
Abstract
Significance:
Neoplasms contain tumor-initiating stem-like cells (TICs) that drive malignant progression and tumor growth with drug resistance. TICs proliferate through a self-renewal process in which the two daughter cells differ in their proliferative potential, with one retaining the self-renewing phenotype and another displaying the differentiated phenotype.
Recent Advances:
Cancer traits (hepatocellular carcinoma) are triggered by alcoholism, obesity, and hepatitis B or C virus (HBV and HCV), including genetic changes, angiogenesis, defective tumor immunity, immortalization, metabolic reprogramming, excessive and prolonged inflammation, migration/invasion/metastasis, evasion of cell cycle arrest, anticell death, and compensatory regeneration/proliferation.
Critical Issues:
This review describes how metabolic reprogramming in mitochondria promotes self-renewal and oncogenicity of TICs. Pluripotency transcription factors (TFs), NANOG, OCT4, MYC, and SOX2, contribute to cancer progression by mitochondrial reprogramming, leading to the genesis of TICs and cancer. For example, oxidative phosphorylation (OXPHOS) and fatty acid metabolism are identified as major pathways contributing to pluripotency TF-mediated oncogenesis.
Future Directions:
Identification of novel metabolic pathways provides potential drug targets for neutralizing the activity of highly malignant TICs found in cancer patients. Antioxid. Redox Signal. 28, 1080–1089.
Introduction
T
First, TICs are found in malignant HCCs. Neoplasms contain distinct subpopulations of cells known as TICs that drive tumor growth and malignant progression combined with drug resistance. The presence of TICs in HCCs is significant since TICs promote recurrence and metastasis, leading to deaths of HCC patients.
Second, the role of pluripotency TFs is discussed in TICs and embryonic stem cells (ESCs). Pluripotency TFs include NANOG, OCT4, SOX2, MYC, and LIN28. These pluripotency TFs reprogram TICs and confer stemness characteristics. Understanding roles of pluripotency TFs in TICs is significant since targeting of these pluripotency TFs becomes a potential therapeutic strategy.
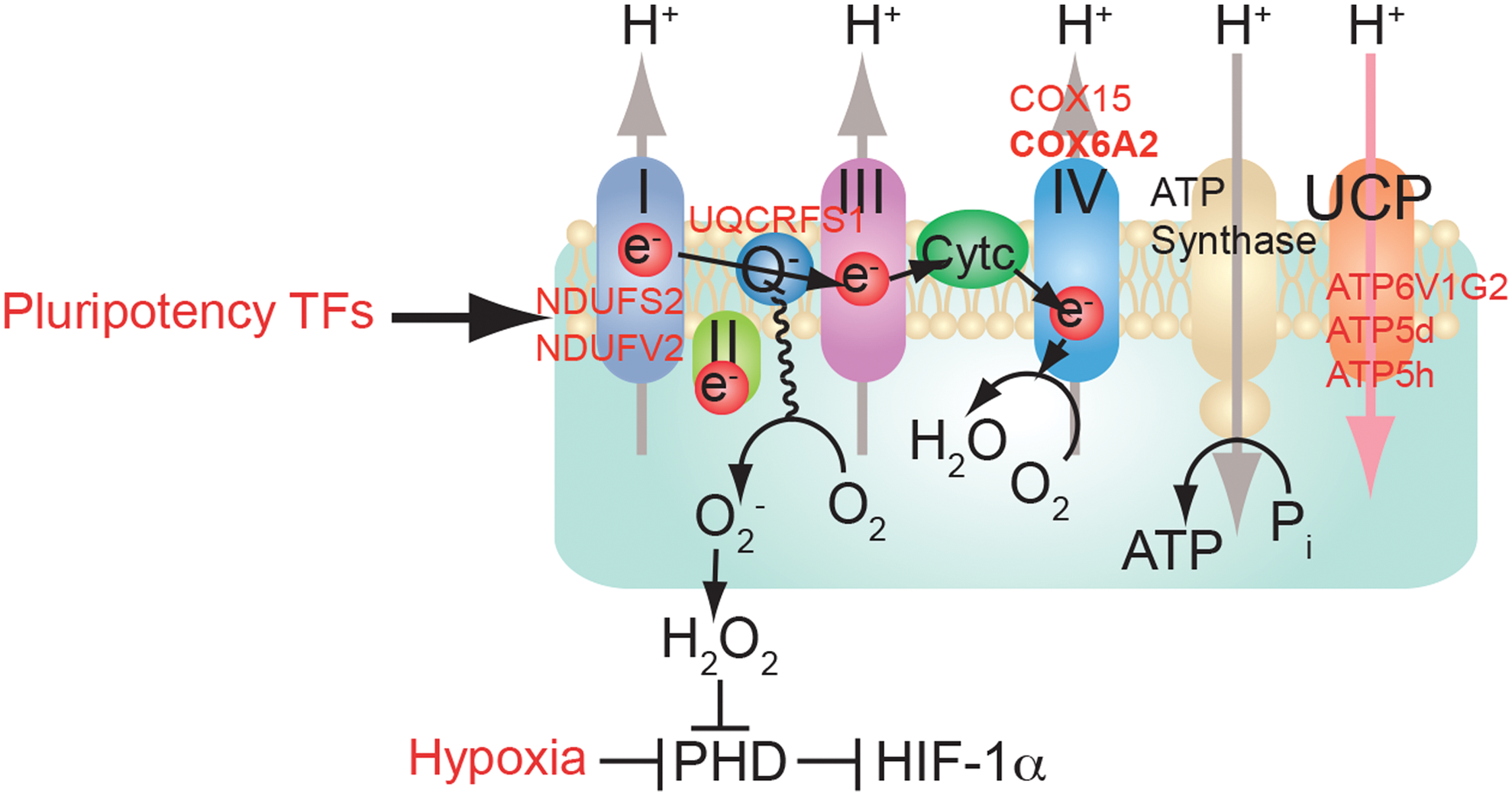
NANOG reduces mitochondrial oxidative phosphorylation (OXPHOS) to prevent mitochondrial ROS production and maintain self-renewal ability (4). Functionality of NANOG site occupancies associated with OXPHOS genes was supported by significant upregulation of genes following NANOG or MYC knockdown with short hairpin RNA (shRNA) in TICs, as determined by quantitative reverse transcription-PCR (Fig. 1) (25). Furthermore, MYC regulates glutaminolysis (63) and OCT4 regulates OXPHOS (65). Therefore, metabolic state is a key determinant for the stem cell renewal stage or differentiation stages. For example, NANOG suppression in whole liver reduces hepatocarcinogenesis. Acyl-coenzyme A dehydrogenase, very long chain (Acadvl) is overexpressed in a small fraction of tumors (8%), but its status in TICs is unknown. Based on this, the NANOG-OXPHOS pathway may play a significant role in cancer cells and not necessarily in TICs (Fig. 1).

Third, low levels of ROS in TICs are discussed. Low levels of ROS confer stemness characteristics in TICs. Regulation of ROS levels in TICs is significant since high levels of ROS deplete stemness characteristics of TICs and normal ESCs. The direct ROS by-product of the mitochondrial respiratory chain is mainly superoxide, which can then be further converted to other ROS. ATP is a key product and various forms of ROS play critical roles in tumorigenicity.
Last, five different metabolic pathways in TICs and ESCs are discussed in the Oxidative Phosphorylation, Fatty Acid Metabolism, Glutaminolysis, Glycolysis Pathways, and Hypoxia sections. Pluripotency TFs and ROS levels rewire metabolic pathways to confer on TICs a survival advantage and assure maintenance of pluripotency in TICs.
Oxidative Phosphorylation
Both p53 inactivation and β-catenin activation lead to metabolic reprogramming similar to NANOG overexpression. This suggests that NANOG synergistically reprograms cellular metabolic pathways in the presence of p53 inactivation and β-catenin activation since p53 promotes glycolysis and OXPHOS (26, 55). β-Catenin disruption impairs OXPHOS with decreased production of ATP. Ethanol induces redox imbalance with inhibition in mitochondrial function, leading to reduced ATP production, OXPHOS, and fatty acid oxidation (FAO) in hepatocyte-specific β-catenin-deficient mice. These changes lead to steatosis and oxidative damage (43).
The Warburg effect promotes glycolysis in embryonic cells and tumor cells in the presence of O2, while OXPHOS is impaired or even shut down. This was demonstrated by analyses combining results of Gas chromatography-mass spectrometry and microarray from undifferentiated and OCT4-depleted human ESCs (hESCs) (1). RNA interference (RNAi)-based OCT4 knockdown induces poised OXPHOS machinery via activation of OXPHOS genes (NDUFC1, UQCRB, and COX), increases tricarboxylic acid (TCA) cycle activity, and decreases lactate metabolism to differentiate ESCs (1). The metabolites of pluripotent stem cells (PSCs) and TICs further rewire metabolic patterns to promote self-renewal and pluripotency (1).
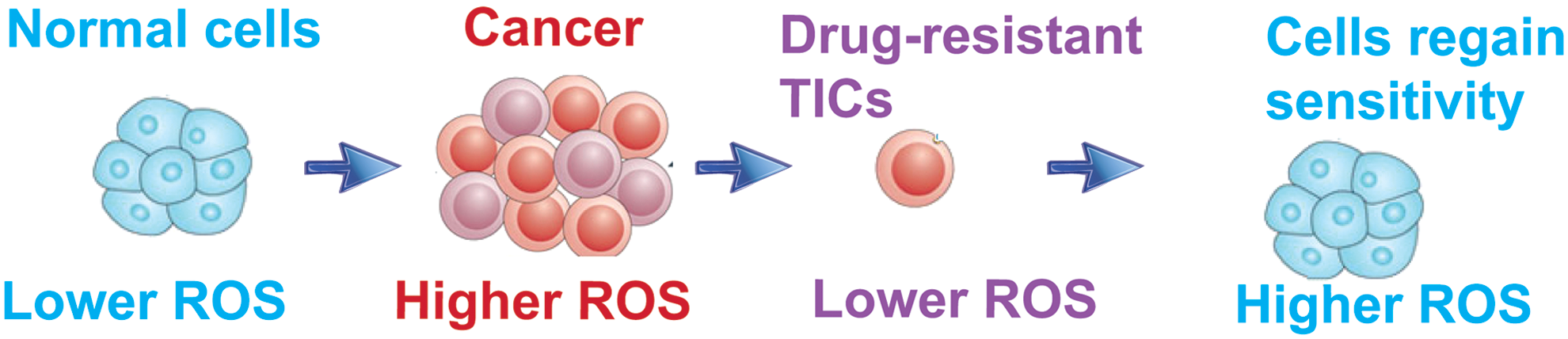
Numerous antitumor agents induce ROS production and activate ROS-dependent apoptotic pathways through a set of master regulatory genes, that is, TP53. However, long-term drug treatment promotes selection of a drug-resistant population by suppression of ROS generation in response to drug treatment, leading to genesis of TICs that are not killed by drugs; this is a hallmark phenotype of TICs (Fig. 2).
Genome-wide approaches (ChIP-seq and RNA profiling) identify the novel role of pluripotency TFs in oncogenesis
The relationship between OXPHOS and TICs
Targeting NANOG-mediated metabolic pathway(s) in TICs increases drug susceptibility of tumors in mice and achieves tumor growth suppression. By developing drug therapies that repress transcription downstream of NANOG, researchers will potentially identify therapies that could be directly applicable for treatment.
Overexpression of OXPHOS genes (e.g., Cox6a2 and Cox15 identified by ChIP-seq; Fig. 1) promotes energy metabolism and inhibits the oncogenic potential of TICs (4). Future investigation is warranted to compare the entire tumor and TIC populations for these possible differences in metabolic activity. We propose that if the NANOG-OXPHOS pathway promotes a more stem-like property of cancer cells in a nonhierarchical manner, then this would result in HCCs developing a more malignant and therapy-resistant phenotype.
This latter hypothesis has been tested by restoration of OXPHOS genes (Ndufs2, Ndufv2, Uqcrfs1, Cox15, Cox6a2, Atp6v1g2, Atp5d, Atp5h, Uqcrfs1a, and Ndufs2) in NANOG-repressed TICs. This resulted in inhibition of self-renewal and restored O2 consumption as demonstrated by serial sphere passaging (4). Thus, overexpression of OXPHOS genes (e.g., Cox6a2 and Cox15) promotes respiration, which inhibits the oncogenic potential of TICs. Pluripotency TF Nanog upregulates other pluripotency factors (e.g., Sox2, Oct4, and CD133) and oncoproteins (YAP1, IGFBP3, and TBC1D15) to generate/maintain TICs. Combined with downregulation of tumor suppressors (p53, TGF-β) this is conducive to the observed clinical properties of advanced HCCs (4).
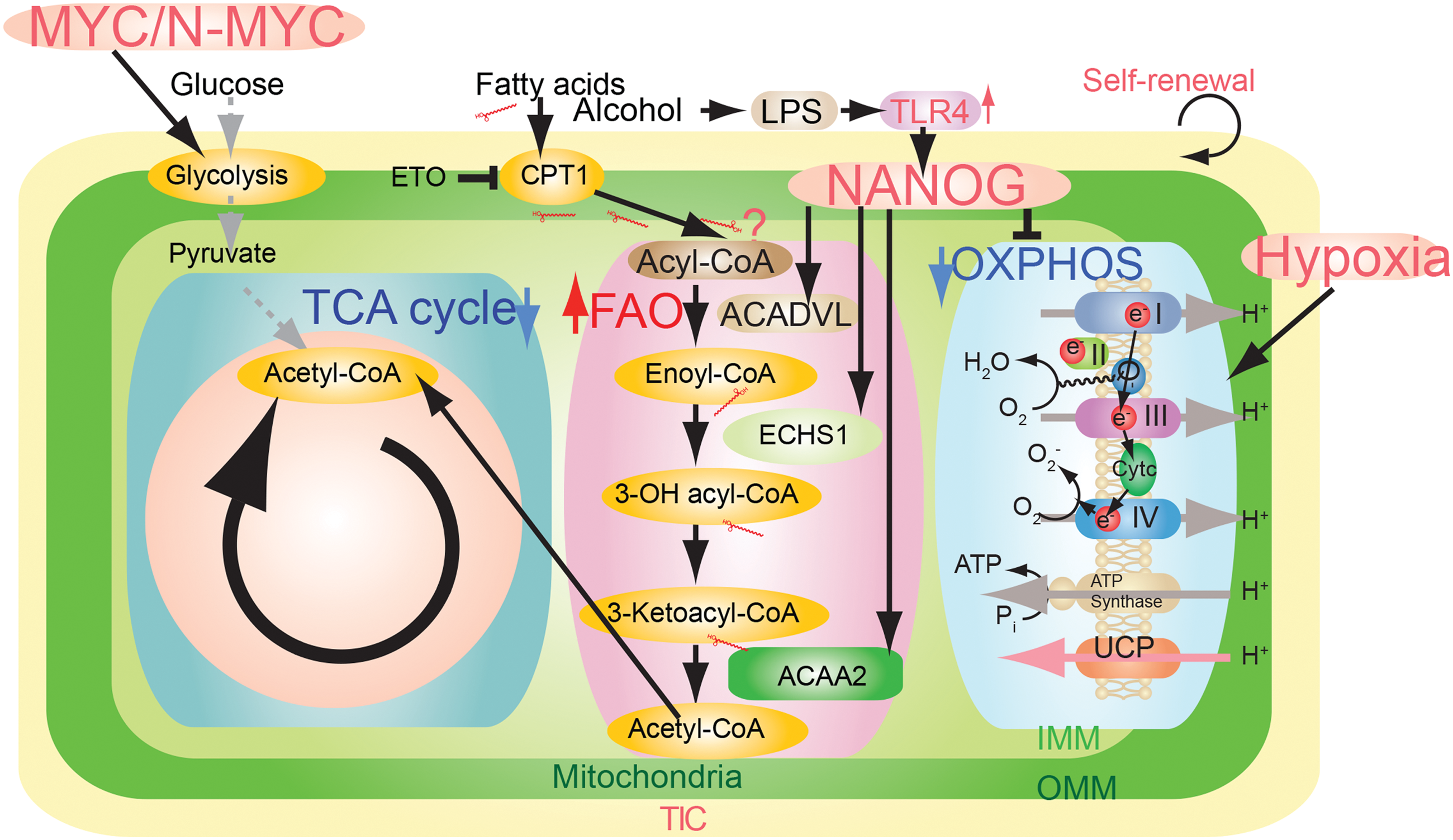
Other predisposing factors contribute to susceptibility to HCC development. Toll-like receptor (TLR) 4 signaling transactivates PSC TF Nanog. Disruption of TLR4 in immune cells reduces the inflammatory response and insulin resistance (10, 57). Furthermore, TLR4 activation and overexpression are detected in HCC sections of alcoholic hepatitis C virus (HCV) or nonalcoholic steatohepatitis (NASH) patients, supporting the clinical relevance of our concept (46). Further example of such an occurrence was observed for a key FAO pathway enzyme: acetyl-CoA acyltransferase (Acaa2), which was upregulated in mouse liver tumor specimens. These results suggest that NANOG is commonly upregulated in several different types of HCCs.
Impact of OXPHOS via inhibition of ROS on TIC self-renewal, growth, and drug resistance
To substantiate the correlation between inhibition of OXPHOS and TIC formation, we probed tumor-bearing mice and their corresponding controls for expression of OXPHOS genes (i.e., Cox6a2 and Cox15). This analysis revealed that even in a tumor-bearing mouse the OXPHOS genes were not completely silent unless the organ region consisted entirely of cancer cells (4); this indicated that TICs have less OXPHOS activity, but perhaps tumor cells express an intermediate level, between normal cells and TICs (Fig. 3). TICs express less expression of OXPHOS genes compared with tumor cells. Suppression of OXPHOS genes leads to a lower level of ROS production in mitochondria (4). In other examples, levels of PKC-θ dictate the amount of ROS production and this phenotype is asymmetrically distributed in leukemia-initiating cell (LIC)-depleted cell fractions (13).
Nanog-silenced (shRNA) TICs have higher ROS production, suggesting that NANOG suppresses production of mitochondrial ROS (Fig. 1). The mitochondrial ROS inhibits the self-renewal ability of TICs, which was demonstrated by the experiment TICs treated with the ROS inducer, Paraquat (4); this results in significantly reduced tumor spheroid formation of TICs (Fig. 1), thus indicating that induction of ROS suppresses self-renewal ability.
In vivo validation of NANOG-targeted OXPHOS gene expression was tested by overexpressing Cox6a2 in human TICs xenografted in mice. We observed that these cells became sensitized to sorafenib and exhibited inhibition of tumor growth (4). NANOG expression reduces mitochondrial oxygen consumption and ROS production, in turn protecting TICs from drug-induced cell death (4). In support of such a process, Nanog silencing restored OXPHOS, mitochondrial ROS generation, mitochondrial cytochrome c release, and apoptosis (4), thus providing a permissive state for a positive outcome from such chemotherapeutic treatment(s). As we showed, NANOG downregulated OXPHOS genes (i.e., Cox6a2 and Cox15), but upregulated FAO gene expression (i.e., Acadvl and Echs1) (4); therefore, inhibition of NANOG-dependent effects on OXPHOS and FAO genes may reduce drug resistance in NANOG+ TICs (4).
As many experiments are performed upon NANOG overexpression or silencing, future physiological analysis of TICs, including mitochondrial function or FAO activity, is warranted. Hypoxic conditions activate hypoxia-inducible factor (HIF) 1α, which promotes glycolysis and HIF-1α-dependent pyruvate dehydrogenase kinase (PDK) 2 and PDK4 activation. Together, these prevent pyruvate from entering the TCA cycle by suppressing the pyruvate dehydrogenase (PDH) complex, thus blocking mitochondrial respiration (68). Such hypoxic conditions are deemed crucial for stem cell maintenance. TICs do not rely on PDH or OXPHOS; instead TICs use the pyruvate carboxylate pathway. Therefore, under hypoxic conditions, the metabolic pattern of TICs becomes ideal for maintenance of stem cell characteristics.
Fatty Acid Metabolism
Impact of FAO via inhibition of ROS on TIC self-renewal, growth, and drug resistance
Oxygen consumption was dependent on mitochondrial respiration, which was shown by using the complex V ATP synthase inhibitor oligomycin (4). This addition reduced the oxygen consumption rate (OCR) in all cells (Fig. 3). Upon addition of the electron transport chain decoupler FCCP, maximal respiratory capacity was observed in all cells. This analysis indicated that the spare respiratory capacity was extremely low in TICs and by silencing Nanog, this respiratory capacity was restored. Addition of 2-deoxyglucose (2-DG), a glycolysis inhibitor, further increased the FCCP-induced OCR in all cells, indicating that TICs became more reliant on OXPHOS for energy metabolism when glycolysis was inhibited.
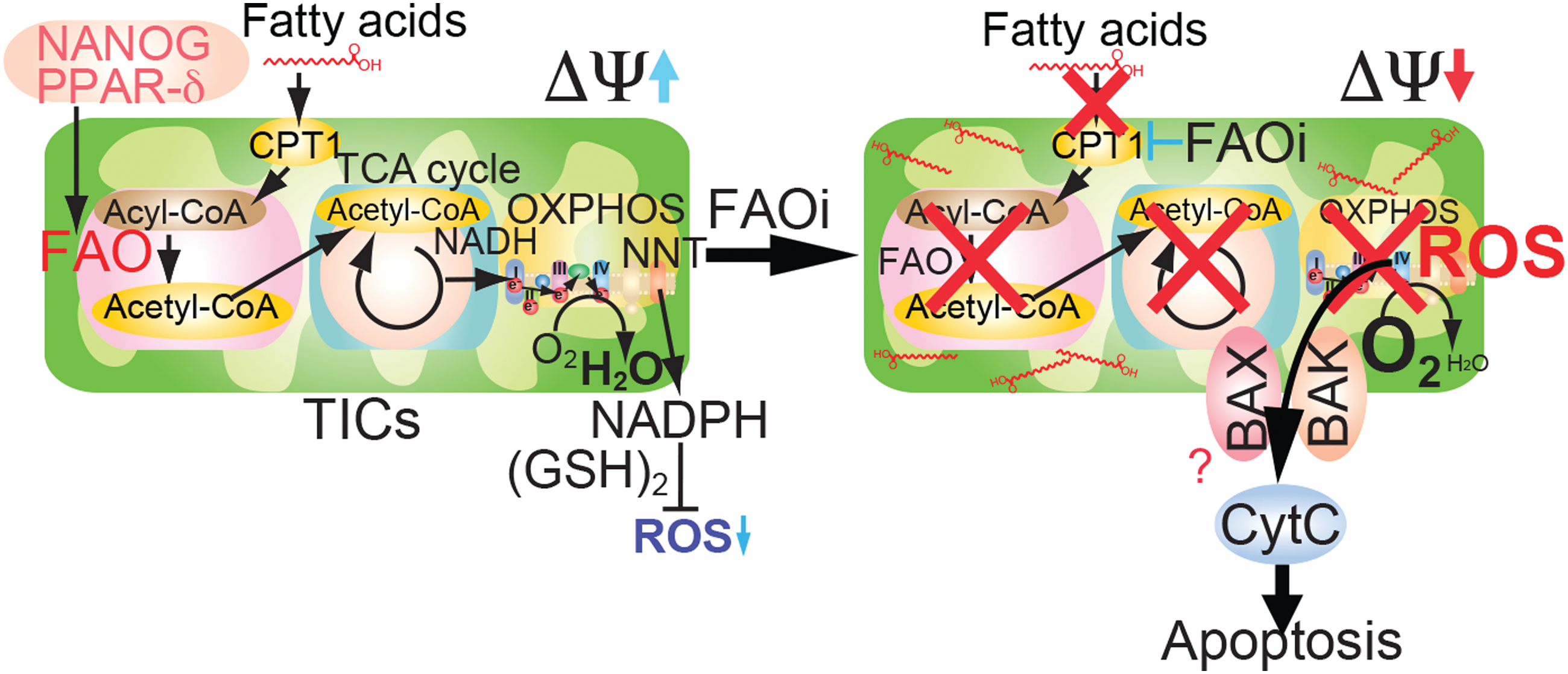
FAO inhibition (Acaa2, Acads, Acadvl, and Echs1) promotes a cytotoxic increase of lipids (58, 71). Another FAO gene repressed by NANOG is Echs1 that encodes enoyl-CoA hydratase, an enzyme that hydrates the double bond between the second and third carbons of acyl-CoA to produce acetyl CoA and energy. For maintenance and function of TICs, TICs rely on active FAO, thus inhibition of FAO could inhibit function of LICs (58). The genetic ablation of another FAO regulatory protein, liver kinase B1 (LKB1), similarly results in depletion of the stem cell pool (12, 18, 49). We experimentally reversed the effects of FAO gene silencing and restored the original TIC phenotype by overexpression of FAO genes in spheroid formation assays (4). Thus, the fate of stem cells is metabolically switched by FAO (21).
The characteristic of self-renewal ability is promoted by elevation of FAO, while de novo FA biosynthesis is inhibited in TICs. Potential mechanisms by which elevation of FAO maintains self-renewal ability include (i) shunting of long-chain FA away from lipid and cell membrane synthesis; (ii) downregulation of ROS through production of NADPH to avoid loss of TICs; and (iii) reduction of metabolic resistance to chemotherapy. By these criteria, NANOG function could be construed to serve as a gatekeeper for FAO activity (Fig. 2). Notably, to date, no role has been ascribed to NANOG with respect to FAO inhibition in TICs. It is interesting that LIN28, a stem cell reprogramming factor and regulator of microRNAs (miRNAs), was also seen as commonly upregulated in the three models of environmentally induced HCCs, along with NANOG. LIN28 may have the same end effects on rewiring cell metabolism as does NANOG in this model system of TICs.
The major by-products of the mitochondrial respiratory chain, besides ATP, are various ROS, which play a critical role in tumorigenicity (73). ROS inhibits stemness genes and self-renewal ability via activation of the p38 MAPK pathway (23), leading to polycomb suppressor protein complex I (BMI) protein degradation and FOXO3 activation (59). This pathway is subject to regulation by alcohol and HCV (69). Related with the FOX family TF, in the role of E2F1 in the activation of NANOG expression in liver cancer, FoxM1 drives expression of NANOG and other pluripotency genes and is required for TICs (34). Overexpression of BMI promotes chemoresistance (64) through changes in the cell cycle, immortalization, and intracellular glutathione (antioxidant molecule in mitochondria) levels in stem cells and TICs (72). As another example, disruption of ATM promotes ROS production and leads to stem cell depletion (22). Antagonism of NANOG in TICs enhances the efficacy of sorafenib in tumor-bearing mice and achieves ∼90% suppression of tumor growth (20). These results demonstrated that NANOG inhibition of mitochondrial respiration was related to ROS production for maintenance of self-renewal ability. Therefore, general ROS production depletes the stem cell compartment and intrinsic self-renewal ability of these cells, indicating that low ROS is of primary importance for TIC maintenance.
Role of PPARs on TIC self-renewal ability
PPAR TFs (in particular, PPARδ and PPARα) have effects on all aspects of lipid metabolism (nutrient sensing and metabolic reprogramming); however, in the development of HCCs, it is also important for differentiation, that is, a release from stemness. The novelty of this system is that lipid metabolism is involved in the transition from a normal cell to a transformed cell type to TICs. These pathways impact the activation of the mitochondrial and peroxisomal FAO transcriptional program (Fig. 4). In particular, changes occur in FA metabolism, ketogenesis, triglyceride turnover, gluconeogenesis, and bile synthesis/secretion (31). For example, PPARr agonists inhibit TIC proliferation by inhibition of NANOG and SOX2 (53). Overexpression of PPARα and PPARδ promotes differentiation through FA uptake, utilization, and catabolism, whereas inhibition of PPARα signaling increases expression of pluripotency markers by deletion of Ppard as well as inhibition of FAO. Other normal hematopoietic stem cells use fat to prevent exhaustion via PPARδ (41).
Pluripotency TFs upregulate mitochondrial FAO while also reducing respiratory enzyme levels
The metabolomic analyses are consistent with a model whereby Nanog upregulates mitochondrial FAO while also reducing respiratory enzyme levels in these organelles. As a consequence, there is an increase in AMP-activated AMPK, which mediated increased fatty acid uptake for catabolism (Fig. 5). Phosphorylation of acetyl CoA carboxylase (ACC) inhibited the activity of ACC (4), which decreased malonyl-CoA levels, leading to inhibition of CPT1. This increase of FAO would facilitate metabolism of fatty acids, as judged by metabolomic analysis, especially of long-chain unsaturated fatty acids. Inhibition of fatty acid synthase (FASN) reduces OCT4 levels (17) since inhibition of FASN regulates the AKT pathway to promote blastocyst hatching.

Adult MRL mouse is twice larger than other strains and has retained populations of cells that express the stem cell markers, Nanog, Islet-1, and Sox2 (50).
Impact of FAO on TIC self-renewal, growth, and drug resistance
When the mitochondrial membrane potential is high, single-electron reductions of molecular oxygen at complexes I and III promote superoxide production in the mitochondrial electron transport chain (37). Mitochondrial respiration occurs at the mitochondrial respiratory chain, a membrane-associated assemblage comprising multiple enzyme complexes (Fig. 6). Although we demonstrated that overexpression of a single subunit of cytochrome c oxidase (Cox6a2 or Cox15) restored mitochondrial respiration, it remains unclear how overexpression of a single subunit of multisubunit cytochrome c oxidase increased respiratory activity in the presence of active NANOG repressing other respiratory genes. However, to completely understand how overexpression of a single subunit will lead to a global increase in OCR, future in-depth studies are warranted. Some studies can support the subunit role, and to this direction, current studies in our group focus on the mechanism of how restoration of some subunits promotes ROS production and inhibits self-renewal (Fig. 7).

Since FAO-dependent NADPH production promotes survival of leukemia cells (3, 58), the concept of targeting FAO for intervention is of high therapeutic relevance (70). As we showed, Acadvl was repressed by NANOG. Acadvl −/− mice have reduced FAO activity and exhibit mitochondrial dysfunction, leading to hepatic steatosis, diacylglycerol accumulation, and hepatic insulin resistance (2, 40, 77, 79). The mitochondrial proteins, BCL2 and truncated-Bid (tBID), inhibit FAO via fatty acid transporter CPT1 (14, 51). Inhibition of FAO facilitates oligomerization of proapoptotic mitochondrial BCL2 family member molecules, BAK and BAX, leading to apoptotic cell death (58).
Glutaminolysis
The glutaminolysis pathway supplies alternative TCA cycle substrate and produces reducing agents (Fig. 8), including glutathione and NADPH (7, 42), and provides a carbon source that promotes cells to use TCA cycle intermediates and glucose-derived carbon as biosynthetic precursors (7). The WNT-LGR5 (eucine-rich repeat-containing G-protein-coupled receptor 5) pathway or MYC also regulates the glutaminolysis pathway (67).
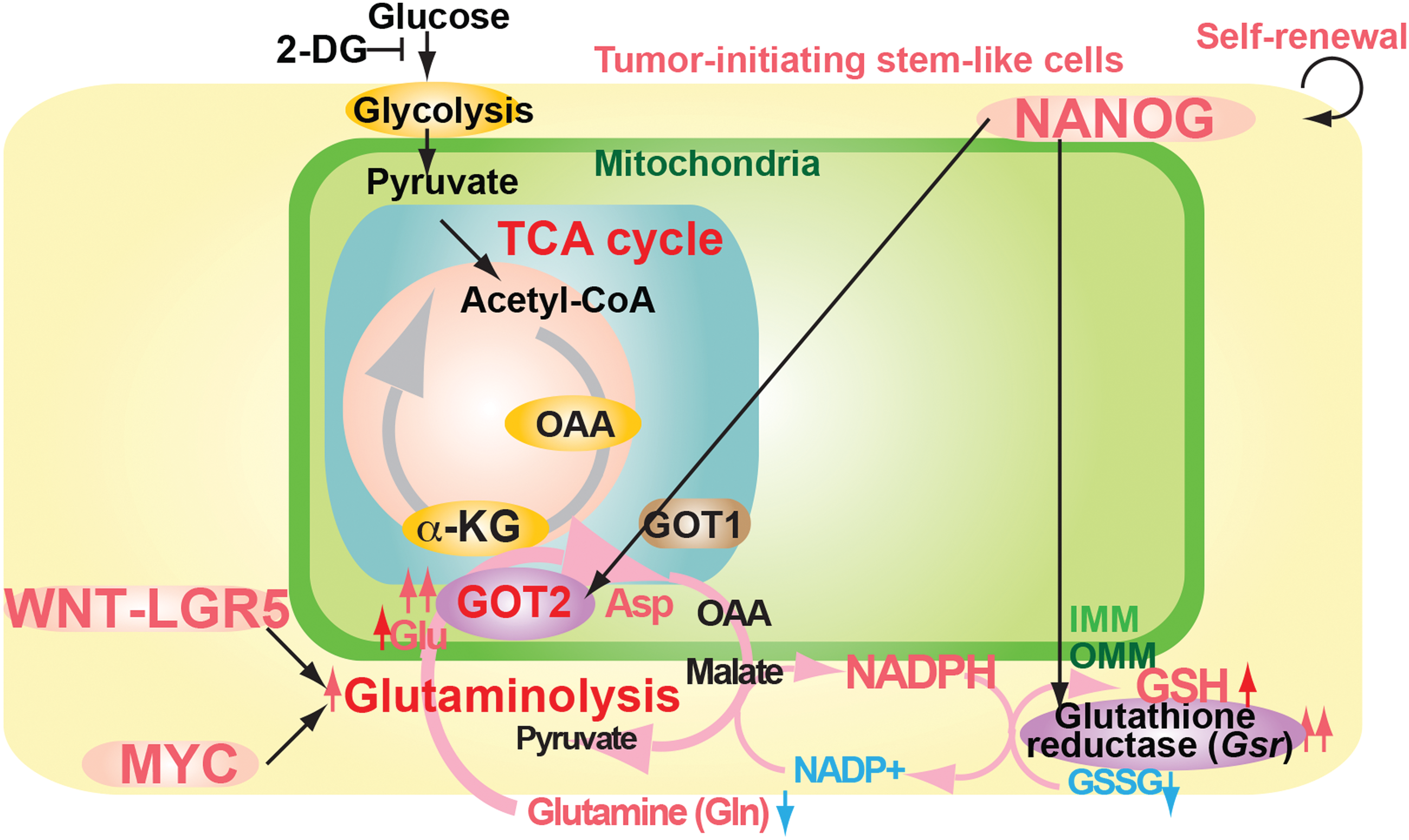
Impact of glutaminolysis on TIC self-renewal, growth, and drug resistance
As TICs have low mitochondrial OXPHOS activity and high glutaminolysis activity, ROS levels may be reduced. Extracellular acidification (dependent on pH) restructures mitochondria to make elongated mitochondria in postmitotic cells (32). Therefore, a single subunit of the electron transfer chain may reorganize the mitochondrial structure or the microenvironment such as hypoxia may promote reorganization of mitochondria.
Besides the OXPHOS pathway, restoration of COX6A2 may also regulate other functions, such as the uncoupling protein response, mitofusion, or restructure of mitochondria since cells from mice deficient in Cox6a2 have constitutive activation of AMPK, and enhanced messenger RNA (mRNA) expression of the mitochondrial inner membrane protein UCP2 (uncoupling protein 2) (33, 56). UCP2 limits free cellular ROS levels, leading to the resting metabolic rate not in adaptive thermogenesis/physiological uncoupling, while UCP1 has thermogenic/uncoupling activities. Disruption of Ucp2 in mice stimulates production of ROS (8). Ectopic UCP2 expression inhibits differentiation of human pluripotent stem cells (hPSCs) (78). Interestingly, UCP2 prevents mitochondrial glucose oxidation and facilitating glycolysis and regulates hPSC energy metabolism via substrate shunting (78). Interestingly, mitochondrial size and perinuclear mitochondria are increased in Cox6a2 −/− muscle cells (56), which are associated with increased expression of a key regulator of mitochondrial biogenesis (peroxisome proliferator-activated receptor γ coactivator, PGC-1α) (76) and regulators of mitochondrial fusion and size (mitofusin-1 [Mfn1] and optic atrophy 1 [Opa1]) (56). In contrast, the mitochondria in hESCs are fragmented or punctate in comparison with those of the well-developed filamentous network of normal cells (78). Future studies are needed to fully understand these phenotypes.
Since Nanog belongs to the molecules that regulate stem cell maintenance and self-renewal capacity, potential side effects of a therapeutic manipulation of Nanog on physiological regenerative capacity need to be further investigated.
Glycolysis Pathways
The glycolysis pathway (Warburg effect) promotes metabolic reprogramming in mitochondria of cancer cells. Dr. Warburg discovered in the 1920s that ascite cancer cells and tumor tissues take up glucose to produce lactate in both normoxia and hypoxic conditions (35). The Warburg effect is a reprogrammed metabolic pathway that depends on aerobic glycolysis in cancer (6, 45). The increase in glycolytic flux promotes glycolytic intermediates to supply subsidiary pathways to fulfill the metabolic demands of proliferating cells (45).
MYC induces many anabolic growth-related genes, including transporters and mitochondrial metabolic enzymes, to regulate glycolysis, fatty acid synthesis, glutaminolysis, and serine metabolism (66). Oncogenes, including Kras, co-opt the physiological functions of PI3K and MYC pathways to promote tumorigenicity. Glycolytic metabolism promotes self-renewal ability in hPSCs via MYC/N-MYC (16, 29).
Loss of p53 increases glycolytic flux to promote anabolism and redox balance, two key processes that promote tumorigenesis (38).
To generate induced pluripotent stem cells (iPSCs), transduction of Oct4, Sox2, and Klf4 (OSK) activates glycolysis via induction of Zic3 and Esrrb to reprogram murine fibroblasts in a manner independent of HIFs (65). In contrast to the suppressive state of OXPHOS in TICs, OXPHOS activity is required for efficient reprogramming. Zic3 represses OXPHOS, whereas Esrrb activates OXPHOS (65). Esrrb restores OXPHOS activity to convert primed PSCs (with Zic3 expression) into the naive state (65). The combinatorial function of TFs coordinates metabolic pathways to induce naive PSCs (65). TICs retain the suppressed state of OXPHOS, while ESCs retain active OXPHOS pathways. Metabolic rewiring may lead to a difference between ESCs and TICs in a manner dependent on the microenvironment and tissue context.
Tumor has a dilemma to balance between glycolysis and OXPHOS (27). Reduction of OXPHOS capacity in different types of cancers contradicted the upregulated state of OXPHOS with dependency of cancer cells on oxidative energy substrates for anabolism (27). Tumor size, hypoxia, and oncogene (i.e., RAS and c-MYC) activation may explain contradictory reports (27). The p53, c-MYC, OCT, and RAS control mitochondrial respiration and glutamine utilization (27). c-MYC and Oct regulate mitochondrial biogenesis and generate ROS to generate iPSCs from oncogene activation (27). Sequential oncogene (i.e., C-MYC, HIF, and RAS) activation reprograms metabolism (27). The cancer cell requires energy in nutrient-depleted conditions and adapts to a metabolic steady state. Therefore, suppression of one oncogene can unbalance this bioenergetic program to impair energy supply and repress the tumor (27).
Hypoxia
Rapid cell division and aberrant blood vessel formation in tumors deprive oxygen (hypoxia) and induce the HIFs (30). Many tumors reside in a low-oxygen environment (hypoxia) ranging from 0% to 2% O2 because the tumor cell proliferation rate exceeds the rate of new blood vessel formation (angiogenesis) (6, 24). HIF-1 coordinates the metabolic adaptation to hypoxia through induction of glycolytic flux genes (62). Various mechanisms display constitutive activation of HIF-1 in some tumors under normoxic conditions, including (i) hyperactivation of mTORC1, (ii) loss of von Hippel–Lindau, (iii) accumulation of ROS, and (iv) accumulation of the TCA cycle metabolites, succinate or fumarate, due to cancer-specific mutations in succinate dehydrogenase (SDH) or fumarate hydratase (FH), respectively (6, 28).
Glycolysis is a physiological response to hypoxia in normal tissues, and ∼4% O2 favors the molecular mechanisms required for maintenance of pluripotency (75). Hypoxia induces pluripotency in primordial germ cells by HIF-1α stabilization and Oct4 deregulation (44). Furthermore, hypoxia-inducing factors promote stemness properties and altered metabolism of cancer- and metastasis-initiating cells (48). HIFs promote tumor progression by altering cellular metabolism (glycolytic enzymes [PGK, ALDA] and glucose transporters [GLUT1]) and stimulating angiogenesis via induction of vascular endothelial growth factor (VEGF) (30, 61). Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor–stroma interplay (11). Cancer cells allocate Warburg metabolism to their corrupted cancer-associated fibroblasts (CAFs), exploiting their by-products to grow in a low-glucose environment, symbiotically adapting with stromal cells to glucose availability (11).
The epithelial–mesenchymal transition enhances cancer invasiveness and confers tumor cells with TIC characteristics. The Snail-G9a-Dnmt1 complex silences the E-cadherin promoter and methylates the promoter of glycolysis enzyme fructose-1,6-bisphosphatase 1 (FBP1) (9). Under the hypoxic condition, loss of FBP1 induces glycolysis, glucose uptake, macromolecule biosynthesis, formation of tetrameric PKM2, and maintenance of ATP production (9). Loss of FBP1 also suppresses mitochondrial complex I activity to inhibit oxygen consumption and ROS production (9); this metabolic reprogramming results in increased stemness and tumorigenicity by enhancing the interaction of β-catenin with T cell factor (9).
Induction of HIF-1α switches from bivalent to glycolytic metabolism during ESC to EpiSC transition (80). Expression of HIF-1α drives ESCs to a glycolytic Activin/Nodal-dependent EpiSC-like stage (80). HIF-1α also shifts to the glycolytic state and upregulates PDK1–3 and PKM2 to reprogram cell fate (54) as remodeling of iPSCs transits from oxidative to glycolytic metabolism, mitochondria, and bioenergetics. Indeed, the ablation of HIF-1α function in matured fibroblasts inhibits reprogramming efficiency (54). Transduction of the four factors upregulates the HIF-1α target PDK and shift to glycolytic state (54). Induction of HIF-1α targets activates the glycolytic program to derive iPSCs (54).
HIFs also activate Notch and Oct4 (30). Notch activation blocks differentiation in hematopoietic bone marrow or T cell progenitor cells and results in T cell acute lymphoblastic leukemia (52). Prolonged Notch activation underlies the developmental defects observed in HIF−/− embryos (19) and in Hif1a −/− adult cells and tissues (60). HIF-1α activates Notch signaling by transactivating c-Myc (30, 74). Disrupted epigenetic silencing derepresses Oct4 in a transformed cell to activate HIF-2α (5). HIF-2α activates c-Myc and Oct4 that cooperatively promote proliferation to transactivate a self-renewal gene (15). Relative expression levels of HIF-1α and HIF-2α regulate c-Myc transcriptional activity (15, 36). HIF expression is correlated with NANOG and OCT4 expression in primary tumors (47). Oct4 dedifferentiates tumors. Dedifferentiation promotes the TIC phenotype (39). Oct4 induces Nanog and Klf4 to dedifferentiate matured cells.
In summary, pluripotency TFs regulate metabolism, including FAO, OXPHOS, glutaminolysis, glycolysis, and hypoxia, while also reducing respiratory enzyme levels in mitochondria to sustain self-renewal and oncogenic activities (Fig. 6).
Footnotes
Acknowledgments
The author appreciates the help of Prof. Stanley M. Tahara at USC for critical reading of the manuscript. This project was supported by National Institutes of Health (NIH) research grants R01AA018857, P50AA011999 (Southern California Research Center for ALPD and Cirrhosis, pilot project, program, animal core, morphology core), Lee-Summer-Project funding, P30DK048522 (USC Research Center for Liver Diseases, pilot project program), Non-Parenchymal Liver Cell Core (R24AA012885), and UO-021898. This research was also supported by a Research Scholar Grant (RSG-12-177-01-MPC); pilot funding from American Cancer Society (IRG-58-007-48); and the Cell and Tissue Imaging Core—USC Research Center for Liver Diseases (P30 DK048522). The project described was supported, in part, by award number P30CA014089 from the National Cancer Institute. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Cancer Institute or the NIH.