
Abstract
Significance:
Amyotrophic lateral sclerosis (ALS) is due to degeneration of upper and lower motor neurons in the anterior horn of the spinal cord and in the motor cortex. Mechanisms leading to motor neuron death are complex and currently the disease is untreatable.
Recent Advances:
Work in genetic models of ALS indicates that an imbalance in the cross talk that physiologically exists between motor neurons and the surrounding cells is eventually detrimental to motor neurons. In particular, the cascade of events collectively known as neuroinflammation and mainly characterized by a reactive phenotype of astrocytes and microglia, moderate infiltration of peripheral immune cells, and elevated levels of inflammatory mediators has been consistently observed in motor regions of the central nervous system (CNS) in sporadic and familial ALS, constituting a hallmark of the disease. Resident glial cells and infiltrated immune cells are considered among the major producers of reactive species of oxygen and nitrogen in pathological conditions of the CNS, including motor neuron diseases.
Critical Issues:
The timing and exact role of oxidative stress-mediated neuroinflammation and damage to motor neurons in ALS are still not fully elucidated.
Future Directions:
It is clear that a major challenge in the next future will be to envisage effective strategies to modulate the neuroinflammatory response in the symptomatic stage of disease, to prevent progression of neurodegeneration through the propagation of oxidative damage. Antioxid. Redox Signal. 29, 15–36.
Introduction
A
Degeneration of motor neurons may follow different mechanisms, according to a “dying-back” or a “dying-forward” model or via “independent degeneration” (81). However, the pathological mechanisms that underlie the development of motor neuron degeneration most probably stem from a complex interplay between genetic and environmental factors, and the disease is thought to be the culmination of a multistep process (3).
Abnormalities of critical molecular and cellular processes, such as mitochondrial dysfunction, oxidative and endoplasmic reticulum stress, impaired RNA regulation, protein misfolding, altered autophagy, dysfunction of the axonal transport system, glutamate excitotoxicity, ion channel dysfunction, and genetic alterations (including epigenetic modifications and a number of (un)identified risk genes for sporadic ALS), contribute to the pathogenesis of ALS (44). Up to date, it is not clear whether the same factors play a pathogenic role in all patients, who are quite heterogeneous as far as age of onset, initial symptoms, and duration of the disease are concerned, and whether these factors are relevant in a determined sequential timing in the disease. Nonetheless, a number of attempts to modify the course of the disease have been made in genetic models of the disease, targeting one or the other of the alteration mentioned above.
Studies on the most commonly used in vivo model, that is, transgenic mice overexpressing a mutant superoxide dismutase 1 (SOD1) typical of a few percent of patients, have indicated “promising” approaches that unluckily failed regularly in clinical trials. Ten years ago we already listed about 30 “more effective” attempts in mutant SOD1 rodents (mostly mice expressing the G93A variant) (29). The number of preclinical studies has steadily grown since then, including work on more recent non-SOD1 genetic models, but there is still only one drug (riluzole) registered for ALS, which slightly increases survival without clearly improving the quality of life or muscle strength of patients (162).
In the continuous attempt to find druggable targets, recent studies have addressed novel facets of the disease, such as the formation of protein aggregates in motor neurons and astrocytes. Indeed several ALS-linked mutant proteins such as SOD1, TAR DNA binding protein 43 (TDP-43), and fused in sarcoma (FUS) do form aggregates, with wild-type TDP-43 often found aggregated in sporadic ALS patients. Protein misfolding may be a direct consequence of the mutation, or a consequence of oxidative stress in the case of wild-type proteins. Accumulation of misfolded proteins in the form of (usually) ubiquitinated inclusions is considered a hallmark of ALS pathogenesis (15, 179) and, as in other neurodegenerative disorders, it may cause—or be a consequence of—an imbalance of the protein degradation pathway or of a defective autophagic flux (38).
Motor neurons seem to be particularly sensitive to alteration of the ubiquitin/proteasome pathways; for instance, it is known that mice where proteasomes, but not the autophagy machinery, are disrupted specifically in motor neurons develop motor neuron degeneration and protein aggregates containing ALS-related proteins such as FUS, TDP-43, and optineurin (223). Furthermore, mutations in proteins that are components of protein quality control (i.e., ubiquilin2 and optineurin, which are proteins containing the ubiquitin association domain) or are involved in autophagy (the autophagic adaptor SQSTM1/p62) are found in some familial ALS cases (224). FUS itself is endowed with SUMO-E3 ligase activity and sumoylation of SOD1, TDP-43, and other proteins may play a role in the pathogenesis of ALS (50).
One example of preclinical studies attempting rescue of protein degradation and removal of aggregates is the modulation through overexpression or chemical induction of the chaperone HSPB8. This approach was successful in reducing the accumulation of aggregates and providing protection in models for SOD1- and TDP-43-mediated toxicity (46 –48) and is probably worth an attempt to translation into clinics. However, past experience indicates that no single-treatment approach may be successful in ALS and that some compounds might be therapeutically beneficial only in combination (111).
Indeed, the main reason for the difficulty to translate preclinical studies to effective treatments (apart from the obvious limitations of mice models) is that ALS is much more complex than what traditionally believed for three reasons.
One problem is that the definition of ALS itself has changed over time. Three clinical phenotypes caused by degeneration of different components of the motor system, that is, ALS, primary lateral sclerosis (upper motor neuron disease), and progressive muscular atrophy (lower motor neuron disease) were once considered separate entities on the basis of clinical symptoms and later grouped as motor neuron disease or ALS (4). Furthermore, evidence that mutations in several “ALS genes” are shared with other diseases has led to the view that ALS represents one end of a continuum with frontotemporal lobar degeneration, a fact supported by the clinical observation that part of ALS patients show also signs of frontotemporal dementia (FTD). Further genetic studies in patients and mounting evidence in models indicate additional possible overlaps, even with unrelated disease entities (Table 1).
A number of other proposed ALS genes with low frequency (such as KIFAP3, DCTN1, EPHA4, and others), which may represent risk factors or disease modifiers and await confirmation by larger studies, have been omitted.
AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; ANG, angiogenin; ATXN2, ataxin 2; CHCHD10, coiled-coil-helix-coiled-coil-helix domain-containing protein 10 gene; CHMP2B, charged multivesicular protein 2B; fALS, familial ALS; FTLD, frontotemporal lobar degeneration; FUS, fused in sarcoma; OPTN, optineurin; PD, Parkinson's disease; PFN1, profilin 1; PGRN, progranulin; sALS, sporadic ALS; SMAJ, late-onset spinal motor neuropathy; SOD1, superoxide dismutase 1; SQSTM1, sequestosome 1 (also called p62); TBK1, TANK binding kinase 1; TDP-43, TAR DNA binding protein 43; UBQLN2, ubiquilin 2; VCP, valosin-containing protein.
A second problem is that many proteins that play a key role both in ALS and in other diseases have a number of different functions and are involved in various cell processes (Table 2). This is well exemplified by proteins (such as TDP-43, FUS, and survival of motor neuron [SMN]) that regulate different steps of mRNA metabolism (117, 194, 226) and are found associated with diverse “ribonucleopathies” (51, 137) but have other functions as well (21, 92). Nonetheless, all these functions seem to converge on a few crucial pathways. For instance, TDP-43, FUS, SMN, ataxin 2 (ATXN2), and angiogenin have all been shown to colocalize with markers of stress granules, which are ribonucleoprotein complexes that form in the cytoplasm in response to various cellular stresses and are regulated by the autophagy pathway (5, 7, 40, 59, 60, 108, 174). Interestingly, even mutant SOD1 (an antioxidant enzyme considered unrelated to mRNA metabolism) is able to affect selected alternative splicing events (138) and stress granule dynamics through its interaction with G3BP1 (78), while FUS (an RNA-binding protein) regulates the expression of another antioxidant enzyme, that is, manganese superoxide dismutase (54).
Some of the genes that have been found mutated in ALS patients, including the expansion in the C9orf72, code for proteins that are involved either directly (•••) or indirectly (ooo) in more than one cell process. Expression of ALS mutant proteins also influences more than one pathway in experimental models. This table does not consider suggested functional interactions between ALS-mutant proteins (e.g., TBK1 interacts with OPT1 and SQSTM1; TDP-43 interacts with ATXN2 and FUS; FIG4 affects SQSTM1).
A third problem is that ALS is not a pure motor neuron disease, as it was once considered. Not only the extent to which each neuronal population is affected varies between individuals (4) but also nonmotoneuronal cell populations are altered in one or another phase of the disease and death of motor neurons is almost unanimously considered a “noncell autonomous” event.
Cells such as interneurons, astrocytes, microglia, Schwann and muscle cells, endothelial cells, and T lymphocytes all seem to contribute to the disease to some extent. There is now evidence, coming from genetic models of the disease, that the contribution of these cell populations to the disease might be the consequence of toxic processes directly engaged by the mutant ALS genes in these same non-neuronal cells (Fig. 1). This may be the case for the contribution of oxidative stress to motor neuron degeneration.
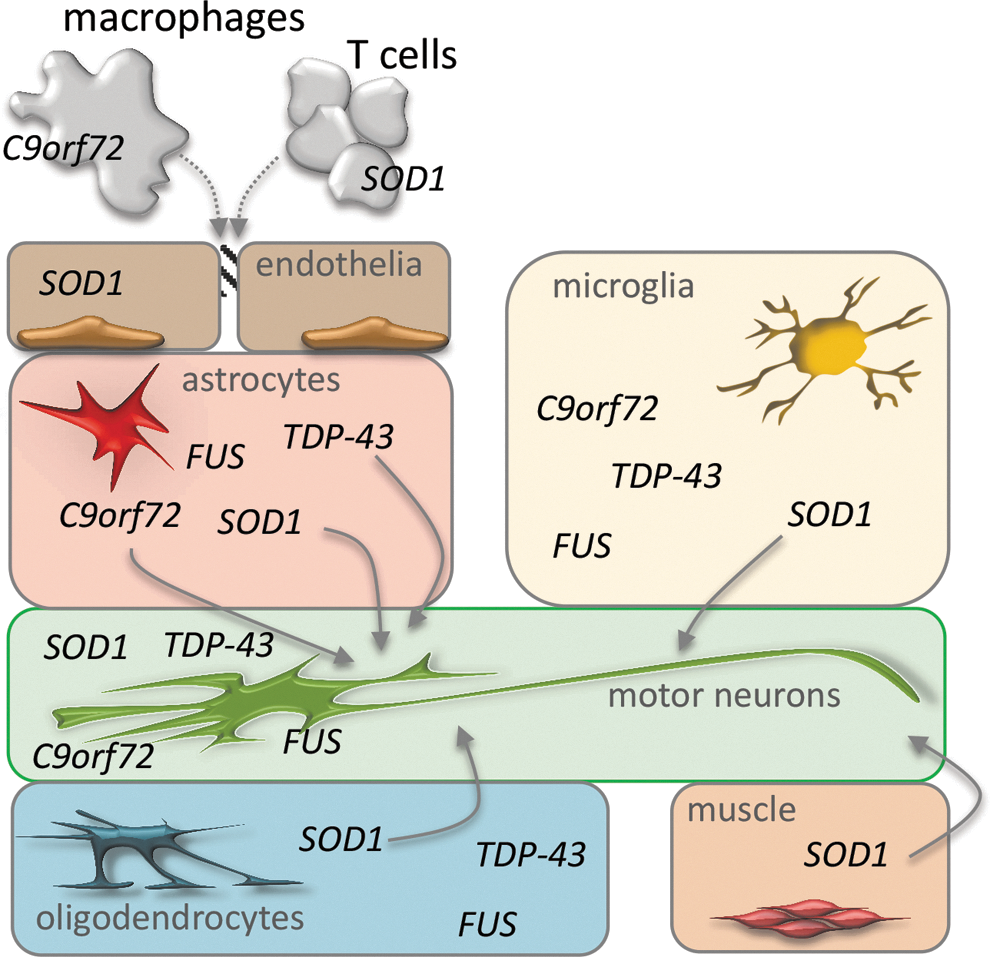
Molecular species such as reactive oxygen species (ROS) and reactive nitrogen species (RNS) have been repeatedly proposed as therapeutic targets in ALS, based on large evidence of the presence of oxidative stress markers in motor neuron degeneration (25). ROS may be directly toxic inside specific compartments of motor neurons; for instance, mice lacking SOD1 show a progressive motor axonopathy that can be reverted by the expression of the protein exclusively in the mitochondrial intermembrane space. These results suggest that oxidative damage initiated at mitochondrial sites is a major determinant of motor axon degeneration (73). However, besides being toxic to neurons, ROS result toxic by causing an imbalance in the cross talk that physiologically exists between motor neurons and the surrounding cells, and that eventually becomes detrimental to motor neurons. Actually, during the adaptive responses to harmful events exerted by immune cells, the release of reactive species is a controlled physiological process, necessary to spread an efficient response and clear the source of damage. However, if the equilibrium between the formation of reactive species and the endogenous antioxidant defense is altered, ROS release leads to oxidative stress and associated damage [see Mittal et al. (163) for a comprehensive review]. Accordingly, the use of antioxidants has the potentiality to limit two disease-contributing mechanisms, that is, oxidative stress and neuroinflammation. For these reasons, it is now clear that studies on possible mediators of the pathological interactions between neurons and surrounding cells in ALS will certainly contribute to the discovery of new treatments.
In the following sections, we will therefore discuss the impact of neuroinflammatory pathways on ALS disease process, with a special focus on current evidence on the role of oxidative stress in neuroinflammation.
Neuroinflammation in ALS
Substantial evidence indicates that neurodegeneration in ALS occurs also because the environment surrounding motor neurons is dysregulated in a cascade of events collectively known as neuroinflammation. This is mainly characterized by a reactive phenotype of astrocytes and microglia, moderate infiltration of peripheral immune cells, and elevated levels of inflammatory mediators in the parenchyma (193). Evidence for gliosis, together with the presence of infiltrated T lymphocytes, has constantly been observed in motor regions of the central nervous system (CNS) in sporadic and familial ALS, constituting a hallmark of manifest disease (184). Different studies have also shown that microglial activation, in particular, occurs formerly or concomitantly with the onset of clinical disease (164). Moreover, by the use of RNA sequencing coupled to specific cell-type selection from isolated spinal cord of mutant SOD1 mice, it was demonstrated that most of the transcripts with increased expression are likewise derived from astrocytes and microglia, whereas those with decreased abundance originate from neurons, likely reflecting the gliosis and neuronal loss that occur with the progression of disease (213).
Crosstalk between neuroinflammation and neurodegeneration in different ALS models
Although the perception that inflammation that commonly associates with neurodegeneration could be instrumental to this process was generally shared, the forthright demonstration that neuroinflammation actively promotes the progression of motor neuron dysfunction in ALS came by several seminal articles. These demonstrated that the selective deletion of mutant SOD1 in microglia and astrocytes in SOD1-G93A mice gives raise to phenotypes with the longest life span ever obtained in preclinical models of the disease (23, 250). In addition, also the protective role of functional T cells in mutant SOD1 mice was similarly demonstrated (17, 35).
These findings established a turning point in ALS research, as they opened the way to an intense examination on the possible contribution of non-neuronal tissues to ALS pathogenesis [see Refs. (44, 185) for review]. Those studies eventually strengthened the concept that ALS is a noncell autonomous disease, and that targeting cells other that motor neurons might represent a valuable strategy to slowdown neurodegeneration and treat ALS. This picture has found significant support more recently, when the role of astrocytes as modifiers of the disease progression in an SOD1 model of ALS was also confirmed by xenotransplantation of human glial progenitors generated from induced pluripotent stem cells (iPSCs) (98, 127). In vitro coculture experiments have further demonstrated that primary astrocytes of human origin or from mice models expressing mutant SOD1 and those differentiated from induced neural progenitor cells derived from mutant SOD1 and C9orf72 patients become toxic for motor neurons (56, 97, 160, 168).
Production of proinflammatory factors, together with reduced glutamate reuptake and metabolic support, has been recognized as a major cause of neurotoxicity (185). Furthermore, changing the immune profile of microglia by selective inhibition of the master control gene nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) or through a gene therapy approach to increase the levels of interleukin (IL)-10 significantly prolongs the duration of the disease in mutant SOD1 mice. Conversely, the constitutive activation of microglial NF-κB is sufficient to induce pathological hallmarks of ALS, such as astrogliosis and motor neuron death (76, 88). Importantly, recent genetic advances have shown that the TANK binding kinase 1 (TBK1), an NF-κB-activating kinase known for its important functions in the production of proinflammatory cytokines, is mutated in ALS-FTD (39, 77). Altogether, these findings reinforce the concept that the neuroinflammatory response plays a crucial role in disease progression and extend also to sporadic ALS what has clearly emerged from studies on SOD1-ALS models and patients.
Whether this is also true for the other genetic forms of ALS that have been described so far is still under investigation. Mutations in the RNA binding proteins FUS and TDP-43, as well as expansions of GGGGCC repeat in C9orf72 gene, account for a major portion of familial ALS cases. Accordingly, new animal models of FUS-, TDP-43-, and C9orf72-linked ALS have been produced to mimic the pathological phenotypes, which are not limited to motor neuron degeneration. In the case of FUS and TDP-43, in particular, mice expressing human wild-type proteins or ALS-associated mutations have been generated by either transgenic overexpression or knockin homologous recombination, to avoid concerns inherent to overexpression in animal models. This offers a precious opportunity to test the contribution of the inflammatory compartment to the noncell autonomous degeneration of motor neurons in non-SOD1 ALS disease. In most of these models [thoroughly reviewed in Refs. (173, 186)], clear signs of gliosis are readily reproduced, indicating that astrocytes and microglia are recruited during disease course also in these contexts.
However, at variance with SOD1-linked ALS, the expression of TDP-43 and FUS in motor neurons is sufficient to determine their degeneration in a cell autonomous manner. In the case of FUS, in particular, the selective expression of mutant human FUS in motor neurons is enough to drive their degeneration as well as muscle denervation, thereby pointing to a cell autonomous mechanism (208). A similar conclusion can be drawn from recently described mutant FUS knockin mice, where the reduced motor neuron number and enhanced motor neuron apoptosis, which characterizes mice at birth, are rescued by CRE-mediated cell-specific expression of wild-type FUS within motor neurons (204).
As discussed before, it is well established that astrocytes expressing mutant SOD1 are toxic to motor neurons expressing wild-type or mutant SOD1, both in vitro and in vivo (56, 97, 168, 180, 250). Strikingly, this was not reproduced in models of TDP-43 toxicity, where the effects of mutant TDP-43 astrocytes on motor neuron viability were analyzed. Indeed, iPSC-derived astrocytes expressing the M337V ALS-linked mutant TDP-43 are not toxic to wild-type or M337V TDP-43 cocultured motor neurons, although the expression of mutant TDP-43 is sufficient to significantly reduce astrocyte viability (207). Differentiated astrocytes from glial-restricted precursors from TDP-43 mice with an A315T mutation, as well as from TDP-43 knockout mice, are equally unable to induce the degeneration of wild-type cocultured motor neurons. Similarly, they do not affect motor neuron viability when engrafted into rat spinal cord where they normally establish interactions with host motor neurons (96). Since astrocytes derived from SOD1-ALS mice are toxic to motor neurons in the same experimental setting, the contribution of astrocytes in ALS pathogenesis might depend on the gene mutation involved. Yet, the restricted expression of the same M337V TDP-43 into astrocytes was shown to be sufficient to cause motor neuron degeneration in rats, which, on the contrary, points to a noncell autonomous contribution of astrocytes to ALS disease, as in the case of SOD1 (227).
Although other investigations are needed to clarify this point, these observations do not rule out the central and dual neuroprotective and neurotoxic activity that neuroinflammation might exert on the onset and progression of the disease.
Mechanisms of neuroinflammation
It has been postulated that the neuroinflammatory events occurring in ALS might result into two distinct phases. The first is an early anti-inflammatory or neuroprotective compensatory response of glia and immune cells, essentially governed by supportive microglia and astrocytes as well as by T helper 2 (Th2) cells and CD4+FoxP3+ regulatory T lymphocytes (Tregs) that secrete neurotrophic factors in the attempt to decrease neuronal stress. Late in the course of the disease, as more motor neurons are being damaged, a second phase takes place, characterized by a cytotoxic response of glia and immune cells (107). In the first phase, Tregs produce high levels of the anti-inflammatory factor IL-4, thus inducing microglia polarization to the neuroprotective M2 phenotype and eventually influencing astrocyte behavior, leading to the release of IL-10, IL-6, glial cell-derived neurotrophic factor (GDNF), and insulin-like growth factor (IGF-1). In the second phase of the disease, the Treg response is suppressed and T helper 1 (Th1) cells produce high levels of interferon-gamma (IFNγ) that, together with activating factors released by damaged neurons, promotes the microglial M1 cytotoxic phenotype (mainly via activation of NF-κB) and a sustained astrogliosis (69, 88, 102, 255).
These events are characterized by secretion of tumor necrosis factor α (TNFα), IFNγ, IL-1β, transforming growth factor beta (TGFβ), and production of monocyte chemotactic protein-1 (MCP-1) (61, 62, 146, 203). This in turn might promote the infiltration of macrophages/microglia into affected tissues and upregulation of reactive species producing enzymes such as inducible nitric oxide synthase (iNOS), NADPH oxidase 2 (NOX2), and cyclooxygenase 2 (COX2), an event that is also associated to a decrease of antioxidant molecules such as glutathione (105, 141, 144, 189).
Despite the copious literature sustaining this theory [for a comprehensive review see Hooten et al. (107)], increasing evidence is leading to reconsider this net binary categorization of the neuroinflammatory response in ALS. An important study using deep RNA sequencing of acutely isolated spinal cord microglia and aimed at defining their transcriptional profile in different phases of the disease has reported the expression of both pro- and anti-inflammatory genes at the same time (36). This would suggest that in ALS microglia, there is no consistent predisposition toward M1 versus M2 phenotypes and this might reflect the highly complex inflammatory mechanisms occurring in vivo. The balance between harmful and trophic phenotypes in microglia is likely affected by both intrinsic and extrinsic factors, among which those released by dying motor neurons, astrocytes, and infiltrating T cells.
Moreover, through high-throughput RNA sequencing with the translating ribosome affinity purification methodology, it has been shown that astrocytes isolated from mutant SOD1 mice before overt phenotypic symptoms are already enriched in genes linked to toxic or proapoptotic effects, including chemokine C-X-C motif chemokine 10 (CXCL10) and insulin-like growth factor-binding protein 7 (IGFBP7). Furthermore, they display a downregulation of peroxisome proliferator-activated receptor gamma coactivator 1 α (PGC1α) a transcriptional coactivator of nuclear receptors and other transcription factors that plays a key role in inducing ROS detoxifying enzymes (219). This suggests that astrocytes may display a harmful reactive species imbalance even in early stages of the disease.
Role of the blood-CNS barrier in neuroinflammation
Regardless of the precise mechanism and timing of glial reactivity in the neuroinflammatory response in ALS, it is believed that this response is strictly linked to the integrity of blood-CNS barrier (B-CNS-B). In healthy conditions, astrocytes in particular secrete and provide key factors to endothelial cells, including TGFβ, GDNF, and fibroblast growth factor, which modulate the B-CNS-B to control the passage of most molecules (1). Several studies have shown an increase in total protein levels, IgG, and altered serum albumin in the cerebrospinal fluid of ALS patients that, together with a clear infiltration of T lymphocytes, local activation of immune cells in affected tissues, and loss of capillary endothelial integrity, collectively argue for an altered B-CNS-B permeability (79). Progressive B-CNS-B damage has been indeed demonstrated, both at functional and structural levels, particularly in mutant SOD1 models of ALS, starting from early stages of the disease, even before evident neuron degeneration and gliosis (80, 256).
In these animal models, endothelial cell degeneration and disarrangements of their tight junctions, as well as swollen end feet of astrocytes, perivascular edema, and microhemorrages have all been demonstrated, suggesting the contributions of these events to motor neuron death (79). Notwithstanding, there is no evidence so far that any of the ALS-associated genes might encode for proteins directly involved in maintaining the integrity of the barriers. Rather, it is possible that primary causes of ALS, such as accumulation of misfolded proteins (130), may also account for alterations in the B-CNS-B. For instance, cytoplasmic mislocalization of TDP-43 has been found in astrocytes (207), and this may possibly interfere with their attachment to the endothelial sheet. Moreover, early oxidative stress, likely due to mitochondria dysfunctioning in endothelial cells and astrocytes, as well as increased proinflammatory cytokine levels in the nearby environment, may lead to damages to the B-CNS-B, suggesting a mutual interaction between neuroinflammatory events and CNS-blood barrier integrity (135, 177).
Glia and Immune Cell Contribution to Oxidative Stress in ALS
Resident glial cells and infiltrated immune cells are considered among the major producers of ROS and RNS in pathological conditions of the CNS (147, 251). High levels of both reactive species are indeed produced by these cells during neurodegenerative events (37) and can damage surrounding neurons and B-CNS-B (135). These may act through a direct oxidation of proteins and lipids, as well as through an indirect mechanism, where they function as signal transduction mediators leading to the release of proinflammatory, cytotoxic factors (74). Mitochondrial dysfunctions, elevated expression, and activation of reactive species-forming enzymes (e.g., NOX, iNOS, xanthine oxidase, and COX2), together with high levels of chemokines, cytokines, and immunoglobulin complexes, seem to exert a primary role in the dysregulation of glial oxidative metabolism taking place in neurodegeneration. The interplay between oxidative stress and the neuroinflammatory response is accordingly an autoregenerating mechanism: proinflammatory factors induce the formation of reactive species and, at the same time, free radicals lead to signaling pathways that often converge in the activation of NF-κB, with a consequent induction of an array proinflammatory genes (166, 239).
Protein oxidation, nitration, and carbonylation, together with lipid peroxidation, were found in neurons and glial cells in familial and sporadic ALS patients and in models of the disease (22, 31, 209), indicating that oxidative stress might be relevant in the noncell autonomous pathogenesis of ALS (30). Oxidative stress may not only increase the net production of ROS and RNS but also affect protein conformation and structure, possibly leading to the accumulation of abnormal protein inclusions, which indeed are extensively found in ALS mouse models and patient-derived tissues, as mentioned above. Conversely, protein aggregation can induce reactive species production both in the cytosol and in mitochondria (25, 68, 109, 145), generating in this way a harmful loop. Since the production of reactive species and proinflammatory molecules is an indissoluble process (24, 166), we may therefore speculate that the neuroinflammatory events participate in creating a misfolding-prone environment.
Although the presence of oxidative and nitrosative damage in the degenerating areas of the CNS in ALS is undeniable, the primary source for the production of reactive species is still unclear. Nonetheless, the contribution of non-neuronal cells in the creation of this pro-oxidative and detrimental environment seems to have a prominent role. Neuroinflammatory events and oxidative stress appear therefore to be intimately linked and interdependent in the etiopathogenesis of ALS, and we will now discuss their interplay in glial and immune cells in disease-affected tissues.
Microglia
Microglia represent the innate immune cells of the CNS, involved both in the defense of neurons whenever an injury occurs and in the removal of pathogens and damaged cells by their phagocytic functions (124). They are considered as the main producers of reactive species and proinflammatory cytokines in the CNS (199). It has been repeatedly demonstrated that the generation of oxidative stress and cell reactivity in these cells are two indissoluble events, relying one on each other, that progressively lead to a harmful cytotoxic state (146, 248). In this respect, one of the major sources of ROS in the CNS is represented by the transmembrane enzyme NOX2, whose expression is restricted to microglial cells in healthy CNS of mice and humans (20, 253). On activation through a GTP-Rac1-dependent mechanism, NOX2 transfers electrons from NADPH to molecular oxygen, generating O− 2, which in turn can give rise to other ROS, such as H2O2 (24). Activation of NOX2 in microglia consistently contributes to the rising of their toxic properties toward neurons (146) and, importantly, the enzyme was found upregulated both in familial and sporadic ALS patients and in animal models of the disease (8, 10, 247).
A function for microglial NOX2 in the etiology of ALS was provided almost 10 years ago by important studies showing that NOX2 inhibition in NOX2−/−/SOD1-G93A mice dramatically slows disease progression, significantly extending mice survival (153, 247). However, a recent article has provided results that seem in disagreement with previous observations, as they indicate that the deletion of either NOX2 or NOX1 in SOD1-G93A mice is not able to modify their survival (206). Moreover, pharmacological inhibition of NOX2 is effective in rescuing motor neurons from the toxicity of microglia bearing mutant SOD1 in culture, while it provided conflicting results when NOX2 inhibitors were administered to SOD1-G93A mice (101, 206, 228). Although the reasons for such discrepancies are not clear, variability in the results from these studies might well originate from the use of mice with distinct backgrounds, by the use of NOX2 inhibitors (such as apocynin) with only modest specific activity, and/or by their incomplete and poorly documented CNS penetrance. The genetic ablation of NOX2 specifically in microglia, which would provide more explicit results about NOX2 contribution in ALS pathogenesis, has not been documented so far.
However, a direct relationship between mutant SOD1 expression and NOX2 activation was convincingly demonstrated. In glial cells expressing SOD1-G93A, NOX2 activation seems to directly rely on the presence of mutant SOD1 by a mechanism where SOD1 binds to Rac1, locking it in an overactive, GTP-bound state and stimulating thereby an excessive production of ROS (101) (Fig. 2). As a consequence, in microglia-neuron cocultures, ROS accumulation affects the inflammatory profile of microglia, leading to NF-κB activation, TNFα secretion, and an overall neurotoxic effect (144) (Fig. 2). An increase in NOX2 has been also demonstrated in different models of ALS. In particular, wild-type and mutant forms of TDP-43 are able to induce microglial reactivity by interaction with the CD14 receptor and upregulation of NOX2, TNFα, and IL-1β, with consequent neuronal toxicity (254) (Fig. 2). Furthermore, also soluble factors, possibly released by injured neurons, may result in further exacerbation of the NOX2 activity and ROS production. For instance, extracellular adenosine triphosphate (ATP) has been shown to increase intra- and extracellular ROS levels through the Rac1-NOX2 axis in SOD1-G93A microglia, contextually enhancing its neurotoxic potential (10) (Fig. 2). Moreover, the inhibition of the microglia-activating P2X7 receptor for extracellular nucleotides in SOD1-G93A mice through the administration of the P2X7 antagonist brilliant blue G, ameliorates the spinal cord pathology, likely interfering with a series of proinflammatory events, including the activation of NOX2 (9).

In addition to the production of high levels of ROS, ALS microglia are also the source of consistent amounts of nitrogen species, mainly by an increased expression and activation of the iNOS enzyme (202) (Fig. 2). While the role of microglia-derived RNS is less defined, it might be an important aspect to be considered in cell-based therapeutic approaches. Indeed, several studies have shown that stem cell-derived motor neurons are susceptible to damage produced by surrounding disease-linked cells (55, 126, 225). After transplantation into the spinal cords of mutant SOD1-expressing rats, motor neuron precursors undergo degeneration, which is accompanied by signs of oxidative damage (225). Microglia cells are supposed to be among the principal culprits in hastening this process, as the reactive oxygen and nitrogen species that they produce are particularly detrimental to the survival of human neural stem cell-derived motor neurons. These may die by nitroxidative damage and/or by an indirect mechanism that involves the reduction of the neuroprotective function of healthy astrocytes, affecting their capability to intake exceeding glutamate (225).
ROS production in microglia has also been identified as an upstream event in the recruitment of inflammasomes, protein scaffolding complexes that induce the secretion of cytokines IL-1β and IL-18 from their precursor forms, activating caspase-1 (Fig. 2). The inflammasomes are present in most immune cells, including microglia, and are stimulated by diverse damage-associated molecular patterns (DAMPs), as well as pathogen-associated molecular patterns, such as extracellular ATP, bacterial toxins, and particulate material (199). Caspase-1 is one of the inflammatory markers found increased in the CNS of ALS mice and patients, which accordingly display high levels of IL-1β (113, 143, 181). It was demonstrated that, in different ALS models, activation of caspase-1 in microglia is induced by extracellular mutant SOD1 through a mechanism of endocytosis (159) and by extracellular TDP-43 through the activation of CD14 receptor (254). SOD1 and TDP-43 might therefore act as DAMPs and induce the activation of the inflammasome, possibly through an ROS-dependent mechanism (Fig. 2).
Indeed, ROS (in particular those released by mitochondria) have been indicated as plausible activators of inflammasomes (155), since reduction of IL-1β production was observed following treatment with antioxidants such as n-acetylcysteine or diphenyliodonium; conversely, in some cases, H2O2 alone was sufficient to induce inflammasome activation (24). Also, in this case, the relationship between ROS and inflammasome activation is not a one-way mechanism. Actually, IL-1β, released as a consequence of inflammasome activation, is, in turn, a pleiotropic factor involved in the induction of inflammatory and neurotoxic molecules such as COX2 and iNOS (Fig. 2). This mechanism amplifies the production of reactive species of oxygen and nitrogen, highlighting once again the autoregenerative features of oxidative stress and cell reactivity (123).
Astrocytes
Astrocytes provide trophic and metabolic support to neurons, and altered properties of astrocytes are likely to play a crucial role in the propagation of motor neuron injury in SOD1-related ALS. Indeed, reduced metabolic support from astrocyte-derived lactate release and activation of pronerve growth factor-p75 receptor signaling (67), together with dysregulated TGFβ signaling pathways (183), have been reported. Furthermore, astrocytes induce reduction the major histocompatibility complex class I (MHCI) expression in motor neurons (211) and the level of MHCI is directly related to motor neuron survival in a mouse model of ALS (172). Thus, astrocytes expressing mutant SOD1 are toxic for motor neurons essentially via production and secretion of diffusible factors, including reactive species such as nitric peroxide and oxygen radicals, and/or decrease in the production of neuroprotective molecules, such as the antioxidant glutathione (14, 56, 57, 97, 152, 160, 168, 238). Primary astrocytes derived from newborn SOD1-G93A mice (when no signs of motor neuron death are evident) display a neurotoxic phenotype, characterized by inflammatory signals and oxidative stress. Moreover, stimulation of these cultures with TNFα and IFNγ produces a further increase in NO2 − synthesis and TNFα expression (Fig. 3) and an overall dramatic increase in protein carbonylation (104), demonstrating that the imbalance in reactive species production by astrocytes is an early mechanism in ALS (104, 113, 192).
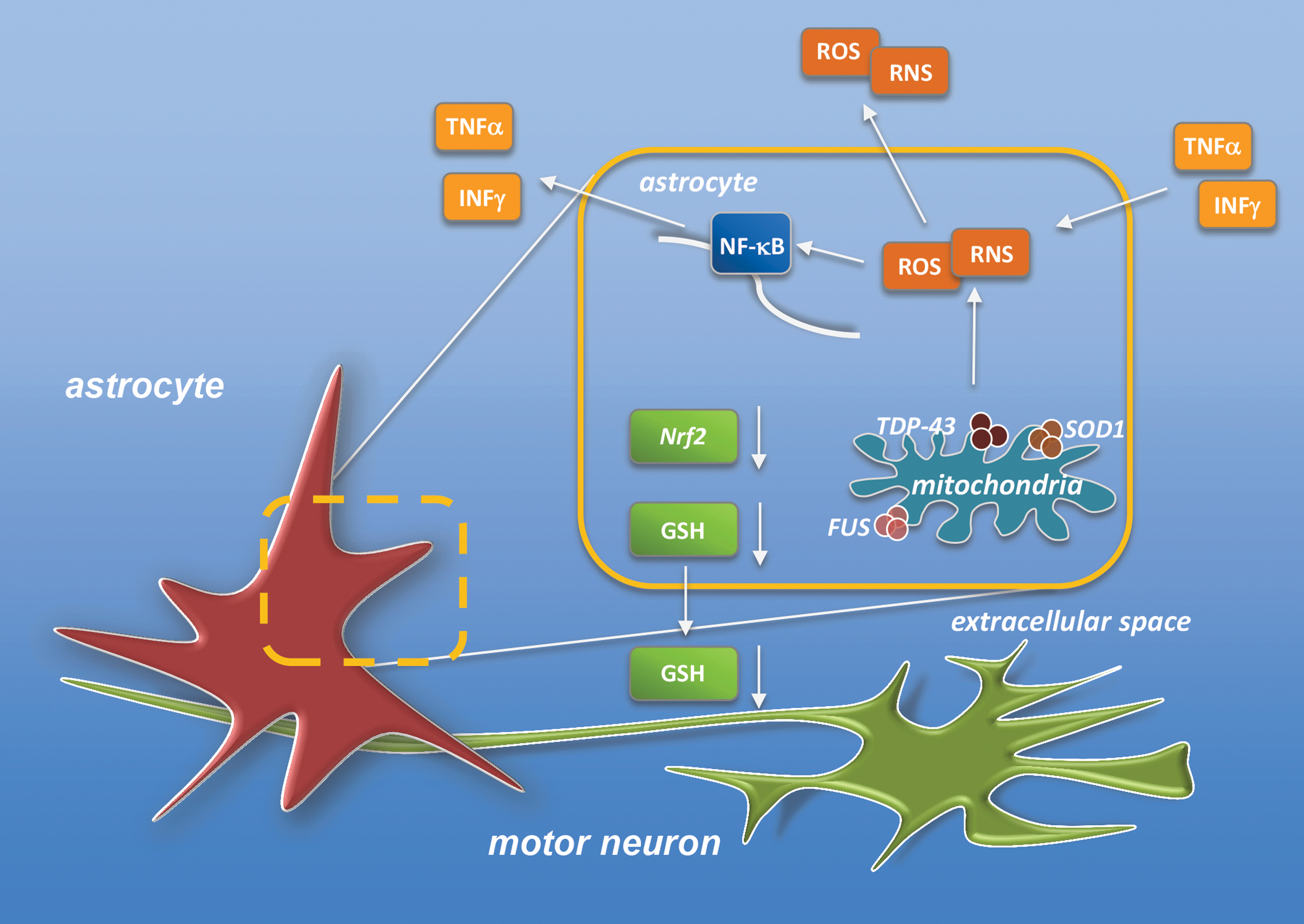
Under this respect, the observations that mitochondrial dysfunctions are major players in the etiology of ALS deserve particular attention, as they are not only an intrinsic cause of motor neuron stress but also a primary cause of altered ROS homeostasis in surrounding glial cells (198). Mitochondria from reactive astrocytes isolated by SOD1-G93A rats show, in fact, increased oxygen consumption and decreased membrane potential, which correlate with increased ROS formation and nitroxidative damage (32). Consistently, mitochondrial-targeted antioxidants, as well as nitric oxide synthase and NOX2 inhibitors, restore astrocytic mitochondrial respiration and prevent the neurotoxic phenotype, increasing motor neuron survival (32, 152). On the same line, other approaches aimed at decreasing oxidative stress in astrocytes have been shown to prevent astrocyte-mediated neuron toxicity. For instance, overexpression of mitochondrial-specific catalase reverts the neurotoxicity of SOD1-G93A astrocytes (182). Similarly, the increase of total and mitochondrial NAD+ content in mutant SOD1 astrocytes (by NAD+ precursor administration or by overexpression of nicotinamide phosphoribosyltransferase) confers resistance against H2O2-induced toxicity, decreases mitochondrial ROS, and completely reverts the neurotoxic phenotype of SOD1-G93A astrocytes (100). Overall, these data highlight the close link existing between SOD1 mutations, mitochondrial dysfunctions, and oxidative stress in astrocytes, on one side, and motor neuron death, on the other. Furthermore, they support the idea that an impaired mitochondrial function in mutant SOD1 astrocytes may lead to reactive species formation, contributing in this way to disease pathogenesis. Consistent with this conclusion is the neuroprotective effect of ROS scavenging in astrocytes. Detoxification and antioxidant enzymes in astrocytes (encoded by a cis-acting regulatory element called the antioxidant response element) are mainly controlled by the nuclear factor erythroid 2 (Nrf2), whose activation confers protection from oxidative insults to neighboring neurons, mainly by increasing the synthesis and secretion of glutathione (237) (Fig. 3). In the specific case of ALS, the selective overexpression of Nrf2 in astrocytes completely protects motor neurons from mutant SOD1 toxicity in coculture paradigms, delays disease onset, and moderately prolongs the survival of SOD1-G93A mice, with a delayed muscle denervation and glial activation (236).
Very recent and intriguing data highlight the role of astrocytic response in drug resistance in ALS, and provide a new perspective on the role of neuroinflammation and oxidative stress in the physiology of B-CNS-B (191). One of the limits for the efficacy of riluzole (and perhaps of other tested drugs) in ALS treatment may be identified in multidrug resistance pump ABCB1 (also named P-glycoprotein [P-gp]), which is known to prevent drugs directed at the CNS to achieve their targets (161). P-gp expression is progressively increased in the spinal cord of SOD1-G93A mice and its localization is confined to capillary endothelial cells (112). Through the use of coculture systems, it has been clearly demonstrated that the upregulation of P-gp in endothelial cells is driven by mutant SOD1-bearing astrocytes by the induction of ROS in endothelial cells and the consequent activation of NF-κB (191), which includes, among its targets, also the P-gp gene (161) (Fig. 4).

Collectively, these findings support an important role for astrocytes in the development of pharmacoresistance in ALS. Indeed, in these circumstances, mutant SOD1-expressing astrocytes might release reactive species and/or proinflammatory chemokines and cytokines that stimulate endothelial ROS levels and/or interfere with their scavenging systems, thus producing endothelial dysfunctions that would influence B-CNS-B permeability (Fig. 4). This conclusion fits well with the knowledge of a tight link between oxidative stress and B-CNS-B integrity. ROS generated by endothelial cells and astrocytes can indeed reduce the strength of tight junctions between cells of the endothelial monolayer and alter in this way the vascular permeability (99) (Fig. 4). The consequent compromised function of the B-CNS-B might provide another daunting obstacle for effective drug delivery into the CNS and highlights again how oxidative stress in non-neuronal cells such as astrocytes may affect different key aspects of disease development and therapeutic treatments.
Infiltrating immune cells
The infiltration of immune cells in the CNS is another hallmark of ALS, and an active dialogue between innate and adaptive immune systems has been firmly established (107, 140). This argues for a possible contribution of immune cells, including infiltrating lymphocytes, macrophages, mast cells, and neutrophils, in ALS pathogenesis.
Being phagocytic cells, by definition macrophages produce high amounts of ROS, mainly by NADPH oxidase complexes, and also nitrogen reactive species via iNOS. For this reason, their contribution to oxidative damage in ALS could be potentially relevant. Yet, no robust supportive data for their presence in ALS-affected CNS tissues have been provided so far. Different observations report their participation in infiltrates in the spinal cord from patients (70, 89, 103, 131). Recently, they were also observed in the spinal cord of a mouse model of the disease. In this context, they were proposed to play a role in the neuroinflammatory process that renders hostile the microenvironment for successful transplantation of neural stem cells (214). Regrettably, a major limitation of these studies is represented by the fact that it is very difficult to distinguish macrophages from parenchymal microglia, since they share the same specific markers for both resting and active states. Therefore, a clear evidence for macrophage presence in ALS-affected tissues still needs to be definitely provided. Also, the presence of mast cells in ALS-affected tissues is still a matter of debate, as they were detected in human tissues (70, 89), but their presence and role in the pathology were excluded in the SOD1-G93A mouse model (216).
Recently, three independent strains of C9orf72 knockout mice have been generated to mimic the haploinsufficiency of C9orf72, which has been proposed to have a role in ALS cases related to mutations in this gene. In these animals, clear signs of massive infiltration of macrophages and lymphocytes in various tissues have been detected. Interestingly, transcriptional profiling of spinal cords from C9orf72 knockout animals shows a clear enrichment in pathways related to inflammation, and these findings are significantly reproduced in brains from C9orf72 patients, but not from sporadic patients. Overall, these results indicate a role of C9orf72 in macrophage function and in immune system regulation. Yet, these results are still not sufficient to support a role of immune cell infiltration in ALS pathogenesis. Indeed, infiltrated immune cells were not clearly present in the brain or spinal cord tissue from these animals. Most importantly, none of these mice shows clear signs of motor neuron degeneration (27, 176, 218). This indicates that the immune phenotypes caused by C9orf72 deficiency are not sufficient to fully cause ALS disease and that haploinsufficiency is unlikely to be the causative factor in C9orf72-ALS.
The direct contribution of infiltrating lymphocytes in altering the oxidative balance of ALS-affected tissues has not been dissected so far. Protective or cytotoxic functions for different T lymphocyte subpopulations in the different phases of the disease have been documented, although the picture of lymphocyte contribution to disease is very complex. Mice resulting from the breeding of mutant SOD1 with mice lacking functional Treg cells (CD4+FoxP3+) showed an accelerated motor neuron disease, accompanied by attenuated morphological markers of gliosis and also by increased levels of mRNA for proinflammatory cytokines and NOX2, and concomitant decreased levels of trophic factors and glial glutamate transporters. Vice versa, reconstituting mice with Treg cells was effective in prolonging survival, suppressing cytotoxicity, and restoring glial activation (17), which argues for a protective role of Treg cells in ALS. Accordingly, during the first, stable phase of the disease, the total number of Tregs is increased in ALS mice (18). However, the deletion of recombination activating gene (RAG) 2 in SOD1-G93A mice, producing colonies lacking mature lymphocytes, significantly delays disease onset. In this case, in early phases of the disease, M2-polarized microglia, expressing Y1m gene, are increased in lumbar sections of the spinal cord (222), indicating that lymphocytes can display adverse effects on the activation of microglia trophic phenotypes. In addition, RAG1−/−/SOD1-G93A mice do not show differences in the onset, progression, and survival with respect to SOD1-G93A mice (215). In coculture experiments, CD4+FoxP3+ Treg lymphocytes are able to reduce the levels of NOX2 and iNOS mRNAs as well as NO production in mutant SOD1-expressing microglial cells, indicating, in this case, that Tregs can suppress the cytotoxic phenotype of microglia. The mediator responsible for this modulatory effect of Tregs is likely IL-4, as the addition of an IL-4 neutralizing antibody has been shown to reverse the levels of NOX2, iNOS, and NO. Moreover, Tregs have also been shown to reduce the proliferation of cytotoxic T cells (Teff), contributing in this way to restrict the damage toward neurons (255). In contrast, during the rapidly progressing phase, the number of Tregs declines while the proliferation of Teffs increases, in turn leading to a synergistic toxic effect by reduction in nitro-oxidative protection and intensification in proapoptotic signals (17, 255). Different cytotoxic T cells, CD81+ and natural killer cells, are indeed found increased in patients with fast progressing disease (102, 151, 196) and they are located in close proximity to dying motor neurons (89), but their role in the pathology is controversial (72, 217). Furthermore, it is unlikely that they contribute directly to the oxidative burst (e.g., via NOX2), as their expression of NOX2 is very low (82).
Finally, while it was recently shown that lower NOX2 activity in peripheral neutrophils of ALS patients positively correlates with a longer survival (154), the general contribution of neutrophils in ALS is still a matter of debate. Affected areas of ALS mice do not show detectable activity of myeloperoxidase (MPO), the most recognized marker of neutrophils, likely reflecting an overall absence of a significant neutrophil infiltration (206). Similarly, the role of B cells in ALS has not been established so far, although immunoglobulins and complement deposition have been reported in the CNS of ALS patients (171).
Conclusions
Studies in models of familial ALS show that animals exhibit pathological abnormalities at presymptomatic stages and long before the appearance of clinical deficits, a fact that suggests that (as in other neurodegenerative diseases) vulnerability and damage might be induced in motor neurons or in other cell populations involved in the disease as early as in the perinatal period also in men (58). This implies that a precise determination of the timing of events in patients is a prerequisite for any attempt to therapy, together with a precise distinction between those alterations that contribute to onset and those that determine the progression of the disease. In 2007, a total of 167 different therapeutic regimens in ALS-SOD1 mice were evaluated in a meta-analysis (19) leading to the conclusion that drugs with an anti-inflammatory mechanism of action and antioxidative agents appeared the most promising for therapeutic trials in patients with familial ALS. The emergent link between oxidative stress and neuroinflammation in ALS outlined above strongly supports the use of antioxidants as anti-inflammatory therapeutics in this disease as in other neurodegenerative conditions (158).
As several cell types contribute to the neuroinflammatory response in ALS, a major challenge in the next future will be to design effective strategies to modulate this response in the symptomatic stage of disease to prevent progression of symptoms through the propagation of oxidative damage. Single treatment with antioxidant or anti-inflammatory molecules has already been tried in patients. A recent example is represented by edaravone, a free radical scavenger of lipid peroxide and hydroxyl radicals (240), which is able to reduce the levels of oxidative damage in patients (169, 252) and has been approved for ALS treatment in 2015 in Japan. Yet, a “combinatorial approach” cotargeting different pathogenic mechanisms in several cell types promises to be a more effective therapeutic strategy for ALS, as indicated by a recent elegant article (75) showing that a simultaneous reduction of mutant SOD1 in motor neurons and astrocytes, combined with suppression of NF-κB in microglia, acts cooperatively to significantly increase the survival of mice.
However, multitarget therapy is more complex in patients than in mice as well exemplified by the case of minocycline, an antibiotic with neuroprotective action and anti-inflammatory properties. Minocycline acts on the activated T cells and on microglia, which results in the decreased ability of T cells to contact microglia and impairment of cytokine production in T cell-microglia signal transduction (85). Failure of a phase III trial with minocycline in ALS patients (87), despite encouraging results in ALS mice (128), may be explained by a number of reasons, including timing of treatment and doses (28, 136), and once more indicates that we still need a detailed knowledge of disease phases to devise a time- and dose-adapted therapeutic approach.
Footnotes
Acknowledgments
M.T.C.'s research is currently supported by a grant from MIUR (PRIN 2015LFPNMN) and by AFM Telethon (21021). M.C.'s research is currently supported by a grant from MIUR (PRIN 2015LFPNMN) and from CNR (Project Interomics).