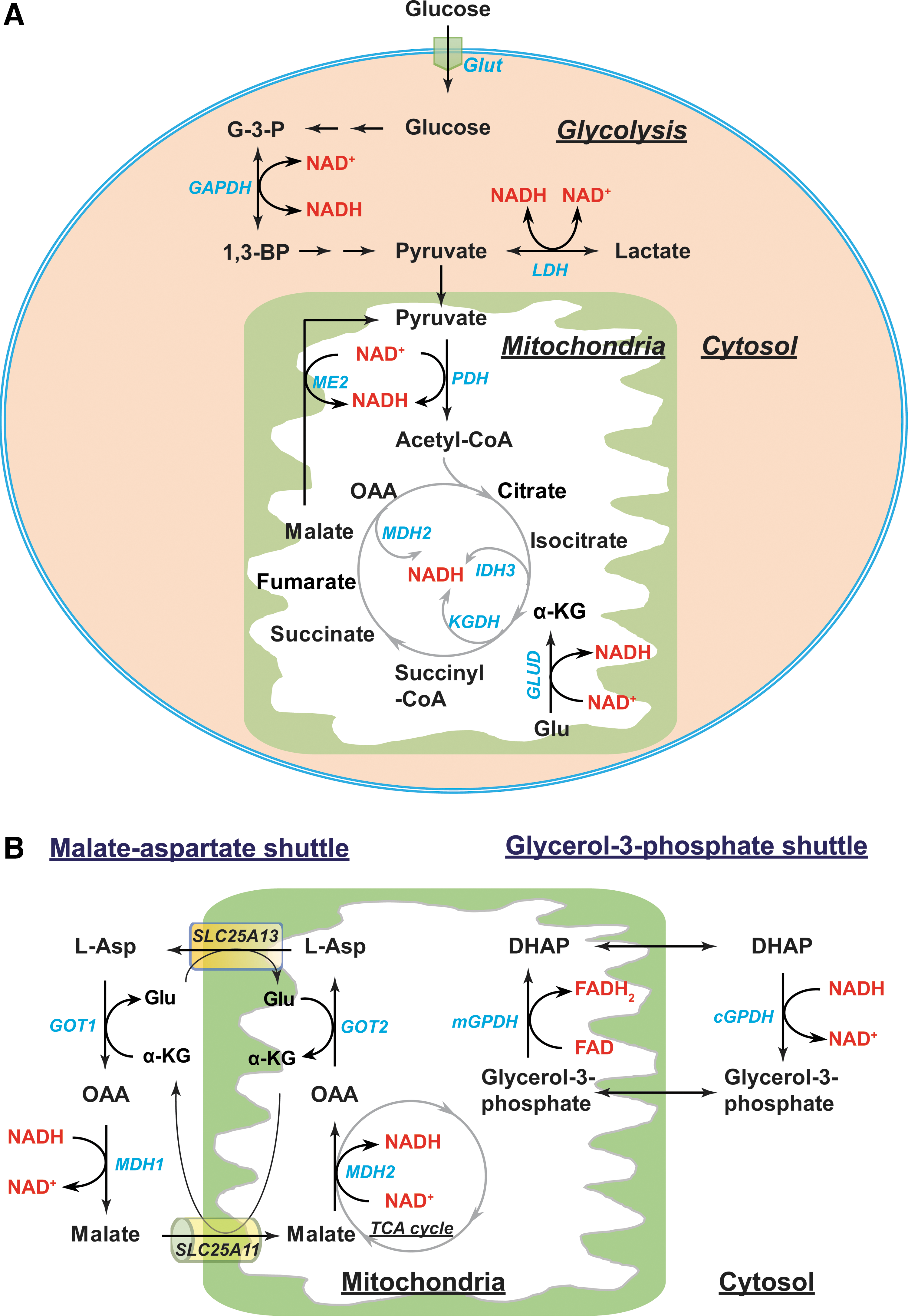
In the cytoplasm, NADH can be generated as a by-product of glycolysis (Fig. 5A). This occurs at the sixth step of glycolysis, where two molecules of G3P are oxidized to two molecules of 1,3-bisphosphoglycerate coupled with the reduction of NAD+ to NADH by glyceraldehyde phosphate dehydrogenase (GAPDH) (87). Cytosolic NADH can also be produced by lactate dehydrogenase (LDH), which catalyzes a reversible conversion between lactate and pyruvate.
In the mitochondria, the TCA cycle can produce eight molecules of NADH per molecule of glucose under well-oxygenated conditions (141). Once the glycolytic end-product pyruvate is transported into mitochondria, the pyruvate dehydrogenase (PDH) complex decarboxylates it into acetyl-CoA and simultaneously reduces NAD+ to NADH (87). Acetyl-CoA then enters the TCA cycle where NAD+ is reduced to NADH by NAD+-dependent isocitrate dehydrogenase 3 (IDH3), α-ketoglutarate dehydrogenase (KGDH), and malate dehydrogenase (MDH2) (Fig. 5A). Moreover, NAD+-linked malic enzyme (ME2) can produce NADH via conversion of malate to pyruvate (111). Glutamate dehydrogenases (GLUD1-2) can also metabolize glutamate into the TCA cycle intermediate α-KG using NAD+ as a cofactor to produce NADH.
In general, the outer mitochondrial membrane is very porous, enabling NADH to diffuse freely into the intermembrane space; however, the inner mitochondrial membrane is impermeable to NADH (24, 102, 141). To circumvent this impediment, two NADH shuttles, the malate–aspartate shuttle and the glycerol-3-phosphate shuttle, can transport NADH into the mitochondrial matrix (Fig. 5B) (66, 90).
In the first shuttle, the electron carrier malate is imported from the cytosol into the mitochondrial matrix through an α-KG/malate antiporter (encoded by SLC25A11 gene) in conjunction with export of α-KG into the cytosol. Once in the matrix, malate is oxidized into oxaloacetate (OAA) by MDH2, transferring electrons to NAD+ forming NADH. OAA is then transaminated into aspartate by mitochondrial glutamate-OAA transaminase (GOT2). Subsequently, the aspartate/glutamate antiporter (encoded by SLC25A13 gene) exports aspartate into the cytosol where cytosolic GOT (GOT1) converts aspartate back to OAA, which is, in turn, reduced to malate in conjunction with oxidizing NADH to NAD+ by cytosolic MDH (MDH1). Thus, the malate–aspartate shuttle is reversible and requires multiple enzymes. In this shuttle, NADH is oxidized to NAD+ in the cytosol and NAD+ is reduced to NADH in mitochondria. NAD+ is used as an electron acceptor during glycolysis, whereas NADH is used by mitochondrial complex I to drive the mitochondrial electron transport chain (ETC).
Unlike the malate–aspartate shuttle, electron transfer via the glycerol-3-phosphate shuttle is irreversible and needs only one enzyme, glycerol-3-phosphate dehydrogenase (GPDH) (Fig. 5B) (90). Cytosolic GPDH reduces the glycolytic intermediate DHAP to glycerol-3-phosphate and simultaneously oxidizes NADH to NAD+ in the cytoplasm. Mitochondrial GPDH catalyzes the reverse reaction by oxidizing glycerol-3-phosphate to DHAP and transferring electrons to FAD forming FADH2, which then donates electrons at mitochondrial respiratory complex II (succinate dehydrogenase; SDH) to reduce ubiquinone (Q) to ubiquinol (QH2). The use of FADH2, rather than NADH, as an electron source yields less energy per mole.
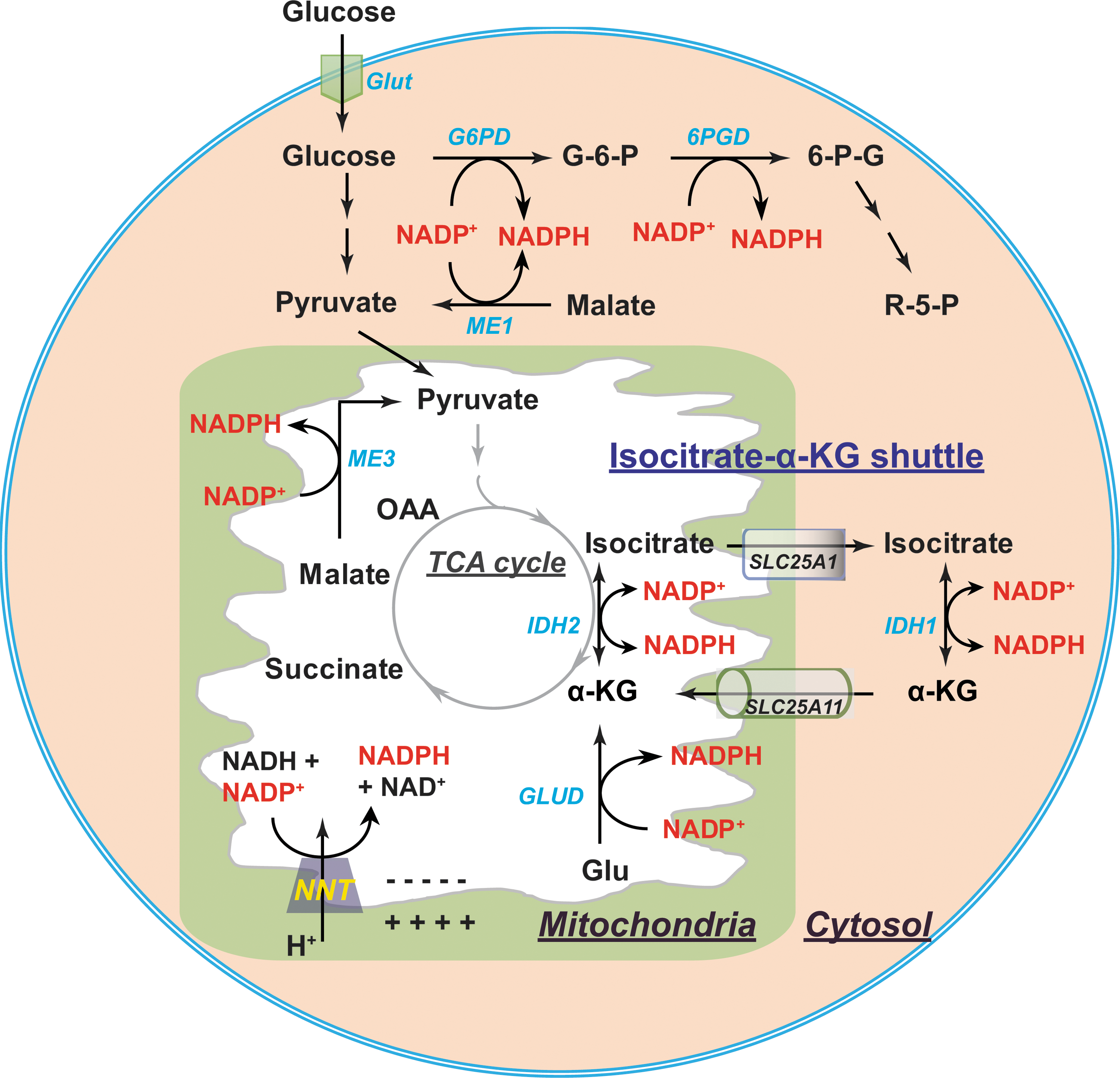
Cytosolic NADPH is primarily generated from the PPP by glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (6PGD) (87, 141). G6PD catalyzes the conversion of glucose-6-phosphate (G6P) into 6-phosphogluconate (6PG), which can be further metabolized into ribose-5-phosphate (R5P) by 6PGD. Both reactions are coupled to the reduction of NADP+ to NADPH (Fig. 6). In addition, other enzymes also contribute to the cytosolic NADPH pool, such as IDHs and MEs (87, 139), all of which have both cytosolic and mitochondrial isozymes. Cytosolic IDH (IDH1) catalyzes the same reaction as mitochondrial IDH3 using NADP+ rather than NAD+ as a cofactor, forming NADPH. Cytosolic ME (ME1) catalyzes oxidative decarboxylation of malate to pyruvate with the reduction of NADP+ to NADPH. The relative contribution of these enzymes to NADPH production remains elusive. Fan and colleagues (38) found in proliferating HEK293T cells that the greatest contributor to cytosolic NADPH is the oxidative PPP. In differentiating adipocytes, Liu et al. (82) showed that ME is the primary source of NADPH; however, in hypoxia the main source is the oxidative PPP.
Mitochondrial NADPH can be produced by mitochondrial isozymes of IDH (IDH2) and ME (ME3) (Fig. 6) (87, 139). In addition, NADP+-dependent GLUDs can also generate NADPH through the conversion of glutamate to α-KG (111). Of note, another significant contributor to mitochondrial NADPH is NAM nucleotide transhydrogenase (NNT), which catalyzes the following reversible reaction: NADH + NADP+ ← → NAD+ + NADPH (66, 113). NNT resides in the mitochondrial inner membrane and is driven by the electrochemical proton gradient. In the presence of a proton gradient, for example, under physiological conditions, the equilibrium of this reaction moves far to the right, which explains the fact that over 95% of mitochondrial NADP+ is reduced and that the redox potential of NADPH/NADP+ (−400 mV) is more negative than that of NADH/NAD+ (−300 mV) in mitochondria (66, 113).
As mentioned earlier, the mitochondrial inner membrane is impermeable to NADP(H) (24, 102, 141). Communication between cytosolic and mitochondrial NADP(H) pools is conducted by the isocitrate-α-KG shuttle (Fig. 6) (66, 100). This NADPH shuttle functions through IDH1 and IDH2 isozymes. In the mitochondrial matrix, NADP+-dependent IDH2 converts α-KG into isocitrate by oxidizing NADPH to NADP+. Isocitrate is then pumped into the cytosol in exchange for malate by the citrate carrier protein (encoded by SLC25A1 gene). In the cytosol, IDH1 catalyzes the reverse reaction by transforming isocitrate to α-KG and NADP+ to NADPH. Subsequently, α-KG is transported into mitochondrial matrix by the α-KG/malate antiporter as a carrier in the malate–aspartate shuttle. Thus, the isocitrate-α-KG shuttle plays a pivotal role in maintaining cellular NADPH levels.
NAD(H) and NADP(H) Regulate Cellular Redox Homeostasis
NAD(P)H and glutathione (GSH) serve as dual-function participants in maintaining cellular redox homeostasis. GSH is a cosubstrate for hydrogen peroxide (H2O2) removal by glutathione peroxidases (GPxs); and NADP(H) functions as an indispensable cofactor for glutathione reductase (GR) and thioredoxin reductases (TRs) that are essential for GPx- and peroxiredoxin (Prx)-mediated peroxide removal, respectively. Paradoxically, excess accumulation of GSH and/or NAD(P)H leads to reductive stress (60), and may directly contribute to the production of O2 •− and H2O2. Excess NAD(P)H, in particular, may be utilized by NADPH oxidases (NOXs) to produce reactive oxygen species (ROS) (59, 116). Since the critical roles of GSH in redox stress (oxidative and reductive stress) have been extensively discussed elsewhere (6, 110, 117), in the context of this review, we primarily focus on the NAD(H) and NADP(H) redox couples.
NAD(H) and NADP(H) as antioxidant cofactors
As mentioned above, NADPH is an essential cofactor of GR and TRs. GR catalyzes the recycling of GSH from its oxidized form (GSSG) (Fig. 7). In this context, NADPH donates two electrons to reduce GSSG to GSH by GR; the recycled GSH can then be used to reduce H2O2 to water by GPxs (21). In the context of TRs, TRs transfer electrons from NADPH to reduce oxidized thioredoxin (Trx-S2) to its reduced form Trx-(SH)2, which serves as a source of reducing equivalents in the enzymatic removal of H2O2 and other organic hydroperoxides by Prxs (21).
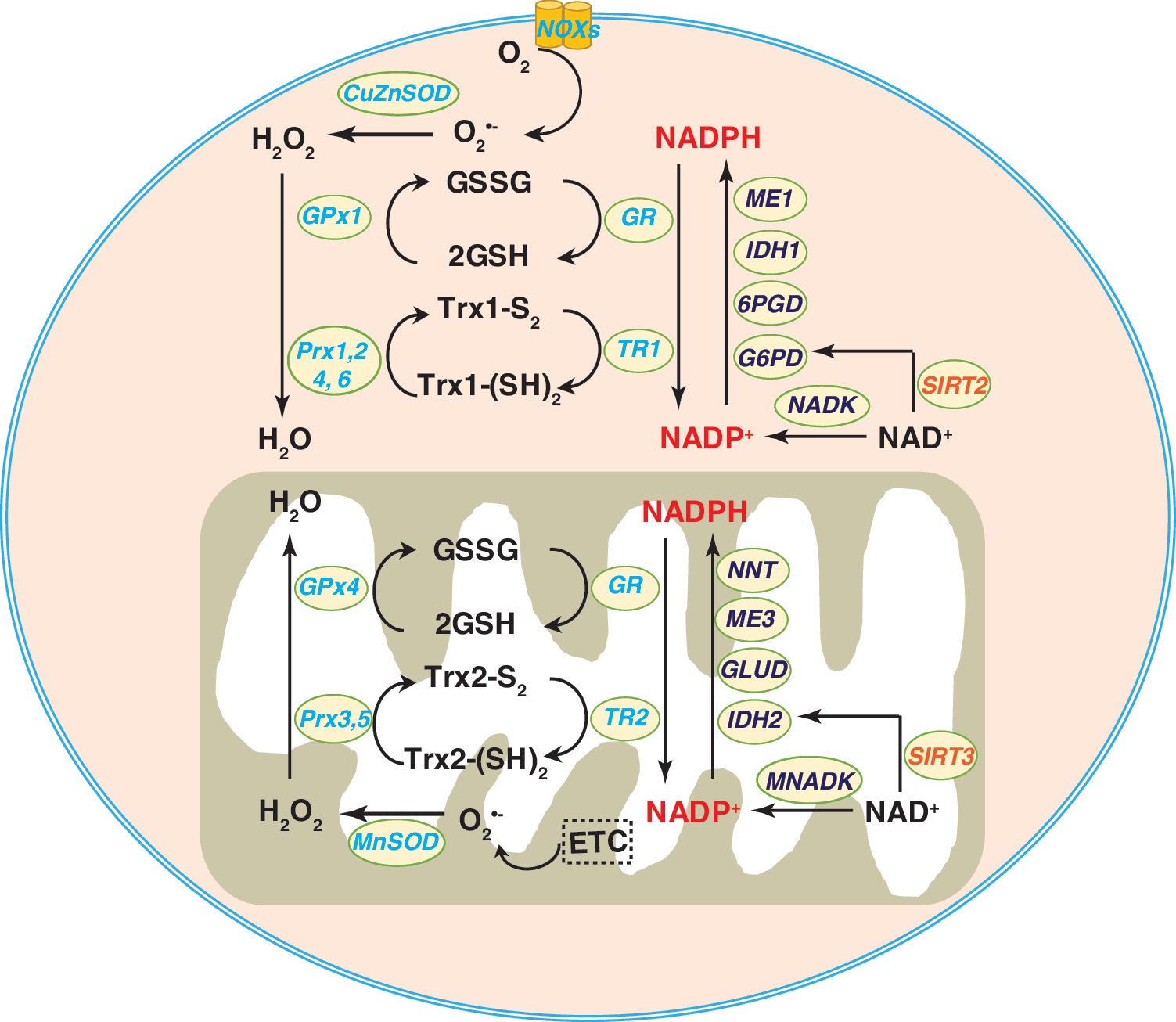
Given that G6PD is the rate-limiting enzyme of the PPP and the principal source of cytosolic NADPH (87, 141), modulation of G6PD activity is expected to affect cellular NADPH levels and consequently cellular redox state and biological functions. Indeed, G6PD-deficient mice showed decreased levels of NADPH and GSH as well as reduced activity of GPx, which contribute to oxidative damage in the renal cortex (135). Similarly, mouse embryonic stem cells lacking G6PD also had lower cellular NADPH and GSH levels, with enhanced sensitivity to oxidative stress-induced cell death due to the shutdown of the PPP (39). Moreover, our group previously reported that G6PD expression and activity are inhibited by aldosterone in mouse aortic endothelial cells and in mouse aorta (77). This inhibition of G6PD is associated with reduced cellular levels of NADPH, GSH, and nitric oxide (NO•), augmented ROS production, and impaired vascular function. Importantly, blockade of aldosterone by a mineralocorticoid receptor antagonist or overexpression of G6PD restored G6PD activity and significantly reversed these adverse effects in vivo (77).
Gain-of-function studies further confirmed the protective role of G6PD. For example, the enhancement of G6PD activity by genetic or pharmacological means elevated cellular NAPDH and GSH pools, promoted ROS detoxification, and increased cell viability in primary vascular endothelial and smooth muscle cells in vitro (34, 78). In transgenic mice, a modest (approximately two-fold) overexpression of G6PD was associated with an increase in NADPH levels in the brain and liver, in protection of these tissues against aging-induced oxidative damage, and an extension of life span (in females) compared to littermates with basal activity of G6PD (95). Thus, these studies clearly support the concept that G6PD is a major source of cytosolic NADPH, and that NADPH is the indispensable reducing agent for ROS elimination and redox homeostasis (Fig. 7).
In the mitochondrion, IDH2 is a major enzyme for NADPH production (Fig. 7) (113); it catalyzes the oxidative decarboxylation of isocitrate producing α-KG and CO2. Gain- and loss-of function studies confirmed the essential functions of IDH2 in maintaining cellular NADPH levels and cellular redox balance. For example, IDH2 knockout mice exhibit decreased NADPH/NADP+ and GSH/GSSG ratios and increased H2O2 levels and oxidative damage. These changes in redox state are associated with cardiac hypertrophy, accelerated heart failure, and apoptotic cell death in cardiomyocytes (74). In other studies, exposure to 7-ketocholesterol, a major oxidation product of cholesterol, inhibited IDH2 expression and activity via upregulation of microRNA-144 (42). The decrease in IDH activity was accompanied by lower NADPH and GSH levels as well as a decrease in NO• production, leading to oxidative stress in human aortic endothelial cells and impaired vascular function in ex vivo murine aortae (42). As expected, IDH2 silencing also decreased NADPH levels (147), while IDH2 overexpression increased NADPH levels and protected against oxidative stress-induced cell death in HEK293 cells (121). Taken together, these findings suggest that IDH2-derived NADPH is required for detoxification of peroxides by NADPH-dependent peroxidases in mammalian mitochondria.
In addition to IDH2, the mitochondrial enzyme NNT is another major source of mitochondrial NADPH that catalyzes the reversible conversion of NADH and NADP+ to NAD+ and NADPH (Fig. 6) (113). The critical role of NNT in regulating redox status has been well described in humans and animals. For example, the left ventricles of heart failure patients displayed lower NNT activity and NADPH levels, which correlated with decreases in GR activity and GSH levels compared to the left ventricles of non-heart failure patients (118). Under a physiological workload, cardiomyocytes from C57BL/6J mice carrying spontaneous loss-of-function mutations of NNT produced markedly higher levels of H2O2 compared to cardiomyocytes from NNT wild-type C57BL/6N mice (93), suggesting that NNT is required for NADPH regeneration to fuel H2O2 detoxifying enzymes. Indeed, concurrent with their decreased levels of NADPH, isolated liver mitochondria of NNT mutant C57BL/6J mice were less effective at removing exogenous peroxide (111). Finally, inactivation of NNT by short hairpin RNA (shRNA) or a chemical inhibitor decreases cellular NADPH and GSH levels, inhibits cell proliferation, and increases H2O2 accumulation and oxidative damage (44, 85, 142). Taken together, these findings indicate that mitochondrial NNT plays an important role in maintaining the necessary pools of cellular reducing equivalents and in preventing mammalian cells from oxidative damage and dysfunction.
It is noteworthy that NADK, the NADP+-producing enzyme, has also been shown to modulate the cellular NADPH pool (Fig. 7). Overexpression of NADK elevated cellular NADPH levels and, thereby, accelerated H2O2 removal in HEK293 cells and rat pancreatic β cells (49, 102). By contrast, silencing NADK reduced NADPH levels leading to H2O2 accumulation and inhibition of glucose-stimulated insulin secretion in rat pancreatic β cells (49).
NAD+ is also involved in regulating the cellular redox state through the actions of SIRT enzymes. Recent studies demonstrate that NAD+-dependent SIRT2 and SIRT5 deacetylate and activate G6PD, thus increasing cellular NADPH and antioxidant capacity in vivo and in vitro (133, 154). The opposite effects were found in HEK293 cells and mouse embryonic fibroblasts lacking SIRT5 activity (154).
Furthermore, like G6PD, IDH2 is regulated by SIRT proteins, with SIRT5 also increasing IDH2 activity by deacetylation. Consequently, silencing SIRT5 diminished IDH2 activity and cellular NADPH and GSH levels, sensitizing cells to paraquat-induced oxidative cytotoxicity (154). Moreover, mitochondrial SIRT3 has been reported to upregulate IDH2 activity through deacetylation in a mouse model of calorie restriction. Elevated IDH2 activity induced by calorie restriction was associated with increases in NADPH levels and the GSH/GSSG ratio in the inner ear, brain, and liver tissues, enhancing the protection of these tissues against oxidative damage (121).
NAD(H) and NADP(H) are pro-oxidants and induce redox stress
Excess levels of cellular NADH and/or NADPH can lead to reductive stress. NAD(P)H fuels cellular ROS production via its role as a substrate for the NOX family proteins (NOX1-7) that produce H2O2 and O2 •− (13). As discussed in the section on NAD(H) and NADP(H) as antioxidant cofactors, G6PD is the major source of the cytosolic NADPH pool. Modulation of G6PD activity can affect cellular NADPH levels and ROS production by NOX proteins. Overexpression of G6PD increased cellular NAD(P)H levels and upregulated NOX gp91phox and p22phox subunit mRNA expression, potentiating ROS production and oxidative damage in mouse pancreatic β cells and thymic lymphoma cells (76, 127).
By contrast, loss of G6PD activity was found to be beneficial in many disease models by opposing high NADPH levels and reductive stress. For example, G6PD deficiency reduced NADPH levels and significantly inhibited angiotensin II (Ang II)-induced O2 •− production and decreased medial aortic thickness (89). Similar effects were observed in pacing-induced heart failure, where inhibition of G6PD activity abrogated elevations in NADPH levels and ROS production in failing hearts (54). Furthermore, mice with cardiac-specific overexpression of the mutant human αB-crystallin (R120G mutant) gene recapitulated the pathology of human protein aggregation cardiomyopathy and exhibited reductive stress in the heart as evidenced by increased GSH levels and increased activities of GR, G6PD, catalase, and GPx1 (107). Notably, these pathological changes and reductive stress were significantly reversed by replacing the wild-type G6PD in the R120G mice with a hypomorphic G6PD mutant (107), suggesting that G6PD-mediated reductive stress contributes to the development of this pathophenotype. Therefore, these pieces of evidence support the notion that NADPH pools produced by G6PD can induce redox stress and cellular dysfunction.
Together, these data suggest that cellular redox homeostasis results from a delicate balance between NADPH-dependent protection against oxidant stress and NADPH-dependent reductive stress. This balance can be influenced by the relative activity of NADPH-dependent NOXs and antioxidant enzymes (e.g., Trxs, Prxs, and TRs) that depend directly or indirectly on NADPH, as well as exogenous/endogenous stimuli in a specific cell type or tissue.
Mitochondrial NADH is oxidized to NAD+ at mitochondrial respiratory complex I (NADH dehydrogenase). Electrons from NADH, in conjunction with electrons from complex II, are relayed through the mitochondrial ETC to reduce molecular oxygen to water at respiratory complex IV (cytochrome C oxidase; COX) (10, 91). Under physiological conditions in mammalian cells, approximately 0.1–0.2% of total oxygen consumed is converted to O2 •− as a result of electron leakage from the ETC. This electron leakage greatly increases under stress or pathological conditions and is associated with enhanced production of mitochondrial O2 •− generation (Fig. 8) (10).

Both complex I and complex III have been described as major sites of mitochondrial O2 •− under physiological and pathological conditions (Fig. 8) (10, 91). Since O2 •− is highly reactive and has a very short half-life, it is rapidly converted to the more stable H2O2 by manganese superoxide dismutase (MnSOD) (or by spontaneous dismutation).
ROS production by complex I requires a high NADH/NAD+ ratio, whereas its dependence on proton motive force is controversial (1, 75, 105, 122, 132). Under basal conditions, isolated mitochondria generate extremely low levels of ROS when exogenous oxidizable substrates are absent (122, 132); however, addition of exogenous complex I substrates, such as glutamate, malate, or α-KG, augments the levels of NADH and, simultaneously, stimulates H2O2 production by ∼10-fold in isolated brain mitochondria (122). Interestingly, subsequent addition of ADP results in oxidation of NADH leading to a >50% reduction in H2O2 production (122). In rat L6 myoblasts, treatment with 1 mM antioxidant N-acetyl-L-cysteine increases the NADH/NAD+ ratio, which correlates with increases in mitochondrial H2O2 levels and free radical leak (119). These biochemical and cell culture studies suggest that a high NADH/NAD+ ratio is required for ROS production at complex I.
This concept is further supported by animal studies (93). Nickel et al. showed that C57BL/6J mice lacking functional NNT activity are protected against cardiac overload-induced heart failure compared to controls (wild-type C57BL/6N mice) (93). Reduced failure of the myocardium in NNT-deficient mice was associated with decreased NADH and H2O2 production as well as reduced oxidative damage (93). These findings suggest that pathological cardiac pressure overload induces reverse flux of NNT to generate NADH at the expense of NADPH resulting in a high NADH/NAD+ ratio, which further promotes ROS generation at complex I leading to oxidative injury and cytotoxicity (Fig. 8B).
Furthermore, complex I can also operate in a reverse electron transfer (RET) mode, which leads to enhanced O2 •− production at complex I compared with the forward mode (1). Under physiological conditions, RET is a minor contributor to ROS production; however, RET-induced ROS production is observed in the heart under an ischemia/reperfusion (IR) challenge (26). During ischemia, succinate is selectively increased in various murine tissues, including the heart, and is generated by the NADH-driven reversal of the SDH reaction. After reperfusion, the accumulated succinate is rapidly reoxidized by SDH, leading to ROS generation at complex I through RET to cause IR injury. Blocking ischemic succinate accumulation by inhibiting SDH attenuates mitochondrial ROS production and heart IR injury (Fig. 8B) (26).
Accumulating evidence demonstrates that hypoxia increases cellular NADH levels and ROS production in most mammalian cells. Under hypoxia, mitochondrial NADH and FADH2 are unable to be oxidized by the ETC leading to a buildup of these reducing equivalents and subsequent reductive stress (28). Reductive stress enables one-electron reduction of oxygen to form O2 •− and thereby underlies the rise in ROS production under hypoxia (28).
Our recent work demonstrates that primary human lung fibroblasts cultured in hypoxia (0.2% O2) produce significantly higher levels of mitochondrial ROS compared with normoxic cells (21% O2) (98). In particular, the elevation in mitochondrial ROS accumulation is associated with an increase in the NADH/NAD+ ratio and an accumulation of L-2-hydroxyglutarate, a reductive metabolite of α-KG (98), whose increase serves to buffer the reductive stress through inhibiting glycolysis and TCA cycle and, thus, NADH production under hypoxia. In bovine coronary artery smooth muscle cells, hypoxia also increased mitochondrial ROS production and NAD(P)H levels, as well as the cytosolic NADH/NAD+ ratio (indicated by the lactate/pyruvate ratio) (Fig. 8B) (45).
It is worthwhile to note that other NADH-related mitochondrial enzymes, such as KGDH and PDH, also contribute to mitochondrial ROS production (123, 131). Two separate studies demonstrate that isolated mitochondrial KGDH and PDH from the brain or heart tissue produce O2 •− and H2O2 in the presence of α-KG and redox cofactors (123, 131). Addition of NAD+ suppresses ROS production by these enzymes by switching KGDH from H2O2 formation mode to catalytic mode (formation of NADH via this reaction: α-KG + NAD+ + CoA → Succinyl-CoA + CO2 + NADH), whereas the addition of NADH stimulates KGDH-mediated ROS production (123, 131). Interestingly, in the presence of its substrates (α-KG and CoA) and different ratios of NADH to total NAD(H) (NADH and NAD+), a higher NADH/NAD(H) ratio is related to more ROS production by KGDH (131). Together, these data suggest that NADH promotes ROS formation by complex I and III of the ETC as well as via the stimulation of the mitochondrial enzymes KGDH and PHD (Fig. 8B).
NAD(H) and NADP(H) and Cellular Metabolism
In addition to their crucial roles in maintaining cellular redox state, the NAD(H) and NADP(H) redox couples are also critical regulators of cellular metabolism (Fig. 9). Typically, NAD+ is necessary for glycolysis and for the biosynthesis of nucleotides and amino acids; NADH provides electrons for mitochondrial oxidative phosphorylation and ATP production (87). NADP+ supports the PPP to generate NADPH that is indispensable for reductive biosynthesis of nucleotides, amino acids, and lipids (87). As described in the Metabolic Sources of NAD(P)H and Cytosolic/Mitochondrial Shuttles section, many metabolic enzymes catalyze the intra/interconversion of NAD(H) and NADP(H). Thus, changes in NAD(H) and NADP(H) levels affect cellular metabolism and vice versa.
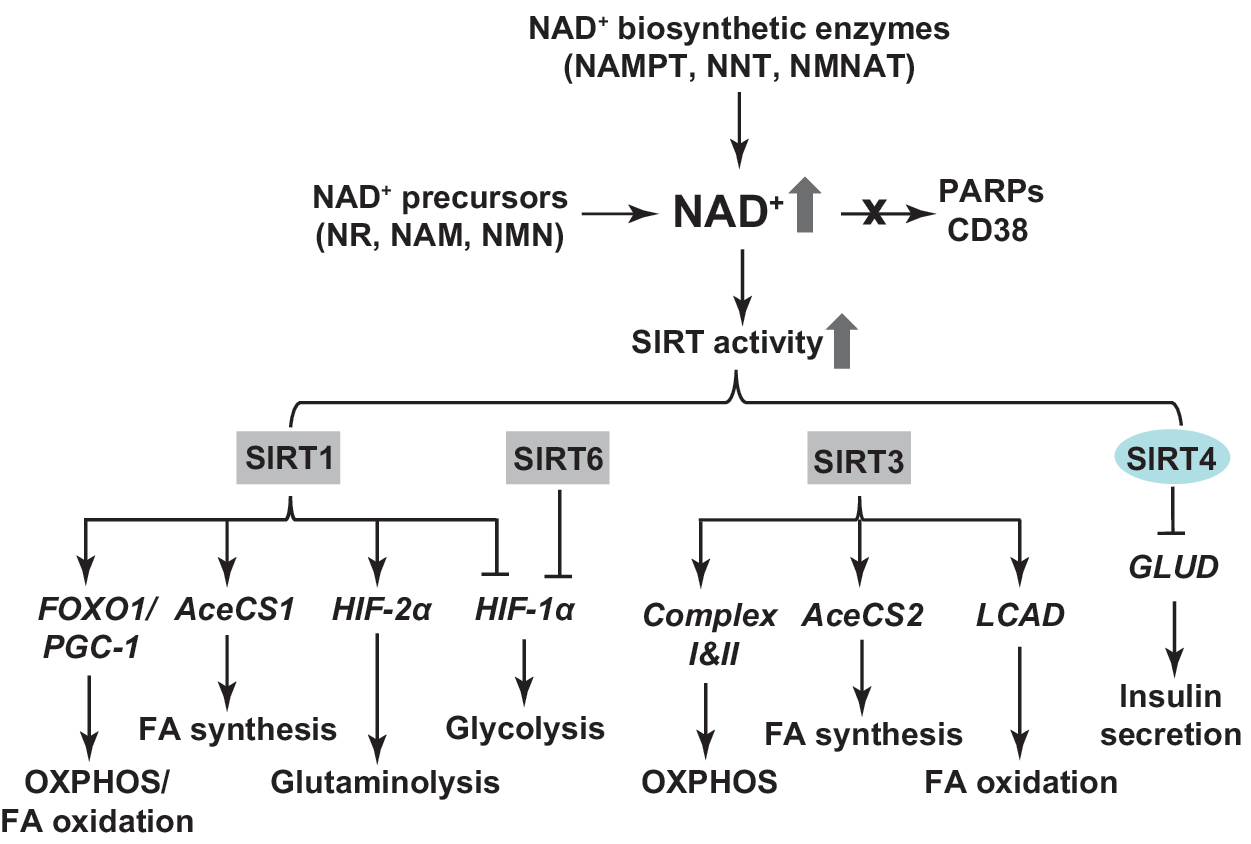
Modulating NAD(H) biosynthesis regulates cellular metabolism
NR, NAM, and NMN are precursors of NAD+ biosynthesis via the salvage pathway (Biosynthesis of NAD+ in Mammals section). Accumulating evidence highlights the importance of these precursors in modulating cellular NAD+ levels and metabolism in various disease models. For example, Canto et al. (23) showed that feeding mice NR elevates cellular NAD+ levels in skeletal muscle and brown adipose tissue and prevents high-fat diet-induced weight gain and obesity, likely due to enhanced insulin sensitivity and energy expenditure. In fact, NR supplementation improves glucose tolerance, mitochondrial biogenesis, and oxygen consumption in mice fed a high-fat diet. Similar protective effects were also observed in prediabetic and diabetic mice that received NR in their diet (129).
In addition, in obese and diabetic mice, NAM supplementation elevates hepatic NAD(H) levels and the NAD+/NADH ratio, resulting in increased mitochondrial content, enhanced mitochondrial glucose metabolism, and improved insulin sensitivity (140). Likewise, administration of NMN ameliorates the decrease in NAD+ levels in the liver and hepatocytes, improves glucose metabolism, and attenuates insulin resistance in high-fat diet-induced type 2 diabetic mice (146). Furthermore, decreases in total and nuclear NAD+ levels are found in skeletal muscles of aged mice, which correlate with compromised mitochondrial function characterized by decreased expression of mitochondrial-encoded respiratory subunits, decreased mitochondrial content, and impairment of ATP production (48). Supplementation of NAM raises cellular NAD+ levels and rescues mitochondrial function in aged mice (48). These lines of evidence suggest that NAD+ precursors can modulate cellular metabolism and reverse metabolic pathophenotypes by promoting NAD+ biosynthesis.
Given that NAD+ biosynthesis from its precursors is governed by a cascade of enzymatic reactions, modulating the activity of rate-limiting enzymes can also alter cellular NAD+ levels and consequently cellular metabolism. Pharmacological inhibition of NAMPT with FK866 reduces cellular NAD(H) levels and NAD+/NADH ratio, which are associated with decreases in oxygen consumption and ATP production and upregulation of glycolytic gene expression, indicating a metabolic shift toward glycolysis in rat primary cardiomyocytes (99). Interestingly, supplementation of NMN, the enzymatic product of NAMPT, is able to abrogate the decrease in NAD(H) levels significantly, preventing the metabolic shift to glycolysis (99). In mice, high-fat diet-induced type 2 diabetes suppresses NAMPT expression in the liver and white adipose tissue leading to reduced NAD+ levels, which correlate with insulin resistance and impaired glucose metabolism (146). Dietary administration of NMN in diabetic mice reverses these phenotypes (146). These findings suggest that NAD+ produced by NAMPT is critical for maintaining glucose metabolism. Notably, NAMPT is a target gene of the transcription factor hypoxia-inducible factor 1α (HIF-1α) (7). HIF-1α signaling is known to reprogram cellular metabolism toward glycolysis (60). Therefore, HIF-1α-mediated upregulation of NAMPT is expected to enhance NAD+ biosynthesis and, thus, cellular NAD+ levels, which is required for glycolysis.
The mitochondrial enzyme NNT is an important source of mitochondrial NAD+ and NADPH (Metabolic Sources of NAD(P)H and Cytosolic/Mitochondrial Shuttles section). Knockdown of NNT results in increased cytosolic NAD+/NADH and NADP+/NADPH ratios and promotes a metabolic switch that utilizes glycolysis rather than glutaminolysis as the major anaplerotic reaction to replenish the TCA cycle with anabolic carbons. NNT knockdown correlates with inhibition of cell proliferation and sensitization of melanoma to glucose deprivation-induced cell death (44). By contrast, cells overexpressing NNT switch back to glutaminolysis as their main energy source (44). Genetic or pharmacological blockade of NNT reduces cellular NAD(P)H levels, depolarizes mitochondrial membrane potential, and inhibits mitochondrial oxidative phosphorylation, thereby increasing the susceptibility to oxidative stress-induced cell death (85, 142). These results suggest that NNT is essential for maintenance of mitochondrial function and redox balance to support cell proliferation and survival.
NMNAT also regulates NAD+ biosynthesis. Silencing NMNAT1 decreases nuclear NAD+ levels and SIRT1 activity, resulting in a metabolic shift from mitochondrial oxidative phosphorylation to aerobic glycolysis owing to impaired mitochondrial function in primary mouse myoblasts (48). Overexpression of NMNAT1 in these cells restores cellular NAD+ levels and completely abrogates these metabolic changes via a SIRT1-dependent mechanism (48).
Manipulating NAD(H) consumption regulates cellular metabolism
In addition to biosynthetic enzymes, cellular NAD(H) levels are also determined by NAD+-consuming enzymes, including SIRT deacetylases (SIRT1-7), PARPs (PARP1-2), and cADP-ribose synthases (CD38 and CD157) (22, 66).
SIRTs are mammalian homologues of the yeast silent information regulator 2 (sir2) and are localized in distinct subcellular compartments: SIRT1, SIRT6, and SIRT7 are primarily found in the nucleus; SIRT3, SIRT4, and SIRT5 are located in mitochondria; and SIRT2 is mainly localized to cytosol (24, 57, 66). SIRTs exhibit both protein deacetylase and mono ADP-ribosyltransferase activity. SIRTs utilize NAD+ as a substrate to catalyze the deacetylation reaction at lysine residue of proteins to produce NAM, deacetylated protein, and 2′-O-acetyl-ADP ribose (24, 57, 66). These enzymes are involved in regulating numerous biological processes, including cellular metabolism. Changes in NAD+ bioavailability can alter the activity of these enzymes and, thereby, affect energy metabolism.
In yeast, calorie restriction lowers cellular NADH levels, an inhibitor of sir2 activity, leading to an increase in the NAD+/NADH ratio and activation of sir2. Concurrently, there is a metabolic shift to mitochondrial oxidative phosphorylation and a resulting extension of life span (80, 81), suggesting that sir2 regulates cellular metabolism in yeast.
Mammalian homologues of sir2 also regulate metabolism (Fig. 9). In a mitochondrial disease model, COX assembly protein knockout/knockin (SCO2 ko/ki) mice exhibit ubiquitous COX deficiency and exercise intolerance (25). NR administration increases the NAD+/NADH ratio in skeletal muscle and improves motor performance compared to vehicle-fed SCO2 ko/ki mice. This protection is associated with SIRT1-mediated deacetylation and activation of forkhead box O1 (FOXO1) and peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), thereby upregulating the expression of genes involved in fatty acid oxidation and mitochondrial oxidative phosphorylation to enhance energy production (25). In addition, SIRT1 has been found to activate acetyl-CoA synthetase 1 (AceCS1) by deacetylation, resulting in a pronounced increase in AceCS1-dependent fatty acid synthesis from acetate in Cos-7 cells (58).
Likewise, mitochondrial SIRT3 is also able to activate mitochondrial AceCS2 (58). Furthermore, SIRT3 has been shown to stimulate ATP production by activating proteins in the mitochondrial respiratory chain. In particular, SIRT3-induced deacetylation of complex I proteins (e.g., NDUFA9) and subunit A (SDHA) in complex II augments the activity of both complexes, increasing ATP production (3, 27). Consequently, SIRT3 expression levels positively correlate with ATP production in murine tissues; and tissue ATP production is lower in SIRT3 knockout mice compared with wild-type littermates (3). Moreover, SIRT3 stimulates mitochondrial fatty acid oxidation by deacetylation and activation of long-chain acyl coenzyme A dehydrogenase in murine liver (64).
Finally, unlike SIRT1 and SIRT3, SIRT4 is an ADP-ribosyltransferase, capable of ADP-ribosylating and inactivating mitochondrial GLUD2 in mouse pancreatic β cells to suppress insulin secretion in response to glucose, glutamine, or leucine (56). Consequently, SIRT4 knockout mice exhibit increased insulin secretion under basal and stimulated conditions. In vitro studies further demonstrate that the restoration of insulin secretion requires the normal activity of GLUD2 in SIRT4-deficient pancreatic β cells (56).
Emerging evidence suggests that SIRT proteins also interact with HIF signaling pathway in regulating cellular metabolism (Fig. 9). It is noteworthy that different SIRTs exhibit distinct effects on HIF activity. Specifically, SIRT1 and SIRT6 are negative regulators of HIF-1α, while SIRT1 activates HIF-2α activity. For example, in skeletal muscle of aged mice, nuclear NAD+ levels and SIRT1 activity are much lower than in skeletal muscle of young mice (48). The decrease in SIRT1 activity is associated with metabolic reprogramming to aerobic glycolysis due to compromised mitochondrial function evidenced by decreases in mitochondrial content, ATP production, and mitochondria-encoded gene expression of the respiratory complexes (48). The metabolic shift and mitochondrial dysfunction are also correlated with pseudohypoxic activation of HIF-1α but not HIF-2α signaling likely through downregulation of von Hippel Lindau (VHL) protein. Importantly, feeding old mice NMN rescues cellular NAD+ levels and SIRT1 activity, and upregulates VHL expression, leading to an inhibition of age-related activation of HIF-1α signaling under normoxia and a normalization of mitochondrial oxidative phosphorylation (48).
Furthermore, cancer cells are known to utilize both glycolysis and glutaminolysis for energy generation. Corbet et al. (29) reported that when human cervical and pharyngeal cancer cells (squamous cell) were cultured in medium at pH 6.5 for 8–10 weeks, they reprogrammed their metabolism from glycolysis to glutaminolysis. This metabolic switch elevated cellular NAD+ levels and enhanced SIRT1 activity in acidic cultures, resulting in deacetylation of HIF-1α and HIF-2α proteins (29). Deacetylation of HIF-1α decreases its activity leading to inhibition of glycolysis, whereas HIF-2α deacetylation boosts its activity to promote glutaminolysis via upregulating glutaminase 1 expression (29). These actions collaboratively ensure the metabolic switch of energy sources to support cell proliferation and tumor growth. Results from these two studies suggest that SIRT1 negatively regulates HIF-1α signaling, but its effects on HIF-2α activity depends on cell context.
Similar to SIRT1, SIRT6 was reported to be a corepressor of HIF-1α signaling; however, SIRT6 accomplished this effect by deacetylating the chromatin of HIF-1α target gene promoters (153). Embryonic stem cells from SIRT6 knockout mice showed normoxic stabilization of HIF-1α protein that led to a metabolic shift toward aerobic glycolysis as demonstrated by increases in glucose uptake, lactate production, and glycolytic gene expression as well as a decrease in oxygen consumption (153).
Cellular NAD+ bioavailability can also be affected by the activity of PARPs and CD38 (Fig. 9). Modulation of PARPs or CD38 activity affects cellular metabolism by altering NAD+, thereby affecting SIRT1 activity. Two different groups reported that skeletal muscle of PARP1 or PARP2 knockout mice exhibited higher NAD+ content and SIRT1 activity than wild-type mice (8, 9). In this context, increased SIRT1 activity deacetylates and activates FOXO1 and PGC-1α, which augments energy expenditure and protects against high-fat diet-induced obesity by increasing mitochondrial biogenesis and oxidative phosphorylation (8, 9). These effects were further confirmed in cultured cells with PARP1 or PARP2 silencing, but were attenuated by simultaneous knockdown of SIRT1 in PARP1- or PARP2-deficient cells (9), suggesting that enhanced mitochondrial function requires NAD+-dependent SIRT1 activity in PARP-deficient cells.
Similarly, tissues from CD38 knockout mice have increased cellular NAD+ levels, which correlate with higher SIRT1 activity in liver nuclear extracts compared to wild-type nuclear extracts (4). By contrast, a decrease in cellular NAD+ levels is found in HEK293 cells overexpressing CD38, resulting in decreased expression of glycolytic genes and attenuated cell proliferation (67). Taken together, these findings highlight the interactions of the three classes of NAD+-consuming proteins in regulating cellular NAD+ levels and energy metabolism.
Concluding Remarks
The NAD(H) and NADP(H) redox couples serve as cofactors or/and substrates for many enzymes to maintain cellular redox homeostasis and energy metabolism. Deficiency or imbalance in cellular NAD(H) and NADP(H) levels perturbs cellular redox state and metabolic homeostasis leading to redox stress, energy stress, and eventually disease states. Thus, maintaining cellular NAD(H) and NADP(H) balance is critical for cellular function. This balance is maintained dynamically and governed by biosynthesis, consumption, and compartmental localization.
Newly identified biosynthetic enzymes, for example, eNAMPT and MNADK, and newly developed genetically encoded fluorescent biosensors have improved our understanding of how compartmentalized NAD(H)/NADP(H) pools inter-relate in response to various physiological and pathological stimuli. The use of these newly developed fluorescent tools has been limited to cell cultures until recently, when xenografts expressing these sensors have been used to monitor the NAD+/NADH redox state in murine models (150, 152). In addition, the metabolism of NAD+ and its precursors in the extracellular compartment and how exogenous NAD+ enters cells have not been well explored and nor understood. Future research on these topics is needed.
The level of cellular NAD(H)/NADP(H) is essential for maintaining redox homeostasis. Deficiency in these redox couples can lead to oxidative or reductive stress, depending on the redox ratio of each. Both oxidative stress and reductive stress are detrimental to normal cell functions. This dual role complicates the use of global antioxidants as rational and effective therapeutic approaches to redox stress disorders.
In addition, it is clear that NAD(H) and NADP(H) are required for maintaining cellular metabolism. Future efforts are still needed to understand how an imbalance of these two redox couples directly affects energy metabolism and how this imbalance alters NAD(H)/NADP(H)-dependent enzymes and, thus, affects their functions in regulating cellular metabolism.
Finally, the emergence of bioactive NAD+ precursors, for example, NR, and of specific pharmacological inhibitors, for example, NAMPT inhibitor FK866, provides promising therapeutic approaches for the treatment of NAD(H)- and NADP(H)-related metabolic disorders through modulating cellular NAD(H) levels.
Footnotes
Acknowledgments
This work is supported by the National Institutes of Health grants HG007690, HL061795, and GM107618 to J.L. We thank Stephanie Tribuna for assistance in preparation of this article and apologize to investigators whose work was not mentioned due to space limits.