
Abstract
Significance:
Type 1 diabetes (T1D) is an autoimmune disease resulting in β-cell destruction mediated by islet-infiltrating leukocytes. The role of oxidative stress in human and murine models of T1D is highly significant as these noxious molecules contribute to diabetic complications and β-cell lysis, but their direct impact on dysregulated autoimmune responses is highly understudied. Pro-inflammatory macrophages play a vital role in the initiation and effector phases of T1D by producing free radicals and pro-inflammatory cytokines to facilitate β-cell destruction and to present antigen to autoreactive T cells.
Recent Advances:
Redox modulation of macrophage functions may play critical roles in autoimmunity. These include enhancing pro-inflammatory innate immune signaling pathways in response to environmental triggers, enforcing an M1 macrophage differentiation program, controlling antigen processing, and altering peptide recognition by oxidative post-translational modification. Therefore, an oxidative environment may act on multiple macrophage functions to orchestrate T1D pathogenesis.
Critical Issues:
Mechanisms involved in the initiation of T1D remain unclear, making preventive and early therapeutics difficult to develop. Although many of these advances in the redox regulation of macrophages are in their infancy, they provide insight into how oxidative stress aids in the precipitating event of autoimmune activation.
Future Directions:
Future studies should be aimed at mechanistically determining which redox-regulated macrophage functions are pertinent in T1D pathogenesis, as well as at investigating potential targetable therapeutics to halt and/or dampen innate immune activation in T1D.
Introduction
Type 1 diabetes
T
Interrogation of the immune system in these mice has shown that the genetics of the NOD mouse confers alterations in innate immune and macrophage responses that impact diabetes progression (183, 184). In particular, proper development of mature macrophages from hematopoietic stem cells in the bone marrow through colony-stimulating factor stimulation was shown to be defective (183), leading to reduced antigen-presentation capacity for inducing regulatory cluster of differentiation (CD)4 T cell responses (184). In another rodent model, the BioBreeding (BB) rat, a specific subset of the strain (diabetes-prone BB [DP-BB]), is susceptible to spontaneous and aggressive T1D onset with the average onset around 12 weeks of age and an overall incidence of ∼85% in both genders (60). Importantly, disease susceptibility in the DP-BB rat is polygenic in nature and similar to humans and the NOD mouse, as many of the disease susceptibility loci involve alterations in immune responses (28, 210).
Although genetic susceptibility is a requirement, it is not completely sufficient for disease onset to occur. This is most evident when studying maternal twins with genetic susceptibility for T1D, as the concordance rate within a 40-year span is only ∼40%, compared with the expected 100% if genetics were fully sufficient for disease to occur (162). In addition, the worldwide incidence of T1D is steadily increasing at a rate that cannot be accounted for by the natural genetic drift of a population. Therefore, it is clear that some environmental factor is also involved in either initiating the autoimmune response or accelerating the process. Rodent models have helped interrogate this interaction as both the NOD mouse and the BB rat can accelerate the onset of diabetes on certain environmental triggers. In particular, we will focus on viral infections and how in the context of genetic perturbations of the immune response, this type of insult can break peripheral tolerance and result in autoimmunity.
As with many chronic inflammatory diseases, oxidative stress is a key component of T1D pathogenesis and complications. Analysis of serum from human T1D patients revealed increased levels of advanced oxidation protein products (AOPP) and peroxidation potential, indicating oxidative stress within these individuals above healthy controls (57). AOPP, the result of plasma proteins to chlorinated oxidants, were initially described as a serum biomarker for oxidative stress and also tightly associated with increased circulating neopterin, a marker of proinflammatory monocyte activation in chronic uremia (213, 214). The study on patients with T1D did not address levels of neopterin, whereas upregulation of circulating antioxidants such as glutathione (GSH) was decreased. Thus, even though other antioxidants such as catalase (CAT) and superoxide dismutase (SOD) were actually increased, the ratio of oxidants to antioxidants was imbalanced, providing evidence that patients with T1D exhibit hallmarks of oxidative stress.
The pancreatic β-cell, the target tissue in autoimmune diabetes, is highly susceptible to free radical-mediated damage (59, 141). A recent proteomics study on NOD mouse-derived NOD insulinoma β-cell line (NIT-1) showed that in response to the cytotoxic cytokines, interferon (IFN)-γ, IL-1β, and tumor necrosis factor (TNF)-α, NIT-1 β-cells failed to upregulate the antioxidants, SOD2 and peroxiredoxin 3, in contrast to cytokine-treated αTC1 α-cells (59). Coinciding with these results, another study comparing basal gene expression profiles of murine pancreatic islets with other murine tissues, including kidney, liver, and lung, revealed dampened expression of transcripts for SOD1/2, glutathione peroxidase (GPX), and CAT (104). This decreased defense against oxidative stress in β-cells has direct consequences in T1D, as alloxan-resistant (ALR) mice, which share common ancestry to the NOD strain, were shown to be resistant against both T cell-mediated and alloxan-induced T1D due to elevated levels and activity of pancreatic SOD1, glutathione reductase, and GPX (119). In addition, islets from ALR mice were also resistant to cytokine cytotoxicity due to their heightened capacity to resolve nitric oxide (NO)-induced cell stress (121).
However, little is known regarding the role of free radicals and oxidative stress in propagating autoimmune dysregulation in T1D. Although not simply involved in the immunopathogenesis of pancreatic β-cells, there is mounting evidence in mouse studies that nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase 2 (NOX2)-derived superoxide produced by immune cells can also influence autoimmune responses. This was suggested by studies using a point mutation in the neutrophil cytosolic factor 1 gene (Ncf1), encoding the p47 phox subunit of NOX2, to completely ablate its function (76). This mutation (Ncf1 m1J ) disables proper expression of p47 phox , which is a crucial subunit for directing the formation of the active NOX2 complex. Without an active NOX2 complex, NOD.Ncf1 m1J mice were significantly protected against the development of spontaneous autoimmune diabetes (196, 202). In addition, deficiency in active NOX2 successfully protected NOD.Ncf1 m1J mice against an aggressive adoptive transfer model of T1D with diabetogenic CD4 T cells (196). As NOX2 expression is the most highly expressed on immune cells, namely macrophages and neutrophils, these results highlight the importance of immune-derived free radicals in driving the pathogenesis of T1D.
Evidence for oxidative stress in promoting autoimmune diabetes has also been suggested in the DP-BB rat, as tissue gene expression profiles revealed an islet-specific reduction in the expression of antioxidants, such as glutathione-S-transferases, SOD, peroxidases, and peroxiredoxins (Prx), when compared between diabetes-resistant (DR)-BB and non-autoimmune-prone Fischer rats (10). Interestingly, treatment of DP-BB rats with the antioxidant, N-acetyl cysteine, was able to significantly delay autoimmune diabetes.
Though beyond the scope of this review article, the inflammatory consequences of oxidative stress within the islet environment are not only relevant in T1D pathogenesis but also of high interest in the development of type 2 diabetes (T2D) and have been extensively reviewed (91, 128, 161). Elevations in glucose and lipids can have detrimental effects on pancreatic β-cell insulin secretion and are correlated with peripheral insulin resistance in T2D (134), but chronic hyperglycemic conditions can elicit enhanced metabolism of β-cells and hyperinsulinemia that will induce oxidative stress mediating β-cell dysfunction and death (135). T2D is also recognized as an inflammatory-mediated disease within the islet microenvironment and during the progression of T2D, islet infiltration of macrophages occurs in both patients with T2D and animal models of T2D (47). Inflammatory mediators that contribute to β-cell decline in T2D include the accumulation of amyloid in the islets that can activate the inflammasome in macrophages to induce the secretion of IL-1β (118) and palmitate, a saturated fatty acid, that can activate the synthesis of free radicals and pro-inflammatory cytokines in islet-resident macrophages to induce β-cell destruction (46). In addition, both T1D and T2D come with long-term consequences of chronic inflammation and oxidative stress due to inconsistent control of blood glucose levels leading to retinopathies, neuropathies, and nephropathies. Therefore, the development of antioxidant-based therapeutics for individuals already with diabetes mellitus may help prevent or relieve the severity of these long-term consequences. However, more research is needed to understand how to target the redox-regulated immune responses in the development of autoimmunity for the potential of preventative therapeutics in T1D-susceptible individuals.
Our review article will describe the importance of free radicals and free radical-mediated signaling in innate immune responses in autoimmune diabetes. T1D is an autoimmune disease exhibiting chronic oxidative stress since islet-infiltrating innate immune cells generate copious amounts of free radicals to elicit pancreatic β-cell damage and unfortunately, low antioxidant defenses by β-cells result in their increased sensitivity to the noxious effects of free radicals. Activation of innate immune cells is pivotal in initiating pancreatic β-cell destruction since they can directly damage insulin-secreting β-cells in the islets of Langerhans, but equally as important, they function to efficiently activate T cells that are the final effector cells in β-cell lysis.
In this first section, we will introduce macrophages as a critical innate immune cell type involved in the progression of T1D. In addition, macrophages are of the highest expressing immune cells for NOX2, a major source of superoxide and its derivatives, during the immune response. In subsequent sections, we will provide a comprehensive overview of the redox-dependent innate immune mechanisms that macrophages utilize to heavily influence T1D pathogenesis, including the activation of redox-dependent signaling pathways, chemokine signaling, antigen presentation, anti-viral responses, and the synthesis of pro-inflammatory effector molecules involved in β-cell destruction. Ultimately, we hope that the reader will develop an appreciation of the role of redox-dependent signaling pathways involved in innate immune responses in T1D and that the development of NOX inhibitors may be efficacious in delaying disease progression.
Macrophages
Macrophages are innate immune cells with many functions, including maintaining tissue homeostasis, phagocytosis of dead or dying cells, mounting a first-line defense against pathogens, activation of the adaptive immune response, and aiding in wound healing after insult. With their multifaceted roles, macrophages respond to their environmental cues by polarizing to specific macrophage subsets. Macrophage polarization was initially defined into two broad subsets: classically activated M1, which elicits the classic inflammatory response, and the alternatively activated M2 macrophages, generally encompassing non-inflammatory macrophage responses. As the field has expanded, the M2 subset has been further divided into Th2-driving (M2a), tolerogenic (M2b), tissue repair (M2c), and deactivated (M2d) phenotypes [as described in detail in the following reviews (52, 113, 117, 131, 187)]. However, in recent years, with the concept of in vivo macrophage plasticity to fit an ever-changing microenvironment, many experts in the field have turned to identifying these different phenotypes based on the combination of environmental signals received by the macrophage (131).
The classically activated M1 macrophage is polarized on interacting with an inflammatory environment, such as sensing IFN-γ, and detection of pathogen-associated molecular patterns, including lipopolysaccharide (LPS, found on gram-negative bacteria), viral RNA/DNA, and various fungal cell wall components. Along with the initial activation of inflammatory nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling through Toll-like receptor (TLR) ligation, these macrophages will fully polarize toward an M1 phenotype through the activation of the signal transducer and activator of transcription (STAT)1 transcription factor via IFN signaling (131). These events will induce an inflammatory response consisting of free radicals, cytokine (TNF-α, IL-1β, IL-12) and chemokine (CXCL10, CCL5) synthesis to combat the perceived pathogen. Following the suggested nomenclature based on environmental cues, the differentiation of non-inflammatory “M2” macrophages listed earlier could be described as M(IL-4), M(IL-10), M(transforming growth factor [TGF]-β), or M(IL-6 + adenosine) phenotypes for M2-a, -b, -c, and–d, respectively (131, 168). Importantly, other signaling cues have also been shown to influence non-inflammatory macrophage responses, including certain immune complexes and glucocorticoids (117). As these immune cells are heavily involved in the development and maintenance of nearly every organ and tissue, in addition to their role in microbial defense, it is likely that many other subtle phenotypes will be described in the future, painting a network of cues taken from the milieu that shapes the fine-tuned macrophage response.
The macrophage is a crucial immune cell in driving pathogenesis of T1D, with multiple roles involving genetic predisposition (182) and the consequences of environmental triggers. As shown in Figure 1, the redox regulation of macrophage responses touches each of these roles for macrophages in T1D pathogenesis. Thus, in this comprehensive review, we will focus on how free radical-mediated macrophage responses lead to diabetogenic consequences in T1D.
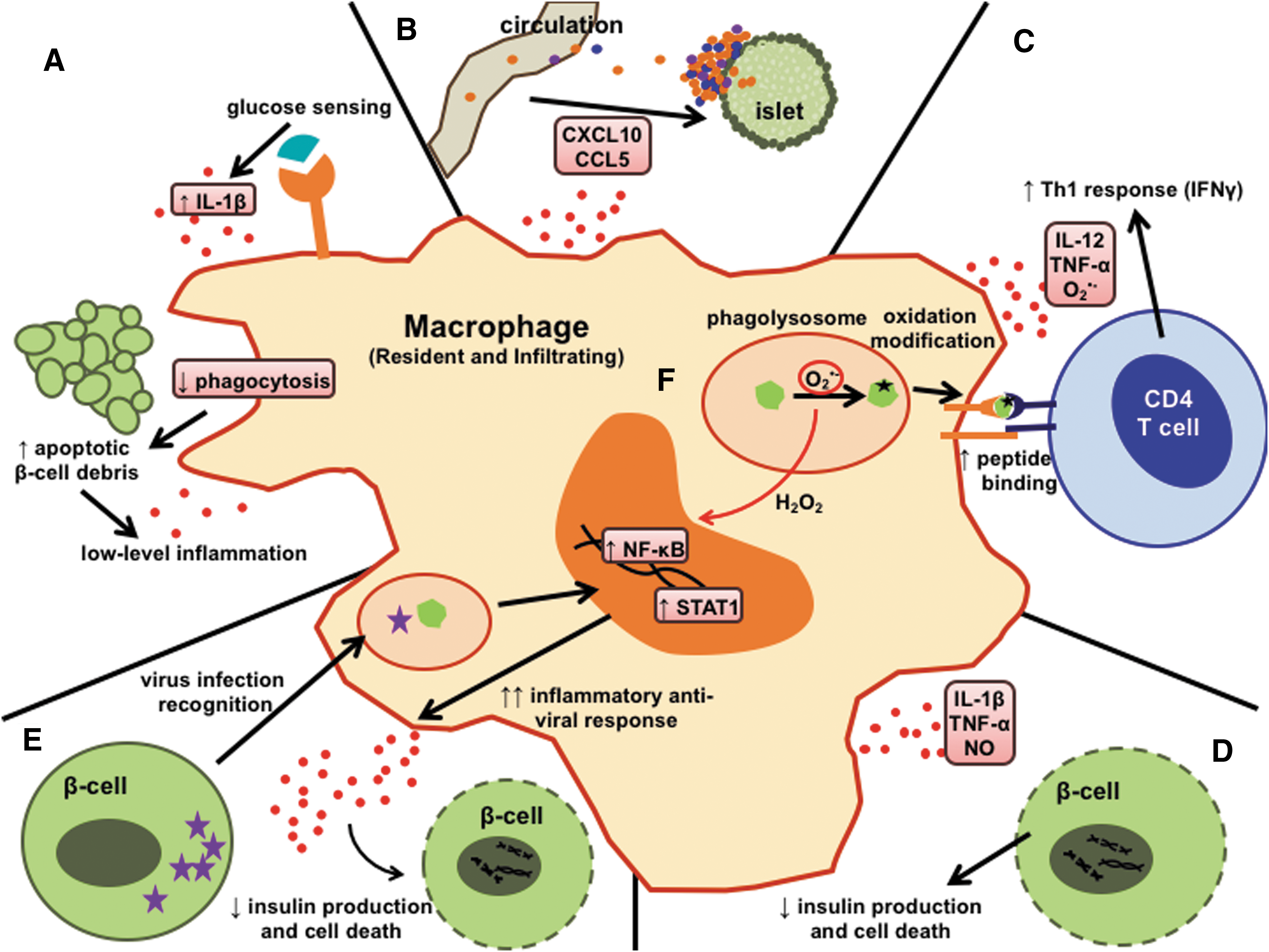
The Multiple Faces of Macrophages in T1D Pathogenesis
A critical innate immune cell type in T1D
Understanding how a break in immunological tolerance against the β-cell occurs is a major focus in defining the immunopathogenesis of T1D, and studies demonstrate that macrophages play a prominent role in this process (16, 86, 101). Much of the advancement in our understanding of T1D pathogenesis comes from studying the NOD mouse, a strain found to have complex polygenic susceptibility that leads to spontaneous development of T1D, in a manner similar to human patients (197). The selective depletion of macrophages from NOD mice, by treatment with silica beads, liposomal dichloromethylene diphosphonate (lip-Cl2MDP), or antibodies, dramatically prevents T1D onset (16, 87, 101, 196). Further investigations into the role of macrophages in driving autoimmunity show that macrophages are one of the first inflammatory immune cells to infiltrate the islet (36), are required for the activation of cytotoxic CD8 T cells that target the β-cell (86), and are major sources of noxious factors for the β-cell, such as pro-inflammatory cytokines (IL-12p70, TNF-α, IL-1β), chemokines (CXCL10, CCL2, CCL5), and free radicals.
Evidence for the multifaceted roles of macrophages in T1D is not restricted to the NOD mouse model and, in fact, has been demonstrated in the spontaneous T1D onset of DP-BB rats. Similar to the NOD mouse, macrophage-depleting silica treatment of DP-BB rats almost completely abrogates onset of spontaneous T1D (142). In addition, tracking the immune infiltration throughout disease progression revealed that macrophages are the initial infiltrating immune cells into the islets of DP-BB rats (65, 102, 209), and that macrophages can also play an effector role during the autoimmune attack (95, 99, 132). In addition to murine models of autoimmune diabetes, histological analyses of pancreata sections from patients with T1D have also demonstrated an influx in macrophage recruitment into the islets (66, 79, 82, 165). These observations further highlight the importance of innate immune cells in T1D and pancreatic β-cell destruction. In these next sections, we will discuss the evidence for redox regulation at each of these macrophage-mediated steps in T1D pathogenesis.
Redox-mediated exacerbation of molecular mechanisms involved in inflammation
In the NOD mouse, Sen et al. reported an intrinsically heightened NF-κB signaling response in macrophages (181). Stimulation of NOD macrophages with LPS, anti-CD40, or TNF-α dramatically enhanced nuclear translocation of NF-κB p50/p65 heterodimer transcription factor complexes and induction of NF-κB-dependent signaling compared with macrophages from non-autoimmune-prone non-obese resistant (NOR) and BALB/c mice (181). Interestingly, this enhanced NF-κB activation did not correspond with any increased capacity of NOD macrophages to activate diabetogenic CD4 T cells over their NOR counterparts, suggesting that this hyper-inflammatory response may be an intrinsic source of the potent macrophage effector function in β-cell-directed cytotoxicity.
A similar exacerbated inflammatory response also occurs in the DP-BB rat, as peritoneal macrophages isolated from DP-BB rats responded to LPS and IFN-γ with significantly greater amounts of TNF-α production as compared with T1D-resistant DR-BB and Wistar rat strains (169). This heightened inflammatory response by macrophages likely coincides with creating an oxidative environment that can be damaging to the β-cell, as DP-BB macrophages have an inherently enhanced capacity to produce excessive amounts of NO on activation (99, 215).
In human patients with T1D, monocytes have been shown to produce significantly more inflammatory IL-1β and IL-6 cytokines, as well as heightened superoxide production, compared with healthy controls (14, 40, 127), suggesting an intrinsic hyper-inflammatory response within human T1D patients, similar to what has been observed in rodent models. Altogether, the sensitivity of macrophages under the genetics of T1D susceptibility to become excessively inflammatory is a likely cause for initiating a chronic inflammatory state throughout T1D pathogenesis, ultimately driving autoimmune activation.
One T1D genetic susceptibility loci that may contribute to enhanced NF-κB activation in macrophages is the SUMO4 (M55V) allele that functions as a negative regulator of NF-κB-dependent signaling [Fig. 2A, and (62, 147)]. Small ubiquitin-related modifier (SUMO) is a group of proteins that participates in post-translational modifications (PTM) by covalent attachment to lysine residues in target proteins, including NF-κB inhibitor alpha (IκBα). Sumoylation of IκBα will prevent degradation and, subsequently, inhibit NF-κB nuclear translocation and transcriptional activation. Patients who have the M55V substitution display a decrease in sumoylation and a concomitant increase in NF-κB transcriptional activation.
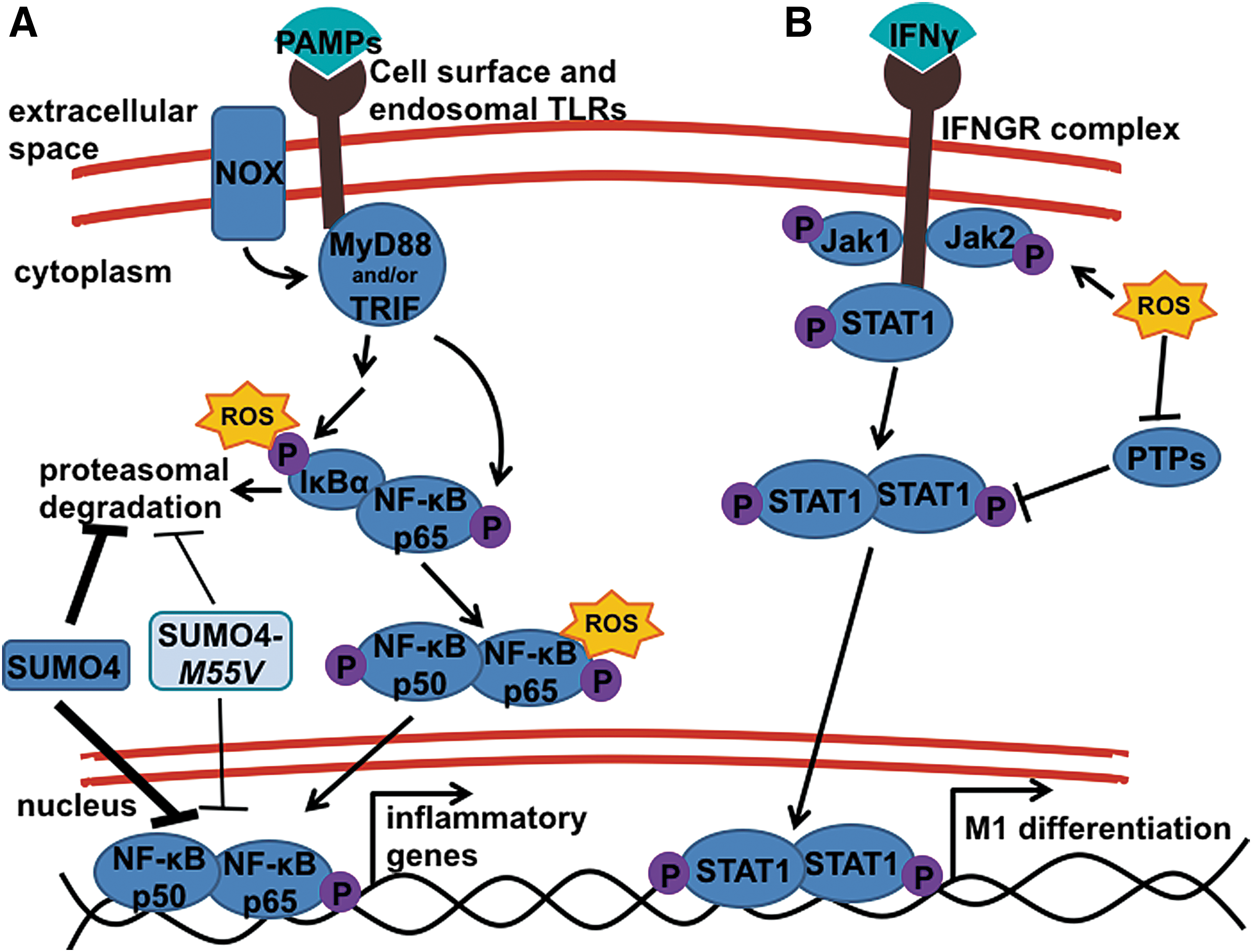
The NF-κB signaling pathway is redox regulated at several points within the pathway [Fig. 2A, and (11, 58, 129, 130, 177, 178)]. The most established mechanism is through the oxidation of IκBα, which triggers its proteolytic degradation by phosphorylation and targeted ubiquitinylation to allow for nuclear translocation of the active NF-κB p50/p65 heterodimer (58, 130). Other studies have suggested that oxidation of the NF-κB p65 subunit within the cytoplasm can enhance transcriptional activation (58). In addition to influencing cell signaling in the cytoplasm, free radicals can also modulate redox signaling within the nucleus. The NF-κB p50 subunit contains a redox-sensitive cysteine amino acid (C62) that needs to be in a reduced state to facilitate efficient DNA binding (123). Oxidation of NF-κB p50 C62 will impair DNA binding and NF-κB-dependent transcriptional activation. Nuclear translocation of antioxidants, such as thioredoxin (TRX)-1, reduces the inhibitory oxidation of the NF-κB p50 subunit, enhances DNA binding, and induces NF-κB activation (69, 129). This relationship between redox status and activation of the NF-κB signaling pathway was recapitulated in a study utilizing a catalytic antioxidant, called AEOL 10113 (201). In this study, Tse et al. (201) reported that treatment of LPS-stimulated macrophages with AEOL 10113 resulted in dampened NF-κB-dependent pro-inflammatory cytokine responses, including TNF-α, IL-1β, and IL-12p70. Interestingly, the ability of AEOL 10113 to suppress NF-κB activation was not at the level of upstream kinases, IκBα phosphorylation, IκBα degradation, or NF-κB p50/p65 nuclear translocation, but it was due to the ability of AEOL 10113 to function as an oxidoreductase and to oxidize the NF-κB p50 subunit to decrease DNA binding.
Another redox-regulated mechanism of the NF-κB pathway is the upstream initiating signaling events after the detection of microbial pathogens. In particular, several NOX isoforms have been shown to directly interact with TLRs to enhance downstream signaling (Fig. 2A). On LPS stimulation, TLR4 was shown in the human embryonic cell line, HEK293T, to directly interact with the NOX4 isoform, an interaction necessary to elicit NF-κB activation (146). The authors also showed that this interaction with NOX4 was not necessary for NF-κB activation in LPS-stimulated U937 cells, a human monocyte cell line. In addition to NOX4, other research groups have shown a role for the NOX2 isoform, which is highly expressed on macrophages and responsible for superoxide production and oxidative burst of activated immune cells, in potentiating TLR-induced inflammatory responses through direct TLR interaction (216, 217). Using a physiological stimulation with Mycobacterium tuberculosis (Mtb), one study showed that macrophages undergo a TLR2-dependent NOX2-derived oxidative burst, after a TLR2/NOX2 direct interaction, which is essential for signaling downstream of NF-κB and mitogen-activated protein kinase pathways to elicit inflammatory responses against the Mtb infection (217). This same group went on to show that this feature of direct interactions of NOX2 with TLRs could be recapitulated under TLR3 stimulation (216). As TLR3 recognizes viral infection through sensing viral dsRNA, superoxide production induced by direct association of the NOX2 complex and activated TLR3 was critical for not only NF-κB signaling but also anti-viral interferon regulatory factor (IRF)3 and STAT1/STAT2 signaling pathways.
In addition to the influence of free radicals on NF-κB activation, a potential link between oxidative stress and STAT1 activation has been reported. Using the NOD mouse background, Stanley et al. revealed that detectable oxidative stress in the pre-diabetic islets of NOD mice is associated with increased oxidation of several PTPs (191). Their study further showed that oxidation of these PTPs leads to enhanced STAT1 activation within the β-cell and within whole islets. Although this study limited their analysis to β-cell specific signaling events, the whole islet responses from the study indicate that responses of resident and early infiltrating macrophages may also involve this oxidative stress and STAT1 activation axis (Fig. 2B). Oxidants may also influence macrophage STAT1 signaling through the activation of upstream kinases, as hydrogen peroxide treatment of both fibroblast and epidermal carcinoma A-431 cells resulted in STAT1 and STAT3 signaling, with authors also showing the capacity of hydrogen peroxide treatment to increase janus kinase (JAK)2 and tyrosine kinase (TYK)2 activity (188). This redox regulation of STAT activation was also suggested in pancreatic acinar cells, in which treatment with the NOX inhibitor, diphenyleneiodonium (DPI), blunted JAK2 and STAT3 signaling (85). Finally, a study of LPS-stimulated RAW264.7 macrophages showed that treatment with the indirect antioxidant, baicalein (218), leads to suppressed phosphorylation of JAK1, JAK2, STAT1, and STAT3 (156), suggesting that increased free radical production after LPS stimulation leads to JAK/STAT pathway activation in macrophages.
Other studies in the NOD mouse have shown that this relationship between redox signaling and macrophage phenotype plays a major role in T1D pathogenesis. Specifically assessing the role of NOX2-derived superoxide production on T1D development, a recent study revealed that ablation of superoxide in the NOD mouse (NOD.Ncf1 m1J ) skewed the macrophage phenotype from an inflammatory M1 phenotype to a non-inflammatory M2 phenotype (143), contributing to the previously established significant protection of NOD.Ncf1 m1J mice against T1D onset (196, 202). In this article, Padgett et al. (143) examined the macrophage phenotype within the islets of NOD and superoxide-deficient NOD.Ncf1 m1J mice throughout the normal progression of spontaneous diabetes. In the absence of a functional NOX2 response, markers of an inflammatory M1 macrophage were significantly diminished throughout diabetes progression, including Stat1, Ifng, Tnfa, Nox2, Ccl5, and Cxcl10. In addition to dampening M1 polarization, loss of superoxide production through NOX2 allowed for M2-like phenotypes to arise in the NOD.Ncf1 m1J islets, including upregulation of Ccl17, and Stat6 early on, and an impressive upregulation of Retnla maintained through 16 weeks of age. Intriguingly, Retnla negatively regulates Th2 response during parasitic infection (149), indicating that the macrophage phenotype induced by the absence of NOX2-derived superoxide is more directed at resolution of inflammation and/or maintenance of tissue homeostasis. They further highlighted the important role for NOX2 in promoting inflammatory macrophage differentiation, by showing that even on adoptive transfer of NOX2-sufficient diabetogenic CD4 T cells into an NOX2-deficient recipient, the endogenous NOX2-deficient macrophages recruited to the pancreas were significantly less inflammatory, with decreased IL-1β and TNF-α production. Not only was genetic ablation of NOX enzymatic activity able to alter the phenotype of pancreatic macrophages, but also was short-term in vivo treatment with a broad antioxidant sufficient to shift the pancreatic macrophages toward decreased TNF-α production and increased Arginase 1 expression (143).
Mechanistically, the authors demonstrated that induction of a pro-inflammatory M1 phenotype in NOD macrophages was redox dependent and was partly due to the activation of the STAT1 and IRF5 signaling pathways that are necessary for efficient M1 macrophage differentiation (143). In the absence of NOX-derived superoxide, the activation of STAT1 and IRF5 signaling pathways was decreased in NOD.Ncf1 m1J macrophages. This redox-dependent mechanism driving STAT1 activation and M1 differentiation (Fig. 2B) appears to be a critical aspect of the diabetogenicity of the NOD macrophage, as NOD.Ncf1 m1J mice are also protected from virally triggered T1D onset, partly due to a decrease in STAT1 signaling (A.R. Burg and H.M. Tse, unpublished). In this study, in vivo infection with a diabetogenic virus, Coxsackievirus B3 (CB3), macrophages infiltrating the pancreas of NOD.Ncf1 m1J mice displayed decreased TNF-α production in response to the viral infection. This study and others implicate a role for free radicals in how viral infections can be diabetogenic—a topic discussed in further detail.
In the DP-BB rat, macrophages produce excessive amounts of NO that represents a major mechanism to facilitate autoimmunity in this strain (99). When combined with the observed decrease in islet-specific antioxidant gene expression in the DP-BB rat, the excessive NO production likely worsens β-cell dysfunction and induces death during autoimmune diabetes. Further studies are needed to determine whether exacerbated macrophage-derived NO production occurs within the islets to trigger or accelerate β-cell demise. Taken together, the hyper-inflammatory responses of macrophages in NOD mice, the DP-BB rat, and human T1D patients suggest that they can heavily impact T1D pathogenesis by creating an oxidized and pro-inflammatory microenvironment within the islet. These redox-driven consequences will be discussed in the next section.
Initiators of chronic inflammation and β-cell stress
Chronic inflammation is a major aspect of T1D progression. Downstream consequences of unresolved inflammation within the islet include aggravation of the immune response to enhance islet infiltration and to directly mediate β-cell damage. Shown in mouse (30), rat (31), and human (32, 159) pancreatic islets, the combination of immune cell-derived cytokines IL-1β, TNF-α, and IFN-γ can cause severe β-cell dysfunction by dampening the synthesis and release of insulin (9, 18, 111, 155, 158), an effect dependent on cytokine-induced production of NO (32). Prolonged exposure to these cytokines, particularly IL-1β, will lead to apoptosis of the β-cell, through the accumulation of dsDNA strand breaks in an NO-independent manner (37). A recent report addressing the susceptibility of β-cells to cytokine cytotoxicity has pointed to the β-cell being particularly unprepared to handle oxidative stress induced by these cytokines (59). In this study, proteomic analysis of cytokine-treated NOD islets, α-cells and β-cells revealed that the largest difference in cell-specific responses to cytokines was with antioxidant defenses, in which β-cells failed to significantly upregulate SOD2 and Prx3. Therefore, local non-specific inflammation could inadvertently target the free radical-susceptible β-cells.
Within the pro-inflammatory islet microenvironment, the noxious effects of free radicals and cytokines can exacerbate endoplasmic reticulum (ER) stress in pancreatic β-cells that may contribute to triggering autoimmune responses in T1D (48, 109, 115). Similar to oxidative stress, insulin-secreting β-cells are highly sensitive to ER stress (59, 116, 195). It has been hypothesized that a culmination of these biochemical processes can facilitate heightened pancreatic β-cell antigenicity and concomitant autoreactive T cell responses by inducing the activation of the Ca2+-dependent transglutaminase-2 enzyme to elicit PTMs of autoantigens, including chromogranin A (116, 124). In addition to transglutamination, other PTMs of putative autoantigens that can enhance antigenicity include the palmitoylated form of glutamic acid decarboxylase (GAD)65 in human T1D (151). The consequences of ER stress and implications in T1D pathogenesis were not only demonstrated in mouse models of T1D but also observed in isolated islets from patients at the onset of T1D (71, 114). Finally, therapies that target the ER stress pathway may prove to be beneficial in T1D, as the treatment of T1D mouse models with tauroursodeoxycholic acid, an inhibitor of ER stress, was effective in delaying pancreatic β-cell destruction and maintaining β-cell responses (50).
Similar to the NOX complex in immune cells, the mitochondrial respiratory chain in pancreatic β-cells is a major source of superoxide generation via complexes I and III in the mitochondrial membrane (135, 203). The pancreatic β-cell will transport glucose into the cell for proper insulin secretion by activating glycolysis and the tricarboxylic acid cycle for ATP generation (107, 145, 198). After a rise in glycolytic flux, an increase in mitochondrial-derived superoxide generation will ensue in pancreatic β-cells. During hyperglycemic conditions in both T1D and T2D, the continuous increase in glycolytic flux may increase oxidative stress in the β-cell and compromise function that is partly due to an inherently low antioxidant defense mechanism (59, 141). Current evidence suggests that loss of first-phase insulin release, β-cell dysfunction, and ER stress predates insulitis in T1D and importantly, the pancreatic β-cell is more than an innocent bystander in T1D (22, 78, 80, 114). Mitochondrial redox balance and dysfunction in T1D has been demonstrated with the mitochondrial encoded gene NADH dehydrogenase subunit 2 (mt-Nd2) (64), whereby a cytosine to adenine transversion mutation [mt-Nd2(a)] elicits T1D protection at the β-cell level (24) in both T1D mouse models and human translational studies (120, 205). The protective mt-Nd2(a) allele elicits a decrease in reactive oxygen species (ROS) production in β-cells and provides evidence that heightened mitochondrial-derived ROS synthesis contributes to mitochondrial dysfunction in T1D (63, 121). Although these studies were focused on pancreatic β-cells, little is known regarding mitochondrial dysfunction in islet-resident and islet-infiltrating macrophages during T1D progression. Recently, it was demonstrated that autoreactive T cells from patients with T1D display mitochondrial hyperpolarization and are dysfunctional in contrast to healthy controls or patients with T2D (23). The observed mitochondrial hyperpolarization was associated with elevations in pro-inflammatory cytokine synthesis, mitochondrial-derived superoxide, and lower ATP levels. Therapies that can effectively dissipate mitochondrial-derived superoxide may be beneficial in delaying T1D and abrogating dysregulated autoimmune responses in T1D.
Although the source of the initial inflammation is still unknown, there is certainly a crucial role of macrophages in the production of these cytotoxic cytokines. In one study, the exposure of isolated rat islets to LPS and IFN-γ stimulated intra-islet production of IL-1β, leading to dampened β-cell insulin secretion (6). When macrophages were depleted by antibody treatment, the damaging effects of LPS and IFN-γ were abrogated, suggesting a role for islet-resident macrophages in initiating the inflammation-induced dysfunction of β-cells in T1D. Notably, a recent report shows that macrophages produce IL-1β as a postprandial response to help stimulate insulin secretion and promote glucose clearance (45). In addition to aiding in glucose uptake, these glucose-induced signaling events also enhanced pro-inflammatory responses by the macrophages. Therefore, combining this physiological response with T1D susceptibility could lead to exacerbated postprandial inflammation by the macrophages, in turn creating a chronic low level of inflammation that has been proposed to underlie T1D pathogenesis.
Within T1D-susceptible NOD mice, macrophages have a severe defect in their ability to phagocytose apoptotic cells (139, 140). Therefore, lack of clearance of apoptotic β-cells during this chronic inflammatory state could potentiate inflammation and autoimmune activation. This is also of particular interest during the early post-natal pancreatic islet remodeling period, an event in which macrophages play an important role (56). During this developmental remodeling event, a wave of β-cell apoptosis occurs around 2 weeks of age for rats, and 1 week of age for mice (175, 200). Interestingly, compared with the non-autoimmune-prone BALB/c mouse strain, NOD post-natal islets contained significantly greater apoptotic cells by terminal deoxynucleotidyl transferase (TdT) deoxyuridine triphosphate (dUTP) nick-end labeling (TUNEL) staining (200). However, the authors suggested that this could be due to a lack of apoptotic cell clearance rather than increased apoptosis and, in fact, mathematical modeling of the TUNEL staining data suggested that NOD islets have a 50% decreased rate of apoptotic cell clearance compared with BALB/c (200). This postulation was later confirmed in both the NOD mouse and the DP-BB rat, in which macrophages had significant defects in phagocytosis of apoptotic cells (138 –140). Interestingly, the NOD macrophages maintained their capacity to phagocytose non-specific microspheres, indicating a specific deficiency toward apoptotic cell clearance (140). Prolonged accumulation of apoptotic cells can lead to secondary necrosis, eliciting an inflammatory response, rather than the tolerogenic response characteristic of phagocytosis of apoptotic cells (94, 122, 208). This inflammatory consequence is compounded in T1D by the heightened TNF-α and IL-1β response of NOD macrophages, in comparison to C57BL/6 and NOR strains, on phagocytosis of cellular debris (192). Therefore, determining the role of free oxygen radicals in the phagocytic response of T1D macrophages is highly relevant to understanding these early and potentially inflammatory insults in T1D progression.
The role for free radicals in the inflammatory response is fairly evident, as the exacerbated inflammatory responses of macrophages stem, in part, from hyper-activation of the NF-κB pathway and potentially the STAT1 pathway, which, as discussed earlier, is heavily regulated by free radicals. However, collective past evidence, as well as recent advances, has shown that free radicals can also influence the response and functions of macrophages aside from simply enhancing signaling pathways. Heightened levels of circulating C-reactive protein (CRP) have long been a marker for systemic inflammation seen in many patients with autoimmunity. In a long-term prospective study, called the Diabetes and Autoimmunity Study in the Young, where those at risk for T1D were followed to identify early events in disease progression, increased serum CRP was detected in the serum of those individuals who eventually became diabetic (21). This inflammation occurring downstream of CRP induction may likely be linked to activation of hyper-responsive macrophages, as treatment of DP-BB rats with human CRP was shown to enhance the macrophage oxidative burst and inflammatory responses (84). An interesting recent publication suggests that the effect of CRP on enhancing inflammation may be redox regulated upstream of inflammatory signaling events, involving the oxidation of CRP itself (189). In this biochemical study, Singh et al. (189) show that CRP can exist in an oxidized form that allows for binding to multiple different ligands, suggesting that under highly oxidative conditions, the oxidized CRP may be a stronger activator of macrophages to push forward an inflammatory response.
Studies on macrophage phagocytosis have also revealed multiple ways in which free radicals are able to influence the uptake of apoptotic/necrotic cell bodies. The release of free radicals by the apoptotic cells themselves can oxidize “eat-me” signals that end up on their outer surface, promoting its phagocytosis. This is particularly the case for the clearance of apoptotic neutrophils, where oxidation of plasma membrane-bound phosphatidylserine by the dying neutrophil enhances uptake and clearance by macrophages (51, 204). The production of free radicals by the macrophages themselves, particularly the production of superoxide by the NOX2 complex, can also impact the phagocytic process depending on the type of material being engulfed (15). NOX2 deficiency in macrophages, generated through genetic knock-out of the critical subunit gp91 phox , actually enhanced the capacity of macrophages to engulf fluorescein isothiocyanate-labeled yeast. However, when cultured with labeled apoptotic neutrophils, the same NOX2 deficiency dramatically decreased phagocytosis by the macrophages. Brown et al. suggest that the role for NOX-induced uptake of apoptotic neutrophils was in the formation of oxidized surface molecules on the macrophage cell surface through extracellular superoxide and hydrogen peroxide production, as human monocytes cultures treated with both SOD and CAT effectively recapitulated the mouse gp91phox deficiency (15).
Calling in reinforcements: chemokine and chemokine receptor expression
In addition to cytotoxic cytokine production driving T1D pathogenesis, secretion of pro-inflammatory chemokines by macrophages is also important for the trafficking of inflammatory immune cells to the pancreatic islet. Similarly, an exacerbated inflammatory cytokine response of monocytes, heightened oxidative stress, and increased expression of the inflammatory chemokine, CXCL10, have been detected within the islets of recent-onset T1D patients (166, 207). A study by Sarkar et al. examining a panel of chemokines found CXCL10 to be the major chemokine expressed within the islets from both pre-diabetic NOD mice and patients with recent-onset autoimmune diabetes (173). Circulating levels of CXCL10 were also found in newly diagnosed children with T1D (5). Although these data suggest CXCL10 to be the dominant chemokine in T1D pathogenesis, several studies suggest that a myriad of other chemokines are also produced during progression of disease, which may altogether culminate in driving T1D (5, 17, 173, 207, 220).
The same study described earlier by Sarkar et al. (173) also identified strong upregulation of chemokine (C–C motif) ligand [(CCL)5, also known as RANTES] and CXCL9 expression within the islets of pre-diabetic NOD mice. Although CCL5 and CXCL9 expression was not detected within the islets of recent-onset patients with T1D, they may play a role in the initiating events of immune cell infiltration into the islet. SNPs within the CCL5 gene corresponded to a decrease in serum CCL5 levels and were able to significantly confer protection against disease onset in T1D-prone individuals (220). The synthesis of pro-inflammatory chemokines is directly regulated by the NF-κB transcription factor and by the redox environment. Therefore, it is plausible that oxidative stress within the islet microenvironment can trigger exacerbated NF-κB-dependent chemokine production to initiate insulitis in T1D-susceptible individuals.
Another equally important aspect in immune cell trafficking is chemokine receptor expression on immune cells. Redox status and the generation of free radicals can influence chemokine receptor expression, as human monocytes demonstrate an increase in CCR5 expression after hydrogen peroxide treatment that was due to induction of the NF-κB pathway (103). Additional studies in human monocytes also demonstrated that chemokine receptor expression was redox regulated as CCR2, CCR5, and CXCR4 synthesis was shown to be upregulated after hydrogen peroxide or xanthine/xanthine oxidase treatment and decreased in the presence of the antioxidant, pyrrolidine dithiocarbamate (172). To date, very little is known regarding the role of free radicals and chemokine receptor expression in T1D. Cantor and Haskins showed that adoptive transfer of T1D with the diabetogenic Barbara Davis Center (BDC)-2.5 CD4 T cell clone recruited pancreas-infiltrating macrophages by upregulating CCR5, CXCR3, and CCR8 expression (20). How free radicals contribute to chemokine receptor expression in this model warrants further study since NOD.Ncf1 m1J mice that lack the ability to synthesize NOX-derived superoxide are highly resistant to transfer of autoimmune diabetes with BDC-2.5 (196). Interestingly, some studies have focused on the impact of free radicals on the oxidation of cell surface thiols on macrophages [recently reviewed here, (186)]. One study demonstrated that mitochondrial-associated thiol oxidative stress in macrophages increased their sensitivity to respond to the inflammatory chemokine CCL2 (157), highlighting the possibility that thiol oxidation of the receptor CCR2 could enhance the signaling cascade that initiates macrophage migration to inflammatory sites. These results further highlight the synergy of oxidative stress on macrophage chemotaxis and show how redox-mediated events can enhance chemokine receptor signaling. It would be of great interest to examine whether oxidation of surface chemokine receptors alters the activation of islet-infiltrating macrophages and signaling cascades during spontaneous autoimmune diabetes.
Antigen-presenting cells to initiate autoimmunity
The role of macrophages in driving T1D pathogenesis may also extend toward their antigen-presentation capacity. Not only can they reside in the pancreatic islet and be one of the early infiltrating immune cells during T1D progression, but also their hyper-inflammatory responses during antigen presentation could be the perfect storm for breaking peripheral tolerance. In addition to the previously discussed role of NOX-derived superoxide in fueling this exacerbated inflammatory response, there is also building evidence that activation of NOX2 also plays a critical role in the processing and presentation of autoantigens in the phagolysosome (Fig. 3A). In dendritic cells, low production of superoxide by NOX2 within the early phagosome results in alkalization of the environment to allow preservation of peptides for presentation to T cells (174). In macrophages, NOX2 activation is much more potent (112) and appears to have a different role in peptide processing (Fig. 3B). Instead of controlling the pH of the phagosome, NOX2 can negatively regulate the proteolytic process through oxidative inhibition of phagocytic cysteine cathepsins [Fig. 3A, and (171)]. This mechanism of controlling peptide degradation may also play a role in autoimmunity, as a study in experimental autoimmune encephalomyelitis, a mouse model for multiple sclerosis, revealed that NOX2-deficient macrophages did not efficiently present the autoantigenic myelin oligodendrocyte glycoprotein (MOG) peptide to MOG-specific autoreactive T cells when given intact MOG protein (2). However, if pulsed with the specific MOG35–55 peptide, macrophages had restored presentation capacity, suggesting a role for NOX2-derived superoxide in controlling antigen processing (2). Therefore, if β-cell-specific autoantigens in T1D contain cathepsin cleavage sites, enhanced NOX2 activation may skew the peptide pool of presented antigens toward autoantigenic peptides.

Additional evidence that antigen processing in autoimmune diabetes is regulated by redox status was shown by Piganelli et al., as they demonstrated that NOD macrophages were compromised in their ability to present antigen in contrast to diabetes-resistant NOR macrophages (153). NOD macrophages were less efficient in stimulating antigen-specific T cells, and this defect was partly attributed to a significant decrease in the cytoplasmic levels of GSH in contrast to NOR macrophages (Fig. 3C). During the processing of antigenic peptides in the lysosome, a reduction of disulfide bonds is essential and requires intracellular GSH and cysteine (29). GSH reduces disulfide bonds within proteins, allows for efficient protein unfolding, and enables proteolytic digestion of proteins into peptides to be bound by class II major histocompatibility complex (MHC) molecules for antigen presentation (83). The authors postulate that inefficient antigen processing and the presentation of autoantigens in a non-tolerogenic manner by NOD macrophages may partly explain the aberrant activation of autoreactive T cells in T1D (Fig. 3B, D). Interestingly, this same defect in antigen processing and presentation in human dendritic cells was also observed when intracellular GSH was depleted with 4-hydroxyifosfamide (4-OH-IF) and allogeneic T cell responses exhibited a decrease in proliferation and IFN-γ synthesis (98). Conversely, T cell responses could be restored when GSH levels in dendritic cells were increased with glutathione monoethyl ester.
Another mechanism by which peptides presented by macrophages could be altered to enhance their antigenicity is through direct oxidation of the peptide itself. This has been reported to occur in multiple autoimmune diseases, including systemic lupus erythematosus, rheumatoid arthritis, and celiac disease (4, 170). Addressing this hypothesis, Trigwell et al. tested whether antibodies from T1D patients showed enhanced reactivity toward oxidative modification of GAD65 (199). In this study, treatment of rat islets with copper sulfate/hydrogen peroxide resulted in the formation of multiple post-translationally modified GAD65 proteins, as detected by Western blot. Remarkably, serum from T1D patients, but not healthy controls showed preferential reactivity toward the modified GAD65 proteins (199). The effects of oxidative modification on GAD65 were confirmed in another biochemical study that showed that hydroxyl radical treatment successfully oxidized GAD65, which, in turn, enhanced the recognition by autoantibodies from patients with T1D (92). Further experiments also showed that the binding affinity of autoantibodies to oxidized GAD65 was increased nearly a log-fold above the unmodified protein. A more recent study has also shown this phenomenon to hold true for oxidative modification of insulin itself (193). Similar to GAD65, the oxidized form of insulin resulted in higher binding by circulating antibodies from T1D patients. Importantly, Strollo et al. also noted that more than a one-third of insulin autoantibody-negative T1D patients tested were actually positive for reactivity to the oxidized form, implicating that current methods for detecting individuals with circulating autoantibodies are highly under-representative (193). These studies clearly show that an oxidative environment can alter β-cell-specific proteins to create new autoantigenic epitopes, often referred to as neoantigens. However, it is currently unknown when these proteins become modified during pathogenesis. Given the extensive role for macrophages and NOX-derived superoxide in driving T1D pathogenesis, it is highly probable that they play a role in generating and presenting these oxidized neoantigens to enhance autoimmunity (Fig. 3B, E). This happens especially with the observation that macrophages engulf apoptotic β-cells or β-cell debris in an inflammatory microenvironment that results in aberrant increases in free radicals either during antigen uptake or within the phagocytic lysosomal compartment during antigen processing. Further studies are highly warranted, as controlling the exposure of T1D-susceptible patients to these oxidized epitopes may potentially curb a more aggressive adaptive immune response or progression of disease (Fig. 3E).
Major cytotoxic effector cells during β-cell attack
Early in vitro studies demonstrated that macrophages were effective in directly destroying pancreatic β-cells or β-cell lines due to the generation of pro-inflammatory cytokines and free radicals [Fig. 4 and (72, 90, 97)]. Neutralization experiments revealed the macrophage-derived factor found to be cytotoxic for co-cultured islet cells, was actually not TNF-α or IL-1β, but production of NO, as removal of NO by
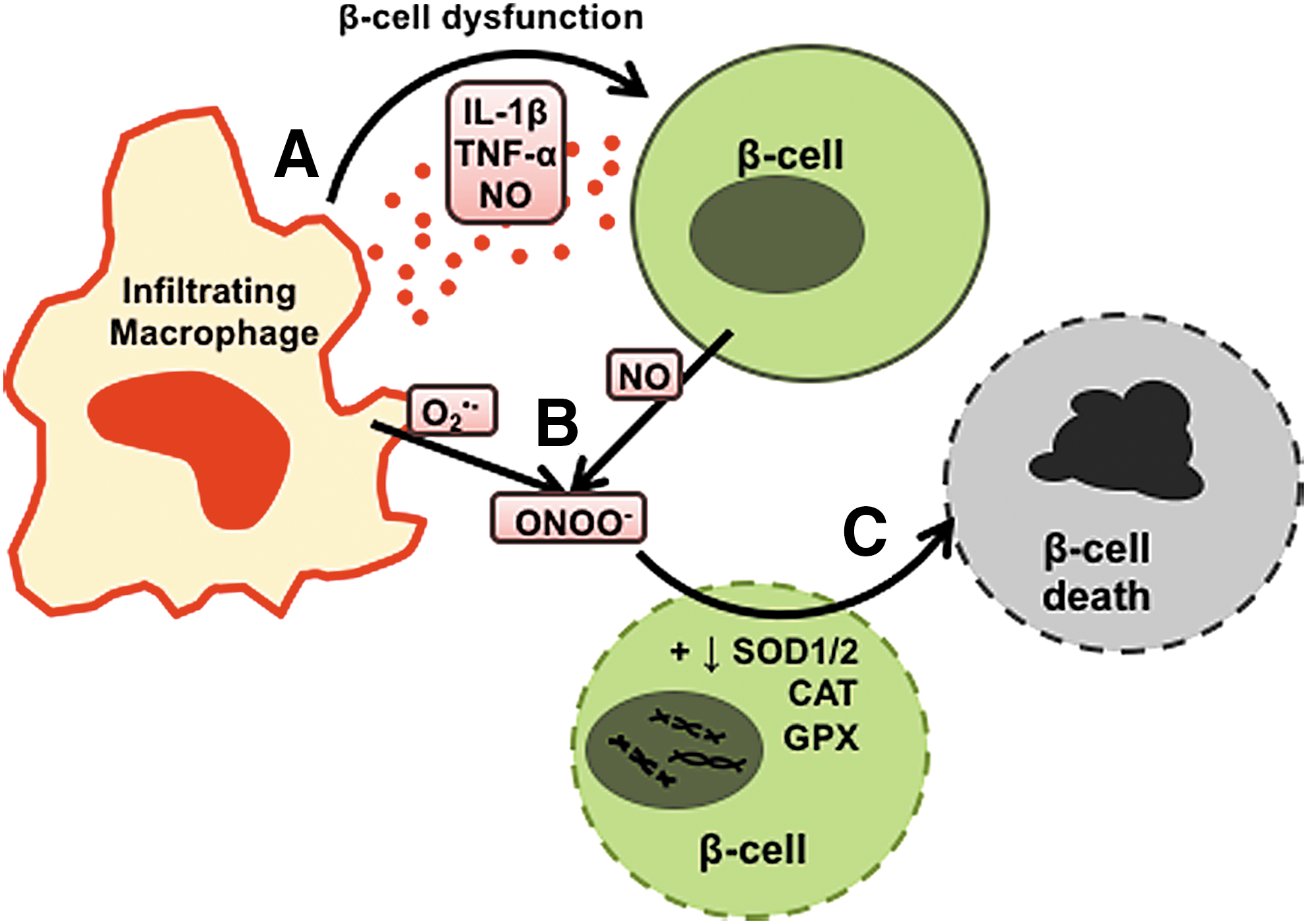
This potent role for macrophages as effector cell types in T1D has also been shown by several studies utilizing the BDC-2.5 diabetogenic CD4 T cell clone (16, 20). The adoptive transfer of BDC-2.5 CD4 T cells into NOD.scid recipients, which usually results in rapid and aggressive onset of diabetes, could be prevented by macrophage depletion using lip-Cl2MDP, whereas neutrophil and natural killer cell depletions are unable to do so (16). The study also showed that isolated CD11b+ macrophages from diabetic pancreata had cytolytic capacity when co-cultured with β-cells. Together, these results suggested that the aggressive diabetogenicity of BDC-2.5 CD4 T cells was due to their ability to recruit and activate cytotoxic macrophages to the islet. Cantor and Haskins expanded on these observations to show that diabetogenic CD4 T cells recruited pro-inflammatory IL-1β-, TNF-α-, and NO-producing macrophages by a CCL1 chemotaxis gradient to the pancreas (20). Both studies concluded that the BDC-2.5 diabetogenic CD4 T cell clone initiates diabetes by recruiting inflammatory macrophages to the pancreas, acting as late-stage effectors in the direct killing of β-cells.
Macrophages play a variety of different roles when driving T1D and there is plenty of evidence to suggest that production of free radicals, particularly NOX2-derived superoxide, may regulate macrophage function at every step. Understanding the importance of the redox sensitivity to specifically drive diabetogenic macrophage responses is critical in uncovering potentially targetable pathways using antioxidant therapies to dampen autoimmunity. Of equal importance is identifying what redox-dependent events initiate autoimmune activation during T1D progression. In this next section, we will cover a major suspect in triggering the onset of T1D, viral infections, and how redox-dependent macrophage responses to this insult may turn diabetogenic.
Free Radicals, Macrophages, and Viral Triggers of T1D
Environmental triggers of T1D
The initiating events in T1D onset have remained elusive as the activation of autoimmunity and the slow progression of β-cell destruction are likely at play for months or years before the clinical diagnosis of hyperglycemia. However, evidence such as discordant onset between twins (162), the massive age range for actual onset (birth to 40 years of age), and the steep rise in T1D incidence in developed countries that cannot be accounted for by genetic shifts underscore the importance of environmental factors in the initiation or progression of the disease. These environmental influences range from microbial infections to exposure to certain dietary antigens. By far, the most evidence for a single environmental factor being involved in T1D pathogenesis points to viral infections as a likely culprit.
Over the decades, T1D has been associated with viruses ranging from Epstein-Barr and Cytomegalovirus (CMV) (DNA viruses), to many different RNA viruses, including Mumps, Rotavirus, Rubella, human endogenous retroviruses, as well as a slew of members of the enterovirus genus (77, 88, 176). The studies on the enterovirus genus of viruses has shown the closest connection to the onset of disease, with a meta-analysis of 24 case-controlled clinical association studies concluding a significant association between enterovirus infection and T1D (219). A recent report from the Type 1 Diabetes Prediction and Prevention study in Finland revealed higher frequencies of enterovirus infection, as detected within the stool, in T1D-susceptible children as compared with controls (70), which occurred more than a year before the initial detection of serum autoantibodies. These results suggest that viral infections may initiate the slow progression toward autoimmunity rather than a sudden trigger. This concept coincides with an early report from The Environmental Determinants of Diabetes in the Young study, in which analysis of blood samples from children who experienced rapid onset of T1D showed no relationship with detection of viremia (100). Excitingly, two independent prospective reports have recently suggested that anti-viral responses precede the activation of autoimmunity in T1D-susceptible individuals (53, 89). In one study, peripheral blood mononuclear cells from susceptible children exhibited an anti-viral Type 1 IFN gene signature that was detected before the clinical detection of autoantibodies, but not present in well-established T1D patients (53). In another report, whole blood RNA transcriptome analysis revealed an upregulation of innate immune responses, namely Type 1 IFN responses, that could be detected before autoantibody seroconversion (89). Taken together, the role for enteroviruses in promoting T1D progression remains a significant probability.
Enteroviruses, by definition, have a major tropism for the gastrointestinal tract, whereas particular members such as Coxsackie B viruses (CBVs) are also pancreas tropic and can successfully infect human pancreatic islets (54). In vitro infections focusing on the CB4 strain revealed that all sub-strains tested were capable of infecting human islet cells, with each conferring a different phenotypic change in the islets. Some strains efficiently replicated to high titers during the culture, and of those, some strains managed to avoid causing extensive cytopathic effect (54). These results suggest that some strains of CBVs are capable of infecting pancreatic islets without damaging the cells, which may allow for persistent infection. Importantly, biopsies from both cadaveric recent-onset patients (41) and living recent-onset patients (96) have shown a significant detection of enterovirus infection within pancreatic islets over healthy controls.
Using a human islet transplant model in β-cell-depleted immunodeficient mice, Gallagher et al. showed that in vivo infection with CBV leads to pronounced infection of human islets, severely compromising their insulin production, which leads to triggering diabetes onset (55). Gaining insight into the mechanisms of viral triggers in T1D, researchers established that CBVs are also capable of infecting murine pancreata. Infection of older pre-diabetic NOD mice with either CB3 or CB4 strains resulted in the acceleration of T1D onset (43, 73, 185). This was shown to be enterovirus specific, as general viral infections with Vaccinia virus, mouse CMV, or Theiler's murine encephalitis virus (a close relative to enteroviruses) did not result in any induction of T1D in the diabetogenic T cell receptor (TCR)-transgenic,

Autoimmune diabetes can be triggered on viral infection in the diabetes-resistant BB rat. In this strain, infection with the DNA parvovirus, Kilham Rat Virus (KRV) causes a 30% penetrance of autoimmune diabetes onset within 4 weeks post-infection, which can be enhanced to nearly 100% when co-administered with polyinosinic-polycytidylic acid [poly(I:C)], a mimic of viral dsRNA (27, 61). An intriguing aspect of this interaction is that the virus infection is not pancreas tropic. This concept of viral infections outside the pancreas impacting a β-cell-specific autoimmune disease, and the synergistic effect of poly(I:C) treatment, suggests a critical role for innate immune responses in driving autoimmunity. In fact, the treatment of DR-BB rats with lip-Cl2MDP (to systemically deplete macrophages) before co-treatment with KRV and poly(I:C) leads to complete protection against virus-induced diabetes (27). In the same study, Chung et al. (27) used an adoptive transfer model of autoimmune diabetes using in vitro-activated splenocytes from KRV-infected DR-BB rats into DP-BB recipients to show that macrophages are responsible for directly priming autoreactive T cells (Fig. 5C). This interaction includes the production of inflammatory mediators such as IL-12p70, TNF-α, and IL-1β, which then exceeds the threshold maintained by peripheral tolerance, and thus activates a Th1-driven diabetogenic T cell response.
Further studies linking viral triggers to the onset of autoimmune diabetes in the BB rat have been extended to several other viral infections. Infection of DP-BB rats with rat Cytomegalovirus (RCMV) results in a significant acceleration of spontaneous T1D (68). Although some latent virus was detected within the pancreas, again authors found that both exocrine and endocrine cells of the pancreas were not permissive to RCMV infection. Therefore, similar to KRV infection, RCMV appears to trigger a cascade of heightened inflammatory immune cell activation and subsequent autoimmune progression. In fact, similar to the pathogenesis of KRV, depletion of macrophages was able to eliminate RCMV-accelerated disease onset (68). Interestingly, this study found that this effect was dependent on depleting macrophages from the peritoneum during the first few days of infection, and since RCMV infection was by intraperitoneal injection, this suggested that the initial innate response to viral infection was the causative trigger, not a prolonged response to chronic infection (68).
In another model of virus-induced T1D in the BB rat, a picornavirus called Ljungan virus was found in a wild rodent population of Scandinavian voles and lemmings to be associated with diabetes onset, and it was also shown to impact diabetes within the DP-BB rat (136). Recently, in an interesting preventative treatment strategy, Niklasson et al. treated pre-diabetic rats with a combination of two picornavirus-specific anti-viral agents, pleconaril and an anti-picornavirus kinase inhibitor (APO-N039), and found that although rats were on the dual-therapy regimen they were completely protected against the development of autoimmune diabetes (136). This suggests that early intervention in human patients with the right combination of anti-viral agents may help in delaying or preventing the triggering of T1D. However, more is needed to understand the mechanisms at play for the viruses that actually trigger T1D in human patients. Another approach to blocking the triggering event induced by viral infections is potentially through modulation of redox-regulated events and understanding how redox signaling contributes to exacerbated innate immune responses and could uncover targetable therapeutic approaches. Therefore, the next subsection will provide evidence that anti-viral immune responses are redox regulated, particularly in the context of triggering autoimmunity.
Redox regulation during viral triggers of T1D
Many of the studies on virus-induced T1D discussed earlier indicate that the activation of the innate anti-viral response is a key driver of diabetogenicity rather than consequences from direct β-cell infection itself. Initiation of the anti-viral response begins with the recognition of virally infected cells through several different pattern recognition receptors (Fig. 6A, B). Some, such as TLRs, are located on the endosomal membrane, and they are poised to recognize viral infection on phagocytosis of virally infected cells or cellular debris. These include TLR3 (for dsRNA recognition), TLR7 (for ssRNA), TLR8 (for ssRNA in humans only), and TLR9 (for CpG-containing DNA). On recognition of viral RNA or DNA, these receptors will initiate a signaling cascade through either TIR-domain-containing adapter-inducing interferon-beta (TRIF) or MyD88 to activate the NF-κB pathway (for inflammatory gene upregulation), as well as IRF3 (for IFN-β production) and IRF7 (for IFN-α production) pathways [extensively reviewed in ref. (144)].
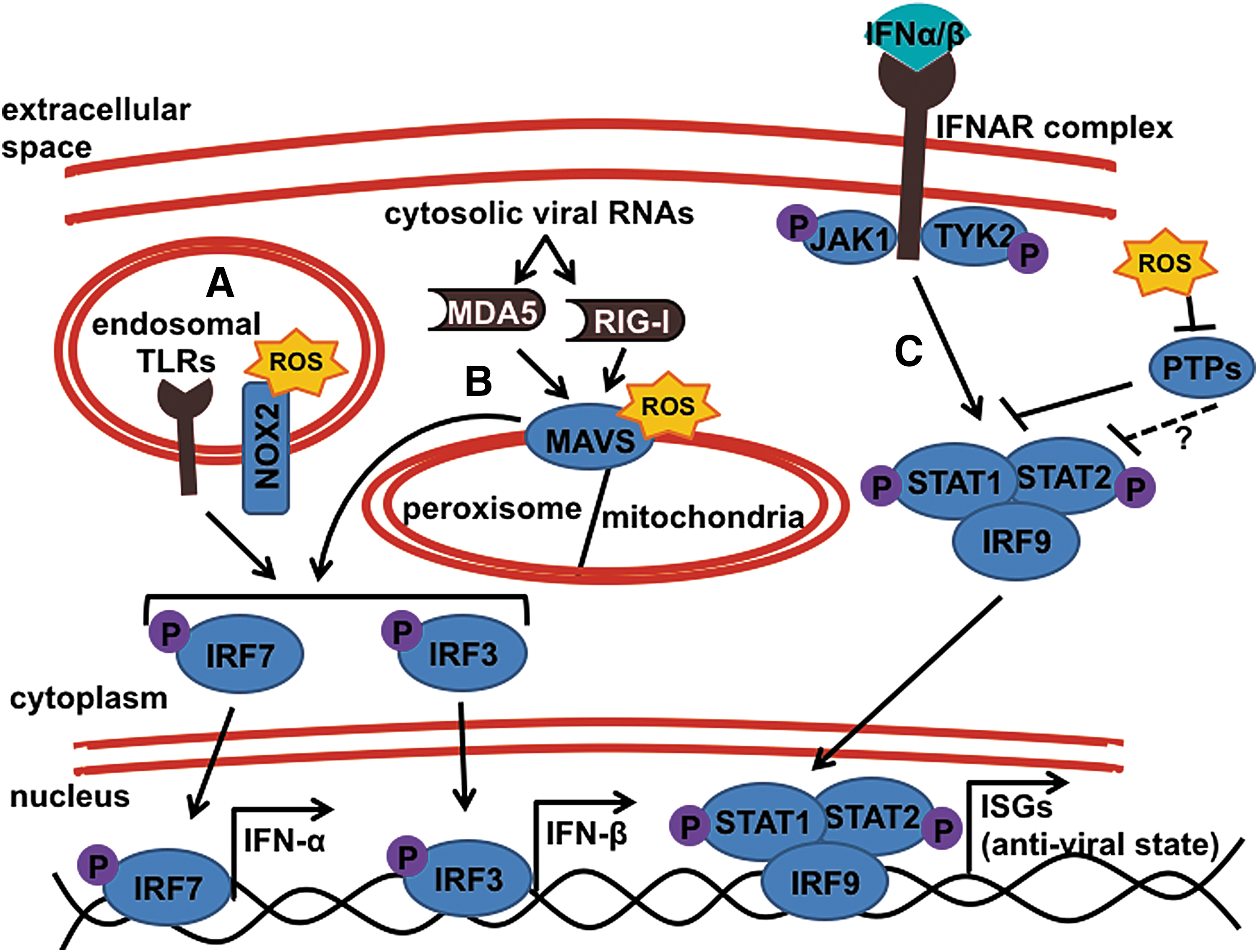
Another class of viral sensors are the retinoic acid-inducible gene protein I (RIG-I)-like receptors (RLRs), which reside within the cytoplasm to detect actively replicating virus. RLRs include RIG-I and MDA5, which are helicases recognizing different forms of single-stranded and double-stranded RNA. On activation, these RLRs will signal through the intermediate signaling protein, mitochondrial antiviral-signaling protein (MAVS, also referred to as IPS-1) to also initiate NF-κB, IRF3, and IRF7 pathways, creating an anti-viral state (144). The recognition of foreign nucleic acids through TLRs and RLRs is suggested to play a role in conferring susceptibility to T1D, as certain polymorphisms in the genomic regions of both TLR3 and MDA5 have shown significant association with T1D individuals, and others were found to confer protection in otherwise-susceptible individuals (7, 42, 133, 212).
The role of free radicals in heightening inflammatory immune responses during autoimmunity is pronounced, and there is clear evidence that NOX-derived superoxide is essential for combating bacterial and fungal infections. However, the role for the NOX-derived oxidative burst during viral infections has not been widely studied. Chronic Granulomatous Disease patients, who harbor mutations within the NOX2 complex, leaving them unable to generate superoxide, are severely susceptible to bacterial and fungal infections (179). Interestingly, there is no clinical evidence to suggest that loss of NOX2 function impairs the immune system's ability to combat and clear viral infection. Though not essential for viral clearance, some evidence suggests that NOX-derived superoxide still plays a role in enhancing the anti-viral response. Mechanistic studies in the A549 airway epithelium cell line utilizing NOX2 inhibitors, DPI and apocynin, as well as an NOX2 RNAi revealed that NOX2 activation enhances the RIG-I viral RNA recognition pathway through transcriptional upregulation of the intermediate signaling protein, MAVS [Fig. 6B, and (190)]. Another study found that Duox2-derived free radicals produced during Influenza A virus infection induced expression of both RIG-I and another cytosolic viral RNA sensor, MDA5, to aid in the clearance of Influenza A virus from both human nasal mucosal epithelium and murine nasal mucosal tissue (93).
However, within macrophage responses to viral infection, activation of NOX2 appears to have an integrated role in establishing the anti-viral response in a TLR3-dependent manner (180, 216). The role of free radicals enhancing anti-viral responses also extends to macrophage responses within the T1D-susceptible NOD mouse, as deficiency in NOX2 via the Ncf1 m1J mutation results in dampened production of inflammatory anti-viral TNF-α and IFN-β, as well as in diminished Type 1 IFN signaling as indicated by significantly lower transcription of interferon-stimulated gene 15 (Isg15) (180). Interestingly, although addition of xanthine oxidase to the culture was able to rescue the dampened TNF-α production in the NOD.Ncf1 m1J macrophage cultures, production of IFN-β was not rescued, suggesting either an NOX2-independent role for the p47 phox subunit (which harbors the mutation) in enhancing IFN-β production or that the specific localization of NOX2-derived superoxide production is necessary for its action on IFN upregulation. The latter suggestion coincides with the previously described study, as transient physical interactions occur between the NOX2 complex and TLR3 during the early signaling responses to poly(I:C)-stimulation [Fig. 6B, and (216)].
The interaction between free radicals and anti-viral signaling was recently more directly tested for relevance in viral triggers of T1D (A.R. Burg and H.M. Tse, unpublished data). Utilizing the CBV infection model for acceleration of T1D in the NOD mouse, Burg and colleagues demonstrated that NOX2-derived superoxide production is a critical component for this triggering event to occur, as CB3-infected NOD.Ncf1 m1J mice remained protected against T1D onset during both the CB3-induced acceleration phase and throughout the rest of the spontaneous onset time-course. Surprisingly, these NOX2-deficient mice maintained a similar viral load and clearance capacity as their NOD counterparts, suggesting that the viral acceleration is an immune-mediated event. As such, NOD.Ncf1 m1J pancreas-infiltrating macrophages had dampened capacity for TNF-α production and in vitro studies revealed that M1 macrophage polarization through STAT1 activation and, consequently, downstream anti-viral responses, including upregulation of viral RNA sensors MDA5 and RIG-I, were heavily redox dependent. This redox-dependent upregulation of MDA5, RIG-I, and activated STAT1 was shown through rescue assays using xanthine oxidase treatment to replenish superoxide within CB3-infected NOD.Ncf1 m1J macrophages. Intriguingly, this study also revealed that the STAT1, MDA5, and RIG-I signaling pathways could be activated simply under exposure to superoxide, even in the absence of CBV infection. These results further highlight the synergy of free radicals and oxidative stress on innate immune activation in macrophages, and they warrant further investigation into whether free radical production is mechanistically acting on JAK1, TYK2, STAT1, or STAT1-inhibiting PTPS (as discussed in the Redox-mediated exacerbation of molecular mechanisms involved in inflammation section), to enhance anti-viral and diabetogenic STAT1 signaling (Fig. 6C).
As mentioned earlier, macrophages from DP-BB rats produce excessive levels of NO on activation. This increased oxidative stress during inflammation plays a role in the interaction of KRV infection and autoimmune activation within the DR-BB rat. On infection with KRV, inducible nitric oxide synthase (iNOS) has been shown to be highly upregulated during the innate response to the viral infection, and most importantly, blockade of iNOS activity by treatment of rats with aminoguanidine (AG) was able to protect KRV-infected DR-BB rats from developing autoimmunity (126). In line with the evidence that this virus-induced phenomenon is based on innate-driven activation of Th1 responses (27), this article found that AG treatment led to selective reductions in Th1-asssociated immune responses, including production of IFN-γ, CCR5, CXCR3, and CXCR4 expression. This was in addition to general immune suppression through dampened MHC-II and IL-2Rα expression, which corresponded to dampened cytotoxic CD8 T cell activation. Interestingly, the synergistic effects of poly(I:C) treatment on the induction of T1D in the DR-BB rat may also be NO regulated. A study examining the response of rat islets to poly(I:C) treatment found that dual treatment with IFN-γ resulted in severe β-cell dysfunction, which was NO dependent (67). However, this effect was independent of islet-resident macrophage responses, suggesting that some redox mechanisms of virus-induced T1D may directly impact β-cell function and the resulting cytotoxicity.
Altogether, these studies provide evidence that anti-viral responses in macrophages are redox regulated, and they highlight the importance of these responses in shaping the pathogenesis of T1D, including the interaction with environmental triggers. Further studies are needed to test whether exacerbated redox-dependent immune responses are a common denominator for other environmental triggers, such as bacterial infections and dietary antigens.
Antioxidant Therapies in T1D: Manipulation of Macrophages for the Good
Antioxidant-based therapies for T1D
Understanding the diabetogenic mechanisms governed by redox regulation is crucial for being able to implement any therapeutic or intervening approaches for T1D. This has been highlighted in past clinical trials in which antioxidant-based therapies were unable to show any efficacy, including metabolic control or indications of preserving β-cell function (108). However, despite the failed improvement for the recent-onset children on this therapy, the study by Ludvigsson et al. (108) importantly noted that this treatment also had no negative side effects. There is still promise for successful antioxidant treatment for Type 1 diabetics, as antioxidant supplements such as Vitamin E have shown associations with decreased complications that result from chronic inflammation seen in both Type 1 and Type 2 diabetics [reviewed in ref. (167)].
In fact, a clinical trial using combinatorial therapy of nicotinamide and vitamin E showed preservation of β-cell function, as measured by C-peptide secretion, particularly in pre-pubertal recent-onset children, as far out as 2 years after diagnosis (33). Although the evidence for antioxidants as potential therapeutics is minimal, its safety profile shown in studies thus far and the potential for having efficacy at earlier time-points (during the process of immune cell activation) warrant continued investigation. In this last section, we propose the therapeutic potential of repurposing antioxidant treatment approaches to protect islets after isolation and islet transplantation. In addition, we will discuss the potential of NOX inhibitors to modulate autoimmune responses in T1D and finally, we will describe how redox modulation can reprogram macrophage differentiation and pro-inflammatory phenotypes in patients with T1D or T1D-susceptible individuals.
Treating oxidative stress during islet transplantation
Another potential therapeutic application for antioxidant treatments is during β-cell replacement therapies, or islet transplantation. In this respect, antioxidants have shown major promise to aid in curbing inflammatory immune responses and in helping maintain β-cell function after islet transplantation (160). In earlier studies using a manganese-porphyrin-based catalytic antioxidant, Bottino et al. showed that treatment of human islets postisolation significantly aided in protecting the islets against cell death and even improved β-cell function (13). This was important as the stress induced during the islet isolation process and culture creates an incredibly inflammatory and oxidative environment. This protective effect of antioxidant treatment was further shown to be efficacious in enhancing the success of islet cell transplantation into diabetic mice. The same study also showed that in vivo treatment significantly protected the transplanted islets from redox-driven ischemia-reperfusion injury, as well as from immune-driven transplant rejection. This form of antioxidant-based therapeutic has also been tested more recently in the context of islet encapsulation. Incorporating the antioxidant, tannic acid, into the layers of polymer, Kozlovskaya and colleagues (150) described an islet encapsulation process that allowed for enhanced islet viability and effectively dampened autoimmune responses in vitro. A very recent paper by the same group expanded their studies to show that the antioxidant-incorporated coating results in decreased inflammatory cell migration, and transplantation of these encapsulated islets successfully provided diabetic mice with long-term glycemic control (150). Overall, these studies provide evidence that incorporating antioxidant-based methods in either therapeutic strategies or curative measures, such as islet transplantation, potentially offers a safe and effective way to modulate the excessive immune-driven inflammatory responses that T1D patients are plagued with.
Immuno-modulation using NOX inhibitors
Broad-range antioxidants have demonstrated promise in ameliorating autoimmune diabetes in mouse models (38, 39, 152), whereas human translational studies will require the use of selective NOX inhibitors to specifically target islet-resident macrophages and autoreactive T cells to demonstrate efficacy in delaying T1D. There has been substantial progress in the development of unique NOX2 inhibitors encompassing peptides, biologics, and small-molecule compounds that have selectivity for the NOX2 isoform [extensively reviewed in ref. (3)]. The best characterized NOX2 inhibitor is the 18-amino acid NOX2ds-tat fusion peptide that was originally named gp91ds-tat (81, 105, 163). This peptide is cell permeable due to the presence of a nine-amino acid tat peptide from human immunodeficiency virus (26) and will bind the p47 phox subunit and prevent assembly of the NOX2 complex without compromising NOX1 and NOX4 function (35). In addition to in vitro inhibition of superoxide production (163), NOX2ds-tat has proved to be effective in several in vivo animal models when delivered intravenously or with adenoviral constructs (81, 105, 163). Other specific inhibitors of NOX2 include monoclonal antibodies targeting the extracellular loops of NOX2 that inhibit in vitro NOX2 activity (19), but whether these antibodies are efficacious in vivo is still unknown. Ultimately, the use of pharmacological inhibitors for NOX2 may be beneficial in delaying autoimmune diabetes in patients susceptible to T1D by suppressing pro-inflammatory innate immune responses from islet-resident macrophages. However, there are several issues that need to be met before the use of NOX inhibitors, including identifying patients (improved biomarkers of T1D prediction) while they still exhibit substantial β-cell mass and function, determining environmental triggers that activate the innate immune responses, and designing specific NOX2 inhibitors that target the pancreas or islets without causing global immunosuppression.
Although the focus of this review article is on NOX2 and effects on autoimmune responses in T1D, there is evidence that the NOX1 isoform can also contribute to pancreatic β-cell dysfunction in T2D (211). Treatment of islets with pro-inflammatory cytokines induced 12-lipoxygenase activity and the production of 12-hydroxyeicosatetraenoic acid that was capable of stimulating NOX1-derived superoxide. Translational studies with islets from human Type 2 diabetic donors demonstrated an elevated expression in NOX1 and provided further evidence that an increase in oxidative stress in the islet microenvironment can also contribute to β-cell dysfunction in T2D. Murine pancreas and islets also express the NOX4 isoform (25, 206), but the role of NOX1 and NOX4 in T1D autoimmune responses and pancreatic β-cell responses is not known. Similar to T1D, the treatment of Type 2 diabetic patients with NOX inhibitors may be efficacious in dampening inflammation in the islet microenvironment, preventing hyperinsulinemia, and restoring insulin sensitivity in peripheral tissues.
Redox modulation of macrophage differentiation and responses in T1D
In T1D, since macrophages have an exaggerated redox-regulated NF-κB signaling pathway (as discussed in detail in the Redox-mediated exacerbation of molecular mechanisms involved in inflammation section), resulting in heightened inflammatory responses, this observation would suggest that macrophages from genetically susceptible individuals for T1D are more poised to differentiate to a pro-inflammatory M1 phenotype on activation. Recent studies have shown that shifting the macrophage response away from an M1 phenotype profile is able to alter disease progression in the NOD mouse. A single adoptive transfer of in vitro polarized M2 macrophages with an IL-4/TGF-β/IL-10 cocktail was sufficient to protect pre-diabetic NOD mice from T1D onset (148). The ability to manipulate the polarization of macrophages in vivo was observed with chronic helminths infection (164). Liu et al. were able to successfully protect NOD mice from T1D onset when chronically infected at 5 weeks of age. The delay in autoimmune diabetes was partly attributed to an induced M2-polarized macrophage response (106).
Polarization of M1 and M2 macrophages appears to be distinct, and evidence suggests that maintenance of these two broad phenotypes involves free radical signaling. Along with promoting M1 differentiation (as discussed extensively earlier), regulating NOX-derived superoxide production appears to also be used as a counter-measure during M2 differentiation. One of the earliest studies looking at the effects of IL-4, a strong M2-inducing cytokine, noted that IL-4-treated macrophages effectively inhibited the oxidative burst and pro-inflammatory responses (1), suggesting that targeting superoxide production may inhibit M1 differentiation. A short 1-h incubation of macrophages with IL-4 was able to confer long-term changes in the capacity for macrophages to undergo superoxide production as long as 4 days later. In addition, a study by El Hadri et al. (49) reported that exposure of macrophages to the antioxidant TRX1 during in vitro M2 polarization with IL-4 treatment significantly enhanced anti-inflammatory macrophage differentiation via expression of CD206 and production of IL-10. In contrast, TRX1 treatment under M1-polarizing conditions with LPS stimulation decreased TNF-α production of macrophages. Importantly, this study showed that long-term treatment of TRX1 in vivo was able to alter the phenotype of macrophages within atherosclerotic vessels, effectively dampening the development of atherosclerotic lesions in ApoE2.Ki mice (49). Finally, treatment of BDC-2.5-transferred NOD.Rag mice with MnTE-2-PyP, an SOD mimetic, was efficient in enhancing an M2 phenotype and a concomitant decrease in an M1 macrophage phenotype in the pancreas (143). This result was also corroborated when BDC-2.5 was adoptively transferred into NOD.Rag.Ncf1 m1J mice, and an increase in pancreas-infiltrating M2 macrophages was observed. The skewed macrophage phenotype and response may explain the delay in adoptive transfer of T1D in recipient mice that lack the ability to generate NOX-derived superoxide (196).
Overall, the successes in treating T1D with M2-skewing therapeutics and the power of antioxidants to control the switch from inflammatory M1 to non-inflammatory M2 macrophages provide great potential for manipulating the innate immune response during T1D pathogenesis (Fig. 7). This is especially true given that in skewing away from the hyper-inflammatory response of the M1 phenotype, we can elicit an M2 phenotype that is less inflammatory, but also supports tissue healing and promotes islet function (34). Therefore, combining these approaches may provide a strong therapeutic strategy for overcoming the inflammatory environment of pre-diabetic individuals driven by hyperactive macrophages, to curb the onset of autoimmunity.

Concluding Remarks
In this extensive review, we have highlighted the importance and ever-growing complexities of oxidative stress and redox regulation of innate immunity that drives T1D progression. Macrophages play a central role in this process. The heightened inflammatory response and free radical production by macrophages of T1D-susceptible individuals and rodents put this cell type at the epicenter of initiating and enhancing autoimmune responses. The evidence presented in this review highly suggests that free radical production and redox signaling are central to the macrophage's diabetogenic role, from exacerbating inflammation to the potential oxidative PTMs of autoantigenic peptides. These features of redox regulation in macrophages extend to bridging the diabetogenicity of environmental factors such as viral infections. Overall, as the field continues to uncover the intricacies of redox regulation of macrophage differentiation and pro-inflammatory immune responses in T1D, the potential use of antioxidant therapies in curbing innate immune activation and initiation of T1D may become more of a reality.
Footnotes
Acknowledgments
The authors would like to apologize to researchers whose primary observations that form the basis of current knowledge in the field could not be cited due to space limitations, or have been acknowledged indirectly, by referring to current reviews. Our work was supported by grants from the American Diabetes Association (7-12-CD-11), the Juvenile Diabetes Research Foundation (1-SRA-2015-42-A-N), and the National Institutes of Health (DK099550, AI007052).